

Abstract
Objectives
Capillarisin (Cap), an active component of Artemisia capillaris root extracts, is characterized by its anti‐inflammatory, anti‐oxidant and anti‐cancer properties. Nevertheless, the functions of Cap in prostate cancer have not been fully explored. We evaluated the potential actions of Cap on the cell proliferation, migration and invasion of prostate carcinoma cells.
Materials and methods
Cell proliferation and cell cycle distribution were measured by water‐soluble tetrazolium‐1 and flow cytometry assays. The expression of cyclins, p21, p27, survivin, matrix metallopeptidase (MMP2 and MMP9) were assessed by immunoblotting assays. Effects of Cap on invasion and migration were determined by wound closure and matrigel transmigration assays. The constitutive and interlukin‐6 (IL‐6)‐inducible STAT3 activation of prostate carcinoma cells were determined by immunoblotting and reporter assays.
Results
Capillarisin inhibited androgen‐independent DU145 and androgen‐dependent LNCaP cell growth through the induction of cell cycle arrest at the G0/G1 phase by upregulating p21 and p27 while downregulating expression of cyclin D1, cyclin A and cyclin B. Cap decreased protein expression of survivin, MMP‐2, and MMP‐9 and therefore blocked the migration and invasion of DU145 cells. Cap suppressed constitutive and IL‐6‐inducible STAT3 activation in DU145 and LNCaP cells.
Conclusions
Our data indicate that Cap blocked cell growth by modulation of p21, p27 and cyclins. The inhibitory effects of Cap on survivin, MMP‐2, MMP‐9 and STAT3 activation may account for the suppression of invasion in prostate carcinoma cells. Our data suggest that Cap might be a therapeutic agent in treating advanced prostate cancer with constitutive STAT3 or IL‐6‐inducible STAT3 activation.
1. INTRODUCTION
Prostate cancer is the most prevalent cancer and is the third leading cause of cancer death in American men.1 Certain alterations in androgen receptor signalling contribute to the development and progression of a lethal and drug‐resistant form of prostate cancer, castration‐resistant prostate cancer (CRPC).2 Currently, therapeutic options for relapses of prostate cancer are limited, especially for patients who have already received androgen depletion therapy (ADT). One possible explanation for the development of CRPC in patients who have had ADT involves activation of signal transducer and activator of transcription 3 (STAT3) in prostate cancer cells.3
Signal transducer and activator of transcription 3 is a signal transduction protein activated by many growth factors and cytokines, including interleukin‐6 (IL‐6).4 By acting like an oncogenic transcription factor, STAT3, upon phosphorylation at tyrosine 705, dimerizes and translocates into the nucleus where it modulates target genes in relation to cell proliferation, survival, invasion, angiogenesis and chemo‐resistance in prostate cancer.5 STAT3 activation is transient and tightly controlled in normal cells, whereas, constitutively activated STAT3 occurs frequently in a variety of malignant tumours and promotes cancer cell development and progression.6 Previous studies indicated that IL‐6 functions as a paracrine growth factor for androgen‐dependent prostate carcinoma LNCaP cells and as an autocrine growth factor for androgen‐independent prostate carcinoma DU145 cells.7 The growth stimulation by IL‐6 is accompanied by activation of the STAT3 signalling transduction pathway.8 Therefore, targeting STAT3 could be a promising therapeutic strategy for treatment of androgen‐responsive and androgen‐resistant prostate cancers.3
Artemisia capillaris (AC) is an edible plant. Its young shoots are consumed daily as a vegetable in China and it is regarded as an herbal remedy for various inflammatory diseases such as hepatitis, cholestatic jaundice, and urinary tract infection in Asia.9 Komiya et al10 first recognized the molecular structure of Capillarisin (Cap), an active element extracted from AC, as 2‐(p‐hdroxyphenoxy)‐6‐methoxy‐5,7‐dihydroxychromoneas (C16H12O7) with strong choleretic activity. Previous studies indicated that Cap has anti‐inflammation, anti‐oxidation and anti‐cancer biological activities.11, 12, 13 Cap induces apoptosis and cell cycle arrest in osteosarcoma cancer cells.14 Furthermore, it has been reported to suppress cancer cell invasion via inhibition of NFκB‐dependent matrix metallopeptidase (MMP)9 expression in MCF‐7 cells and inhibition of the STAT3 signalling cascade in multiple myeloma cell lines.15, 16 Nevertheless, the functions and effects of Cap in prostate carcinoma cells are still unexplained, and Cap‐mediated inhibition of STAT3 activation in the prostate carcinoma cells remains unexplored.
In this study, we determined the effects of Cap on cell proliferation and invasion/migration in prostate carcinoma cells. The inhibitory effect of Cap on constitutive and IL‐6‐inducible STAT3 activation, which may account for the suppression of migration and invasion of prostate carcinoma cells, was also investigated.
2. MATERIALS AND METHODS
2.1. Chemicals
Capillarisin was purchased from Wako (Osaka, Japan), RPMI 1640 media from Life Technologies (Rockville, MD, USA), and recombinant human IL‐6 from Peprotech (Rocky Hill, NJ, USA). The 4′ 6‐diamidino‐2‐phenylindole (DAPI) and other chemicals of reagent grade used in this study were obtained from Sigma Chemical (St. Louis, MO, USA).
2.2. Cell cultures
Androgen‐independent prostate carcinoma DU145 cells and androgen‐dependent prostate carcinoma LNCaP cells were obtained from the Bioresource Collection and Research Center (BCRC, Hsinchu, Taiwan) and maintained in culture medium RPMI 1640 (Gibco, Grand Island, NY, USA) containing with 10% foetal calf serum (FCS; Gibco), 100 U mL−1 penicillin, 100 μg mL−1 streptomycin and 0.25 μg mL−1 amphotericin B (Gibco) in a 5% CO2 atmosphere at 37°C as described previously.17
2.3. Cell growth assay
DU145 cells were seeded at 1 × 104 cells per well in a 24‐well plate in culture medium for 24 hours. After refreshing the medium with 5% FCS, cells were treated with various concentrations of Cap for 24, 48, or 72 hours. To determine cell growth rate, 50 μL water‐soluble tetrazolium‐1 (WST‐1; abcam, Cambridge, UK) was added to each well at the end of the incubation time, and the absorbance of each sample was measured using a microplate reader at a wavelength of 450 nm.
2.4. Flow cytometry
In response to the Cap treatment, cells were serum starved for 24 hours and then cultured in RPMI‐1640 medium with 10% FCS and various concentrations of Cap as indicated for another 24 hours. Cell cycle analysis was performed using the FACSCalibur E6147 Cytometer and CELLQuest Pro Software (BD Biosciences, San Jose, CA, USA); the data were analysed using ModFit LT Mac 3.0 Software, as previously described.18
2.5. Annexin V‐FITC apoptosis detection
The cell pellets were harvested after cells were treated with or without Cap for 48 hours. Apoptosis detection and quantification were performed after treatment with Annexin V‐FITC (BioVision Inc, Milpitas, CA, USA) and propidium iodide (PI) for 1 hour using the FACSCalibur E6147 Cytometer (BD Biosciences) as previously described.19
2.6. Cell migration assay
For cell migration analysis, a culture insert (ibidi GmbH, Martinsried, Germany) was placed on a culture dish, and DU145 cells were seeded into each reservoir. After 24 hours incubation at 37°C, the insert was removed and the medium was refreshed with the same medium only containing 5% FCS. Then, cells were treated without or with Cap at indicated concentrations for another 24 hours. The wound closure (the gap width) was photographed under an optical microscope with a digital camera. The quantification of cell migration was determined within a defined area using the Image J program (NIH, Bethesda, MD, USA).
2.7. Matrigel invasion assay
The matrigel invasion assay was performed as described previously.20 Briefly, 500 μL RPMI 1640 medium with 10% FCS was added to the lower chamber of a 24‐well plate, and 200 μL of cell suspension (1 × 105 cells mL−1) with no serum was added onto the upper well in the absence or presence of Cap at indicated concentrations. The plates were placed in an incubator with a 5% CO2 atmosphere at 37°C for 24 hours. The non‐invading cells were removed from the upper surface of the membrane by scrubbing with a cotton tip. Cells that migrated into the matrigel‐coated transmembrane containing 8 μm pores were fixed in 4% paraformaldehyde and then stained with 0.1% crystal violet solution for 30 minutes. The results were recorded by a digital camera connected to an inverted microscope (IX71, Olympus, Tokyo, Japan). The membrane was then soaked in 10% acetate acid and agitated at 37°C for 1 hour. The acetate acid solution was then read by a spectrophotometer at 635 nm (DU640; Beckman, Fullerton, CA, USA).
2.8. Immunoblot analysis
DU145 or LNCaP cells were lysed in lysis buffers (containing 0.2% sodium dodecyl sulphate, 1% NP‐40, 5 mmol L−1 ethylenediaminetetraacetic acid, 1 mmol L−1 phenylmethylsulphonyl fluoride, 10 μg mL−1 leupeptin, and 10 μg mL−1 aprotinin), and then analysed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis. After being transferred onto a nitrocellulose membrane, antigens were analysed by specific antibodies to STAT3 (79D7), phospho‐STAT3 (Tyr 705), cyclin D1 (DCS6, Cell Signaling, Danvers, MA, USA), cyclin A (C‐19), cyclin B1 (D‐11), cyclin E (13A3), survivin (D8, Santa Cruz Biotechnology, Santa Cruz, CA, USA), MMP2 (ab86607) and anti‐MMP9 (ab7299; abcam). The housekeeping proteins, β‐actin (MAS1501, Millipore, Temecula, CA, USA) and Lamin B (M20, Santa Cruz Biotechnology), were detected as internal controls. Antigen‐antibody complexes were detected using horseradish peroxide‐labelled rabbit anti‐mouse immunoglobulin G and an enhanced chemoluminescence detection system (Pierce, Rockford, IL, USA).
2.9. Reporter assay
The reporter vector, pSTAT3‐TA‐Luc was purchased from Clontech Laboratories, Inc (Mountain View, CA, USA). Cells were plated on 24‐well plates at 1 × 104 cells per well for 24 hours before transfection. Cells were transiently transfected using TransFast™ transfection reagent with 1 μg per well reporter vector and 0.5 μg per well pCMVSPORT‐gal (Invitrogen, Carlsbad, CA, USA), as described previously.21 Transfected cells were treated with IL‐6 and/or Cap as indicated in RPMI 1640 medium with 10% FCS for an additional 24 hours. Luciferase activity was adjusted for transfection efficiency using the normalization control plasmid pCMVSPORT‐gal.
2.10. Statistical analysis
Data were presented as mean ± SE. Statistical significance was determined by Student's t test and one‐way ANOVA using the SigmaStat program for Windows, version 2/03 (SPSS Inc, Chicago, IL, USA). Statistical significance was defined as *P < .05 and **P < .01.
3. RESULTS
3.1. Cap inhibits cell growth without induction of cell apoptosis
To explore the anti‐proliferative effect of Cap on prostate carcinoma DU145 cells, we compared and analysed the inhibitory effects and the cell‐cycle distribution in Cap‐treated cells. WST‐1 assays revealed that inhibition of DU145 cell growth occurred initially at 100 μmol L−1 Cap for 24 hours, increasing in a dose‐ and time‐dependent manner. Cap at 200 μmol L−1 significantly blocked 44%, 79% and 84.7% of cell proliferation in DU145 cells after treatment for 24, 48 and 72 hours, respectively (Figure 1A). The EC50 for 48 and 72 hours of treatments were 80.35 μmol L−1 and 50.34 μmol L−1, respectively. The immunoblot assays demonstrated that Cap did not induce cell apoptosis in DU145 cells even after 72 hours of treatment since the poly (ADP‐ribose) polymerase (PARP) cleavage form, an apoptotic marker, did not present in the Cap‐treated DU145 cells (Figure 1B). We further used Annexin V‐FITC in conjunction with PI staining to distinguish early apoptotic, late apoptotic and necrotic cells. Results of fluorescence intensity for Annexin V‐FITC and PI in DU145 cells after treatment with or without Cap for 48 hours revealed that Cap did not induce apoptosis significantly (Figure 1C).
Figure 1.
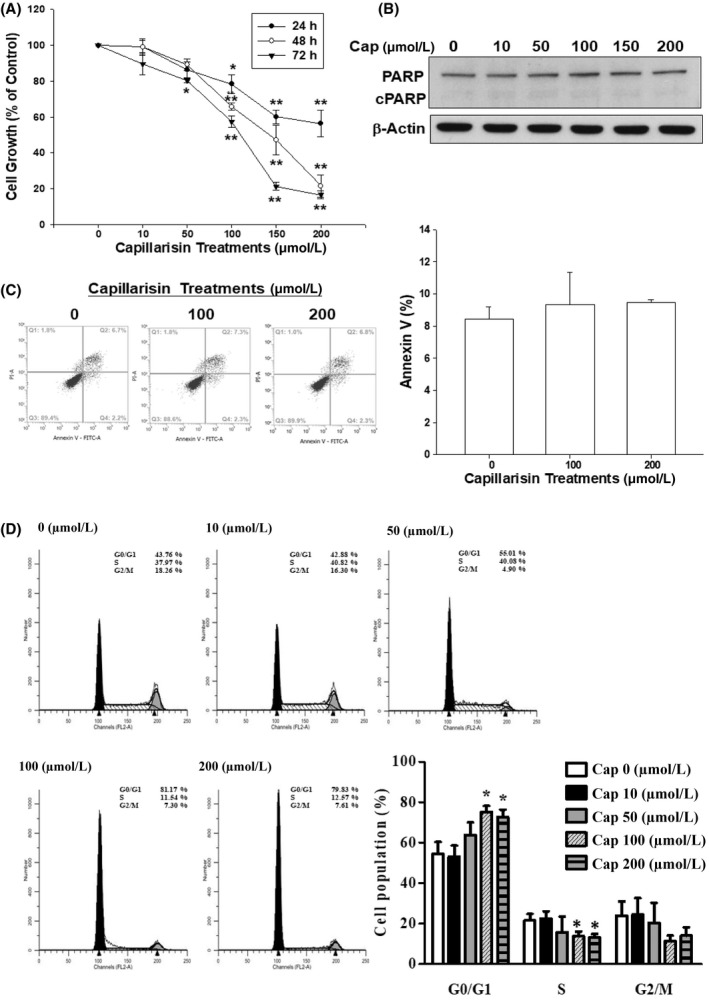
Effects of cap on growth and distribution of DU145 cells. DU145 cells were treated with various concentrations of Capillarisin (Cap) for indicated time periods. (A), After treatment, cell growth was determined by WST‐1 assay. Cell growth of the vehicle‐treated group is set as 100% and data obtained from three independent experiments are expressed as the mean ± SE. (B), Protein expression of PARP and cleaved PARP after treatment with various concentrations of Cap as indicated for 72 hours was analysed by immunoblotting assays. (C), The fluorescence intensity for Annexin V‐FITC and PI in DU145 cells after treatment with or without Cap for 48 hours. (D), The cell cycle distribution after DU145 cells were treated with various concentrations of Cap as indicated for 24 hours
3.2. Cap inhibits cell cycle progression in DU145 cells
In the flow cytometric analysis, we found that 100‐200 μmol L−1 Cap induced a 22% increase in G0/G1 arrest together with a decrease in G2/M phase and S phase cells after 48 hours of incubation (Figure 1D), confirming retardation of the growth rate by Cap treatment for 48 hours owing to cell cycle arrest.
3.3. Cap inhibits migration and invasion of DU145 cells
To evaluate cell mobility after Cap treatment in DU145 cells, wound closure (Figure 2A) and matrigel invasion (Figure 2B) assays were performed. Both assays revealed that Cap dose‐dependently inhibited cell migration and invasion. Cap 200 μmol L−1 significantly blocked 51% of cell migration and 82% of cell invasion compared with the vehicle treatment group in DU145 cells.
Figure 2.
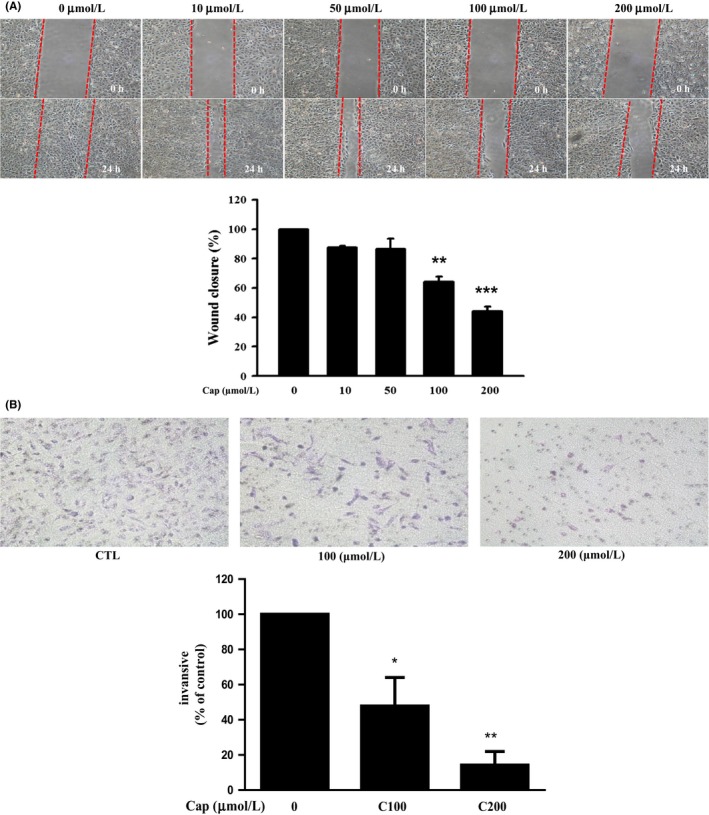
Effects of Capillarisin (Cap) on migration and invasion of DU145 cells. (A) Cells were treated with Cap at indicated concentrations for 24 hours after a wound gap was formed. The wound closure was quantified using Image‐J software (NIH, Bethesda, MD, USA). The results are expressed as percentage of control and depicted in the bar chart (in lower panel). (B) Matrigel invasion assay was performed using a transwell method. Representative image shows that cells adhered to the bottom of the matrigel coated filters (upper panel), and quantitation of the results is depicted in the bar chart (lower panel)
3.4. Cap modulates protein expression involving in cell growth and invasion of DU145 cells
Results of the immunoblot assays showed that Cap downregulated cyclin D1, cyclin A, cyclin B, survivin, MMP2 and MMP9 in DU145 cells. However, Cap treatment did not significantly affect the protein expression of cyclin E (Figure 3). Results of the quantitative analysis are illustrated in Figure 3B,D,F.
Figure 3.
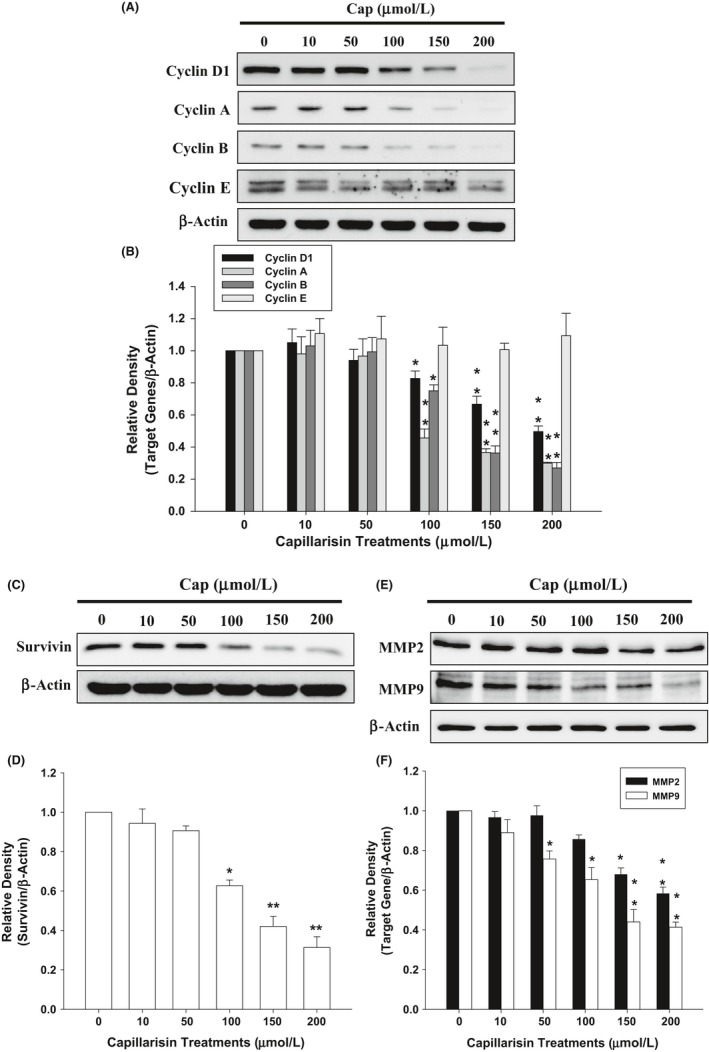
Capillarisin (Cap) modulation of cell cycle and invasion regulatory proteins in DU145 cells. Cells were treated with various concentrations of Cap as indicated for 24 hours. Expression of (A) cyclin D, cyclin A, cyclin B, cyclin E, (C) survivin, (E) matrix metallopeptidase (MMP)2, and MMP9 was determined by immunoblotting assays. The fold‐induction data are expressed as the intensity of the protein bands produced from the target gene/β‐actin (±SE; n = 3) relative to that of the vehicle‐treated group (B,D,F)
3.5. Cap inhibits cell growth of LNCaP cells via downregulation of cyclin A and cyclin D1 but upregulation of p21 and p27
We observed that Cap inhibited LNCaP cell growth in both dose‐ and time‐dependent ways (Figure 4A). The EC50 for 48 and 72 hours of treatments were 82.75 and 52.81 μmol L−1, respectively. Then, we detected the cell cycle distribution with flow cytometry and cell cycle regulatory proteins by immunoblotting in cells with or without Cap treatment. Cap obviously increased in a 12% cell accumulation at the G0/G1 phase together with a decrease in S phase but not G2/M phase cells after 48 hours of incubation (Figure 4B). Consistently, with Cap treatment, the cell cycle regulatory proteins (cyclin A and cyclin D1) were decreased, whereas cyclin‐dependent kinase inhibitors (p21 and p27) were increased (Figure 4D). The results of the quantitative analysis presented in Figure 4E, indicate that Cap inhibits LNCaP cell growth through arresting the cell cycle at the G0/G1 phase by modulating cell cycle regulatory proteins and cyclin‐dependent kinase inhibitors.
Figure 4.
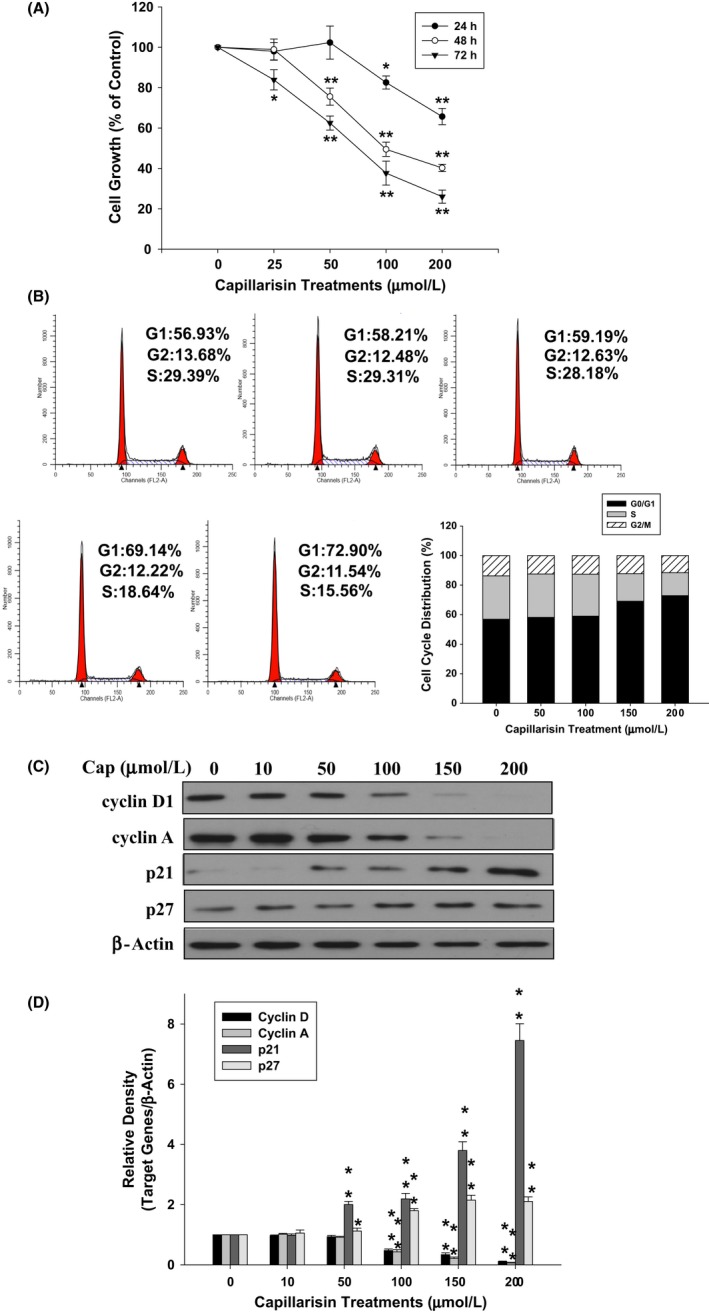
Capillarisin (Cap) modulation of growth and distribution of LNCaP cells. (A) Cells were treated with various concentrations of Cap at indicated time periods, and cell growth was determined by water‐soluble tetrazolium‐1 (WST‐1) assays. Cell growth of the vehicle‐treated group is set as 100% and data obtained from three independent experiments are expressed as mean ± SE. (B) The cell cycle distribution of LNCaP cells after cells were treated with various concentrations of Cap as indicated for 24 hours. (C) Cells were treated with various concentrations of Cap as indicated for 24 hours. Expression of cyclin D1, cyclin A, p21, and p27 was determined by immunoblotting assays. (D) The fold‐induction data are expressed as the intensity of the protein bands produced from the target gene/β‐actin (±SE; n = 3) relative to that of the vehicle‐treated group
3.6. Cap inhibits constitutive and inducible phospho‐STAT3 in prostate carcinoma DU145 cells
As shown in Figure 5A, Cap treatment for 24 hours apparently inhibited protein levels of phospho‐STAT3 but not STAT3 in a dose‐dependent manner in DU145 cells. Moreover, Cap treatment (100 μmol L−1) blocked STAT3 activation in a time‐dependent manner (Figure 5B). Further immunoblot assays using cell lysis of the nuclear fraction from DU145 cells revealed that Cap downregulated protein levels of the phosho‐STAT3 in the nuclear fraction indicating that Cap blocked the activation of STAT3 (Figure 5C). Results of reporter assays using the STAT3 specific reporter vector (pSTAT3‐TA‐Luc) containing the STAT3 binding site further indicated that Cap downregulated STAT3 activity (Figure 5D). When DU145 cells were cotreated with 20 ng mL−1 IL‐6 and various concentrations of Cap for 24 hours, the protein levels of phosph‐STAT3 and MMP2 were upregulated by IL‐6, while Cap reversed the effects of IL‐6 (Figure 5E). Results of the quantitative analysis are shown in Figure 5F.
Figure 5.
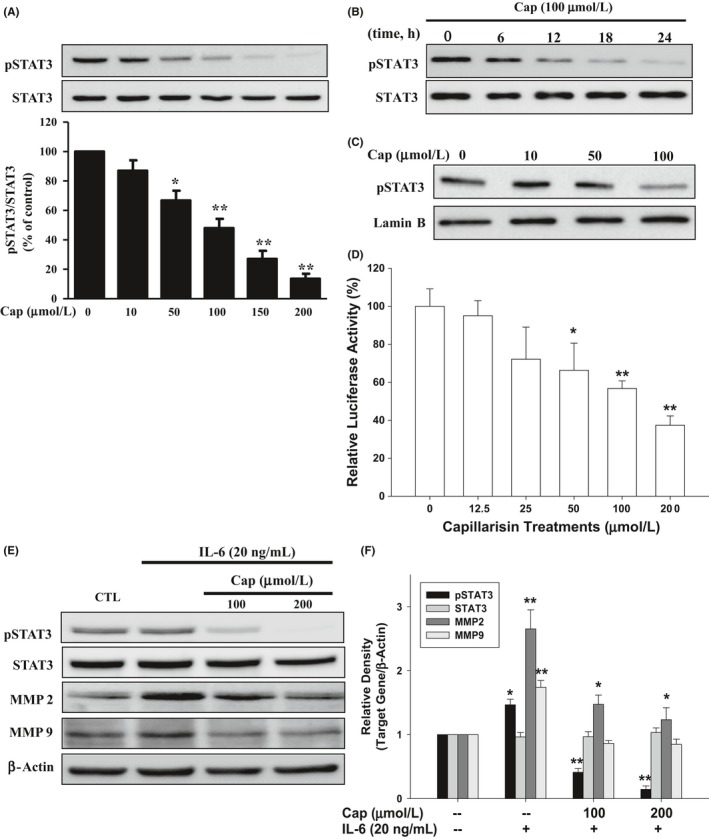
Effects of Cap on constitutive and interlukin‐6 (IL‐6) ‐inducible signal transducer and activator of transcription 3 (STAT3) activation in prostate carcinoma DU145 cells. DU145 cells were treated (A) with Cap at indicated concentrations for 24 hours; (B) with 100 μmol L−1 Cap for the indicated period of time. (C) DU145 cells were lysed after treatments with various concentrations of Cap, and then the nuclear and cytoplasmic fractions were separated. The protein levels of pSTAT3 and lamin B in the nuclear fraction were determining by immunoblotting assays. (D) The pSTAT3‐TA‐Luc‐transfected DU145 cells were treated with various concentrations of Cap as indicated for 24 hours. Data are presented as mean percentage ±SE (n = 6) of the luciferase activity in relation to the vehicle‐treated group. (E) DU145 cells were cotreated with 20 ng mL−1 interlukin‐6 (IL‐6) and Cap at indicated concentrations for 24 hours. The protein levels of STAT 3, phosph‐STAT3, matrix metallopeptidase (MMP)2, and MMP9 were analysed by immunoblotting assays. (F) The fold‐induction data are expressed as the intensity of the protein bands produced from the target gene/β‐actin (±SE; n = 3) relative to that of the control group
3.7. Cap inhibits IL‐6‐inducible phospho‐STAT3 in prostate carcinoma LNCaP cells
Results of the immunoblot assays showed different expression patterns of phospho‐STAT3 and STAT3 in DU145 and LNCaP cells, indicating that STAT3 was constitutively activated in DU145 cells but not in LNCaP cells (Figure 6A). We further determined the effect of Cap on the inducible activation of STAT3 by IL‐6 in LNCaP cells. Consistently, Cap showed a dose‐dependent reduction in IL‐6‐induced STAT3 phosphorylation in LNCaP cells (Figure 6B). Similar results found in the reporter assays using the reporter vector, pSTAT3‐TA‐Luc, revealed that IL‐6 upregulated STAT3 activity while Cap treatment blocked IL‐6‐induced STAT3 reporter activity in LNCaP cells (Figure 6C).
Figure 6.
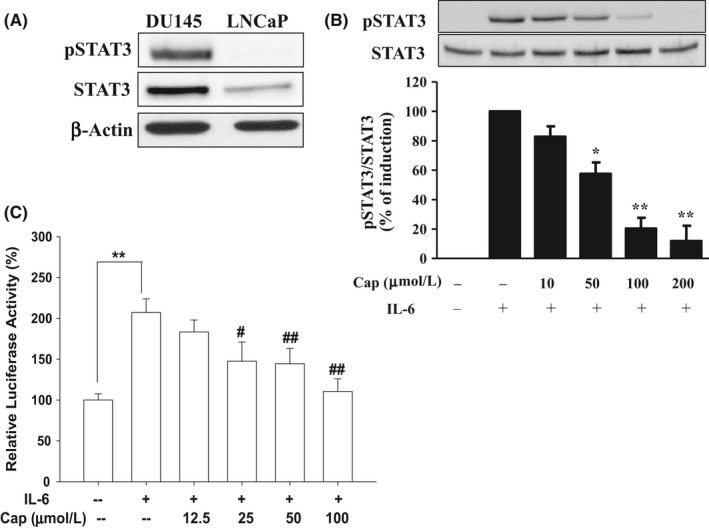
Effects of Cap on interlukin‐6 (IL‐6) inducible STAT3 activation in prostate carcinoma LNCaP cells. (A) Protein levels of STAT 3 and phosph‐STAT3 in DU145 and LNCaP cells were determined by immunoblotting assays. (B) LNCaP cells were cotreated with 20 ng mL−1 interlukin‐6 (IL‐6) and various concentrations of Cap as indicated for 24 hours. The protein levels of STAT 3 and phosph‐STAT3 were analysed by immunoblotting assays. Data from quantitative analysis are expressed as fold‐induction ±SE (n = 3). (C) The pSTAT3‐TA‐Luc‐transfected LNCaP cells were treated as indicated. Data are presented as mean percentage ±SE (n = 6) of the luciferase activity in relation to the vehicle‐treated group
4. DISCUSSION
Prostate cancer is one of the most frequently diagnosed malignancies and the leading cause of cancer‐related deaths in men worldwide.1, 22 In this study, we demonstrated that Cap suppressed cell growth without inducing apoptosis in androgen‐independent DU145 cells since the Cap did not significantly induce poly (ADP‐ribose) polymerase (PARP) cleavage form, an apoptotic marker, which is similar to a previous study in androgen‐dependent LNCaP cells.19 Moreover, the Cap‐inhibited cell growth of prostate carcinoma cells was due to its induction of cell cycle arrest at the G0/G1 phase (Figures 1 and 4). These results were consistent with other studies in human breast carcinoma cells, MCF7, osteosarcoma and B‐lymphoblast cells, although some of them showed cell apoptosis was induced with higher Cap concentrations.14, 15, 16 The EC50 between DU145 and LNCaP after Cap treatments for 48 and 72 hours has shown no significant differences; however, recent study revealed that Cap blocks androgen activation on AR‐mediated prostate specific antigen expression in LNCaP cells.19 Whether Cap has the same effect on prostate normal epithelial cells still needs further investigations. Our report is the first study to discover the potential mechanisms of Cap on the anti‐proliferative effects of both androgen‐dependent and ‐independent prostate carcinoma cells.
Prior study showed that downregulation of cyclin D1 and cyclin A but upregulation cyclin E, p21 and p27 induced G1 cell cycle arrest in prostate carcinoma cells.23 Therefore, to explore the potential mechanisms of Cap on cell proliferation, we determined the protein expressions of cyclin D1, cyclin A, cyclin B, cyclin E, p21 and p27 after DU145 or LNCaP cells were treated with Cap. Our immunoblot assays showed that Cap downregulated cyclin D1, cyclin A and cyclin B, but not cyclin E in DU145 cells (Figure 3). These results were in agreement with a previous study which indicated that Cap induced expression of cyclin D1 in human multiple myeloma U266 cells.16 Our study found that Cap treatment not only downregulated cyclin D1 but also upregulated p21 and p27 protein expression in LNCaP cells (Figure 4). Study has indicated that down modulations of both p21 and p27 enhance the aggressive type of prostate carcinoma cells in vitro and in vivo.24 Our results demonstrated that Cap enhanced protein expression of p21 and p27, suggesting that Cap is an effective agent in the prevention of prostate cancer.
We continued to verify whether Cap has effects against the migration and invasion of prostate carcinoma DU145 cells. As shown in Figure 2, results of wound closure and matrigel invasion assays demonstrated that Cap dose‐dependently inhibited cell migration and invasion (Figure 2). Our immunoblot assays showed that Cap downregulated protein levels of survivin, a potential mediator of prostate cancer metastasis (Figure 3C). Survivin was found to be overexpressed in prostate cancer tissues in vivo. Ectopic overexpression of survivin in prostate carcinoma cells upregulated cell invasion in vitro.25 Another study in U266 cells also found that Cap downregulated survivin expression.16 Immunoblot assays in this study indicated that Cap downregulated protein expression of MMP‐2 and MMP‐9, which may account for the inhibition of migration and invasion (Figure 3). MMP‐2 and MMP‐9 are well‐known promoters of invasion in prostate cancer,26 and blocking of MMP‐2 and MMP‐9 downregulated cell migration and invasion in prostate carcinoma cells.27 These results are in agreement with other studies which suggested that Cap blocks MMP‐9 expression to decrease the invasion of U266 and MCF7 cells.15, 16 Interestingly, our results demonstrated that Cap not only downregulated protein expression of MMP‐2 and MMP‐9 but also blocked the activation of IL‐6 on MMP‐2 and MMP‐9 expression in androgen‐resistant DU145 cells with constitutively activated STAT3 (Figure 5).
Interlukin‐6 is a multifunctional cytokine known to activate STAT3 and participates in the malignant progression of prostate cancer.28 Results from our previous in vitro studies have shown that both IL‐6 and IL‐6 receptor are expressed in DU145 cells, and IL‐6 enhances the expression of prostate‐specific antigen in LNCaP cells.29, 30 These results are consistent with early studies indicating that IL‐6 functions as a paracrine growth factor for androgen‐dependent prostate carcinoma LNCaP cells and as an autocrine growth factor for androgen‐independent prostate carcinoma DU145 cells.7 As shown in Figure 5, Cap apparently not only inhibited protein levels of phospho‐STAT3 in a time‐ and dose‐dependent manner but also blocked the IL‐6‐induced STAT3 activation. Moreover, our results showed that IL‐6 enhanced protein expression of MMP2 and MMP2, while Cap treatment downregulated the activation of IL‐6 on MMP2 and MMP9 expression (Figure 5). These results indicated that Cap blocks migration and invasion possibly through targeting STAT3 activation in DU145 cells. The constitutive or IL‐6‐induced activation of the STAT3 pathway has been reported to stimulate cell proliferation and to promote metastatic progression of advanced prostate cancer cells; therefore, the IL‐6/STAT3 signal pathway is considered promising in targeted therapy for hormone‐resistant prostate cancer.31 In this study we demonstrated that Cap inhibits the constitutive and inducible phosphor‐activation of STAT3 in prostate carcinoma DU145 cells.
Increased STAT3 activation by IL‐6 as an autocrine growth factor is also considered a potential mechanism to enhance androgen receptor reactivation in a ligand‐independent manner after androgen deprivation, and thereby promote transition of androgen‐sensitive prostate cancer to castration‐resistant status.5 Here, we showed that Cap apparently inhibited the IL‐6‐induced activation of STAT3 in LNCaP cells. Since cyclin D1, survivin, p21, p27, and MMP9 are the target genes of STAT3 in prostate carcinoma cells,32, 33 Cap may have benefits in the prevention of biological functions mediated by sustained and inducible STAT3 activation, such as cell growth and metastasis, in prostate carcinoma cells in vitro.
Our results demonstrated that Cap upregulated p21 and p27 but downregulated cyclin D1, cyclin A, cyclin B, survivin, MMP2 and MMP9 expression concomitant with suppression of growth, migration, and invasion in androgen‐dependent and androgen‐independent prostate carcinoma cells. Cap also inhibits constitutive and inducible STAT3 activation. These findings provide evidence suggesting that Cap has potential as an agent in treating prostate cancer with constitutive or IL‐6‐inducible STAT3 activation.
ACKNOWLEDGEMENTS
This work was supported by grants from the Taiwan Ministry of Science and Technology (MOST‐104‐2314‐B‐182A‐140‐MY3 and MOST‐105‐2320‐B‐182‐020‐MY3), and Chang Gung Memorial Hospital (CRRPD1F0041‐2, CMRPD3E0041‐2, CMRPD1F0141‐3, CMRPG3F0801‐3, CMRPG3E0151‐3 and CMRPG3D0311‐3). The authors declare no conflicts of interest.
Tsui K‐H, Chang Y‐L, Yang P‐S, et al. The inhibitory effects of capillarisin on cell proliferation and invasion of prostate carcinoma cells. Cell Prolif. 2018;51:e12429 10.1111/cpr.12429
Tsui and Chang contributed equally to this study.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Coutinho I, Day TK, Tilley WD, Selth LA. Androgen receptor signaling in castration‐resistant prostate cancer: a lesson in persistence. Endocr Relat Cancer. 2016;23:T179‐T197. [DOI] [PubMed] [Google Scholar]
- 3. Sciarra A, Gentilucci A, Salciccia S, et al. Prognostic value of inflammation in prostate cancer progression and response to therapeutic: a critical review. J Inflamm (Lond). 2016;13:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Calò V, Migliavacca M, Bazan V, et al. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol. 2003;197:157‐168. [DOI] [PubMed] [Google Scholar]
- 5. Pencik J, Wiebringhaus R, Susani M, Culing Z, Kenner L. IL‐6/STAT3/ARF: the guardians of senescence, cancer progression and metastasis in prostate cancer. Swiss Med Wkly. 2015;145:w14215. [DOI] [PubMed] [Google Scholar]
- 6. Huang H. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin Cancer Res. 2007;13:1362‐1366. [DOI] [PubMed] [Google Scholar]
- 7. Okamoto M, Lee C, Oyasu R. Interleukin‐6 as a paracrine and autocrine growth factor in human prostatic carcinoma cells in vitro. Cancer Res. 1997;57:141‐146. [PubMed] [Google Scholar]
- 8. Lou W, Ni Z, Dyer K, Tweardy DJ, Gao AC. Interleukin‐6 induces prostate cancer cell growth accompanied by activation of stat3 signaling pathway. Prostate. 2000;42:239‐242. [DOI] [PubMed] [Google Scholar]
- 9. Jang E, Kim BJ, Lee KT, Inn KS, Lee JH. A survey of therapeutic effects of artemisia capillaris in liver diseases. Evid Based Complement Alternat Med. 2015;2015:728137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Komiya T, Tsukui M, Oshio H. Capillarisin, a constituent from Artenisiae capillaris herba. Chem Pharm Bull. 1975;23:1387‐1389. [DOI] [PubMed] [Google Scholar]
- 11. Yu Z, Tang L, Chen L, et al. Capillarisin suppresses lipopolysaccharide‐induced inflammatory mediators in BV2 microglial cells by suppressing TLR4‐mediated NF‐kappaB and MAPKs signaling pathway. Neurochem Res. 2015;40:1095‐1101. [DOI] [PubMed] [Google Scholar]
- 12. Khan S, Shehzad O, Chun J, et al. Anti‐hyperalgesic and anti‐allodynic activities of capillarisin via suppression of inflammatory signaling in animal model. J Ethnopharmacol. 2014;152:478‐486. [DOI] [PubMed] [Google Scholar]
- 13. Han S, Lee JH, Kim C, et al. Capillarisin inhibits iNOS, COX‐2 expression, and proinflammatory cytokines in LPS‐induced RAW 264.7 macrophages via the suppression of ERK, JNK, and NF‐kappaB activation. Immunopharmacol Immunotoxicol. 2013;35:34‐42. [DOI] [PubMed] [Google Scholar]
- 14. Chen NJ, Hao FY, Liu H, Zhao H, Li JM. Capillarisin exhibits anticancer effects by inducing apoptosis, cell cycle arrest and mitochondrial membrane potential loss in osteosarcoma cancer cells (HOS). Drug Res. 2015;65:422‐427. [DOI] [PubMed] [Google Scholar]
- 15. Lee SO, Jeong YJ, Kim M, Kim CH, Lee IS. Suppression of PMA‐induced tumor cell invasion by capillarisin via the inhibition of NF‐kappaB‐dependent MMP‐9 expression. Biochem Biophys Res Comm. 2008;366:1019‐1024. [DOI] [PubMed] [Google Scholar]
- 16. Lee JH, Chiang SY, Nam D, et al. Capillarisin inhibits constitutive and inducible STAT3 activation through induction of SHP‐1 and SHP‐2 tyrosine phosphatases. Cancer Lett. 2014;345:140‐148. [DOI] [PubMed] [Google Scholar]
- 17. Tsui KH, Chung LC, Feng TH, et al. Divergent effect of liver X receptor agonists on prostate‐specific antigen expression is dependent on androgen receptor in prostate carcinoma cells. Prostate. 2015;75:603‐615. [DOI] [PubMed] [Google Scholar]
- 18. Lee JC, Chiang KC, Feng TH, et al. The iron chelator, Dp44mT, effectively inhibit human oral squamous cell carcinoma cell growth in vitro and in vivo. Int J Mol Sci. 2016;17:1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsui KH, Chang YL, Feng TH, et al. Capillarisin blocks prostate‐specific antigen expression on activation of androgen receptor in prostate carcinoma cells. Prostate 2017. 10.1002/pros.23463. [DOI] [PubMed] [Google Scholar]
- 20. Tsui KH, Lin YH, Chung LC, et al. Prostate‐derived ets factor represses tumorigenesis and modulates epithelial‐to‐mesenchymal transition in bladder carcinoma cells. Cancer Lett. 2016;375:142‐151. [DOI] [PubMed] [Google Scholar]
- 21. Chiang KC, Yeh CN, Huang CC, et al. 25(OH)D is effective to repress human cholangiocarcinoma cell growth through the conversion of 25(OH)D to 1α,25(OH)2D3 . Int J Mol Sci. 2016;17:1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomarkers Prev. 2016;25:16‐27. [DOI] [PubMed] [Google Scholar]
- 23. Mukhopadhyay I, Sausville EA, Doroshow JH, Roy KK. Molecular mechanism of adaphostin‐mediated G1 arrest in prostate cancer (PC‐3) cells. J Biol Chem. 2006;281:37330‐37344. [DOI] [PubMed] [Google Scholar]
- 24. Srirupa R, Rana P, Singh C, et al. Down‐regulation of both p21/Cip1 and p27/Kip1 produces a more aggressive prostate cancer phenotype. Cell Cycle. 2008;7:1828‐1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang M, Coen JJ, Suzuki Y, et al. Survivin is a potential mediator of prostate cancer metastasis. Int J Radiat Oncol Biol Phys. 2010;78:1095‐1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brehmer B, Biesterfeld S, Jakse G. Expression of matrix metalloproteinases (MMP‐2 and ‐9) and their inhibitors (TIMP‐1 and ‐2) in prostate cancer tissue. Prostate Cancer Prostatic Dis. 2003;6:217‐222. [DOI] [PubMed] [Google Scholar]
- 27. Moroz A, Delella FK, Almeida R, et al. Finasteride inhibits human prostate cancer cell invasion through MMP2 and MMP9 downregulation. PLoS ONE. 2013;8:e84757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Culig Z. Proinflammatory cytokine interleukin‐6 in prostate carcinogenesis. Am J Clin Exp Urol. 2014;2:231‐238. [PMC free article] [PubMed] [Google Scholar]
- 29. Tsui KH, Lin YF, Chen YH, Chang PL, Juang HH. Mechanisms by which interleukin‐6 regulates prostate specific antigen gene expression in prostate LNCaP carcinoma cells. J Androl. 2011;32:383‐393. [DOI] [PubMed] [Google Scholar]
- 30. Tsui KH, Feng TH, Chung LC, et al. Prostate specific antigen gene expression in androgen insensitive prostate carcinoma subculture cell line. Anticancer Res. 2008;28:1969‐1976. [PubMed] [Google Scholar]
- 31. Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin‐6 signaling pathway in targeted therapy for cancer. Cancer Treatment Rev. 2012;38:904‐910. [DOI] [PubMed] [Google Scholar]
- 32. Saha A, Blando J, Silver E, et al. Shogaol from dried ginfer inhibits growth of prostate cancer cells both in vitro and in vivo through inhibition of STAST3 and NF‐kappaB signaling. Cancer Prev Res. 2014;7:627‐638. [DOI] [PubMed] [Google Scholar]
- 33. Kwon GT, Jung JI, Song HR, et al. Piceatannol inhibits migration and invasion of prostate cancer cells: possible mediation by decreased interleukin‐6 signaling. J Nutr Biochem. 2012;23:228‐238. [DOI] [PubMed] [Google Scholar]