

Abstract
Objectives
To reveal whether B‐myb is involved in preventing senescence of vascular endothelial cells, and if so, to identify possible mechanisms for it.
Materials and methods
C57/BL6 male mice and primary human aortic endothelial cells (HAECs) were used. Bleomycin was applied to induce stress‐related premature senescence. B‐myb knockdown was achieved using an siRNA technique and cell senescence was assessed using the senescence‐associated β‐galactosidase (SA‐β‐gal) assay. Intracellular reactive oxygen species (ROS) production was analysed using an ROS assay kit and cell proliferation was evaluated using KFluor488 EdU kit. Capillary tube network formation was determined by Matrigel assay. Expressions of mRNA and protein levels were detected by real‐time PCR and western blotting.
Results
B‐myb expression significantly decreased, while p53 and p21 expressions increased in the aortas of aged mice. This expression pattern was also found in replicative senescent HAECs and senescent HAECs induced by bleomycin. B‐myb knockdown resulted in upregulation of p22phox, ROS accumulation and cell senescence of HAECs. Downregulation of B‐myb significantly inhibited cell proliferation and capillary tube network formation and activated the p53/p21 signalling pathway. Blocking ROS production or inhibiting p53 activation remarkably attenuated SA‐β‐gal activity and delayed cell senescence induced by B‐myb‐silencing.
Conclusion
Downregulation of B‐myb induced senescence by upregulation of p22phox and activation of the ROS/p53/p21 pathway, in our vascular endothelial cells, suggesting that B‐myb may be a novel candidate for regulating cell senescence to protect against endothelial senescence‐related cardiovascular diseases.
1. Introduction
Cellular senescence is a state of stable cell cycle arrest in response to diverse stresses.1 It can be caused by various factors and can be classified into replicative senescence and stress‐induced premature senescence according to the type of stress.2 Replicative senescence is induced by extended cell replication and mediated through the shortening of telomeres.3, 4 However, stress‐induced premature senescence is induced by DNA damage,5 oxidative stress6 and oncogene activation,7 which is independent of telomeres. Cellular senescence is considered an essential contributor to the ageing process. Senescent cells can secret certain inflammatory cytokines and change its microenvironment to induce senescence their neighbour cells via gap junction‐mediated cell‐cell contact.8 Inhibition of proliferative ability in senescent cells can further impact tissue repair and reduce organ functions.
Senescent cells exhibit phenotypic alterations that include enlarged and flattened morphology,9 as well as positive staining for senescence‐associated β‐galactosidase (SA‐β‐gal) activity. SA‐β‐gal is a widely used marker of senescence in both cells and tissues.10 In addition, certain proteins have been identified as markers of cellular senescence, including p53, p21, p16, pRb and cyclin D1.9, 11, 12 The p53 pathway is a crucial mediator of cellular senescence response to many stimuli in normal somatic cells.13 The stressors, from exogenous and endogenous sources of the cells, engage various cellular signalling cascades and activate p53.14 The activated p53 can activate p21, which is an important cell cycle inhibitor.15, 16 Inactivation of p53 can reverse the senescent growth arrest.17 Although reactive oxygen species (ROS) are normal products of cellular metabolism, excessive accumulation of ROS can provoke oxidative damage of diverse cellular macromolecules, such as DNA, RNA, and proteins, and thereby accelerate cellular senescence.18 It has been reported that excessive ROS production can suppress the transcription of genes involved in cellular growth and mitochondrial functions19 and induce the upregulation of p53 and p21.20 ROS generation is controlled by NADPH oxidases that comprise a cytochrome b558 component consisting of gp91phox and p22phox embedded in membranes. The p22phox catalytic unit is an essential component of NADPH oxidases that stabilize the large subunit providing a docking for the cytosolic factors.21
B‐myb is a member of the MYB family of transcription factors and is broadly expressed in all proliferating cells.22 Accumulating evidence implicates that B‐myb plays an essential role in cell division, cell cycle progression, cell development, DNA replication and maintenance of genomic integrity.23, 24 It has been reported that B‐myb expression is required for cell entry into S‐phase and can overcome growth inhibitory signals.25 B‐myb not only promotes S‐phase through interacting with polymerase delta‐interacting protein 1 during cell cycle progression26 but also promotes G2/M‐phase by the activation of a large number of genes including PLK1, Aurora A, Cyclin A and CyclinB1/2.27 It has recently emerged that B‐myb acts as a potential candidate molecule for regulating cell entry into senescence. On one hand, B‐myb deficient can induce cellular senescence in primary fibroblasts28, 29, 30; on the other hand, overexpression of B‐myb can reverse cellular premature senescence in primary mouse embryonic fibroblasts.31 High levels of B‐myb expression can bypass p53‐induced G1 arrest.32 Although more and more evidences have been discovered, till now, the molecular mechanisms underlying cellular senescence are complicated and still obscure.
Vascular endothelial cells are important to form an endothelial monolayer between circulating blood and the rest of the vascular wall. In addition to its important role as the barrier between the circulating blood and underlying tissues, the endothelium is a key regulator of cardiovascular homeostasis and provides protection against vascular diseases.33 Endothelial cell senescence can lead to endothelial dysfunction which is an independent risk factor for the development of hypertension and atherosclerosis.34 However, the mechanism is unclear, especially with regard to whether and how B‐myb is involved in vascular endothelial cell senescence.
This study demonstrates the effects and possible mechanisms of B‐myb involved in the context of cellular senescence. The results indicate that B‐myb expression is decreased not only in the aortas of aged mice but also in replicative senescent endothelial cells, even in stress‐senescent endothelial cells induced by bleomycin. More interestingly, the downregulation of B‐myb induced cellular senescence via the upregulation of p22phox to activate the ROS/p53/p21 pathway. Taken together, the findings indicate B‐myb has an essential role in regulating senescence of vascular endothelial cells.
2. Materials and methods
2.1. Materials
Bleomycin and p53 inhibitor Pifithrin‐α (PFTα) were obtained from Selleck Chemicals (Houston, TX, USA). N‐acetyl‐l‐cysteine (NAC) was purchased from Enzo (Farmingdale, NY, USA). SA‐β‐gal assay kit and ROS assay kit were obtained from Beyotime (Haimen, China). KFluor488 EdU kit for detection cell proliferation was purchased from KeyGEN (Nanjing, China). Matrigel was obtained from Becton Dickinson Biosciences (Bedford, MA, USA). Antibody for detecting B‐myb was purchased from Merck Millipore (Darmstadt, Germany). Antibodies for detecting p21, p53 and GAPDH were obtained from Proteintech (Chicago, IL, USA). Anti‐phospho‐Histone H2AX (Ser139) and anti‐phosphorylated p53 (Ser15) antibody were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies for detecting cyclin D1, p16, anti‐phospho‐Rb (pRb) and p22phox were purchased from Abcam (Cambridge, UK). The goat anti‐rabbit conjugated‐HRP secondary antibody was obtained from Jackson (West Grove, PA, USA). Alexa Fluor 594‐conjugated goat anti‐rabbit IgG antibody was purchased from Invitrogen (Grand Island, NY, USA).
2.2. Animal and cell culture
C57/BL6 male mice were obtained from the Central Animal Facility of Tongji University (Shanghai, China). Two groups of mice were evaluated: young group (aged at 3 months) and agedness group (aged at 24 months). Mice were anesthetized with sodium pentobarbitone (40 mg/kg; ip) and killed by cervical dislocation. The aortas were harvested and cleaned of surrounding tissue in ice‐cold PBS. All studies involving mice were carried out after the approval from the Ethics Committee for Animal Experimentation. Primary cultured human aortic endothelial cells (HAECs) were purchased from PriCells (Wuhan, China) and maintained in endothelial cell medium containing 1% endothelial cell growth supplement (ScienCell, USA) with 5% foetal bovine serum at 37°C in an atmosphere of 5% CO2 and cultured until the experiment.
To induce replicative senescence, HAECs were maintained under subconfluent conditions at all times and passaged every 4‐5 days. Cell numbers were counted when seeding and harvesting from each passage for calculating population doubling (PDL) as described35: PDL=(log10 F−log10 I)/0.301, where F is the number of cells at the end of 1 passage and I is the number of cells that were seeded at the beginning of the passage. All experiments were carried out with in PDL20, otherwise be indicated in experiment.
2.3. Determination of SA‐β‐gal activity
The degree of senescence in cultured HAECs was evaluated by quantifying the activity of SA‐β‐gal by using the SA‐β‐gal assay kit following the instruction book. Briefly, cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature and then incubated with SA‐β‐gal staining solution for 12‐16 hours at 37°C (no CO2). The percentage of SA‐β‐gal‐positive cells was calculated by counting the cells in five random fields under microscope as previously described.10
2.4. RNA isolation and quantitative PCR
Total RNA was extracted from cells or tissue samples with RNAiso Reagent (Takara, Dalian, China) according to the manufacturer's instructions. cDNA was synthesized with PrimeScript® RT Master Mix (Perfect Real Time) Kit (Takara). Real‐time PCR was performed with SYBR Premix Ex Taq II (Tli RNaseH Plus) Kit (Takara) in the ABI Prism 7500 Detection System (Applied Biosystems, Foster City, CA, USA). The primer sense and antisense sequences are as follows: 5′‐CGC AAG TCT CTG GCT CTC‐3′ and 5′‐GAC ACT GGT TGG CAA GGA‐3′ for mouse B‐myb (NM_008652.2); 5′‐TGG AAG ACA GGC AGA CTT‐3′ and 5′‐ACT TGT AGT GGA TGG TGG TA‐3′ for mouse p53 (NM_011640.3); 5′‐TGT CCA ATC CTG GTG ATG T‐3′ and 5′‐CAA CTG CTC ACT GTC CAC‐3′ for mouse p21 (NM_007669.4); 5′‐GCC CAT CAC CAT CTT CCA‐3′ and 5′‐GTA GAC TCC ACG ACA TAC TCA G‐3′ for mouse GAPDH (XM_011241212.1); 5′‐CCG AGA AGC AGA AGA GGA A‐3′ and 5′‐ACA ATG TCA AGA GCC AGA GA‐3′ for human B‐myb (NM_001278610.1); 5′‐GCG TGT GGA GTA TTT GGA TGA C‐3′ and 5′‐ATG TAG TTG TAG TGG ATG GTG GTA‐3′ for human p53 (NM_000546.5); 5′‐GAA GTG AGC ACA GCC TAG‐3′ and 5′‐TGC CTT CAC AAG ACA GAG‐3′ for human p21 (NM_000389.4); 5′‐CAT CAA GAA GGT GGT GAA‐3′ and 5′‐TGT TGA AGT CAG AGG AGA‐3′ for human GAPDH (NM_002046.5). The cycle number represents the relative quantity of the specific template when the fluorescence of the amplified gene product first reached a preset threshold (Ct). The gene relative expression levels were calculated by the double delta Ct method36 and normalized to the internal control GAPDH.
2.5. Protein extraction and immunoblotting
For total protein extraction, samples were lysed in RIPA buffer (Beyotime, Haimen, China) and centrifuged for soluble proteins. The extracted protein was separated by SDS‐PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). Blots were blocked with TBS‐T buffer containing 5% BSA for 60 minutes at room temperature and then incubated for an additional 18 hour at 4°C with the primary antibody indicated in the experiments. Washed with TBS‐T, the blots were then incubated with associated secondary antibody for 60 minutes at room temperature. Finally, antibody‐bound proteins were detected using the ECL system (Millipore).
2.6. Immunohistochemical staining
Sections with 5 μm thick, from formalin‐fixed mouse aorta embedded in paraffin, were processed for immunohistochemistry staining as previously described.37 Briefly, sections were blocked in 3% H2O2 for 10 minutes at room temperature to remove endogenous peroxidases and then incubated in TBS with 0.1% Triton X‐100 plus 10% normal goat serum at 37°C for 60 minutes. Sections were incubated with a mouse monoclonal anti‐p16 antibody for overnight after antigen retrieval, and then the slides were incubated with secondary antibody conjugated to biotin for 30 minutes. After rinsing, the sections were incubated with avidin‐biotinylated horseradish peroxidase, stained with DAB (diaminobenzidine tetrahydrochloride) substrate and counterstained with haematoxylin. The images of brown positive nuclei were directly observed under a Nikon microscope.
2.7. Immunofluorescence
DNA damaged foci were examined by using immunofluorescence microscopy essentially as previously described.38 Cells grown on glass coverslips were washed with PBS twice and fixed with 4% paraformaldehyde for 15 minutes after the cells' treatment. Then, the cells were permeated with 0.3% Triton X‐100 for 20 minutes and blocked with 5% BSA for 60 min. The primary antibody was applied for overnight at 4°C following incubating with fluorescent‐labelled antibody for 60 minutes at room temperature. The cells were then counterstained with DAPI. The positive fluorescent signals were directly observed under an inverted fluorescence microscope.
2.8. RNA interfering experiments
Small interfering RNAs for B‐myb, p22phox and the negative control were designed and synthesized by Invitrogen. HAECs were transfected with 50 nM siRNAs for B‐myb (siB‐myb) or negative control (siNC), respectively, by using HiPerFect transfection reagent (Qiagen, Hilden, Germany) according to the manufacturer's protocol. The sequences of siRNA targeting B‐myb (NM_002466.3) are as follows: 5′‐CAC CAG AAA CGA GCC UGC CUU ACA A‐3′ and 5′‐UUG UAA GGC AGG CUC GUU UCU GGU G‐3′. The sequences of siRNA targeting p22phox (NM_000101.3) are as follows: 5′‐GCU GUU CGG GCC CUU UAC CAG GAA U‐3′ and 5′‐AUU CCU GGU AAA GGG CCC GAA CAG C‐3′. The RNA interfering efficiency was analysed by real‐time PCR and western blotting. Then, the siRNA transfected HAECs were further used depending on the experiment design.
2.9. Reactive oxygen species measurements
Detection of intracellular ROS production was performed by using ROS assay kit according to the manufacturer's instructions (Beyotime, Haimen, China). Briefly, after treatment, the cells were incubated with 10 μmol/L DCFH‐DA probes for 30 minutes at 37°C with 5% CO2. The positive fluorescent signals images were directly captured with an inverted fluorescence microscope. The percentage of DCFH‐DA‐positive cells was calculated by counting the cells in three random fields.
2.10. Cell proliferation assay
Cell proliferation assay was performed as described in the specifications of KFluor488 EdU kit (KeyGEN Biology, Nanjing, China). Briefly, HAECs were grown on glass coverslips and treated with 30 μmol/L EdU for 3 hours. The cells were then fixed in 4% paraformaldehyde for 10 minutes followed by permeating with 0.5% Triton X‐100 for 20 minutes. Cellular nuclear was stained with DAPI. The positive fluorescent signals were directly observed under an inverted fluorescence microscope. The percentage of EdU‐labelled cells was determined by counting at least 400 nuclei in five random selected fields per slide.
2.11. Capillary tube network formation assay
Human aortic endothelial cells were transfected with control siRNA or B‐myb siRNA for 7 days. After transfection, the cells were seeded on top of the Matrigel‐coated wells and incubated for 6 hours at 37°C as described with modifications.39 Then, cells were fixed with 4% paraformaldehyde and stained with crystal violet. Tubule formation was viewed under a Nikon inverted microscope. Images were captured with NIS Elements software, and the number of formatted tubes was manually counted in five random selected fields per well.
2.12. Statistical analysis
All study procedures were repeated at least three times. The data were expressed as mean ± SEM. Statistical comparisons between different groups were performed by paired Student's t test. A P value <.05 was considered statistically significant.
3. Results
3.1. B‐myb was downregulated in the aortas of aged mouse and replicative senescent HAECs
An increasing boy of evidence indicates that B‐myb plays an essential role in attenuating senescence. It has been reported that low level of B‐myb expression was found in aged liver tissue,40 senescent HeLa28 and embryonic fibroblasts cells.29, 41 However, B‐myb expression pattern and its role in the ageing vascular system remain unclear. To address this question, first, the expression mRNA and protein levels of B‐myb in aortas from aged mice (24 months) and young control (3 months) groups were detected. It clearly shows that both mRNA and protein levels of B‐myb were dramatically reduced, while the age‐related markers of p53, p21, p16 and cyclin D1 were significantly increased and pRb was significantly decreased in aged group compared with young group (Figure 1A,B). Furthermore, immunostaining results show that the p16‐positive signal in aortic cell nuclei was significantly increased in the aged aortas. From Figure 1C, it is clearly shown in aged samples that p16‐positive nuclei were observed not only in vascular smooth muscle cells but also found in endothelial cells. As we known, vascular endothelial cell is a key regulator of cardiovascular homeostasis. To clarify the expression pattern of B‐myb in senescent endothelial cell in vitro, the expression level of B‐myb was monitored during replicative senescence in primary HAECs. The SA‐β‐gal staining assay was used for identifying replicative senescence during PDL raise. As shown in Figure 1D, the rate of senescent cells was augmented with the increasing PDL number. Meanwhile, the expression mRNA and protein levels of B‐myb were significantly dropped, while p53, p21 and p16 were remarkably increased, and pRb was diminished at high PDLs (Figure 1E,F). These results indicate that B‐myb was downregulated in the aged aortas and replicative senescent HAECs.
Figure 1.
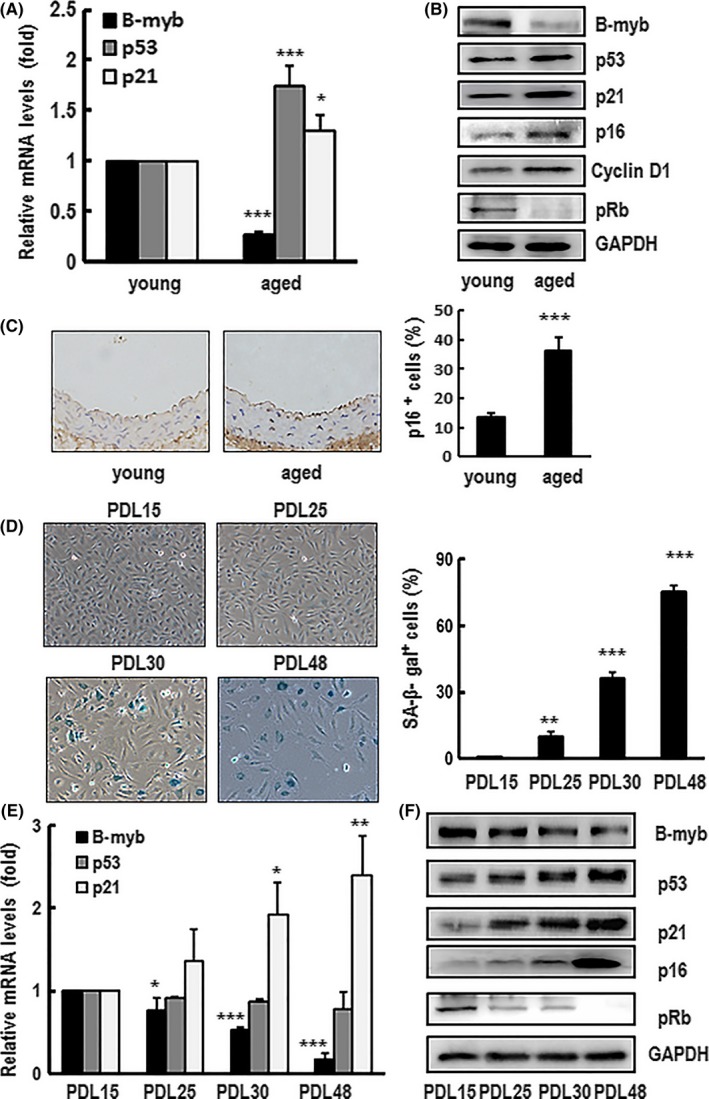
B‐myb was downregulated in the aortas of aged mice and human aortic endothelial cells (HAECs) undergoing replicative senescence. (A, B) The mRNA and/or protein expression levels of B‐myb, p53, p21, p16, pRb and cyclin D1 in the aortas of young (3 months) and aged (24 months) mice groups were analysed by real‐time PCR and western blotting. Each relative mRNA value was justified to the housekeeping gene GAPDH before normalization with that of the controls. Data are presented as mean ± SEM of three independent experiments. ∗ and ∗∗∗ indicate P<.05 and P<.001, respectively, between the two groups. A typical group of blots is shown and similar results were obtained in three separate experiments. GAPDH was used as a loading control. (C) Representative image of immunohistochemistry analysis performed with p16 antibody. A typical group of immunostaining is shown and similar results were obtained from young and aged groups. The percentage of senescent cells marked with p16 was calculated. Data are presented as mean ± SEM of three different samples. *** indicates P<.001 between the two groups. (D) Primary cultured HAECs at 15, 25, 30 and 48 population doubling (PDL) were stained by senescence‐associated β‐galactosidase (SA‐β‐gal). The SA‐β‐gal‐positive cells were directly observed under Nikon inverted microscope, and the representative images were shown (×100 magnification). After analysing SA‐β‐gal‐positive cells, the percentage rate is presented as mean ± SEM of three independent experiments. ** and *** indicate P<.01 and P<.001, respectively, compared with PDL15 cells. (E, F) The mRNA and/or protein expression levels of B‐myb, p53, p21, p16 and pRb in different PDLs cells were detected by real‐time PCR and western blotting. Each relative mRNA value was justified to the housekeeping gene GAPDH before normalization with that of the controls. Data are presented as mean ± SEM of three independent experiments. *, ** and *** indicate P<.05, P<.01and P<.001, respectively, compared with PDL15 cells. A typical group of blots is shown, and similar results were obtained in three separate experiments. GAPDH was used as a loading control
3.2. B‐myb was downregulated in premature senescence cells induced by bleomycin
Besides replicative senescence, cells can also undergo stress‐induced senescence in response to DNA damage, oxidative stress or oncogenic stimuli. A suitable senescent inducer bleomycin was employed to verify whether B‐myb expression level changed in this microenvironment. As known, DNA damage is an important contributor to ageing and causes many cell types to undergo senescence.42, 43 It has been found that H2AX histone becomes rapidly phosphorylated to form γH2AX in response to initial DNA strand breakdown.44 So, γH2AX is often used as an early indicator of DNA damage.45, 46, 47 In this study, γH2AX was used as an indicated marker to confirm whether bleomycin could induce DNA damage in primary HAECs. As shown in Figure 2A, bleomycin could cause γH2AX activation in a dose‐dependent and time‐dependent manner in the cells. Furthermore, γH2AX foci staining also showed that γH2AX positive cells were significantly increased after bleomycin treatment (Figure 2B). These results clearly indicate that bleomycin can result in DNA damage in a dose‐ and time‐dependent response manner in HAECs. To ascertain whether bleomycin could induce primary HAECs senescence, cells were treated with bleomycin for 60 minutes followed by culturing in normal culture medium for a further 4 days. Interestingly, HAECs displayed enlarged size, flattened morphology as premature senescence‐like phenotype. The rate of senescent cells was significantly increased in bleomycin‐treated group compared with the control group (Figure 2C). Meanwhile, the expression levels of p53, p21 and p16 were significantly increased and pRb was dramatically decreased, while B‐myb expression was sharply dropped at mRNA and protein levels compared with the controls (Figure 2D). Taken together, these observations certify that bleomycin treatment not only significantly upregulates p53, p21 and p16 expression but also dramatically downregulates B‐myb and pRb expression in primary HAECs.
Figure 2.
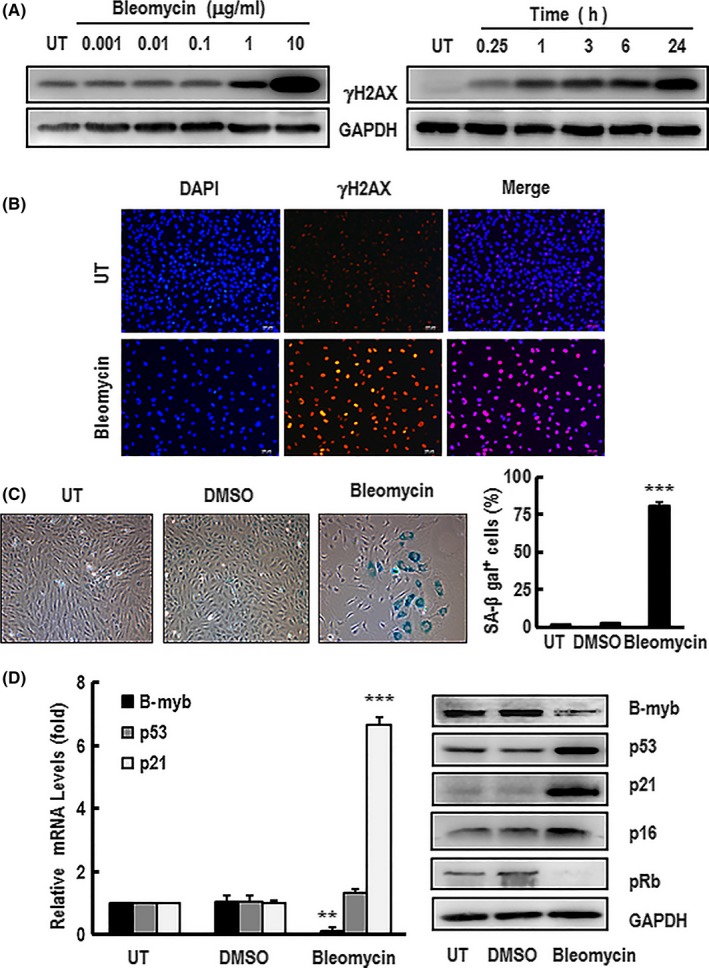
HAEC premature senescence induced by bleomycin influenced the expression of B‐myb, p53, p21, p16 and pRb. A, The dose‐response curves and time course of activation of γH2AX were detected by western blotting after HAECs were treated with bleomycin in the indicated concentrations for 24 h or 10 μg/mL bleomycin for the time period indicated. GAPDH was used as a loading control. These blots were obtained from one of three independent experiments. B, Cells were stimulated with 10 μg/mL bleomycin for 24 h before immunostaining with antibodies against γH2AX (red) and counterstaining with DAPI (blue). A representative group image of stained cells was observed under fluorescence microscope (×100 magnification). HAECs were treated with 10 μg/mL bleomycin for 60 min following replacement of normal culture medium for 4 d. C, The SA‐β‐gal‐positive cells were observed under inverted microscope (×100 magnification) after the treated cells were stained with senescence‐associated β‐galactosidase (SA‐β‐gal). The percentage rate of SA‐β‐gal‐positive cells was analysed. Data are presented as mean ± SEM of three independent experiments. *** indicates P<.001 compared with the control. D, The expression mRNA and/or protein levels of B‐myb, p53, p21, p16 and pRb in the treated cells were detected by real‐time PCR and western blotting, respectively. GAPDH was used for normalization. The mRNA data are presented as mean ± SEM of three independent experiments. ** and *** indicate P<.01 and P<.001, respectively, compared with the control. A typical group of blots for analysing the expression proteins levels of B‐myb, p53 and p21 in the cells is showed from one of three independent experiments. GAPDH was used as a loading control
3.3. Inhibition of B‐myb triggered HAECs senescence and upregulated p53/p21
Above findings distinctly demonstrate that B‐myb is involved in the progression of senescence. It has been reported that the inhibition of B‐myb induces senescence in primary fibroblasts.28, 29, 30 Increasing B‐myb expression can also rescues Ras‐induced premature senescence in primary mouse embryonic fibroblasts.31 Nevertheless, how B‐myb effects in vascular endothelial cell is unknown. To investigate the function of B‐myb in endothelial cell senescence, RNAi technique was employed to knockdown B‐myb gene to investigate the role of B‐myb in HAECs. Primary cultured HAECs, transiently transfected with siB‐myb, could successfully depress B‐myb expression at mRNA and protein levels (Figure 3A). Importantly, the downregulated B‐myb cells showed morphological change including a large‐flat shape, binucleation and polyploidy after 7 days. B‐myb‐silenced cells also displayed an increase in the proportion of SA‐β‐gal‐positive cells compared with the control (Figure 3B). Interestingly, the protein levels of p53 and p21 were significantly increased in B‐myb‐silenced cells compared with that of the controls. However, there was no obvious effect on p16 and pRb expression. These results are consistent with the above animal study that the downregulation of B‐myb expression was negatively correlated with p53 and p21 expression in senescent cells, which confirms that the inhibition of B‐myb can trigger cellular senescence and upregulate p53/p21 in HAECs.
Figure 3.
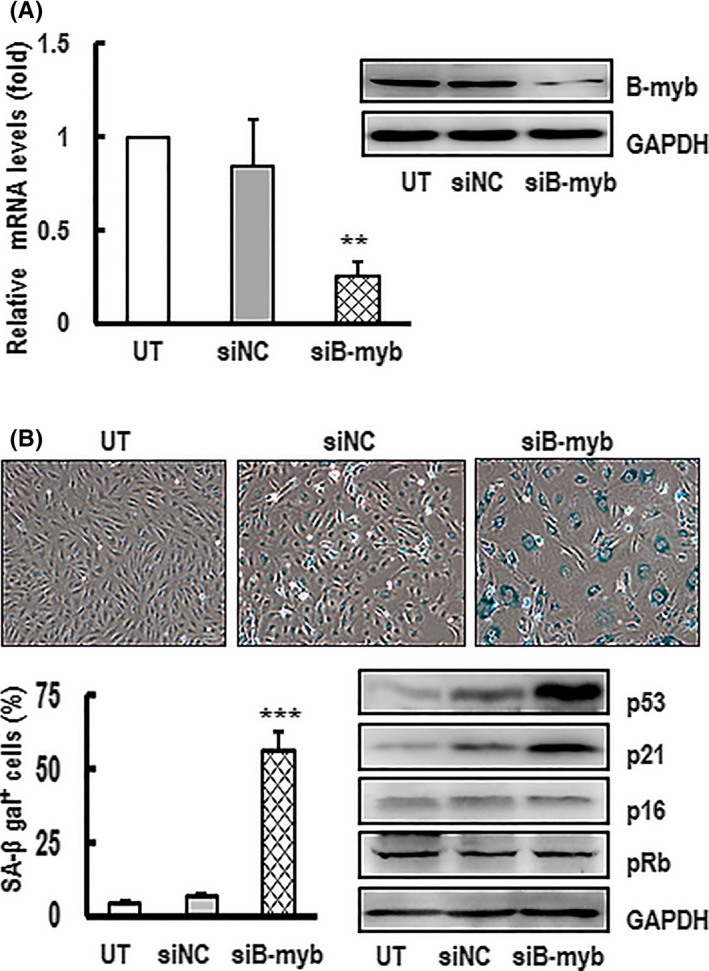
Inhibition of B‐myb expression promoted HAECs undergoing premature senescence and upregulating p53 and p21 expression. A, HAECs were transfected with siRNA against B‐myb (siB‐myb) or the controls (siNC) for 3 d. The knockdown efficiencies of B‐myb mRNA and protein levels were confirmed by real‐time PCR and western blotting. GAPDH was used for normalization. The mRNA data are presented as mean ± SEM of three independent experiments. ** indicates P<.01 compared with the control. A typical group of blots was shown from one of three independent experiments. GAPDH was used as a loading control. B, The percentage rate of SA‐β‐gal‐positive cells and the expression protein levels of p53, p21, p16 and pRb were analysed after the cells were transfected with siB‐myb or siNC for 7 d. The SA‐β‐gal‐positive cells were observed under inverted microscope (×100 magnification) after the treated cells were stained with senescence‐associated β‐galactosidase (SA‐β‐gal). A representative group image of stained cultures is shown. The percentage rate of SA‐β‐gal‐positive cells was analysed. Results are presented as the mean ± SEM of three independent experiments. *** indicates P<.001 compared with the control. A typical group of blots for analysing the expression protein levels of p53 and p21 in the cells is shown from one of three independent experiments. GAPDH was used as a loading control
3.4. Cell premature senescence induced by silencing B‐myb through the ROS/p53/p21 pathway
As known, p21 is considered to be one of the hallmarks of cellular senescence, while p53 is a major intracellular regulator of senescence. The above results show that the downregulation of B‐myb causes upregulation of p53 and p21 (Figure 3B). To further clarify whether p53 pathway is involved in senescence induced by B‐myb knockdown, a p53‐specific inhibitor PFTα was used. As shown in Figure 4A, the upregulation of p‐p53, p53 and p21 in B‐myb knockdown cells could be abolished in the presence of PFTα. PFTα could also obviously attenuate the upregulation of SA‐β‐gal activity in B‐myb knockdown cells (Figure 4B). These results indicate that p53 pathway is involved in senescence induced by B‐myb knockdown.
Figure 4.
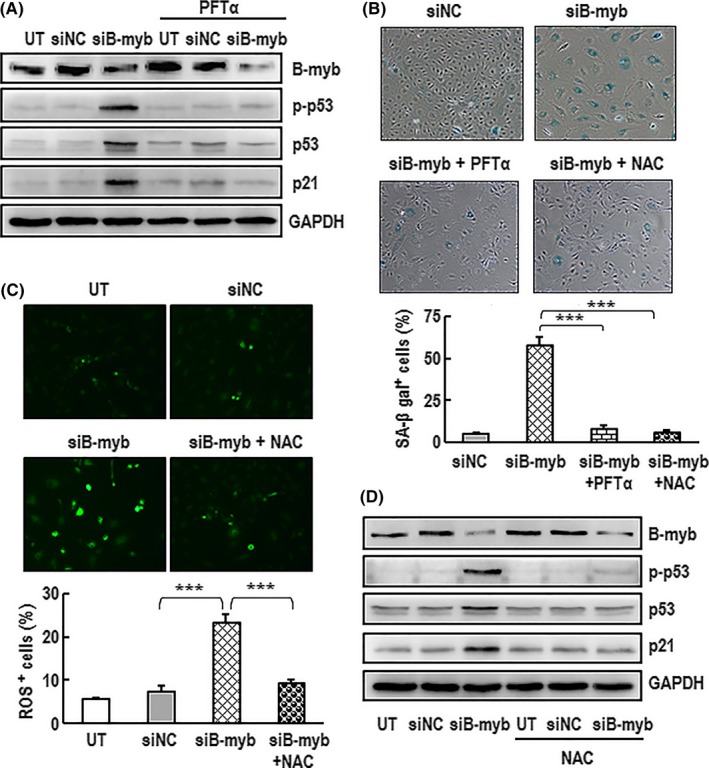
Silenced B‐myb induced cell premature senescence through the ROS/p53/p21 pathway. A, HAECs were per‐incubated with PFTα (3 μmol/L) for 60 min followed by transfected with siB‐myb or siNC in the presence of PFTα (3 μmol/L) for 7 d. The expression levels of B‐myb, p‐p53, p53 and p21 proteins were analysed by western blotting. GAPDH was used as a loading control. A typical group of blots is shown from one of three independent experiments. B, Cells were incubated with PFTα (3 μmol/L) or NAC (5 mmol/L) for 60 min followed by transfection with siB‐myb in the presence of PFTα (3 μmol/L) or NAC (5 mmol/L) for 7 d. The cells were then stained with SA‐β‐gal. A representative group image of stained cultures is shown (×100 magnification). The percentage rate of SA‐β‐gal‐positive cells was analysed. Data are presented as mean ± SEM of three independent experiments. *** indicates P<.001 compared between two groups. C, Cells were per‐incubated with NAC (5 mmol/L) for 60 min followed by transfection with siB‐myb in the presence of NAC (5 mmol/L) for 7 d. The production of intracellular ROS in the cells was determined with ROS indicator DCFH‐DA. A representative group image of stained cultures is shown (×100 magnification). The percentage rate of ROS‐positive cells was analysed. Data are presented as mean ± SEM of three independent experiments. *** indicates P<.001 compared between two groups. D, Cells were incubated with or without NAC (5 mmol/L) for 60 min followed by transfection with siB‐myb or siNC in the presence of NAC (5 mmol/L) for 7 d. The expression levels of B‐myb, p‐p53, p53 and p21 protein in the cell were analysed by western blotting. GAPDH was used as a loading control. A typical group of blots is shown from one of three independent experiments
Activation of ROS can induce premature cellular senescence,48 and the inhibition of ROS accumulation has been previously shown to protect against senescence.49 To test whether ROS is involved in senescence induced by B‐myb knockdown, the levels of intracellular ROS were analysed. As shown in Figure 4C, the production of intracellular ROS was significantly increased in B‐myb‐silenced cells. NAC, a ROS inhibitor, could effectively scavenge the ROS induced by B‐myb knockdown. Surprisingly, B‐myb‐silenced cells incubated with NAC could significantly diminish the upregulation of p‐p53, p53 and p21 (Figure 4D). The presence of NAC could also attenuate SA‐β‐gal activity in senescence induced by B‐myb knockdown (Figure 4B). These results demonstrate that the inhibition of B‐myb induces premature senescence possibly via the ROS‐p53 pathway in HAECs. The accumulation of ROS plays a critical role in senescence induced by B‐myb cut‐down.
NADPH oxidases are the major enzymes with the primary function of generating ROS. A membrane‐bound p22phox is the major component of NADPH oxidase and responsible for enzyme stability and activity.21 To explore whether p22phox was involved in senescence induced by B‐myb knockdown, RNAi technique was employed to knockdown p22phox and B‐myb genes. As shown in Figure 5A, the downregulation of B‐myb causes upregulation of p22phox. However, downregulation of p22phox can decrease the expression of p53 and p21 but has no effect on B‐myb expression (Figure 5B). The ROS generation induced by B‐myb knockdown was significantly reduced by downregulation of p22phox (Figure 5C). The presence of sip22phox can also attenuate SA‐β‐gal activity in senescence induced by B‐myb knockdown (Figure 5D). These results demonstrate that the inhibition of B‐myb induces premature senescence possibly via the upregulation of p22phox to activate the ROS/p53/p21 pathway in HAECs.
Figure 5.
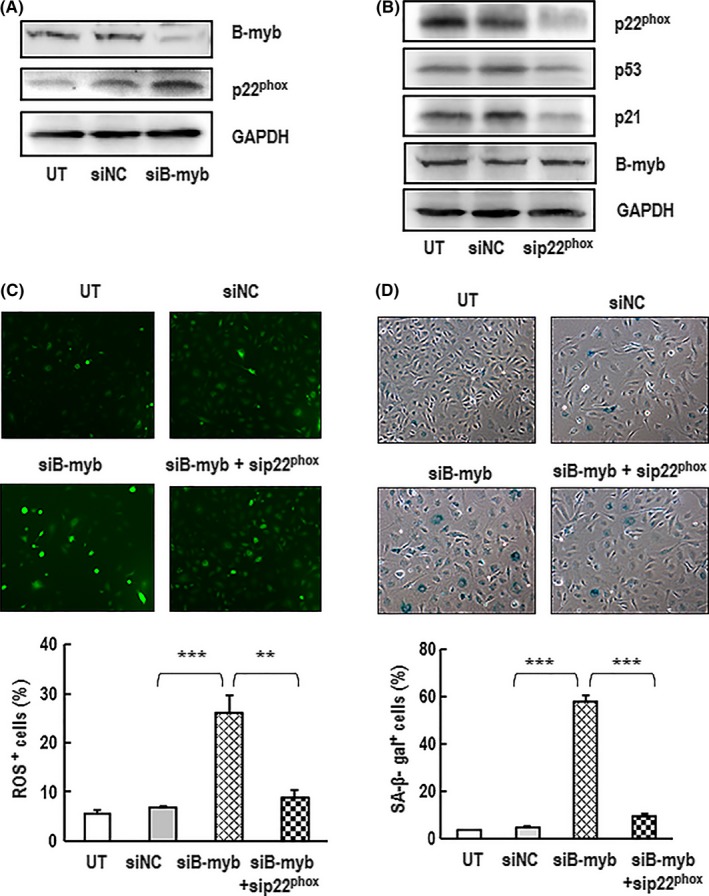
NADPH oxidase was involved in B‐myb silencing induced cell premature senescence. A, HAECs were transfected with siB‐myb or siNC for 7 d. The protein expression levels of B‐myb, p22phox were analysed by western blotting. GAPDH was used as a loading control. A typical group of blots is shown from one of three independent experiments. B, Cells were transfected with sip22phox or siNC for 7 d. The protein expression levels of B‐myb, p53, p21 and p22phox in the cells were analysed by western blotting. GAPDH was used as a loading control. A typical group of blots is shown from one of three independent experiments. C, Cells were transfected with siB‐myb and sip22phox for 7 d. The productions of intracellular ROS in the cells were determined with ROS indicator DCFH‐DA. A representative group image of stained cultures was shown (×100 magnification). The percentages rate of ROS‐positive cells was analysed. Data are presented as mean ± SEM of three independent experiments. ** and *** indicate P<.05 and P<.001, respectively, between the two groups. D, Cells were transfected with siB‐myb and sip22phox for 7 d. The cells were then stained with SA‐β‐gal. A representative group image of stained cultures was shown (×100 magnification). The percentages rate of SA‐β‐gal‐positive cells was analysed. Data are presented as mean ± SEM of three independent experiments. *** indicates P<.001 between the two groups
3.5. Downregulation of B‐myb expression in HAECs inhibited cell proliferation and the formation of capillary tube networks
Lack of proliferative capacity is a fundamental step in achieving senescence.1 It has been reported that the proliferation of cells is impaired in the ageing brain50 and the prostate gland of aged rats.51 The proliferation signalling pathways are involved in preventing cellular senescence.52, 53 B‐myb is a cell‐cycle‐regulated transcription factor that has an essential role in cell cycle progression and promotes cells proliferation.54, 55, 56 However, the effects of B‐myb on the proliferation of vascular endothelial cells have not yet been clarified. To assess the effects of B‐myb on HAECs proliferation in the early stage, B‐myb was silenced in HAECs by using RNAi. The cell proliferation was significantly reduced in B‐myb knockdown cells compared with that of the controls (Figure 6A). More interestingly, after B‐myb knockdown for 7 days, the cells formed less developed tubule networks and the number of tubules was significantly decreased (Figure 6B). These results indicate that silencing of B‐myb not only inhibits cell proliferation but also disturbs tubule networks formation, which directly affects vascular system functions.
Figure 6.
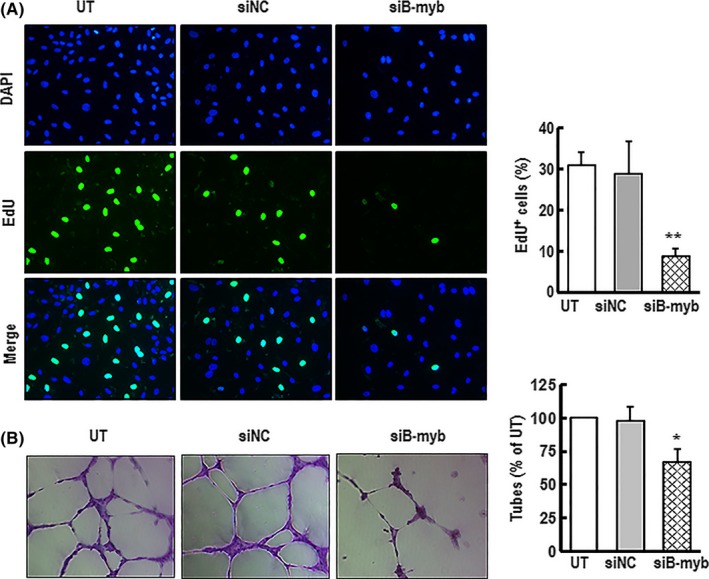
Downregulation of B‐myb expression in HAECs inhibited cell proliferation and the formation of capillary tube networks. A, HAECs were transfected with siB‐myb or siNC for 3 d. The cell proliferation was detected by KFluor488‐EdU. A typical representative cell proliferation images are shown (green) (×200 magnification). The percentage rate of EdU‐positive cells was analysed. Results are presented as mean ± SEM of four independent experiments. ** indicates P<.01 compared with the control. B, After cells transfection with siB‐myb or siNC for 7 d, the cells were seeded on top of the Matrigel‐coated wells and incubated for 6 h at 37°C. Tubular structures were photographed through inverted microscope after the cells were stained with crystal violet. Representative images of formed tubes are shown (×100 magnification). The circumferences of formed capillary‐like tube were measured. Data are presented as mean ± SEM of three independent experiments. * indicates P<.05 compared with the control
4. Discussion
The senescence of vascular endothelial cells is associated with endothelial dysfunction which contributes to the progression of the atherosclerotic process.57 B‐myb is downregulated during senescence and involved in protecting cell from senescence in many cell types.28, 29, 41 Both p53 and p21 are broadly considered to be the major hallmarks in the induction of senescence and ageing.11 In this study, the results show that B‐myb expression was significantly decreased, while p53 and p21 expression levels were enhanced in the aortas of aged mice. These findings further confirmed in vitro studies. The expression level of B‐myb was markedly downregulated; meanwhile, the expression levels of p53 and p21 were significantly increased during replicative senescence and DNA damage‐induced senescence in primary HAECs. The downregulation of B‐myb not only enhanced ROS activation and promoted senescence, which manifested in inhibiting cell proliferation and forming capillary tube networks, but also activated p53/p21 signalling pathway. These findings point out that ROS/p53/p21 signalling pathway was involved in B‐myb suppression‐induced senescence in vascular endothelial cells.
Cellular senescence can be categorized into replicative senescence and stress‐induced senescence. Most somatic cells always undergo replicative stress that is associated with telomere shortening or dysfunction. Besides replicative senescence, cells can also undergo stress‐induced senescence in response to DNA damage, oxidative stress or oncogenic stimuli.5, 6, 7 Bleomycin, as a chemotherapeutic agent, can mediate both single‐stranded and double‐stranded DNA damage, and it causes DNA single‐strand breaks at low doses and double‐strand breaks at high doses.58 Phosphorylation of histone H2AX (γH2AX) has been identified as an early event and sensitive cellular response to the presence of DNA double‐stranded breaks.59 A recent report suggests to target γH2AX as a non‐invasive imaging method to monitor DNA damage induced by bleomycin or other stresses in vivo.60 Previous studies have also shown that bleomycin could cause γH2AX foci formation rapidly in many cell types, including embryonic fibroblasts, hepatoblastoma cells and prostate cancer cells.61, 62, 63 The present data confirm that bleomycin can stimulate DNA damage in a dose‐ and time‐dependent response manner in primary HAECs. DNA damage is a major cause of inducing senescence and ageing. It has been reported that bleomycin can also induce senescence and ageing in vivo and in vitro.64 The effect of bleomycin in cellular senescence has been examined to be associated with genomic instability induced by DNA strand breaks, which blocks both the G1/S and G2/M transition in cell cycle.65 This study shows that bleomycin could induce premature senescence‐like phenotype in HAECs, which is similar to replicative senescence as characterized by a significant increasing activity of SA‐β‐gal and that the cells display enlarged size, flattened morphology and irreversible growth arrest.
The mechanism of endothelial cells senescence remains incompletely understood. B‐myb is a highly conserved member of the Myb family of transcription factors and expressed in most proliferating cells.22 It has been reported that B‐myb regulates diverse cellular functions, including cell cycle progression,66 cell survival67 and development.68 An increasing body of evidence has demonstrated that B‐myb is inhibited during senescence.28, 29, 40, 41 However, the role of B‐myb during senescence events remains largely unknown, especially in vascular system. The present results show that B‐myb has been depressed in HAECs not only during replicative senescence but also in stress‐induced senescence. It has been reported that B‐myb plays a major role in impeding cellular senescence. Loss of B‐myb may have a causative role in senescence.69 Recent studies have revealed that B‐myb is a growth‐regulated gene which could promote cell cycle progression by relaxing the inhibitory effects of p107 on mitotic cyclins.67 B‐myb has essential functions in cell cycle progression from G1 into S‐phase. The inhibition of B‐myb expression causes reduction of cell proliferation in fibroblasts.70 Consistent with previous observations, the data obtained from this study demonstrate that the suppression of B‐myb resulted in the inhibition of cell proliferation and capillary tube formation.
Persistent activation of the p16/pRb and p53 signalling pathways are broadly considered to be the key mechanisms involved in the induction of senescence. B‐myb could be influenced in the progress. Its effects may be through two possible pathways. One way in which B‐myb represses senescence is via the inhibition of the p16/pRb pathway. B‐myb could bind to that negative regulatory element located in the region of p16 promoter and suppress p16 promoter activity. It has been reported that knockdown of B‐myb can upregulate p16 expression and promote cellular senescence process.29 The other possible way in which B‐myb represses senescence is via the inhibition of the p53 pathway.69 High levels of ectopic B‐myb expression can bypass p53‐induced G1 arrest.32 B‐myb is required for recovery from the DNA damage, which plays an essential role in the expression of G2‐M phase genes in p53 mutant cells.71 This study shows for the first time that the loss of B‐myb results in premature senescence in vascular endothelial cells. In addition, the knockdown of B‐myb can significantly upregulate p53, p21 and the phosphorylation of p53 but has no effect on the expressions of p16 and pRb. Importantly, B‐myb silencing induced cellular senescence could be suppressed by using PFTα, a p53 inhibitor. These findings indicate a pivotal role of p53 in B‐myb knockdown‐induced senescence.
ROS can damage many cellular components and lead to oxidative stress promoting cellular senescence.72 ROS accumulation is considered to be one of major cause contributing to senescence or ageing and age‐related diseases. Both p53 and ROS have been shown to be involved in senescence. The relationship between p53 and ROS is very complex. Previous studies have shown that ROS can activate p53 pathway and induce senescence.20 However, p53 can regulate the level of intracellular ROS.73 It has been reported that p53 can alleviate oxidative stress, in responding to acutely stressed or physiological conditions, and exacerbate oxidative stress in responding to prolonged stress.74, 75 There is a positive feedback loop between p53 and ROS that drives cellular senescence.76 The link between senescence induced by B‐myb knockdown and ROS accumulation is still unclear. Based on the previous reports and findings from this study, it can be hypothesized that senescence induced by B‐myb silencing might be mediated by ROS. The present data show B‐myb knockdown increased ROS accumulation and p53 expression in endothelial cells, which can be inhibited by NAC, a ROS inhibitor. These results drop a hint that ROS was involved in B‐myb knockdown‐induced cellular senescence through activating p53 pathway.
Both p21 and ROS are broadly considered to be involved in senescence with a feedback loop between p21 and ROS. Increased expression of p21 has been shown to lead an increasing production of ROS and subsequent senescence.77 Elevation of ROS can upregulate p21 expression and induce cellular senescence.20 Consistent with these findings, the present study shows that treatment with the ROS scavenger NAC blocks p21 activation in B‐myb knockdown cells. The level and duration of ROS are the determining factors in the initiation of p53's transcription of p21 leading to senescence.78 Meanwhile, the upregulation of p21 sustains ROS accumulation and activation of p53‐p21‐ROS feedback loop. Therefore, p53‐p21‐ROS feedback loop might be involved in the senescence induced by abatement of B‐myb.
NADPH oxidases are a family of enzymes that generate ROS. Because the primary catalytic function, this property sets NADPH oxidases apart from all other ROS generating enzymes, either as a by‐product of their normal catalytic activity or as a result of aberrant functioning in disease.79 NAPDH oxidases are composed by p22phox and gp91phox membrane subunits and the cytosolic proteins p40phox, p47phox, p67phox and Rac.80 These molecules are expressed at much lower levels during normal cellular metabolism. However, in pathophysiological conditions, NAPDH oxidase activity is markedly increased, leading to an overproduction of ROS. The p22phox is the main catalytic unit expressed in most mammalian cell types including vascular endothelial cell.21 Previous studies have reported that there is an association between p22phox and vascular ageing.81, 82 This study showed that B‐myb silencing could induce senescence and upregulate the expression of p22phox in HAECs. Interestingly, the ROS generation induced by abatement of B‐myb decreased with the downregulation of p22phox. Furthermore, B‐myb silencing induced cellular senescence could be blocked by p22phox knockdown. The expression of p53 and p21was reduced with downregulation of p22phox. These results suggest that p22phox was involved in B‐myb silencing induced senescence. Probably, the upregulation of p22phox promotes ROS generation and then activates p53/p21 signalling pathway.
In summary, this study demonstrates that B‐myb was significantly downregulated during replicative senescence and DNA damage‐induced premature senescence in HAECs. More importantly, the inhibition of B‐myb resulted in upregulation of p22phox and ROS accumulation, activation of the p53/p21 pathway and further inhibition of cell proliferation, which results in cellular senescence. The upregulation of p53 and p21 can be blocked by NAC in B‐myb knockdown cells (Figure 7). These findings from this study suggest that downregulation of B‐myb is involved in endothelial cell senescence and triggers senescence by upregulation of p22phox and activating the ROS/p53/p21 signalling pathway. These observations provide a better understanding of endothelial cell senescence as well as a probable strategy to ameliorate atherosclerosis.
Figure 7.
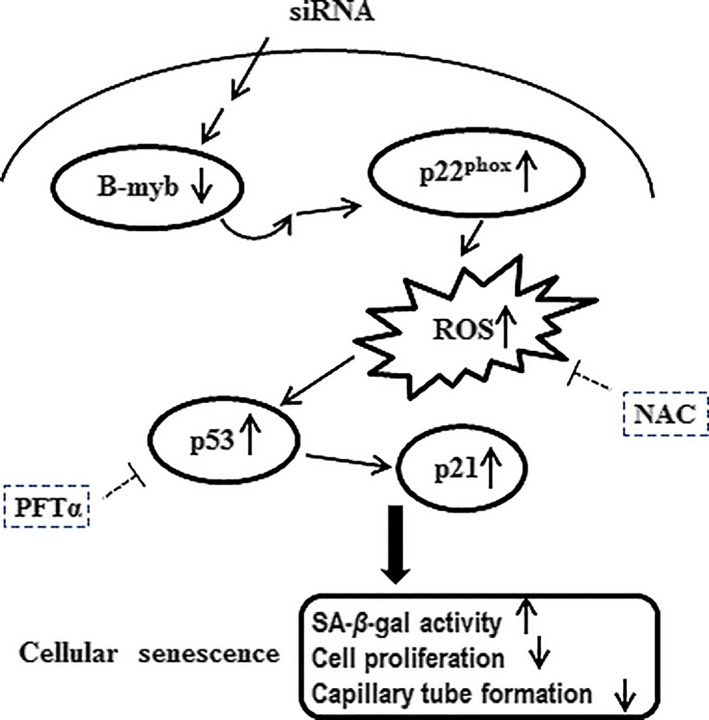
Schematic summary of the signalling pathway underlying the senescence of HAECs induced by downregulation of B‐myb. Knockdown of B‐myb by siB‐myb upregulates p22phox, which can promote ROS production in primary cultured HAECs. Excessive ROS accumulation can activate p53/p21 pathway which is the major pathway of senescence, causing further cellular senescence. The upregulation of p53 and p21 can be blocked by ROS scavenger NAC or specific p53 inhibitor PFTα in B‐myb knockdown‐induced senescence. These findings suggest that activation of ROS/p53/p21 signalling pathway is involved in B‐myb knockdown‐induced senescence in HAECs
Disclosure
The authors declare that they have no conflicts of interest.
Acknowledgement
This study was supported by the National Basic Research Program of China (973 Program, grant no. 2013CB530700).
References
- 1. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harley CB, Futcher AB, Greider CW. Telomeres shorten during aging of human fibroblasts. Nature. 1990;345:458–460. [DOI] [PubMed] [Google Scholar]
- 4. Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. [DOI] [PubMed] [Google Scholar]
- 5. Yu Q, Katlinskaya YV, Carbone CJ, et al. DNA‐damage‐induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015;11:785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Volonte D, Zou H, Bartholomew JN, Liu Z, Morel PA, Galbiati F. Oxidative stress‐induced inhibition of Sirt1 by caveolin‐1 promotes p53‐dependent premature senescence and stimulates the secretion of interleukin 6 (IL‐6). J Biol Chem. 2015;290:4202–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang P, Han L, Shen H, et al. Protein kinase D1 is essential for Ras‐induced senescence and tumor suppression by regulating senescence‐associated inflammation. Proc Natl Acad Sci USA. 2014;111:7683–7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nelson G, Wordsworth J, Wang C, et al. A senescent cell bystander effect: senescence‐induced senescence. Aging Cell. 2014;11:345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. [DOI] [PubMed] [Google Scholar]
- 10. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muñoz‐Espín D, Cañamero M, Maraver A, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–1118. [DOI] [PubMed] [Google Scholar]
- 12. Burton DG, Sheerin AN, Ostler EL, et al. Cyclin D1 overexpression permits the reproducible detection of senescent human vascular smooth muscle cells. Ann N Y Acad Sci. 2007;1119:20–31. [DOI] [PubMed] [Google Scholar]
- 13. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. [DOI] [PubMed] [Google Scholar]
- 14. van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell. 2000;102:55–66. [DOI] [PubMed] [Google Scholar]
- 16. Tan Y, Chen Y, Yu L, et al. Two‐fold elevation of expression of FoxM1 transcription factor in mouse embryonic fibroblasts enhances cell cycle checkpoint activity by stimulating p21 and Chk1 transcription. Cell Prolif. 2013;43:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beauséjour CM, Krtolica A, Galimi F, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ro SH, Nam M, Jang I, et al. Sestrin2 inhibits uncoupling protein 1 expression through suppressing reactive oxygen species. Proc Natl Acad Sci USA. 2014;111:7849–7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chiarugi P, Buricchi F. Protein tyrosine phosphorylation and reversible oxidation: two cross‐talking posttranslation modifications. Antioxid Redox Signal. 2007;9:1–24. [DOI] [PubMed] [Google Scholar]
- 20. Dimozi A, Mavrogonatou E, Sklirou A, Kletsas D. Oxidative stress inhibits the proliferation, induces premature senescence and promotes a catabolic phenotype in human nucleus pulposus intervertebral disc cells. Eur Cell Mater. 2015;30:89–102. [DOI] [PubMed] [Google Scholar]
- 21. Cave A. Selective targeting of NADPH oxidase for cardiovascular protection. Curr Opin Pharmacol. 2009;9:208–213. [DOI] [PubMed] [Google Scholar]
- 22. Martinez I, Dimaio D. B‐Myb, cancer, senescence, and microRNAs. Cancer Res. 2011;71:5370–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campanini EB, Vandewege MW, Pillai NE, et al. Early evolution of vertebrate Mybs: an integrative perspective combining synteny, phylogenetic, and gene expression analyses. Genome Biol Evol. 2015;7:3009–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baker SJ, Ma'ayan A, Lieu YK, et al. B‐myb is an essential regulator of hematopoietic stem cell and myeloid progenitor cell development. Proc Natl Acad Sci USA 2014;111:3122–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sala A, Casella I, Bellon T, Calabretta B, Watson RJ, Peschle C. B‐myb promotes S phase and is a downstream target of the negative regulator p107 in human cells. J Biol Chem. 1996;271:9363–9367. [DOI] [PubMed] [Google Scholar]
- 26. Werwein E, Schmedt T, Hoffmann H, et al. B‐Myb promotes S‐phase independently of its sequence‐specific DNA binding activity and interacts with polymerase delta‐interacting protein 1 (Pdip1). Cell Cycle. 2012;11:4047–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klein DK, Hoffmann S, Ahlskog JK, et al. Cyclin F suppresses B‐Myb activity to promote cell cycle checkpoint control. Nat Commun. 2015;6:5800. [DOI] [PubMed] [Google Scholar]
- 28. Johung K, Goodwin EC, DiMaio D. Human papillomavirus E7 repression in cervical carcinoma cells initiates a transcriptional cascade driven by the retinoblastoma family, resulting in senescence. J Virol. 2015;81:2102–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang Y, Wu J, Li R, et al. B‐MYB delays cell aging by repressing p16 (INK4α) transcription. Cell Mol Life Sci. 2011;68:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hauser S, Ulrich T, Wurster S, Schmitt K, Reichert N, Gaubatz S. Loss of LIN9, a member of the DREAM complex, cooperates with SV40 large T antigen to induce genomic instability and anchorage‐independent growth. Oncogene. 2012;31:1859–1868. [DOI] [PubMed] [Google Scholar]
- 31. Masselink H, Vastenhouw N, Bernards R. B‐myb rescues ras‐induced premature senescence, which requires its transactivation domain. Cancer Lett. 2001;171:87–101. [DOI] [PubMed] [Google Scholar]
- 32. Lin D, Fiscella M, O'Connor PM, et al. Constitutive expression of B‐myb can bypass p53‐induced Waf1/Cip1‐mediated G1 arrest. Proc Natl Acad Sci USA. 1994;91:10079–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol Metab. 2014;25:452–463. [DOI] [PubMed] [Google Scholar]
- 34. Herrera MD, Mingorance C, Rodríguez‐Rodríguez R, Alvarez de Sotomayor M. Endothelial dysfunction and aging: an update. Ageing Res Rev. 2010;9:142–152. [DOI] [PubMed] [Google Scholar]
- 35. Bai B, Liang Y, Xu C, et al. Cyclin‐dependent kinase 5‐mediated hyperphosphorylation of sirtuin‐1 contributes to the development of endothelial senescence and atherosclerosis. Circulation. 2012;126:729–740. [DOI] [PubMed] [Google Scholar]
- 36. Liu W, Peng Y, Yin Y, Zhou Z, Zhou W, Dai Y. The involvement of NADPH oxidase‐mediated ROS in cytokine secretion from macrophages induced by Mycobacterium tuberculosis ESAT‐6. Inflammation. 2014;37:880–892. [DOI] [PubMed] [Google Scholar]
- 37. Szabova L, Yin C, Bupp S, et al. Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res. 2012;72:4141–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell. 2004;14:501–513. [DOI] [PubMed] [Google Scholar]
- 39. Tie L, An Y, Han J, et al. Genistein accelerates refractory wound healing by suppressing superoxide and FoxO1/iNOS pathway in type 1 diabetes. J Nutr Biochem. 2013;24:88–96. [DOI] [PubMed] [Google Scholar]
- 40. Shi L, Ko S, Kim S, et al. Loss of androgen receptor in aging and oxidative stress through Myb protooncoprotein‐regulated reciprocal chromatin dynamics of p53 and poly(ADP‐ribose) polymerase PARP‐1. J Biol Chem. 2008;283:36474–36485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Song J, Sandoval R, Pilkinton MA, Tian X, Raychaudhuri P, Colamonici OR. ARF‐induced downregulation of Mip130/LIN‐9 protein levels mediates a positive feedback that leads to increased expression of p16Ink4a and p19Arf. Oncogene. 2010;29:1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53‐dependent G1 arrest and long‐term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8:2540–2551. [DOI] [PubMed] [Google Scholar]
- 43. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. d'Adda di Fagagna F, Reaper PM, Clay‐Farrace L, et al. A DNA damage checkpoint response in telomere‐initiated senescence. Nature. 2003;426:194–198. [DOI] [PubMed] [Google Scholar]
- 45. Kiran S, Oddi V, Ramakrishna G. Sirtuin 7 promotes cellular survival following genomic stress by attenuation of DNA damage, SAPK activation and p53 response. Exp Cell Res. 2015;331:123–141. [DOI] [PubMed] [Google Scholar]
- 46. Jeon HJ, Kim YS, Park JS, et al. Age‐related change in γH2AX of Drosophila muscle: its significance as a marker for muscle damage and longevity. Biogerontology. 2015;16:503–516. [DOI] [PubMed] [Google Scholar]
- 47. Valdiglesias V, Giunta S, Fenech M, Neri M, Bonassi S. & #x03B3;H2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat Res. 2013;753:24–40. [DOI] [PubMed] [Google Scholar]
- 48. Panieri E, Gogvadze V, Norberg E, Venkatesh R, Orrenius S, Zhivotovsky B. Reactive oxygen species generated in different compartments induce cell death, survival, or senescence. Free Radic Biol Med. 2013;57:176–187. [DOI] [PubMed] [Google Scholar]
- 49. Marazita MC, Dugour A, Marquioni‐Ramella MD, Figueroa JM, Suburo AM. Oxidative stress‐induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: implications for age‐related macular degeneration. Redox Biol. 2016;7:78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Romine J, Gao X, Xu XM, So KF, Chen J. The proliferation of amplifying neural progenitor cells is impaired in the aging brain and restored by the mTOR pathway activation. Neurobiol Aging. 2015;36:1716–1726. [DOI] [PubMed] [Google Scholar]
- 51. Vaz CV, Marques R, Maia CJ, Socorro S. Aging‐associated changes in oxidative stress, cell proliferation, and apoptosis are prevented in the prostate of transgenic rats overexpressing regucalcin. Transl Res. 2015;166:693–705. [DOI] [PubMed] [Google Scholar]
- 52. Khor SC, Mohd Yusof YA, Wan Ngah WZ, Makpol S. Tocotrienol‐rich fraction prevents cellular aging by modulating cell proliferation signaling pathways. Clin Ter. 2015;166:e81–e90. [DOI] [PubMed] [Google Scholar]
- 53. Kang SW, Kim J, Shin DY. Inhibition of senescence and promotion of the proliferation of chondrocytes from articular cartilage by CsA and FK506 involves inhibition of p38MAPK. Mech Ageing Dev. 2016;153:7–13. [DOI] [PubMed] [Google Scholar]
- 54. Arsura M, Introna M, Passerini F, Mantovani A, Golay J. B‐myb antisense oligonucleotides inhibit proliferation of human hematopoietic cell lines. Blood. 1992;79:2708–2716. [PubMed] [Google Scholar]
- 55. Lorvellec M, Dumon S, Maya‐Mendoza A, Jackson D, Frampton J, García P. B‐Myb is critical for proper DNA duplication during an unperturbed S phase in mouse embryonic stem cells. Stem Cells. 2010;28:1751–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Joaquin M, Watson RJ. The cell cycle‐regulated B‐Myb transcription factor overcomes cyclin‐dependent kinase inhibitory activity of p57(KIP2) by interacting with its cyclin‐binding domain. J Biol Chem. 2003;278:44255–44264. [DOI] [PubMed] [Google Scholar]
- 57. Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544. [DOI] [PubMed] [Google Scholar]
- 58. Chen J, Stubbe J. Bleomycins: towards better therapeutics. Nat Rev Cancer. 2005;5:102–112. [DOI] [PubMed] [Google Scholar]
- 59. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double‐stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. [DOI] [PubMed] [Google Scholar]
- 60. Cornelissen B, Kersemans V, Darbar S, et al. Imaging DNA damage in vivo using gammaH2AX‐targeted immunoconjugates. Cancer Res. 2011;71:4539–4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Watters GP, Smart DJ, Harvey JS, Austin CA. H2AX phosphorylation as a genotoxicity endpoint. Mutat Res. 2009;679:50–58. [DOI] [PubMed] [Google Scholar]
- 62. Rahmutulla B, Matsushita K, Satoh M, et al. Alternative splicing of FBP‐interacting repressor coordinates c‐Myc, P27Kip1/cyclinE and Ku86/XRCC5 expression as a molecular sensor for bleomycin‐induced DNA damage pathway. Oncotarget. 2014;5:2404–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen CS, Wang YC, Yang HC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double‐strand breaks by targeting Ku70 acetylation. Cancer Res. 2007;67:5318–5327. [DOI] [PubMed] [Google Scholar]
- 64. Aoshiba K, Tsuji T, Nagai A. Bleomycin induces cellular senescence in alveolar epithelial cells. Eur Respir J. 2003;22:436–443. [DOI] [PubMed] [Google Scholar]
- 65. Kaufmann WK, Kies PE. DNA signals for G2 checkpoint response in diploid human fibroblasts. Mutat Res. 1998;400:153–167. [DOI] [PubMed] [Google Scholar]
- 66. Joaquin M, Watson RJ. Cell cycle regulation by the B‐Myb transcription factor. Cell Mol Life Sci. 2003;60:2389–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sala A. B‐MYB, a transcription factor implicated in regulating cell cycle, apoptosis and cancer. Eur J Cancer. 2005;41:2479–2484. [DOI] [PubMed] [Google Scholar]
- 68. Zhan M, Riordon DR, Yan B, et al. The B‐MYB transcriptional network guides cell cycle progression and fate decisions to sustain self‐renewal and the identity of pluripotent stem cells. PLoS ONE. 2012;7:e42350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mowla SN, Lam EW, Jat PS. Cellular senescence and aging: the role of B‐MYB. Aging Cell. 2014;13:773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Santilli G, Schwab R, Watson R, Ebert C, Aronow BJ, Sala A. Temperature‐dependent modification and activation of B‐MYB: implications for cell survival. J Biol Chem. 2005;280:15628–15634. [DOI] [PubMed] [Google Scholar]
- 71. Mannefeld M, Klassen E, Gaubatz S. B‐MYB is required for recovery from the DNA damage‐induced G2 checkpoint in p53 mutant cells. Cancer Res. 2009;69:4073–4080. [DOI] [PubMed] [Google Scholar]
- 72. Yang KE, Jang HJ, Hwang IH, et al. Phenyl 2‐pyridyl ketoxime induces cellular senescence‐like alterations via nitric oxide production in human diploid fibroblasts. Aging Cell. 2016;15:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Johnson TM, Yu ZX, Ferrans VJ, Lowenstein RA, Finkel T. Reactive oxygen species are downstream mediators of p53‐dependent apoptosis. Proc Natl Acad Sci USA. 1996;93:11848–11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jiang L, Hickman JH, Wang SJ, Gu W. Dynamic roles of p53‐mediated metabolic activities in ROS‐induced stress responses. Cell Cycle. 2015;14:2881–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wei Z, Guo H, Liu Z, et al. CUL4B impedes stress‐induced cellular senescence by dampening a p53‐reactive oxygen species positive feedback loop. Free Radic Biol Med. 2015;79:1–13. [DOI] [PubMed] [Google Scholar]
- 77. Masgras I, Carrera S, de Verdier PJ, et al. Reactive oxygen species and mitochondrial sensitivity to oxidative stress determine induction of cancer cell death by p21. J Biol Chem. 2012;287:9845–9854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fitzgerald AL, Osman AA, Xie TX, et al. Reactive oxygen species and p21Waf1/Cip1 are both essential for p53‐mediated senescence of head and neck cancer cells. Cell Death Dis. 2015;6:e1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Simão S, Gomes P, Pinto V, et al. Age‐related changes in renal expression of oxidant and antioxidant enzymes and oxidative stress markers in male SHR and WKY rats. Exp Gerontol. 2011;46:468–474. [DOI] [PubMed] [Google Scholar]
- 81. Kim KI, Na JE, Kang SY, et al. Impact of NAD(P)H oxidase p22 phox gene polymorphism on vascular aging in Korean centenarian and nonagenarian. Int J Cardiol. 2011;123:18–22. [DOI] [PubMed] [Google Scholar]
- 82. Zarzuelo MJ, López‐Sepúlveda R, Sánchez M, et al. SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: implications for vascular aging. Biochem Pharmacol. 2013;85:1288–1296. [DOI] [PubMed] [Google Scholar]