

Abstract
Objectives
The reprogramming of cancer cells into induced pluripotent stem cells or less aggressive cancer cells can provide a modern platform to study cancer‐related genes and their interactions with cell environment before and after reprogramming. Herein, we aimed to investigate the reprogramming capacity of murine melanoma B16F10 cells.
Materials and methods
The B16F10 was transfected using non‐viral circular DNA plasmid containing the genes Sox‐2, Oct4, Nanog, Lin28 and green fluorescent protein (GFP). These cells were characterized by immunofluorescence, analysis RT‐PCR and cell cycle.
Results
Our results demonstrated for the first time that reprogramming of B16F10 may be induced using non‐viral minicircle DNA containing the four reprogramming factors Oct4, Sox2, Lin 28, Nanog (OSLN) and the GFP reporter gene. The resulting clones are composed by epithelioid cells. These cells display characteristics of cancer stem cells, thus expressing pluripotent stem cell markers and dividing asymmetrically and symmetrically. Reprogrammed B16F10 cells did not form teratomas; however, they showed the suppression of tumourigenic abilities characterized by a reduced tumour size, when compared with parental B16F10 cell line. In contrast to parental cell line that showed accumulation of the cells in S phase of cell cycle, the cells of reprogrammed clones are accumulated in G1 phase. Long‐term cultivation of reprogrammed B16F10 cells induces regression of their reprogramming.
Conclusions
Our data imply that in result of reprogramming of B16F10 cells less aggressive Murine Melanoma Reprogrammed Cancer Cells may be obtained. These cells represent an interesting model to study mechanism of cells malignancy as well as provide a novel tool for anti‐cancer drugs screening.
1. INTRODUCTION
In recent years, different research groups focused on identification of genetic changes related to carcinogenesis, possible epigenetic mechanisms and chromosomal alterations responsible for cell transformation, tumour initiation and progression.1, 2 Reversion of cancer cells into induced pluripotent stem cells (iPSC) or into a less aggressive cancer cell population is a challenge that has also been discussed during last decades. Due to highly heterogeneous nature of cancer cells, such transformation involves many genetic and epigenetic factors,3 which are specific for each type of tumour.4, 5 Different methods of cancer cells reprogramming have been established6, 7 and demonstrate a possibility to obtain less aggressive8 or even normal cells. These methods, however, are quite complex, thus a simpler and efficient method of reprogramming is still required. As soon as iPSC technology, which demonstrated the capacity to reprogram terminally differentiated cells into embryonic stem cells (ESC)‐like,9, 10 was developed, it strongly attracted the attention of researches, opening new perspectives for stem cell personalized therapies and offering a powerful in vitro model for drug screening. Currently, it was suggested to be used for cancer cells reprogramming,11 thus providing a modern platform to study cancer‐related genes and the interaction between these genes and cell environment before and after reprogramming, in order to elucidate the mechanisms of cancer occurrence and progression.7 Using this novel dedifferentiation technique, reprogrammed cancer cells with or without cancer properties can be produced.12
Heterogeneity is an intrinsic characteristic of melanoma cells that contribute to the vast phenotypic and genotypic variety of these tumours.13, 14, 15, 16 An interesting way to modulate this phenomenon is the reprogramming of these tumourous cells, followed by check out of what this entails in terms of expression of tumour markers and cancer stem cells (CSC) markers17, 18, 19 as well. Thereby, the tumour cells reprogramming is mostly an interesting strategy to understand which phenomenon leads to heterogeneity.20
Commonly retroviral or lentiviral vectors are used to generate iPSC, however such plasmids may integrate into the genome of the host cells.10, 21 This aleatory integration may result in malignant transformations caused by mutagenesis, which can increase the instability in tumoural cells that have already accumulated mutations.22, 23 Moreover, during reprogramming, the cells increase their intolerance to different types of DNA damage that may occur due to different reasons, including viral integration. Therefore, it is of a great importance to test non‐viral methods to obtain transgene‐free cancer cells‐derived iPSC.
Herein, we used non‐viral minicircle DNA, which contained the four reprogramming factors Oct4, Sox2, Lin 28, Nanog (OSLN), and the green fluorescent protein (GFP) reporter gene in order to reprogram murine melanoma B16F10 cells, which was previously employed to generate transgene‐free iPSC from adult human cells.24 We also aimed to investigate the reprogramming capacity of these tumour cells in order to establish a model for studying the mechanisms of loss of malignancy through reprogramming of tumour cells into cancer iPSC. This technique is advantageous in translation studies, once it allows verifying the tumoural cell answer after reprogramming in the absence of genomic modification, viral sequences, effectively mitigating safety concerns.
2. MATERIALS AND METHODS
2.1. Cell culture
Murine melanoma (B16F10) cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with: 10% foetal bovine serum (Atlanta Biologicals, Lawrenceville, GA, USA), 100 IU/mL penicillin and 100 μg/mL streptomycin (MP Biomedicals, Solon, OH, USA). The cell cultures were maintained in 5% CO2 at 37°C, in a fully humidified incubator. Primate mES medium combine knockout DMEM, 20% (v/v) ES cell FBS, 0.1 mmol/L non‐essential amino acids, and 0.1 mmol/L 2‐mercaptoethanol and 10³ U/mL LIF (ESGRO Merk Millipore, Darmstadt, Germany). The cells were cultivated into feeder‐free conditions on Matrigel (BD Biosciences, Franklin Lakes, NJ, USA; diluted 1:100 in DMEM/F12).
2.2. Reprogramming method
B16F10 cells were cultured under OPTI‐MEM medium (Gibco ‐ Life Technologies, Carlsbad, Califórnia, USA) and transfected with non‐self‐replicating minicircle DNA (Stemcircles™—StemCell Technologies, Vancouver, British Columbia, Canada) containing the four reprogramming factors Oct4, Sox2, Lin 28, Nanog (OSLN), and the GFP reporter gene. Cells were transfected using the reagent Lipofectamine 2000 (Invitrogen). After the transfection, the cells were switched to DMEM/F12 medium (Gibco) supplemented with 20% knockout serum (Gibco) and LIF. GFP+ cells were seen in microscopy 18 hours after transfection. At day 4, the cells were then seeded into feeder‐free conditions on Matrigel (BD Biosciences; diluted 1:100 in DMEM/F12) on 6 cm dishes at ~0.5 × 105 cells per well. Culture medium was refreshed every 2‐3 days. Colonies with morphologies similar to hESC colonies were clearly visible by 1 week after transfection. At day 12‐18 after transfection, GFP‐positive cells colonies were individually picked for further expansion and analysis, this expression was transient.
2.3. Cell cycle analysis
Synchronization of B16F10 and derived cells has been performed through deprivation of serum for 24 hours, which followed by the induction of cell cycle in these cells by serum addition. Next, the cells were harvested by enzymatic digestion and fixed in cold 70% ethanol, and stored at −20°C. For cell cycle analysis, the cells were washed twice in phosphate‐buffered saline (PBS) and re‐suspended in the same solution following by incubation at 37°C for 45 minutes with 10 mg/mL RNAse. After this, 1 mg/mL propidium iodide (Sigma, St. Louis, MO, USA) was added. Flow cytometry analysis was performed using a FACSCalibur (Becton Dickinson, San Jose, CA, USA). Cell DNA content in the different cell cycle phase was determined using ModFit LT software (Verity Software House, Topsham, ME, USA) and Prism 5 (GraphPad Prism Software, CA, USA).
2.4. Phalloidin staining
The actin cytoskeleton was visualized using fluorescently labelled phalloidin which binds to and stabilizes f‐actin.25 Cells were washed twice with PBS and then fixed using 1 mL of 4% paraformaldehyde (Sigma‐Aldrich Chemie GmbH, Munich, Germany). After washing twice with PBS, cells were permeabilized with 1 mL 0.1%‐Triton X‐100 (Sigma‐Aldrich Chemie GmbH) for 10 minutes at room temperature. Besides, again washing twice with PBS and after the cells were incubated with FICT‐phalloidin (Sigma‐Aldrich) for 1 hour at 37°C. Co‐stainings with Hoechst (Invitrogen) were performed as described above. Specimens were embedded in Vectashield and sealed with cover slips.
2.5. Immunofluorescence
The B16F10 and their derived cells were grown on chamber slides and were fixed in 4% paraformaldehyde for 15‐30 minutes at room temperature for immunofluorescence preparation. Cells were washed with PBS and permeabilized with 0.1% Triton X‐100, and after 5% BSA blocked for 40 minutes at room temperature. Slides were then incubated with anti‐Oct4 (Abcam, Cambridge, UK) (diluted 1:600), anti‐Nanog (Santa Cruz Biotechnology, Dallas, TX, USA) (diluted 1:100) and anti‐Sox2 (Abcam) (diluted 1:100) overnight, at 4°C washed in PBS. Appropriate fluorophore labelled secondary antibody was added at a dilution of 1/500 and incubated for 1 hour at room temperature, and after washing in PBS. Cells were mounted in Vectashield with DAPI (Vector Labs, Burlingame, CA, USA) to reveal nuclear DNA. Immunofluorescence was visualized in a Nikon Eclipse Ni (Tokyo, Japan) microscope.
2.6. RNA isolation and PCR
Total RNA was extracted from one well of 50‐70% confluent six‐well plate containing established reprogrammed clones, using the Qiagen RNeasy mini kit following the manufacturer's instructions. Synthesize cDNA with the ImProm‐II Reverse Transcription System Kit (Promega, Fitchburg, WI, USA). PCR amplification was performed using GoTaq Green Master Mix (Promega). Primers used in RT‐PCR are listed in Table 1. PCR reactions were performed by initially denaturing cDNA at 95°C for 5 minutes followed by 30 cycles of denaturing at 95°C for 30 seconds, annealing at 58‐62°C for 1 minute, extension at 72°C for 1 minute, and a final 10 minutes extension at 72°C. PCR products were loaded into 1.2% agarose gels containing 0.6 lg/mL ethidium bromide and run in Tris‐acetate‐ethylenediaminetetraacetic acid buffer. The Uvitec 2.0 (Cambridge, UK) gel documentation station was used to observe PCR products.
Table 1.
Primers details
| Primer name | Sequence (5′ to 3′) | Sense | Fragment size (pb) | Tm (°C) |
|---|---|---|---|---|
| Oct4 | CCTGGGCGTTCTCTTTGGAA | F | 123 | 57.6 |
| GCTTCCTCCACCCACTTCTC | R | 57.7 | ||
| Nanog | TGGAAGCCACTAGGAAAGC | F | 115 | 57.2 |
| GCCCAGATGTTGCGTAAGTC | R | 56.3 | ||
| Sox2 | TTTGTCCGAGACCGAGAAGC | F | 146 | 57.1 |
| CTCCGGGAAGCGTGTACTTA | R | 56.4 | ||
| β‐actin | GCTCCGGCATGTGCAAAG | F | 114 | 59.8 |
| CCTTCTGACCCATTCCCACC | R | 60.0 |
2.7. Tumour formation and histological analysis
The cells were harvested by triple (Invitrogen) treatment, collected into tubes, and centrifuged, and the pellets were suspended in RPMI, and 5 × 105 cells was injected subcutaneously to dorsal flank of a C57BL/6J mice (Charles River). Twenty days after the injection, tumours were surgically dissected from the mice. Samples were weighed, fixed in PBS containing 10% formaldehyde, and embedded in paraffin. Sections were stained with haematoxylin and eosin. This procedure was approved by Butantan Institute Ethics Committee for Use of Animal Experimentation (CEP 250/06).
3. RESULTS
3.1. Transfection assay
We used the minicircles DNA containing GFP in order to induce the pluripotency in B16F10 cells. In the next day after the transfection, multiple cells already showed GFP expression, confirming the presence and expression of minicircles (Figure 1A‐A2). At day third, small juxtaposed colonies GFP+ can be observed (Figure 1B‐B2). After 4 days, GFP+ cells were harvested by trypsinization and plated on Matrigel. These small colonies grew rapidly, achieving iPSC‐like morphology (Figure 1C‐C2). These reprogrammed cells denominated MMRCs demonstrated GFP+ expression in the nucleus and cytoplasm (Figure 1D‐D2). Accordingly, B16F10 control cells were GFP−, once they received only lipofectamine without minicircles DNA (Figure 1E,E1). Additionally, murine immortalized fibroblasts (3T3) and mouse embryonic fibroblasts (MEF) were used as controls in transfection assay and both of them demonstrated very limited transfection capacity (Figure 1F,F1, G,G1) compared to B16F10 cells (Figure 1A‐A2). Eighteen hours after addition of non‐viral vector in B16F10 cells, they present the highest number of GFP+ cells (~80%) when compared with 3T3 (~10%) and MEF (~1%) that received the same vector (Figure 1H).
Figure 1.
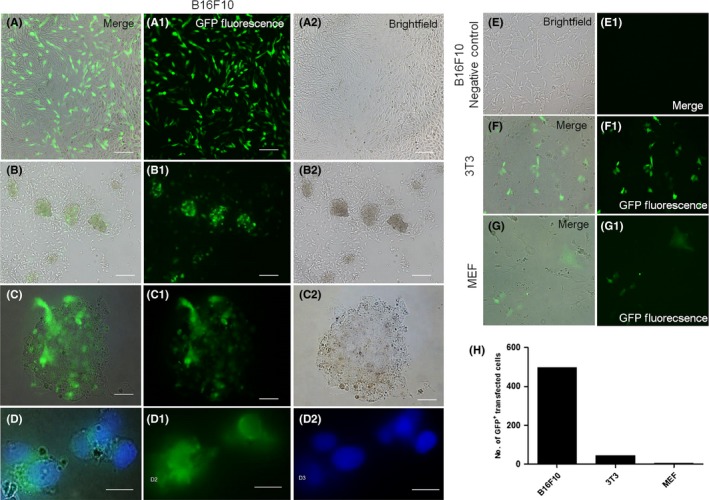
Transfection assay. Light microscopy and fluorescent imaging of the B16F10 cell line transfection procedures. A‐A2, GFP + cells highlighted in green fluorescence 18 h after the transfection. B‐B2, It is possible to see GFP + colonies formation 72 h after de transfection. C‐C2, iPSC‐like colony morphology, in which not all cells are GFP +. D‐D2, GFP + in the cell nucleus. E‐E1, Negative control of B16F10 for GFP −. Comparison of transfection efficiency between the B16F10 cells and fibroblasts (F and G) 18 h after the transfection, showing the GFP + cells. In (H), a comparative bar graph of the number of GFP + transfected cells from the B16F10, 3T3 and MEF, calculated by Wimasis Software
After a few passages, the MMRCs green fluorescence gradually disappeared, suggesting minicircles loss.
Approximately, one week after MMRCs were plated on Matrigel, different cells and colonies showing pluripotent‐like cells morphologies appeared (Figure 2A). It is of knowledge that stem cells divide asymmetrically, thus producing two daughter cells with different cellular fates: one is a copy of the original stem cell, while second is a daughter programmed to differentiate into a non‐stem cell fate.26 After reprogramming, B16F10 cells demonstrate both asymmetrical and symmetrical division (Figure 2A1,B,B1).
Figure 2.
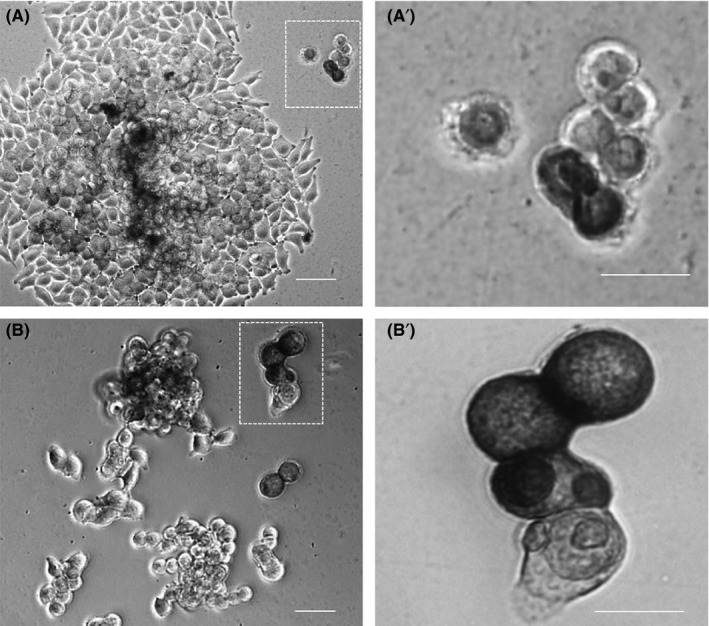
Incompletely reprogrammed cells asymmetric division. Light microscopy image of the different colonies morphology during growing on matrigel plate for 1 wk after transfection in (A) and (B). The zoom in (A′) and (B′) showed the asymmetric division between the cells, demonstrating the heterogeneity generated after reprogramming these cells
3.2. Morphology of MMRC clones
Three colonies of different morphologies were selected for further analysis. The morphology of the parental cell line B16F10 and of three MMRC colonies is presented in Figure 3A‐D. In order to demonstrate cytoskeleton rearrangement, the cells were additionally stained by phalloidin (Figure 3A1‐D1). The Clo1 forms broad colonies (Figure 3B,B1) composed by the cells more similar to parental B16F10 (Figure 3A,A1). The Clo2 and Clo3 form juxtaposed colony (Figure 3C‐D1), resembling to colonies of pluripotent cells, such as of ESC and iPSC. Figure 3A1‐D1 highlight differences in cytoskeleton organization among parental cells and MMRC clones.
Figure 3.
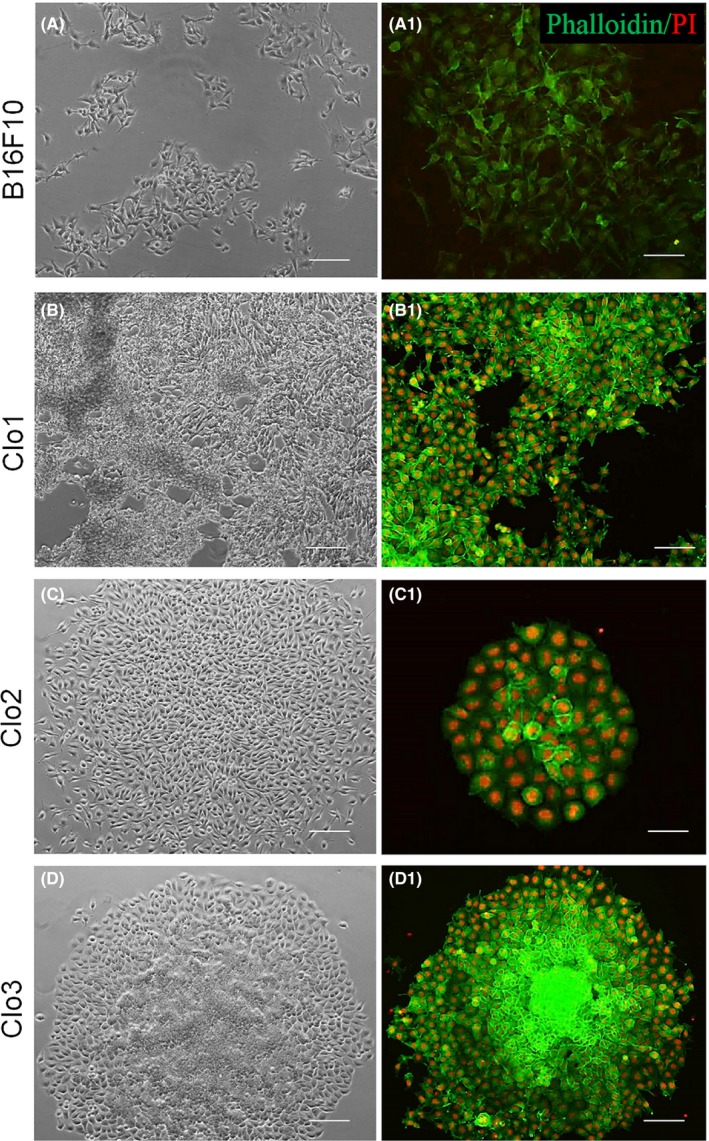
Isolated clones morphology. Light microscopy and fluorescent imaging of the clones, with the parental B16F10 cells in (A‐D). Phalloidin stain highlights differences in the cytoskeleton morphology (A1‐D1). The colonies of Clo1 (B‐B1) are composed by separate cells, resembling parental B16F10 (A‐A1). Meanwhile, the Clo2 (C‐C1) and Clo3 (D‐D1) colonies are juxtaposed, being similar to pluripotent cells colonies
3.3. Expression pluripotent stem cell markers by MMRC clones
After reprogramming, isolated clones showed expression of the three transcription factors (Figure 4A‐C5). However, expression of Oct4, Nanog and Sox2 transcription factors were already observed in a few cells of B16F10 cell line (Figure 4D‐D5) before reprogramming. RT‐PCR analysis of Oct4, Nanog and Sox2 genes confirm their expression in paternal cell line as well as in all three isolated clones (Figure 4E). Murine ESC was used as a positive control (Figure 4E).
Figure 4.
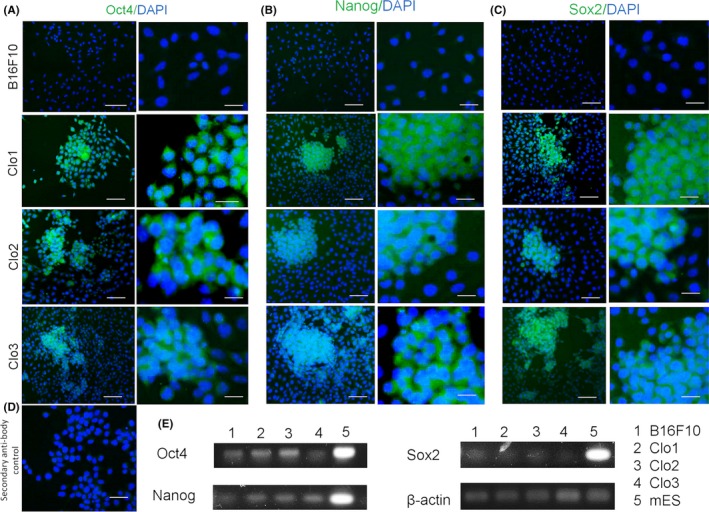
Expression of ESC‐markers in MMRC clones. ESC markers via immunofluorescence in B16F10 cells and MMRC clones. Reprogrammed cells are positive for ESC markers: Oct4 (A), Nanog (B) and Sox2 (C). (D) Secondary antibody negative control. (E) RT‐PCR analysis of ESC‐marker genes in B16F10 cell line, MMRC clones and mES positive control. Primers used for Oct3/4, Sox2, Klf4 and c‐Myc specifically detect the transcripts of the interest genes
3.4. In vivo pluripotency assay
In order to evaluate in vivo reprogramming of MMRC, the B16F10 cells and the clones were subcutaneously transplanted into dorsal flanks of mice. Twenty days after infection, we observed tumour formation. The tumours were then removed and evaluated in respect of their size and cells composition. In Figure 5, tumours formed by three clones and paternal cell line can be observed. Notable, Clo1 produced tumour of intermediate size and Clo2 and Clo3 generate smaller tumours, while B16F10 cells formed bigger size tumour (Figure 5A). Histological analysis of tumours derived from all clones demonstrated less aggressive tumours formation with reduced tissue necrosis and lower cell heterogeneity, when compared with B16F10 (Figure 5B).
Figure 5.
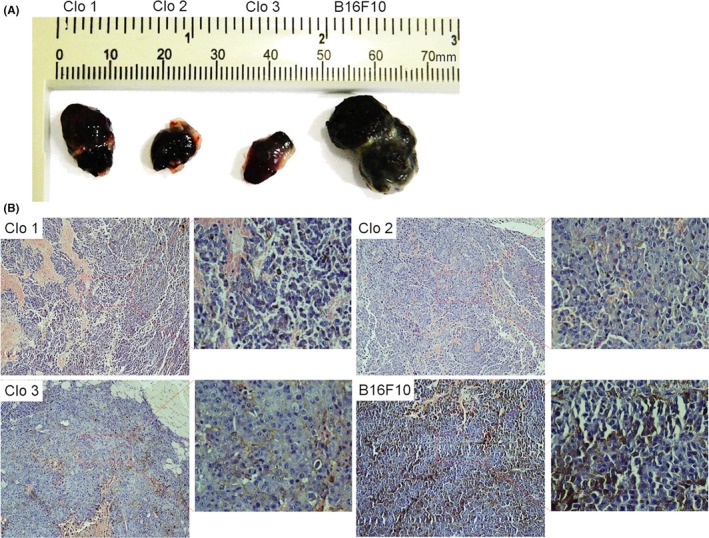
Histological tumour‐derived analysis. The B16F10 cells and the MMRC were subcutaneously transplanted into dorsal flanks of mice (C57Bl6). After twenty days, the animals were euthanized and the tumours were collected at the same day. In (A) comparison of the size tumours derived from MMRC and B16F10 parental cell line. (B) Haematoxylin and eosin staining of tumour derived from MMRC clones and B16F10 cell line. Compared to B16F10, the MMRC tumour‐derived histological analysis suggests lower tumourigenicity, reduced tissue necrosis and decreased cell heterogeneity
3.5. Cell cycle of B16F10 and MMRC
Cell cycle of parental cell line and isolated clones was investigated in order to understand in vivo suppression of tumourigenic abilities of reprogrammed B16F10 cells. Parental cell line cell cycle demonstrated expressive accumulation of the cells in S phase (Figure 6A,F), while MMRC clones showed a high number of cells in G1 phase and significant reduction in cell number in S phase (Figure 6B‐D,F). We also verified whether changes observed in cell cycle occurred in result of reprogramming or clonal selection. Rapidly dividing murine ESC that present great cell number in S phase27 were used as a control (Figure 6E,F).
Figure 6.
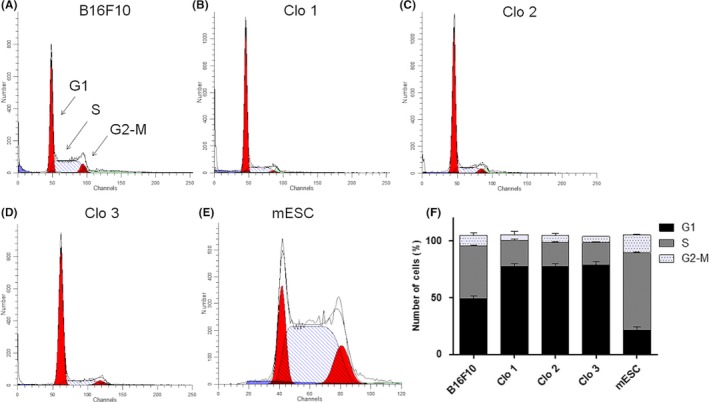
Cell cycle analysis. (A‐E) Histograms of cell cycle of control cells and MMRC, F representative graph for comparison of the cell cycle phases of MMRC and respective controls. B16F10 parental cell line showed accumulation of cells in S phase (A, F), while MMRC demonstrated a high number of cells in G1 phase, besides the reduction of cell number in S phase (B‐D, F). mESC were used as control and shows high number of cells in S phase, due to rapidly cell division (E, F)
4. DISCUSSION
During reprogramming, cells increase their intolerance to different types of DNA damage,28 that may occur due to different reasons, including viral integration. Therefore, we supposed that the use of less invasive non‐viral vector would help to produce more viable cells, thus increasing reprogramming efficiency. This method was successfully used previously for human adipose tissue stem cells reprogramming and for cancer cells.29, 30 Using non‐viral vector, we reprogrammed highly heterogeneous population of melanoma cells into less aggressive Murine Melanoma Reprogrammed Cancer Cells (MMRCC). In fact, eighteen hours after reprogramming, the majority of melanoma cells were alive and 80% of these cells expressed GFP gene reporter. However, overtime the loss of GFP expression was observed, thus indicating loss of minicircles.
Several basic approaches are commonly used to confirm the reprogramming of differentiated cells into less differentiated state, which include: cells morphological changes, expression of pluripotent stem cells markers and teratomas or chimeras formation.31, 32 We showed that MMRCC clones present morphology similar to iPSC and express pluripotent stem cell markers. It is noteworthy that ESC shows a high level of pluripotent markers expression.33, 34, 35 However, this is not a rule for iPSC, because several studies demonstrated that reprogrammed somatic cells, as cancer cells, showed variable and even lower level of pluripotency markers expression, when compared to pluripotent cells. In our study, we also observed that expression of pluripotent stem cell markers in MMRCC was lower than in ESC. Although, endogenous expression levels of pluripotent genes could be relevant to tumour cell malignancy and malignant transformation,36 it is not clear whether these gene products would be translated into functional proteins.
However, neither morphology nor expressions of pluripotent stem cell markers are trustable to conclude whether the cells were in fact reprogrammed. Therefore, all three isolated clones, as well as the cells of B16F10 line, were injected subcutaneously into mice dorsal flanks. We did not observe teratomas formation, which is a proof of concept of complete reprogramming. In contrast, MMRCC formed tumours, however, these tumours showed significantly smaller size, when compared with tumours formed by B16F10 cell line. Histological analysis of tumours derived from MMRCC clones demonstrated less necrosis and less tumour cells phenotypic heterogeneity than paternal B16F10 line. Therefore, although the reprogramming was incomplete it leads to less aggressive tumours formation.
In normal cells, cell cycle control is regulated by a complex series of signalling pathways that also include mechanisms that correction DNA damages. In cancer cell, this regulatory process is defective and results in uncontrolled cell proliferation.37, 38, 39 Therefore, we analysed cell cycles of MMRCC clones, of parental cell line and of clones derived from parental cell line. Accumulation of the cells in S phase occurs due to more active cells proliferation or their arrest (eg, because of DNA damage) in the middle of replicating their DNA.40, 41 After reprogramming, the three MMRCC clones showed significantly decrease in number of cells in S‐phase, compared to cancer B16F10 cell line. It seems that reprogramming may reduce cancer cell proliferation that in turn may lead to tumour formation of smaller size as compared with B16F10 cells. Some studies demonstrated in their analysis that aggressive types of tumours contain higher percentages of cells in S‐phase, while the less aggressive ones have lower percentage of cells in this phase.42, 43
Recent reports have identified asymmetric cell division in various cancers that were characterized by the presence of a subpopulation of cells that share some stem cell‐like properties (CSC), which shows a negative correlation between the frequency of asymmetric division and their proliferative capacity. Based on this, highly proliferative CSC performs more symmetric division than asymmetric.44, 45, 46 Although we did not perform statistic evaluation of symmetric than asymmetric divisions in parental and reprogrammed cells, asymmetric division was mainly observed in reprogrammed clones, which suggest more immature state of these cells.
Our reprogramming was unstable, compared to Zhao et al. (2015),30 which achieved a complete murine melanoma reprogramming, corroborating with studies that include an inefficient and unstable reprogramming of tumour cells.47 The multistep repeated transfections Zhao's protocol, followed by longer time and high cell density generated stable C‐iPSC, the same way as Kaji et al. (2009)48 developed a protocol to induce normal somatic adult cells. Our data, otherwise, open new perspectives to study heterogeneity and asymmetric division of tumour cells. They suggest a new intermediate point in the reprogramming process, which can serve as base to future studies of the cancer biology, the association between pluripotency and tumour cells.
The majority of published works did not mention stability of cancer cells reprogramming. Choong and co‐workers (2014)23 reported that during long‐term in vitro culturing, these cells might regress in reprogramming. We also noted the loss of reprogramming in isolated clones over time. We believe that during long‐term culture in vitro, asymmetric division may contribute to heterogeneity among cancer cells, thus inducing regression of reprogramming.
AUTHORSHIP
DC, AP: data acquisition, analysis, and interpretation, writing the first draft of manuscript, figure and table production, references organization and manuscript revision. AC, RA: contributed with reagents, materials, analysis and participation in the manuscript organization. IK: research design, analysis, and interpretation of data, writing of the manuscript, figure production, and critical revision for important intellectual content.
COMPETING INTERESTS
The authors declare that they have no competing interests.
ACKNOWLEDGEMENTS
This work was supported by FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) grant number: 2010/51051‐6 and CAPES, Brazil. We acknowledged to Dr. Nelson Foresto Lizier and Dr. Paulo de Sá Júnior for technical support.
Câmara DAD, Porcacchia AS, Costa AS, Azevedo RA, Kerkis I. Murine melanoma cells incomplete reprogramming using non‐viral vector. Cell Prolif. 2017;50:e12352 10.1111/cpr.12352
Contributor Information
D.A.D. Câmara, Email: diana.adc@gmail.com
I. Kerkis, Email: irina.kerkis@butantan.gov.br.
REFERENCES
- 1. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789‐799. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 3. Muñoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol. Oncol. 2012;6:620‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem‐like states, and drug resistance. Mol Cell. 2014;54:716‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Diaz‐Cano SJ. Tumor heterogeneity: mechanisms and bases for a reliable application of molecular marker design. Int J Mol Sci. 2012;13:1951‐2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miyoshi N, Ishii H, Nagai K, et al. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci USA. 2010;107:40‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim J, Zaret KS. Reprogramming of human cancer cells to pluripotency for models of cancer progression. EMBO J. 2015;34:739‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang X, Cruz FD, Terry M, Remotti F, Matushansky I. Terminal differentiation and loss of tumorigenicity of human cancers via pluripotency‐based reprogramming. Oncogene. 2013;32(2249–60):2260‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663‐676. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861‐872. [DOI] [PubMed] [Google Scholar]
- 11. Kim JJ. Applications of iPSCs in cancer research. Biomark Insights. 2015;10:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Curry EL, Moad M, Robson CN, Heer R. Using induced pluripotent stem cells as a tool for modelling carcinogenesis. World J Stem Cells. 2015;7:461‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seftor EA, Seftor RE, Weldon DS, et al. Melanoma tumor cell heterogeneity: a molecular approach to study subpopulations expressing the embryonic morphogen nodal. Semin Oncol. 2014;41:259‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zellmer VR, Zhang S. Evolving concepts of tumor heterogeneity. Cell Biosci. 2014;4:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Quintana E, Shackleton M, Foster HR, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;18:510‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schatton T, Murphy GF, Frank NY, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kasai T, Chen L, Mizutani A, et al. Cancer stem cells converted from pluripotent stem cells and the cancerous niche. J Stem Cells Regen Med. 2014;10:2‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brooks MD, Burness ML, Wicha MS. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell. 2015;17:260‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Q, Wennborg A, Aurell E, et al. Dynamics inside the cancer cell attractor reveal cell heterogeneity, limits of stability, and escape. Proc Natl Acad Sci USA. 2016;113:2672‐2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun X‐X, Yu Q. Intra‐tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin. 2015;36:1219‐1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wernig M, Meissner A, Foreman R, et al. In vitro reprogramming of fibroblasts into a pluripotent ES‐cell‐like state. Nature. 2007;448:318‐324. [DOI] [PubMed] [Google Scholar]
- 22. Saha K, Jaenisch R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell. 2009;5:584‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choong PF, Teh HX, Teoh HK, et al. Heterogeneity of osteosarcoma cell lines led to variable responses in reprogramming. Int J Med Sci. 2014;11:1154‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jia F, Wilson KD, Sun N, et al. A nonviral minicircle vector for deriving human iPS cells. Nat Methods. 2010;7:197‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wulf E, Deboben A, Bautz FA, Faulstich H, Wieland T. Fluorescent phallotoxin, a tool for the visualization of cellular actin. Proc Natl Acad Sci USA. 1979;76:4498‐4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morrison SJ, Kimble J. Asymmetric and symmetric stem‐cell divisions in development and cancer. Nature. 2006;441:1068‐1074. [DOI] [PubMed] [Google Scholar]
- 27. Barta T, Dolezalova D, Holubcova Z, Hampl A. Cell cycle regulation in human embryonic stem cells: links to adaptation to cell culture. Exp Biol Med (Maywood). 2013;238:271‐275. [DOI] [PubMed] [Google Scholar]
- 28. Zhang M, Yang C, Liu H, Sun Y. Induced pluripotent stem cells are sensitive to DNA damage. Genomics Proteomics Bioinformatics. 2013;11:320‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Narsinh KH, Jia F, Robbins RC, Kay MA, Longaker MT, Wu JC. Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat Protoc 2011;6:78‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao H, Davies TJ, Ning J, et al. A highly optimized protocol for reprogramming cancer cells to pluripotency using nonviral plasmid vectors. Cell Reprogram. 2015;17:7‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cantz T, Martin U. Induced pluripotent stem cells: characteristics and perspectives. Adv Biochem Eng Biotechnol. 2010;123:107‐126. 10.1007/10. [DOI] [PubMed] [Google Scholar]
- 32. Nordin N, Lai MI, Veerakumarasivam A, Ramasamy R. Induced pluripotent stem cells : history, properties and potential applications. Med J Malaysia. 2011;66:4‐9. [PubMed] [Google Scholar]
- 33. Nichols J, Zevnik B, Anastassiadis K, et al. Formation of pluripotent stem cells in the mammalian embryo dependes on the POU transcription factor Oct4. Cell. 1998;95:379‐391. [DOI] [PubMed] [Google Scholar]
- 34. Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell‐Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17:126‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chambers I, Colby D, Robertson M, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643‐655. [DOI] [PubMed] [Google Scholar]
- 36. Ben‐Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell‐like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297‐308. [DOI] [PubMed] [Google Scholar]
- 38. Maria A, Velez A, Howard MS. Tumor‐suppressor genes, cell cycle regulatory checkpoints, and the skin. N Am J Med Sci. 2015;7:176‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mjelle R, Hegre SA, Aas PA, et al. Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair (Amst). 2015;30:53‐67. [DOI] [PubMed] [Google Scholar]
- 40. Ho A, Dowdy SF. Regulation of G1 cell‐cycle progression by oncogenes and tumor suppressor genes. Curr Opin Genet Dev. 2002;12:47‐52. [DOI] [PubMed] [Google Scholar]
- 41. Vermeulen K, Berneman ZN, Van Bockstaele DR. Cell cycle and apoptosis. Cell Prolif. 2003;36:165‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Powell L, Harty‐Golder B. Percentage of cells in the S phase of the cell cycle in human lymphoma determined by flow cytometry. Cytometry. 1980;1:171‐174. [DOI] [PubMed] [Google Scholar]
- 43. Knudsen KE, Booth D, Naderi S, et al. RB‐dependent S‐phase response to DNA damage. Mol Cell Biol. 2000;20:7751‐7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bu P, Chen KY, Chen JH, et al. A microRNA miR‐34a‐regulated bimodal switch targets notch in colon cancer stem cells. Cell Stem Cell. 2013;12:602‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bu P, Chen K, Lipkin SM, Shen X. Asymmetric division: a marker for cancer stem cells? Oncotarget. 2013;4:4‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bajaj J, Zimdahl B, Reya T. Fearful symmetry: Subversion of asymmetric division in cancer development and progression. Cancer Res. 2015;75:792‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ramos‐Mejia V, Fraga MF, Menendez P. IPSCs from cancer cells: challenges and opportunities. Trends Mol Med. 2012;18:245‐247. [DOI] [PubMed] [Google Scholar]
- 48. Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjem K. Virus‐free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]