

Abstract
Objectives
Bone marrow derived endothelial progenitor cells (BM‐EPCs) are increased in chronic liver disease (CLD). Their role in hepatic fibrosis and regeneration remains an area of intense studies. We investigated the migration and secretory functions of BM‐EPCs in fibrotic mice liver.
Materials and methods
Bone marrow cells from C57BL6‐GFP mice were transplanted into the femur of irradiated C57BL6 mice, followed by CCl4 doses for 8 weeks, to develop hepatic fibrosis (n = 36). Transplanted C57BL6 mice without CCl4 treatment were used as controls. EPCs were analyzed in BM, blood and liver by flow cytometry and immunofluorescence. VEGF and TGF‐β were analysed in the hepatic stellate cells (HSCs) and BM‐EPCs co‐cultures using ELISAs.
Results
There was a significant migration of EPCs from BM to blood and to the liver (P ≤ 0.01). Percentage of GFP + CD31+ EPCs and collagen proportionate area was substantially increased in the liver at 4th week of CCl4 dosage compared to the controls (19.8% vs 1.9%, P ≤ 0.05). Levels of VEGF (533.6 pg/ml) and TGF‐β (327.44 pg/ml) also increased significantly, when HSCs were treated with the EPC conditioned medium, as compared to controls (25.66 pg/ml and 5.87 pg/ml, respectively; P ≤ 0.001).
Conclusions
Present findings suggest that BM‐EPCs migrate to the liver during CCl4‐induced liver injury and contribute to fibrosis.
Abbreviations
- BM
bone marrow
- EPC
endothelial progenitor cells
- BM‐EPCs
bone marrow derived endothelial progenitor cells
- CLD
chronic liver disease
- HSCs
hepatic stellate cells
- PH
partial hepatectomy
- LSECs
liver sinusoidal endothelial cells
- HGF
hepatocyte growth factor
- GFP‐BM‐MNCs
GFP labelled bone marrow mononuclear cells
1. INTRODUCTION
Angiogenesis occurs during liver regeneration after liver injury or after two‐third partial hepatectomy (PH) conditions.1, 2 A cross‐talk among liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs) and hepatocytes is believed to play an important role in the construction of new sinusoids and proliferation of hepatocytes both during physiological and pathophysiological conditions.3, 4 During chronic liver injury, there is a loss of LSEC fenestrae (also known as sinusoidal capillarization) following which, there is an increased resistance to blood flow and oxygen delivery from the sinusoids to the parenchyma, leading to hypoxia and activation of angiogenic mediators.5, 6 Along with LSECs, the proangiogenic functions during liver injury are also mediated by activated HSCs that show an up‐regulation of VEGF, angiopoietins and their receptors.7, 8
Besides LSECs, bone marrow (BM)‐derived endothelial progenitor cells (EPCs) harvested from peripheral blood are also known to participate in postnatal vasculogenesis and revascularization of ischemic tissues.9, 10, 11 Our recent investigations have demonstrated increased endogenous levels of circulating EPCs in patients with cirrhosis as compared to the controls, suggesting enhanced mobilization of these cells in CLD patients. Our study has also revealed that these circulating EPCs interact with resident LSECs via paracine mediators including VEGF and PDGF‐BB and enhance their angiogenic functions in vitro.12 In the current study, we investigated the numbers of endogenous EPCs in BM, blood and the liver during progressive liver injury using green florescent protein (GFP)‐bone marrow transplanted cells in CCL4‐induced liver injury mouse models for up to 8 weeks. Further, we studied the correlation of EPC numbers in the liver with liver fibrosis at different weeks in these mouse models. Moreover, interactions of EPCs with HSCs were also analyzed in the in vitro cultures to determine the role of EPCs in liver fibrosis.
2. MATERIALS AND METHODS
2.1. Development of the fibrotic mouse model
All experiments were conducted as approved by the Institutional Animal Ethics Committee of NII, New Delhi.
Bone marrow cells were isolated from C57BL6‐GFP mice by flushing the femur and tibia with 3% IMDM. Collected cells were passed through 40 μm filter and treated with Gey's solution for 90 s to remove RBCs and suspended in IMDM for transplantation (Figure 1A).
Figure 1.
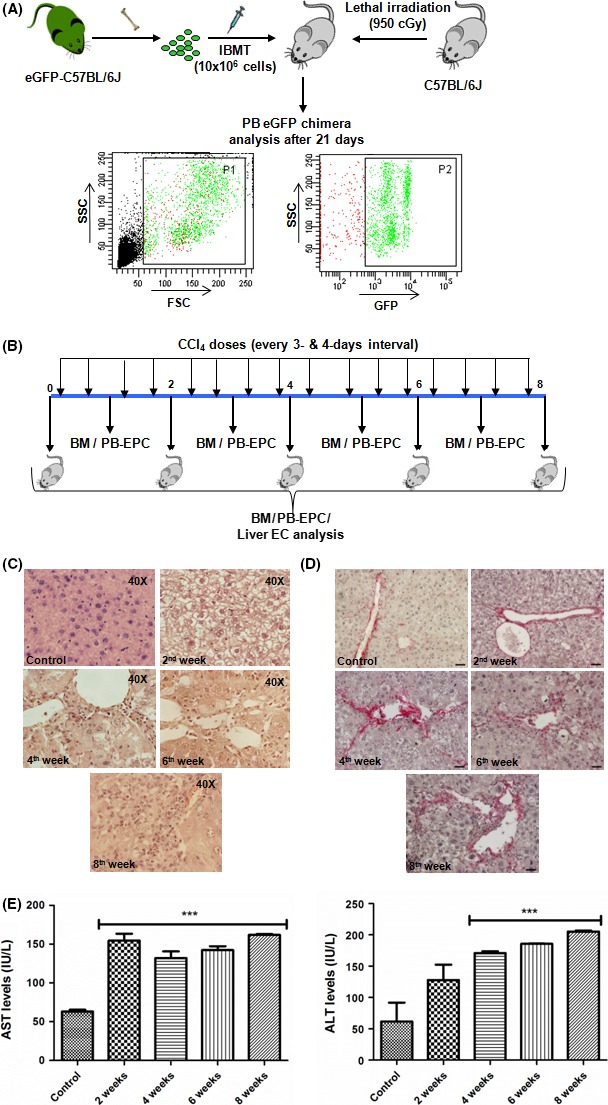
Model development – (A) Schematic for the development of GFP chimeric mice with FACS analysis dot plot of peripheral blood (PB); (B) Schematic for the CCl4 dosage plan and analysis timepoints of EPCs in bone marrow (BM), peripheral blood (PB) and liver; (C) H&E staining (40X) and (D) picrosirius staining at different weeks of CCl4 dosage; scale bar represents 20 X magnification, ***P value is >.0001
Another batch of C57BL6 mice were lethally irradiated (900 cGy) and 10 × 106 GFP bone marrow cells were transplanted through intra‐femoral route. After 21 days, percentage of chimerism was checked in peripheral blood by flow cytometry. Mice with more than 80% chimerism were selected and given intra‐peritoneal 0.4 ml CCl4/kg b.w. in olive oil, twice a week, for 8 weeks. Mice were sacrificed after 48 h of each CCl4 injection for peripheral blood and bone marrow analysis (Figure 1B). Transplanted C57BL6J mice, without any CCl4 dosage were used as controls.
2.2. Analysis of EPCs in bone marrow
At each time point, bone marrow and peripheral blood mononuclear cells were separated. 6 x 106 cells were stained with 5 μg/ml Hoechst and 1 μg/ml pyronin separately for 1 h at 37°C for cell cycle analysis. The cells were stained with the anti‐CD34 and Flk‐1for EPCs and analyzed by BDFACSAria III (BD Biosciences) using DIVA software.
2.3. Analysis of EPCs in peripheral blood
At each time point, peripheral blood was collected by puncturing the retro‐orbital sinus. After plasma removal and RBC lysis, the cells were incubated with the anti‐CD34 (biotinylated rat anti‐mouse, e‐Biosciences; 1:100) and Flk‐1 (APC conjugated rat anti‐mouse, BD; 1:50) in PBS for 30 min at 4°C, followed by incubation with secondary antibody for CD34 (streptavidin anti‐rat APC‐Cy7, BD; 1:400) for 30 min at 4°C. The cells were then fixed with 4% PFA in PBS and analyzed by BDFACSAria III (BD Biosciences) using DIVA software.
2.4. Analysis of EPCs in liver
Liver perfusion was done at 2nd, 4th, 6th and 8th week of CCl4 dosage to isolate single cell suspension of non‐parenchymal cells. The liver was perfused through the heart using HBSS‐EGTA‐HEPES buffer and then digested with 0.025% collagenase in HBSS‐HEPES buffer. The liver was shaken off in 15% RPMI using forceps to release the cells. The cell suspension was passed through 100 μm filter and hepatocytes were removed by centrifugation. The supernatant was again centrifuged at 2000 rpm to collect the non‐parenchymal cells of the liver. The cells were counted and 5 × 106 cells were stained with biotinylated rat anti‐mouse CD31 (marker for mature endothelial cell, 1:50, Biolegend, USA) and secondary antibody, APC‐Cy7 (streptavidin anti‐rat, BD Biosciences, San Jose, CA, USA; 1:400). Cells were analyzed by BDFACSAria III (BD Biosciences) using DIVA software.
2.5. Histopathology and immunofluorescence
At each time point, liver was isolated from the same mouse whose peripheral blood was collected. Half of the tissue was fixed in 10% formalin for embedding in paraffin and the other half was fixed in 4% PFA for cryoembedding in OCT. H&E and picrosirius staining were carried out on 5 μm‐thick paraffin liver sections. Immunofluorescence staining for endothelial cells was carried out on 3 μm‐thick cryosections. The cryosections were permeabilized with 0.5% saponin in PBS for 30 min, blocked with 3% BSA in PBST for 30 min and stained with antibodies for 2 h at room temperature.
Primary antibodies for GFP (mouse monoclonal, Clonetech, Mountain View, CA, USA; 1:200), VCAM and Flk‐1 (purified rat anti‐mouse, e‐biosciences; 1:50) were used. Secondary antibodies for GFP, VCAM and Flk‐1were used (anti‐mouse AF488, 1:400, anti‐rat AF594, 1:200, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Nuclear staining was done using DAPI (1:1000, Sigma) and analyzed on Olympus fluorescence microscope.
2.6. Quantification of degree of fibrosis
The percentage of collagen proportionate area was calculated in 20X images of picrosirius stained liver sections using Image J software (NIH, Bethesda, USA).
2.7. In‐vitro culture of EPCs
Mononuclear cells (MNCs) from the bone marrow of a normal C57BL6J mouse were separated by density gradient centrifugation using histopaque (Sigma). Endothelial progenitor cells (EPCs) were cultured from these MNCs in 20% FBS and Endothelial Growth Medium containing VEGF and FGF (EGM‐2, Lonza, Basel, Switzerland) for 8 days, with a media change in every 2 days. For preparation of EPC conditioned media, EPCs after 8 days of culture were treated with reduced FBS (2%) devoid of any growth factors. The EPC conditioned medium was then collected after 24 h and pooled for further experiments.
2.8. In‐vitro culture of HSCs
Liver non‐parenchymal cells (NPC) from a normal C57BL6J mouse were isolated by perfusion as before. Hepatic stellate cells (HSCs) from the NPC fraction were separated by density gradient centrifugation using 27% histodenz (Sigma). Separated HSCs were collected and cultured in 10% DMEM F‐12 medium for 5 days.
2.9. EPC‐HSC interactions
For the in vitro assay, the HSCs were trypsinized and replated on sterile coverslips in a 24‐well plate, with 10,000 cells in each well. The cells were allowed to grow in 10% DMEM F‐12 medium for 3 days and on the third day, medium was replaced with the EPC conditioned medium, or with serum free endothelial basal media (EBM‐2, Lonza, Basel, Switzerland) as control. The culture medium was collected from each well after 24 h for VEGF and TGF‐β ELISA assays.
For immunocytochemistry, HSCs were permeabilized with 0.5% saponin in PBS for 30 min and then blocked with 3% BSA for 30 min at room temperature. Staining with antibody for α‐SMA (mouse monoclonal, Santa Cruz; 1:200) and the secondary antibody AF594 (anti‐mouse, Invitrogen, MA, USA; 1:200) was done for 2 h at room temperature. Nuclear staining was done using DAPI (1:1000, Sigma). Washing was done using 0.1% triton X‐100 in PBS. The coverslips were removed from each well, mounted and stained cells were photographed on fluorescence microscope.
2.10. Statistical analysis
Results of multiple experiments were reported as mean ± SD (Standard Deviation). Graph pad Prism software, Version 5.02 was used for ANOVA statistical analysis. P < 0.05 were considered significant (* < 0.05; ** < 0.01; *** < 0.005).
3. RESULTS
3.1. Establishment of chronic liver injury model
CCl4 based fibrotic mouse model was made as described earlier.13 To monitor circulating EPCs in BM, PB and liver, we created a BM chimera by directly injecting GFP‐BM‐MNCs in the femurs of lethally irradiated C57BL6 mice. This was done to avoid unintentional engraftment of donor cells in the liver during tail vein injection. Chimerism in transplanted mice was checked by analysing the peripheral blood (Figure 1A). No mortality was observed after the transplantation of GFP bone marrow cells. Mice with more than 80% GFP‐expressing MNCs in the peripheral blood were given repeated doses of CCl4 to induce chronic liver injury. The survival rate of the GFP‐CCl4 mice was between 70 and 80%. The death was due to toxic effect of CCl4. GFP mice without CCl4 treatment were used as controls.
The scheme of the experiments and subsequent analyses are depicted in Figure 1B. Histopathological examinations suggest ballooning of hepatocytes (steatosis) with minute inflammation (changes of acute hepatitis in hepatic acinus) initiated within 2nd week of treatment with CCl4 (Figure 1C). In subsequent weeks, perivenular and periportal inflammation, linking inflammatory bridges, necrosis and inflammation with confluent necrosis was increased (Figure 1C and Supplementary Figure 1A). Distinct liver fibrosis was noticed after administrations of CCl4 for 4 weeks due to deposition of rich fibril‐forming collagen with distinct portal to portal bridging (Figure 1D). Finally, persistent liver injury was marked by the elevated levels of ALT and AST (Figure 1E).
3.2. Enumeration of EPCs in bone marrow (BM) and peripheral blood (PB)
The presence of donor‐derived EPCs in BM and PB was determined by the phenotype, GFP+CD34+Flk‐1+ at different times of CCl4 treatment. Furthermore, cell cycle analysis was performed in BM cells to assess the proliferative response of EPCs with each CCl4 treatment. Cell proliferation was determined on the basis of the percentage of cells belonging to S/G2M phase. The representative dot‐plots for the cell cycle analysis in BM (second week post CCl4 administration) are shown in Figure 2A.
Figure 2.
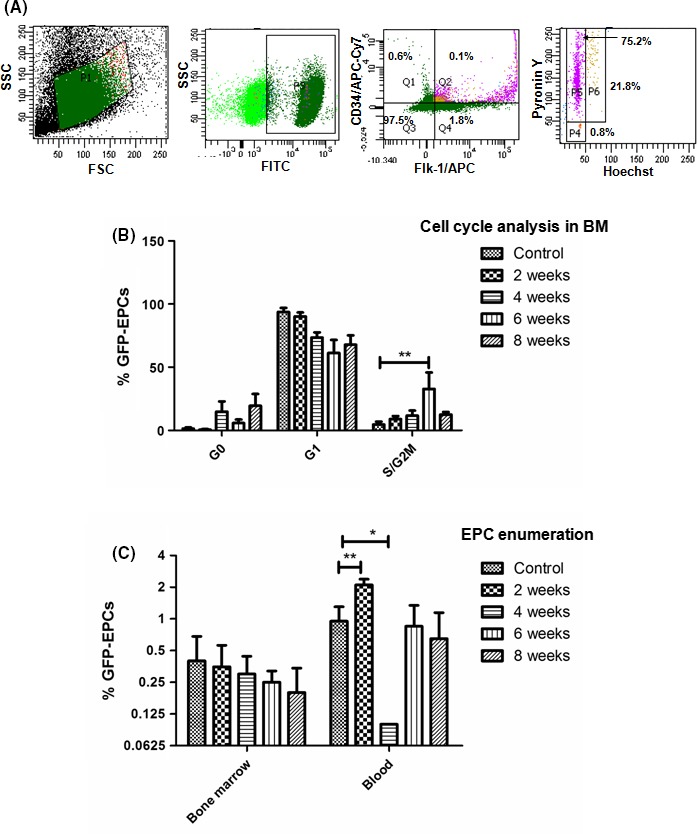
Enumeration of GFP + EPCs in bone marrow (BM) and peripheral blood (PB) at selected timepoints – (A) FACS analysis dot plots showing the cell cycle status of the GFP + CD34+Flk‐1+ EPCs in BM; (B) Enumeration of GFP + EPCs in G0/G1/SG 2M phases of the cell cycle; (C) Enumeration of GFP + EPCs in BM and PB, *P value is >.05, **P value is >.005
In comparison with the controls without CCl4 treatment, there was an increase in the EPC proliferation in the BM right after the administration of CCl4. However, statistically significant increases in the EPC proliferation in the BM were observed only at the 6th week CCl4 treatment (P < 0.01, Figure 2B). However, we did not find any significant difference in the EPC numbers in the BM, despite of the activation of cell cycle (Figure 2C). This may be attributed to the egress of EPCs from the BM to the PB.
In the PB, the number of EPCs was significantly higher at 2 weeks of CCl4 administration compared to the controls, (P < 0.01, Figure 2C). Interestingly, their levels drastically declined during the 4th week. After that, it further increased in the subsequent weeks (Figure 2C). These results suggest that the levels of BM‐derived EPCs in the PB do not remain at a steady value and that they are governed by the number of cells egressing from the BM and the number of cells going to the liver.
3.3. Enumeration of EPC in liver
After CCL4 injury, we anticipated that these mobilized BM‐derived EPCs, at least in part, will home and engraft in the liver. To enumerate the number of BM‐derived EPCs at different times of CCl4 treatment, a representative dot‐plot of 4th week of liver sample is shown in supplementary Figure 1B. Significant increment of BM‐derived EPCs (GFP+CD31+) up to 20 ± 3% was observed in the liver during the 4th week (Figure 3A) as compared to controls. Interestingly, after 4 weeks, we observed a decline of these cells in the liver.
Figure 3.
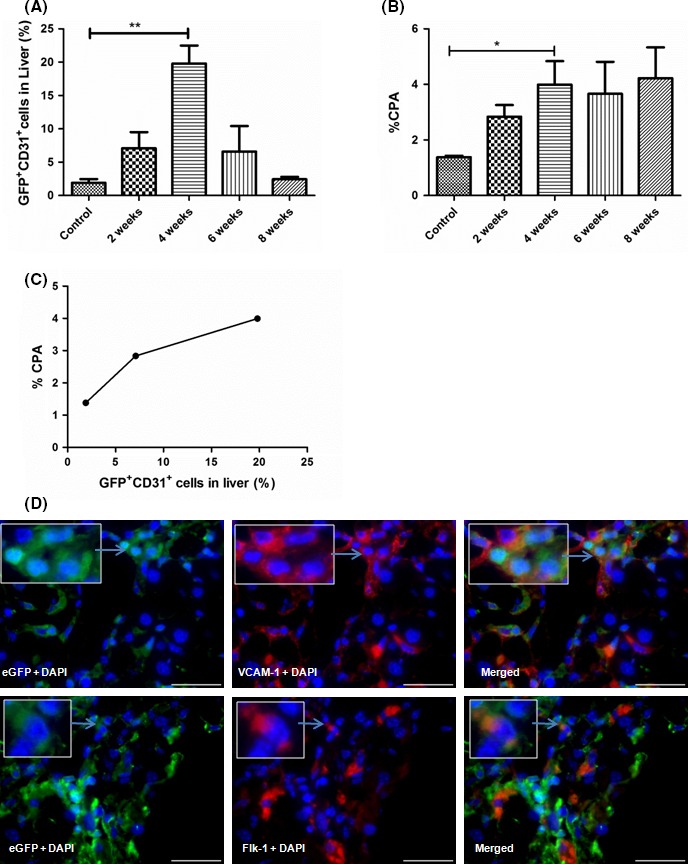
Enumeration of GFP + EPCs in liver at selected timepoints – (A) Enumeration of GFP +/CD31+endothelial cells (ECs) in liver; (B) collagen quantification or percentage of collagen proportionate area (CPA) at different weeks of CCl4 dosage; (C) Correlation plot between the % CPA and % GFP +/CD 31+ ECs in liver till 4th week of CCl4 dosage; (D) immunofluorescence in the liver after 4th week of CCl4 dosage with staining of VCAM‐1 (upper panel) and Flk‐1 for endothelial cells (lower panel); scale bar represents 60 X magnification, *P value is >.05, **P value is >.005
Correlation of GFP+CD31+ endothelial cells in liver with the degree of fibrosis:
Quantitative analysis showed that liver fibrosis, expressed by percentage collagen proportionate area (CPA), gradually increased with CCl4 treatment, with maximum increase during the 4th week of CCl4 treatment as compared to the controls (Figure 3B). Correlation analysis between GFP+CD31+ endothelial cells in liver and % CPA showed positive correlation till 4th week of CCl4 treatment (Figure 3C).
Immunohistochemical analysis in liver: Immunohistochemical analysis of 4th week liver tissues on the basis of VCAM‐1 and Flk‐1, was used to identify GFP‐EPCs that migrated from BM to liver (Figure 3D). Results revealed the presence of GFP+‐ Flk‐1+ and GFP+‐VCAM‐1+ EPCs around the LSECs in the liver tissues. We further observed a co‐localization of α‐SMA and VCAM‐1 positive cells around the LSECs in these liver sections. Furthermore, the location pattern of α‐SMA+ ‐ VCAM‐1+ cells was similar to that of the GFP+‐ Flk‐1+ and GFP+‐VCAM‐1+ EPCs. These results indicated close interactions between BM derived EPCs and HSCs (Supplementary Figure 2A). We also observed cells co‐expressing GFP and α‐SMA during the 4th week of treatment (Supplementary Figure 2B).
3.4. Study of EPC‐HSC interactions in‐vitro
To elucidate the interactions between EPCs and HSCs, in vitro cultures of HSCs with or without EPCs conditioned medium were performed. After 24 h, VEGF and TGF‐β were quantified in the culture medium. VEGF and TGF‐β were significantly increased in HSCs within 24 h of co‐cultures with the EPC conditioned medium (Figure 4A). The treated HSCs also showed an increased expression of α‐SMA, indicating their activation (Figure 4,C). In immunohistochemical analysis, other markers of fibrosis was also included as increased collagen 1 in the bridging necrosis at 2nd week and at 4th week, but Timp 1 expression was found more in 2nd week than in 4th week (Supplementary Figure 2C,D).
Figure 4.
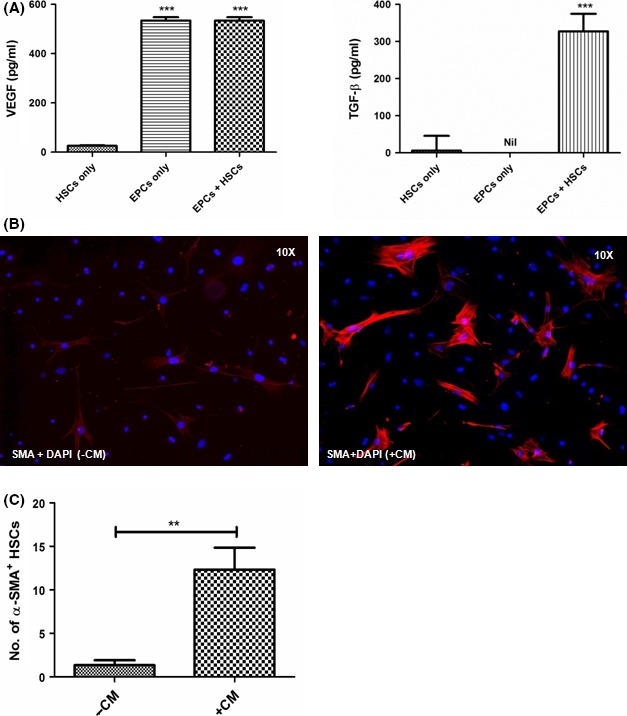
EPC‐HSC interactions in‐vitro – (A) Quantification of levels of VEGF and TGF‐β in the culture medium; (B) Immunofluorescence images showing the inactivated and activated α‐SMA + stellate cells in without (‐CM) and with (+CM) the EPC conditioned medium; (C) Quantitation of α‐SMA + stellate cells in the immunofluorescence images, **P value is >.005, ***P value is >.0001
4. DISCUSSION
Bone marrow chimeric mice have been extensively used to investigate the mechanisms of tissue regeneration, cell migration and disease pathogenesis.14, 15, 16 In this study, we generated GFP chimeric mice to track the migration of EPCs from bone marrow to the peripheral blood and finally their engraftment in the damaged liver.
The study suggested that cell cycle activation of EPCs in the bone marrow and liver injury are closely related. Bone marrow responds to liver injury through a potential autocrine loop, resulting in activation and proliferation of EPCs. The autocrine factor(s) responsible for such physiological changes, however, remain to be identified. Probably, these factors(s) are mitogenic and are secreted in the damaged liver for self‐regeneration. Despite cell cycle activation, we did not observe any significant changes in EPC numbers in the bone marrow with time. This could be due to the dynamic nature of the cells, which proliferate and at the same time egress from the bone marrow, resulting in maintenance of cell number.
In the blood, we observed an increase in circulating EPCs during early stage of the liver injury, which however declined to a lowest value in 4th week. This decline is attributed to the engraftment of cells to the liver during the 4th week of CCl4 treatment. In later time points, EPC levels returned back to original values, potentially due to lack of demand by the regenerating liver. This cyclic nature of EPC levels in blood is entirely due to physiological demand and supply of the cells and not due to the harmful effect of CCl4. The circulating EPC levels are expected to be unaffected, as CCl4 reached liver via mesenteric vessels and portal vein route.17 It is, however, to be noted that with increasing CCl4 mediated injury, there was a sharp decrease in the number of EPCs in the liver at 6th week. There was a substantial positive correlation of EPC numbers with liver fibrosis till 4th week of CCl4 treatment, indicating a potential role of EPCs in liver fibrosis. In our earlier study, we had reported that higher EPC numbers in patients with cirrhosis correlates significantly with the degree of fibrosis in these patients.12
It has been documented that EPCs contribute to postnatal neo‐vasculogenesis by converting into matured form and integrating into growing vessels in cirrhotics and HCC patients.18 In the present study, co‐localization of GFP with VCAM‐1 and GFP with Flk‐1 in cells was observed in few engrafted cells, which were rarely present in the endothelial lining of the liver. These results suggest that EPCs contributes little towards endothelial regeneration during liver injury. It has been earlier shown that the intraperitoneal administration of EPCs into rat model of liver injury by CCl4 enhances liver regeneration and suppresses liver fibrogenesis.19, 20 In the above study, Liu et al. has used ex vivo cultured EPCs after 8 weeks of CCl4 treatment. The study has reported that in comparison to the 8‐week CCl4 treated control group, the levels of ALT and AST are reduced in the EPCs transplanted group. In another study, infusion of enriched population of EPCs in 6 week‐CCl4 treated rats has shown anti‐fibrogenic effect of the cells in terms of biochemical and histological evidences. However, the study has failed to observe any differentiation of transplanted cells into hepatocytes or endothelial cells, indicating that the donor cells function mainly through a paracrine effects to prevent liver fibrosis and regeneration, rather than by direct differentiation.20 In the present study, we observed GFP‐EPCs around LSECs during the 4th week of CCl4 treatment. Since, HSCs are also present around the LSECs, we next analysed whether the migrated EPCs are present near the HSCs. Localization of α‐SMA, a marker for activated HSCs and VCAM‐1 in the cells surrounding the sinusoidal endothelium during 4th week of CCl4 treatment indicated close proximity between liver fibroblasts and endothelial cells. Intriguingly, these cells shared the same pattern of localization in the liver as observed with GFP+Flk‐1+ and GFP+VCAM‐1+, implicating that the endothelial cells might be of the BM origin. Furthermore, the interactions between HSCs and EPCs by in vitro culture assays revealed that in the presence of EPC conditioned medium, HSCs secreted more TGF‐β in the cultures, indicating potential paracrine interactions of EPCs with liver HSCs in vivo too. Also, EPC conditioned media contained high levels of VEGF, which they maintained in the HSC‐EPC co‐cultures indicating their pro‐angiogenic role in liver fibrosis. We, however, did not observe significant amounts of VEGF in the HSC cultures alone, suggesting most of the VEGF is secreted by the EPCs. It is well‐known that similar to LSECs, HSCs assume an angiogenic role during chronic liver insult, however, the underlying mechanisms are poorly understood.21
Our study points towards an important fibrogenic role of BM‐derived endothelial progenitors in activating liver HSCs through paracrine interactions. Previously, our group, including others, has reported a paracrine crosstalk of EPCs with LSECs via VEGF and PDGF instead of actual physical interaction.12, 22, 23 An interaction of EPCs with LSECs may contribute towards endothelial regeneration, but their interaction with HSCs can also aggravate HSC‐mediated angiogenesis and fibrogenesis. Activated HSCs produce excess pro‐fibrotic mediator, TGF‐β, that promotes their differentiation into myofibroblasts and leads to production of excess collagen. This disturbs the balance of fibrolysis process, leading to fibrogenesis and cirrhosis. It is also worth mentioning here that we observed cells co‐expressing GFP and α‐SMA during the 4th week of CCl4 dosage suggesting that a fraction of bone marrow cells also contributed to liver fibrosis, as has been reported by earlier studies.13, 24
In conclusion, CCl4 mediated liver injury causes proliferation and migration of BM‐derived EPCs from the BM to the liver. The migration of EPC seems to have correlation with the degree of liver fibrosis and collagen deposition until the fibrosis is completely established. This study also suggests that BM‐EPCs do not participate in the repair of endothelial cells; however, they secrete VEGF and induce the activation of HSCs and subsequent fibrosis of liver.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by research grant from the Indian Council of Medical Research, New Delhi, India (80/5/2010/‐BMS).
Garg M, Kaur S, Banik A, et al. Bone marrow endothelial progenitor cells activate hepatic stellate cells and aggravate carbon tetrachloride induced liver fibrosis in mice via paracrine factors. Cell Prolif. 2017;50:e12355 10.1111/cpr.12355
REFERENCES
- 1. Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836‐847. [DOI] [PubMed] [Google Scholar]
- 2. Drixler TA, Vogten MJ, Ritchie ED, et al. Liver regeneration is an angiogenesis‐ associated phenomenon. Ann Surg. 2002;236:702‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaur S, Anita K. Angiogenesis in liver regeneration and fibrosis: “a double‐edged sword”. Hep Intl. 2013;959‐968. [DOI] [PubMed] [Google Scholar]
- 4. Ding B‐S, Nolan DJ, Butler JM, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468:310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fernandez M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50:604‐620. [DOI] [PubMed] [Google Scholar]
- 6. Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol. 2010;53:976‐980. [DOI] [PubMed] [Google Scholar]
- 7. Ankoma‐Sey V, Wang Y, Dai Z. Hypoxic stimulation of vascular endothelial growth factor expression in activated rat hepatic stellate cells. Hepatology. 2000;31:141‐148. [DOI] [PubMed] [Google Scholar]
- 8. Novo E, Cannito S, Zamara E, et al. Proangiogenic cytokines as hypoxia‐dependent factors stimulating migration of human hepatic stellate cells. Am J Pathol. 2007;170:1942‐1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawamoto A, Gwon HC, Iwaguro H, et al. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation. 2001;103:634‐637. [DOI] [PubMed] [Google Scholar]
- 10. Fan CL, Gao PJ, Che ZQ, Liu JJ, Wei J, Zhu DL. Therapeutic neovascularization by autologous transplantation with expanded endothelial progenitor cells from peripheral blood into ischemic hind limbs. Acta Pharmacol Sin. 2005;26:1069‐1075. [DOI] [PubMed] [Google Scholar]
- 11. Sakamoto M, Nakamura T, Torimura T, et al. Transplantation of endothelial progenitor cells ameliorates vascular dysfunction and portal hypertension in carbon tetrachloride‐induced rat liver cirrhotic model. J Gastroenterol Hepatol. 2013;28:168‐178. [DOI] [PubMed] [Google Scholar]
- 12. Kaur S, Tripathi D, Dongre K, et al. Increased number and function of endothelial progenitor cells stimulate angiogenesis by resident liver sinusoidal endothelial cells (SECs) in cirrhosis through paracrine factors. J Hepatol. 2012;57:1193‐1198. [DOI] [PubMed] [Google Scholar]
- 13. Baligar P, Mukherjee S, Kochat V, Rastogi A, Mukhopadhyay A. Molecular and cellular functions distinguish superior therapeutic efficiency of bone marrow CD45 cells over mesenchymal stem cells in liver cirrhosis. Stem Cells. 2016;34:135‐147. [DOI] [PubMed] [Google Scholar]
- 14. Alvarez‐Dolado M, Pardal R, Garcia‐Verdugo JM, et al. Fusion of bone‐marrow‐derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425:968‐973. [DOI] [PubMed] [Google Scholar]
- 15. Lagasse E, Connors H, Al‐Dhalimy M, et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med. 2000;6:1229‐1234. [DOI] [PubMed] [Google Scholar]
- 16. de Freitas Souza BS, Nascimento RC, de Oliveira SA, et al. Transplantation of bone marrow cells decreases tumor necrosis factor‐alpha production and blood‐brain barrier permeability and improves survival in a mouse model of acetaminophen‐induced acute liver disease. Cytotherapy. 2012;14:1011‐1021. [DOI] [PubMed] [Google Scholar]
- 17. Turner PV, Brabb T, Pekow C, Vasbinder M. Administration of substances to laboratory animals: routes of administration and factors to consider. J Am Assoc Lab Anim Sci. 2011;50:600‐613. [PMC free article] [PubMed] [Google Scholar]
- 18. Yu D, Sun X, Qiu Y, et al. Identification and clinical significance of mobilized endothelial progenitor cells in tumor vasculogenesis of hepatocellular carcinoma. Clin Cancer Res. 2007;13:3814‐3824. [DOI] [PubMed] [Google Scholar]
- 19. Liu F, Fei R, Rao HY, Cong X, Ha MH, Wei L. The effects of endothelial progenitor cell transplantation in carbon tetrachloride induced hepatic fibrosis rats. Zhonghua Gan Zang Bing Za Zhi. 2007;15:589‐592. [PubMed] [Google Scholar]
- 20. Lian J, Lu Y, Xu P, et al. Prevention of liver fibrosis by intrasplenic injection of high‐density cultured bone marrow cells in a rat chronic liver injury model. PLoS ONE. 2014;9:e103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garbuzenko DV, Arefyev NO, Belov DV. Mechanisms of adaptation of the hepatic vasculature to the deteriorating conditions of blood circulation in liver cirrhosis. World J Hep. 2016;8:665‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu Y, Huang L. Function and modulation of endothelial progenitor cells. Zhonghua Xin Xue Guan Bing Za Zhi. 2007;35:1067‐1069. [PubMed] [Google Scholar]
- 23. Yu D, Chen J, Sun X, Zhuang L, Jiang C, Ding Y. Mechanism of endothelial progenitor cell recruitment into neo‐vessels in adjacent non‐tumor tissues in hepatocellular carcinoma. BMC Cancer. 2010;10:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hep. 2006;21:S84‐S87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials