

Abstract
Objectives
Serum amyloid A (SAA), an acute phase protein, is highly expressed in psoriatic lesions but its function is not fully understood. The aim of this study was to explore its role in activation of keratinocytes.
Materials and methods
Real‐time PCR and immunofluorescence were performed to examine SAA expression in imiquimod (IMQ)‐induced psoriasis‐like mice. In vivo function of SAA was examined by treating psoriasis‐like mice with SAA neutralising antibody. Cell viability was monitored using the CCK‐8 assay. Real‐time PCR was performed to determine expression of genes associated with differentiation and inflammation. Ki67+ percentage and immunological markers were analysed by flow cytometry. Involvement of formyl peptide receptor‐like 1 (FPRL1) in SAA signal transduction was determined by RNA interference. Binding of SAA and FPRL1 was examined by co‐immunoprecipitaion. Western blotting was conducted to assess phosphorylation of downstream signalling molecules.
Results
SAA was highly expressed in skin lesions of IMQ‐treated psoriasis‐like mice and neutralising SAA attenuated epidermal hyperplasia and inflammation. SAA in vitro promoted keratinocyte proliferation and expression of immunological mediators, while inhibiting differentiation. Effects of SAA on keratinocyte proliferation and inflammation were mediated by FPRL1, as well as activation of the PI3K/Akt pathway.
Conclusions
These observations indicate that SAA/FPRL1 contributed to pathogenesis of psoriasis by promoting keratinocyte proliferation and inflammation, thus providing a potential therapeutic target for disease therapy.
Keywords: imiquimod‐induced psoriasis‐like mouse, keratinocytes, psoriasis, serum amyloid A
1. Introduction
Psoriasis is a chronic immune‐mediated inflammatory disease, affecting 2‐3% of the population worldwide.1 It is characterised by histological changes, including epidermal hyperproliferation, infiltration of T cells and dendritic cells, and a distinct increase in skin angiogenesis.2 In psoriasis vulgaris, the epidermal hyperproliferation and inflammation leads to the typical skin lesion characterised by redness, thickness and scaling, which severely impairs patient quality of life.3 Despite the current focus on the role of IL‐23/Th17 immune axis in the pathogenesis of psoriasis, keratinocytes, the most predominant cell type in human epidermis, are hyperproliferative and deviate from the normal differentiation cycle, leading to the formation of plaque with a scaling surface.4, 5 The activated keratinocytes also have a positive feedback impact on the local immune system by producing soluble proinflammatory mediators, including cytokines, chemokines and antimicrobial peptides.6 However, the mechanism of keratinocyte activation in psoriasis is not well understood.
Serum amyloid A (SAA) is one of the most prominent positive acute phase proteins, which are highly elevated in serum due to inflammation, infections, neoplasia and tissue injury.7, 8, 9 SAA is induced in peripheral tissues in response to infection and acute injury, while it can promote inflammation, in part through elicitation of proinflammatory cytokine production and recruitment of granulocytes, monocytes, and T lymphocytes.10, 11 In addition, SAA also plays an important role in regulating the cellular proliferation and apoptosis.12, 13 It has been demonstrated that SAA promotes the proliferation and survival of human fibroblast‐like synoviocytes.14
Recently, we and others have detected increased SAA mRNA and protein expression in psoriatic lesions.15, 16 Rooney et al.16 reported that SAA is able to induce angiogenesis, a pathological hallmark of psoriasis. We demonstrated that SAA stimulated keratinocytes to produce IL‐1β in a NLRP3 inflammasome‐mediated mechanism, thereby providing positive feedback regulation of Th17 responses.15 However, it is unknown whether SAA plays any role in the proliferation and differentiation of keratinocytes.
The aim of the present study was to determine the potential role of SAA in psoriasis. Our data showed that neutralising SAA could ameliorate epidermal hyperplasia and inflammation; we determined that SAA stimulated the proliferation and proinflammatory phenotype of normal human keratinocytes via binding to its receptor, formyl peptide receptor‐like 1 (FPRL1), and that this effect is mediated by the activation of PI3K/Akt signalling pathway. These findings may constitute an attractive target for therapeutic interventions for psoriasis.
2. Materials and methods
2.1. Mice
Female BALB/c mice (8‐ to 11‐week old) were purchased from the Shanghai SLAC Laboratory Animal Center, Chinese Academy of Science. All experiments were performed according to the Animal Care and Use Committee guidelines of Shanghai Skin Disease Hospital.
2.2. Imiquimod‐induced psoriasis‐like mice
The imiquimod (IMQ)‐induced psoriasis‐like mice were constructed following previous protocols.17, 18 The mice received a daily topical IMQ cream (5%) (Aldara; 3M Pharmaceutical, St Paul, MN, USA) or Vaseline (Unilever, London, UK) on the shaved back skin (n=6 for each group). For the antibody treatment, four groups of mice received the following interventions: (i) topical Vaseline with ip injection of control IgG (ab37373; Abcam, Cambridge, MA, USA); (ii) topical Vaseline with ip injection of anti‐SAA antibody (LS‐C150247; LifeSpan BioSciences, Seattle, WA, USA); (iii) topical IMQ with ip injection of control IgG; and (iv) topical IMQ with ip injection of anti‐SAA antibody (n=6 for each group). On day 14, all mice were sacrificed and skin specimens were collected and inspected.
2.3. RNA isolation and real‐time PCR
Total RNA was extracted from mouse skin tissues or cultured keratinocytes using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) in accordance with the manufacturer's instructions. About 1 μg of total RNA isolated was reverse transcribed with the reverse transcriptase kit (Takara, Kusatsu, Shiga, Japan) according to the manufacturer's protocol. FPRL1 cDNA was amplified using ExTaq DNA polymerase (Takara). PCR assay cycles were as follows: 94°C for 5 minutes, 35 cycles of 94°C for 30 seconds, 56°C for 30 seconds and 72°C for 30 seconds. The PCR products were visualised on 2% agarose gels and ethidium bromide staining. To check the mRNA levels of mouse SAA, IL‐17A, CCL20, GAPDH in skin tissues, and the mRNA levels of human keratin 5, keratin 10, filaggrin, loricrin, FPRL1, IL‐1β, CCL20, and DEFB4 and GAPDH in cultured keratinocytes, real‐time PCR was performed in triplicate with a real‐time RT‐PCR system (ABI PRISM 7500; Applied Biosystems, Foster City, CA, USA) using a SYBR detection kit (Takara) according to the standard protocol. The mRNA levels of each target gene were normalised to the levels of mouse or human GAPDH and were represented as fold induction. The primer sequences of for RT‐PCR and real‐time PCR are shown in Table 1.
Table 1.
The sequence of primers used for RT‐PCR and real‐time PCR
| Gene | Primers |
|---|---|
| a GAPDH | 5′‐ACC TGC CAA GTA TGA TGA C‐3′ |
| 5′‐CTG TTG CTG TAG CCG TAT‐3′ | |
| a SAA | 5′‐CAG GAG ACA CCA GGA TGA‐3′ |
| 5′‐TAA TAG GAG GAC GCT CAG TA‐3′ | |
| a IL‐17A | 5′‐TTT AAC TCC CTT GGC GCA AAA‐3′ |
| 5′‐CTT TCC CTC CGC ATT GAC AC‐3′ | |
| a CCL20 | 5′‐GCC TCT CGT ACA TAC AGA CGC‐3′ |
| 5′‐CCA GTT CTG CTT TGG ATC AGC‐3′ | |
| b keratin 5 | 5′‐AGG AGT TGG ACC AGT CAA CAT‐3′ |
| 5′‐TGG AGT AGT AGC TTC CAC TGC‐3′ | |
| b keratin 10 | 5′‐ATG TCT GTT CGA TAC AGC TCA AG‐3′ |
| 5′‐CTC CAC CAA GGG AGC CTT TG‐3′ | |
| b filaggrin | 5′‐TGA AGC CTA TGA CAC CAC TGA‐3′ |
| 5′‐TCC CCT ACG CTT TCT TGT CCT‐3′ | |
| b loricrin | 5′‐ATG TCT TAA CCT ACC TGG AAG‐3′ |
| 5′‐TTA TTG ACT GAG GCA CTG G‐3′ | |
| b FPRL1 | 5′‐CTC CAC TCC TCT GAA TGA AT‐3′ |
| 5′‐GTT GAT GTC CAC CAC GAT‐3′ | |
| b IL‐1β | 5′‐AGC TAC GAA TCT CCG ACC AC‐3′ |
| 5′‐CGT TAT CCC ATG TGT CGA AGA A‐3′ | |
| b CCL20 | 5′‐TGC TGC TAC TCC ACC TCT‐3′ |
| 5′‐GCA AGT GAA ACC TCC AAC‐3′ | |
| b DEFB4 | 5′‐CTC CTC TTC TCG TTC CTC TTC A‐3′ |
| 5′‐GCA GGT AAC AGG ATC GCC TAT‐3′ | |
| b GAPDH | 5′‐AAT CCC ATC ACC ATC TTC C‐3′ |
| 5′‐TTG AGG CTG TTG TCA TAC TTC T‐3′ |
Human genes.
Murine genes.
2.4. Histology and immunofluorescence
Mice back skin tissues were collected. The samples were fixed in 4% formalin‐buffered solution and processed for standard haematoxylin and eosin (H&E) staining. For immunofluorescence, skin tissue sections were cryoprotected before freezing by overnight incubation in 20% sucrose solutions in phosphate‐buffered saline (PBS) and then in 30% sucrose in PBS overnight at 4°C. Tissues were embedded in optimal cutting temperature embedding medium (Sakura Finetek, Torrance, CA, USA) and frozen in liquid nitrogen. Cryosections were cut and collected on glass slides, dried at room temperature, and stored at −80°C until use. The skin cryosections were washed with PBS and incubated with blocking solution (10% goat serum in PBS) at room temperature for 2 hours with anti‐SAA (sc‐20275; Santa Cruz Biotechnology, Dallas, TX, USA) at 4°C overnight. After they were washed with PBS three times, sections were incubated at room temperature for 1 hour with secondary antibodies coupled to Alexa Fluor® 488 (1:1000 dilution; Invitrogen, Carlsbad, CA, USA). Nuclei were counterstained with Hoechst 33342. Sliders were analysed on a scanning laser confocal microscope.
2.5. Cell cultures
The normal human epidermal keratinocytes were cultured in accordance with a previously described procedure.15 In brief, the keratinocytes were grown in EpiLife cell culture medium (Cascade Biologics, Portland, OR, USA) containing 0.03 or 1.2 mmol/L Ca2+ and 1×EpiLife defined growth supplement at 37°C under standard tissue culture conditions. Stock cultures were maintained for up to five passages in this medium with the addition of gentamicin (10 μg/mL) and amphotericin B (0.25 μg/mL). Cells at 60‐80% confluency were stimulated for different time periods with SAA (1‐50 μg/mL; Peprotech, Rocky Hills, NJ, USA).
2.6. Cell viability assay
A CCK‐8 viability assay kit (Dojindo, Kumamoto, Japan) was adopted to analyse cell viability according to the manufacture's instruction. Briefly, cells were cultured in 96‐well plates (Corning, Lowell, MA, USA) and incubated with 10 μL CCK‐8 solution in 100 μL of fresh media for 3 hours at 37°C. The absorbance at 450 nm was detected after incubation.
2.7. Flow cytometry
To analyse the expression of Ki67, keratinocytes were harvested and washed by centrifugation. The cells were then fixed and permeabilised for 60 minutes at 4°C using the fixation/permeabilisation diluent and concentrate (eBioscience, San Diego, CA, USA). Cells were then stained with Alexa Fluor 488‐conjugated anti‐Ki67 (ab197234; Abcam). To analyse the expression of HLA‐DR and ICAM‐1, treated keratinocytes were harvested and washed by centrifugation. Alexa Fluor 488‐conjugated anti‐HLA‐DR (ab187601; Abcam) and FITC‐conjugated anti‐ICAM‐1 (ab27582; Abcam) were added to the cells. After incubation for 30 minutes at 4°C, the cells were washed twice with stain buffer (BD Biosciences, San Jose, CA, USA). FACS data analysis was performed using the Diva (BD Biosciences) and FlowJo (TreeStar, Ashland, OR, USA) software.
2.8. Western blot analysis
After treatment, keratinocytes were harvested and lysed with RIPA buffer (Shenggong, Shanghai, Japan). Cytosol proteins from keratinocytes (40 μg/lane) were separated by electrophoresis in 8‐12% SDS‐polyacrylamide gel and then transferred onto a PVDF membrane (Millipore, Bedford, MA, USA). Subsequently, the membrane was blocked in 2% BSA for 1 hour at room temperature and incubated overnight at 4°C with the primary antibodies against FPRL1 (ab63022; Abcam), phospho‐PI3K (ab182651; Abcam), PI3K (ab40755; Abcam), phospho‐Akt (ab8932; Abcam), Akt (ab25893; Abcam), phospho‐Erk1/2 (ab136926; Abcam), Erk1/2 (ab17942; Abcam), phospho‐NF‐κB p65 (ab28856; Abcam), NF‐κB p65 (ab16502; Abcam) and β‐actin (ab8227; Abcam). After washing with PBST (phosphate buffer solution containing Tween‐20) five times, the membranes were probed with horseradish peroxidase‐conjugated secondary antibodies. The level of β‐actin was examined at the same time as an internal control. The membranes were developed with an enhanced chemiluminescence system from Amersham and exposed to X‐ray film (Fuji Photo Film, Shanghai, China).
2.9. Co‐immunoprecipitation
Keratinocytes were cultured to subconfluency in six‐well plates (Corning). The cells were treated for 10 minutes with 20 μg/mL of SAA. The cells were then harvested, and lysed in PBS with 1% Triton X‐100, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulphate (PBS‐TDS) as described previously. The protein concentration was determined by the bicinchoninic acid assay (BCA protein assay kit; Pierce, Rockford, IL, USA). Dynabeads protein G (25 μL) (Invitrogen) and 2 μg of anti‐FPRL1 (ab63022; Abcam) were added to 500 μg protein samples. After overnight incubation at 4°C on a rotator platform, the immunoprecipitate was collected, and brought down and boiled in the presence of SDS to liberate antigen. And then the immunoprecipitate was analysed by Western blot with an anti‐SAA (sc‐20275; Santa Cruz Biotechnology) antibody following the same procedures as described above.
2.10. Small interfering RNA (siRNA)
To reduce endogenous FPRL1 expression, keratinocytes were transfected with 80 pmol of siRNA oligonucleotides following the manufacturer's instructions. The cells were transfected with either a siRNA oligonucleotide against FPRL1 (sc‐40123; Santa Cruz Biotechnology) or a non‐targeted control siRNA oligonucleotide (sc‐37007; Santa Cruz Biotechnology) and incubated for 7 hours at 37°C under standard culture conditions. At 12 hours post‐infection, cells were washed with PBS and maintained in defined keratinocyte‐serum‐free media. After expansion in culture for 72 hours, the cells were used for in vitro assays.
2.11. Statistical analysis
All results are shown as mean and the standard error of the mean (mean ± SEM). The data were assessed for normal Gaussian distribution with Kolmogorov‐Smirnov test. We used two‐tailed Student's t‐test to determine significances between two groups. We did analyses of multiple groups by one‐way analysis of variance (ANOVA) with Bonferroni post hoc test. A value of P<.05 was considered significant, where *P<.05, **P<.01 and ***P<.001. Analyses and graphical representation were performed using Graph‐Pad Prism 5.01 software (GraphPad Software, La Jolla, CA, USA).
3. Results
3.1. SAA is abundantly expressed in IMQ‐induced psoriatic skin lesions
Given that increased SAA expression was found in human psoriasis skin lesions in our previous report,15 we sought to explore its role in the pathogenesis of psoriasis. First, we established a psoriasis‐like skin disorder in BALB/c mice using IMQ treatment, according to the previous reports.17, 18 Then, we detected the SAA expression profile in the IMQ‐treated mice. Using real‐time PCR, we found that SAA expression in the back skins of IMQ‐treated mice was significantly upregulated (Figure 1A). Moreover, immunofluorescence confirmed the increased SAA expression at protein level, especially in the epidermis of IMQ‐treated mice (Figure 1B).
Figure 1.
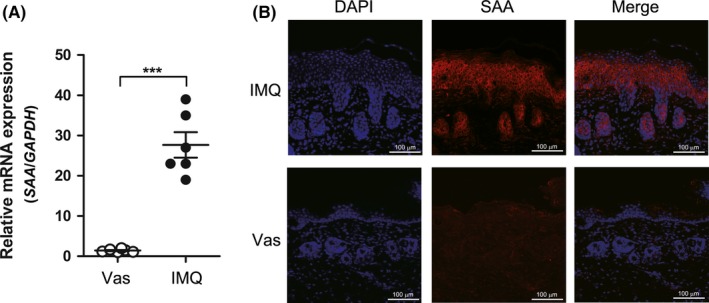
Serum amyloid A (SAA) expression is increased in imiquimod (IMQ)‐induced psoriatic skin lesions. IMQ‐induced psoriasis mouse model was constructed as described in the Materials and methods section. A, SAA mRNA expression in back skins of control (topical Vaseline, n=6) and IMQ‐treated mice (n=6) detected by real‐time PCR. Each point represents a skin sample obtained from a control or IMQ‐treated mice. The data are presented as mean ± SEM of three different experiments (***P<.001, Student's t‐test). B, Representative results of immunofluorescence analysis using anti‐SAA antibody in back skins of control and IMQ‐treated mice are shown. Scale bar=100 μm. The data represent one of three independent experiments
3.2. Neutralisation of SAA ameliorates skin lesions in IMQ‐induced psoriasis‐like mice
To investigate the role of SAA in psoriatic lesion formation, we treated IMQ‐induced psoriasis‐like mice with SAA neutralising antibody. Analysis of H&E‐stained back skin sections revealed that anti‐SAA antibody treatment reduced IMQ‐induced epidermal thickness and the number of infiltrated inflammatory cells in the dermis (Figure 2A‐C). Meanwhile, the expression of psoriasis‐associated cytokines, such as IL‐17A and CCL20, were significantly decreased in anti‐SAA antibody‐treated group (Figure 2D,E). These data suggested that neutralising SAA reduces epidermal hyperplasia and inflammation in skin lesions in vivo, and SAA might play an important role in the development of skin lesions in psoriasis.
Figure 2.
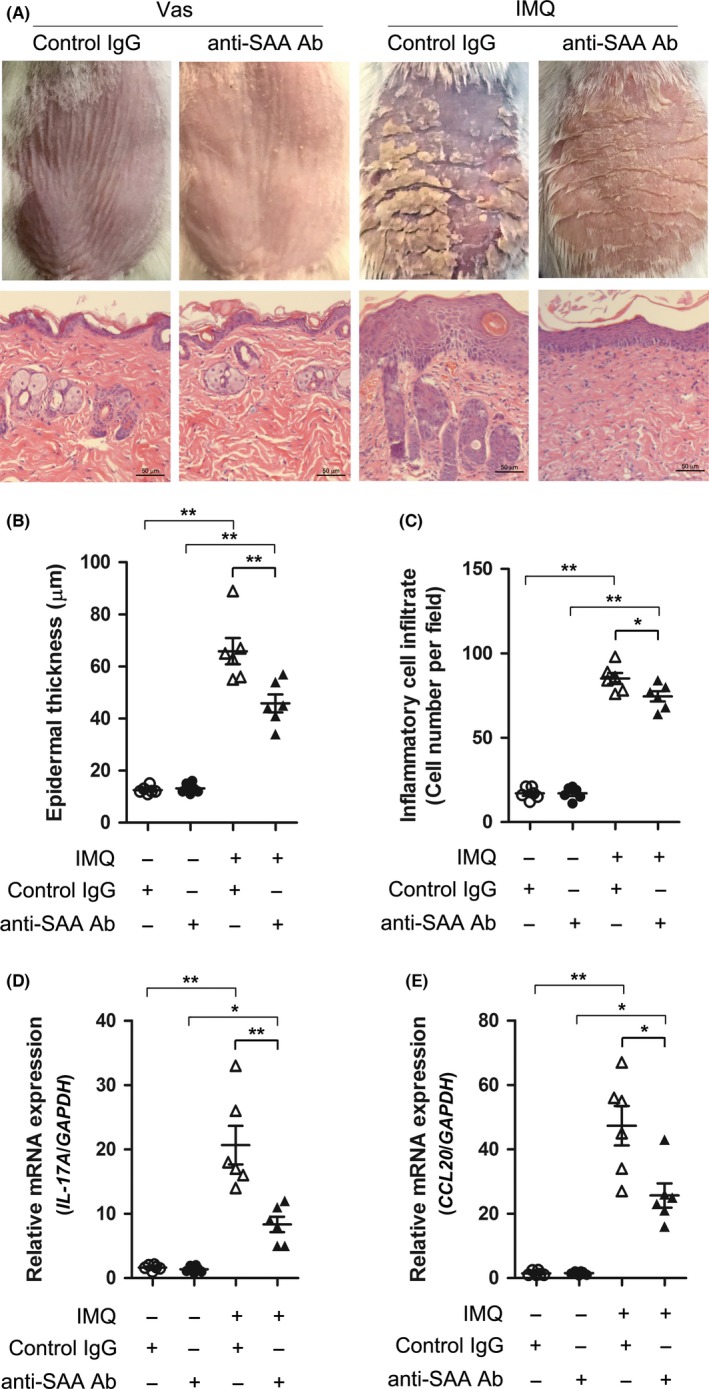
Neutralising serum amyloid A (SAA) alleviates epidermal hyperplasia in imiquimod (IMQ)‐induced mice. IMQ‐induced mice were treated with neutralising anti‐SAA antibody or control IgG. A, Representative clinical pictures (top) and H&E staining (bottom) are shown on skin sections of BALB/c mice treated with topical Vaseline and ip injection of anti‐SAA antibody (n=6), topical Vaseline and ip control IgG (n=6), topical IMQ and ip anti‐SAA antibody (n=6), or topical IMQ and ip control IgG (n=6) at day 14 of treatment. Scale bar=50 μm. B, Epidermal thickness and (C) infiltrated cells in dermis were measured in back skin from the anti‐SAA antibody or control IgG‐treated mice. D, IL‐17A and (E) CCL20 mRNA expression was detected by real‐time PCR. For B‐E, each point represents a skin sample obtained from a mouse. The data represent one of three independent experiments (*P<.05, **P<.01, one‐way ANOVA)
3.3. SAA stimulates keratinocyte proliferation via the inhibition of differentiation genes
Keratinocyte proliferation and differentiation is critical in psoriatic epidermal hyperplasia; therefore, we next investigated whether SAA was involved in the regulation of keratinocyte proliferation and differentiation. When undifferentiated cultured keratinocytes were exposed to SAA, increased proliferation was observed as detected by an increase in cell number (Figure 3A). We also examined the keratinocyte proliferation in response to SAA using Ki67 staining. The percentage of Ki67+ cells increased following SAA treatment in a dose‐dependent manner (Figure 3B). Furthermore, we found that SAA significantly inhibited the expression of the terminal differentiation marker genes keratin 10 (KRT10), filaggrin (FLG), and loricrin (LOR) but not keratin 5 (KRT5) in keratinocytes (Figure 3C). Extracellular calcium is a physiological signal of terminal differentiation in keratinocytes. Thus, we treated keratinocytes with high calcium (1.2 mmol/L Ca2+) or low calcium (0.03 mmol/L Ca2+) in the presence or absence of SAA. After 24 hours, SAA increased cell number in both high and low calcium medium (Figure 3D). Moreover, the Ca2+‐increased expression of LOR mRNA in keratinocytes was abrogated by SAA (Figure 3E). These data suggest that SAA can both increase keratinocyte proliferation and inhibit differentiation, supporting a potential role of SAA in psoriasis.
Figure 3.
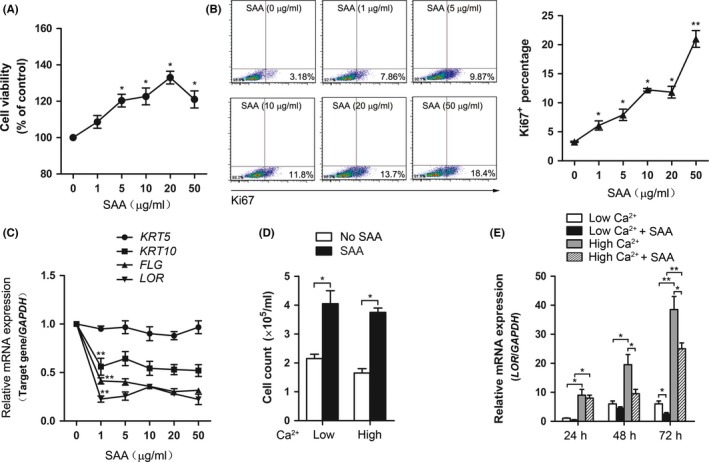
Serum amyloid A (SAA) increased keratinocyte proliferation and inhibited differentiation. Normal human keratinocytes were stimulated with different doses of SAA (0, 1, 5, 10, 20 and 50 μg/mL), after 72 h, (A) cell viability was evaluated with a CCK‐8 assay, (B) and the Ki67+ percentage was assessed by flow cytometry analysis. Representative histograms of three independent experiments are shown. (C) Keratin 5 (KRT5), keratin 10 (KRT10), filaggrin (FLG) and loricrin (LOR) gene expressions were then assessed by real‐time PCR and normalised against the amount of GAPDH mRNA. Gene expression is graphed as mean fold induction over medium control. (D) Keratinocytes were treated with 1.2 mmol/L CaCl2 or 0.03 mmol/L CaCl2 in the presence or absence of 20 μg/mL SAA. After 72 h, cell number was measured. (E) Keratinocytes were treated with 1.2 mmol/L CaCl2 or 0.03 mmol/L CaCl2 in the presence or absence of 20 μg/mL SAA. After 24, 48 and 72 h, gene expression of LOR was assessed by real‐time PCR. The data from (A‐E) represent the mean ± SEM of three experiments (*P<.05, **P<.01, one‐way ANOVA)
3.4. FPRL1 mediates SAA‐induced proliferation of keratinocytes
Having demonstrated the involvement of SAA in the regulation of keratinocyte proliferation and differentiation, we next sought to explore the mechanism by which keratinocytes respond to SAA. FPRL1 is one of the potential receptors for SAA, and has been implicated in regulating cellular proliferation and survival.14, 19 We thereby investigated whether FPRL1 is required for the effects of SAA in keratinocytes. FPRL1 mRNA and protein were observed to be expressed in keratinocytes and treatment with SAA further increased its expression (Figure 4A,C,D). Moreover, the binding of SAA to FPRL1 in keratinocytes was demonstrated by immunoprecipitation of SAA‐treated keratinocytes with FPRL1 antibody and detection with antibody to SAA (Figure 4B). In order to determine the effects of FPRL1 on keratinocyte proliferation and differentiation, we adopted siRNA. Transfection with siRNA efficiently downregulated FPRL1 mRNA and protein expression (Figure 4C,D). Knockdown of FPRL1 inhibited the capacity of SAA to increase cell viability (Figure 4E), and the upregulation of Ki67 induced by SAA was also blocked (Figure 4F). As shown in Figure 4G, silencing of FPRL1 also rescued SAA‐induced LOR downregulation.
Figure 4.
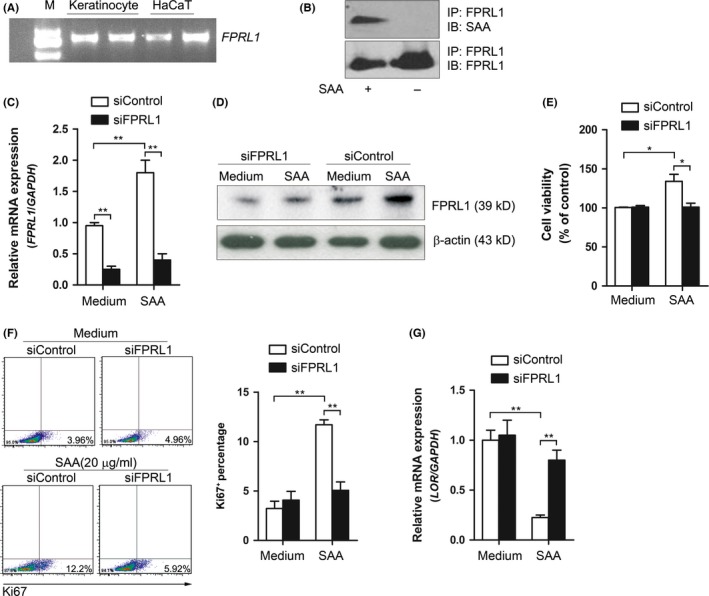
FPRL1 is required in SAA‐induced proliferation of keratinocytes. A, Gene expression of FPRL1 in cultured keratinocytes and immortal HaCaT cells was examined by RT‐PCR. One experiment represents three experiments. B, The binding of SAA to FPRL1 in keratinocytes was analysed by co‐immunoprecipitation. One experiment representative of three experiments. C, Keratinocytes were transfected with siRNA oligos specific for FPRL1 (siFPRL1) or a non‐specific siRNA oligo (siControl), and subsequently stimulated with 20 μg/mL SAA for 24 h, and the (C) mRNA and (D) protein expression of FPRL1 was assessed using real‐time PCR and Western blot. One experiment representative of three experiments. The siRNA‐transfected keratinocytes were stimulated with 20 μg/mL SAA. After 72 h, (E) the cell viability was evaluated with a CCK‐8 assay, (F) and the Ki67+ percentage was assessed by flow cytometry analysis, (G) and the gene expression of LOR was assessed by real‐time PCR. The data from (C, E, F and G) represent the mean ± SEM of three experiments (*P<.05, **P<.01, one‐way ANOVA)
3.5. SAA/FPRL1 stimulates the proinflammatory phenotype in keratinocytes
In psoriasis, keratinocytes produce proinflammatory mediators and upregulate immune‐related surface markers.3, 18 Next, we sought to examine the effect of SAA on the immunologic phenotype of keratinocyte. Treatment with SAA elevated the mRNA levels of IL‐1β, CCL20 and DEFB4 (Figure 5A‐C), as well as the protein levels of HLA‐DR and ICAM‐1 (Figure 5D). Moreover, we showed that silencing of FPRL1 significantly impaired these effects (Figure 5A‐D), suggesting that SAA/FPRL1 enhances the proinflammatory phenotype of keratinocytes, which are associated with psoriasis development. The knockdown of FPRL1 did not completely abrogate the cytokine‐inducing ability of SAA, implying the remaining function of FPRL1. However, possibility exists that other potential receptors for SAA might also be responsible for its proinflammatory effects.
Figure 5.
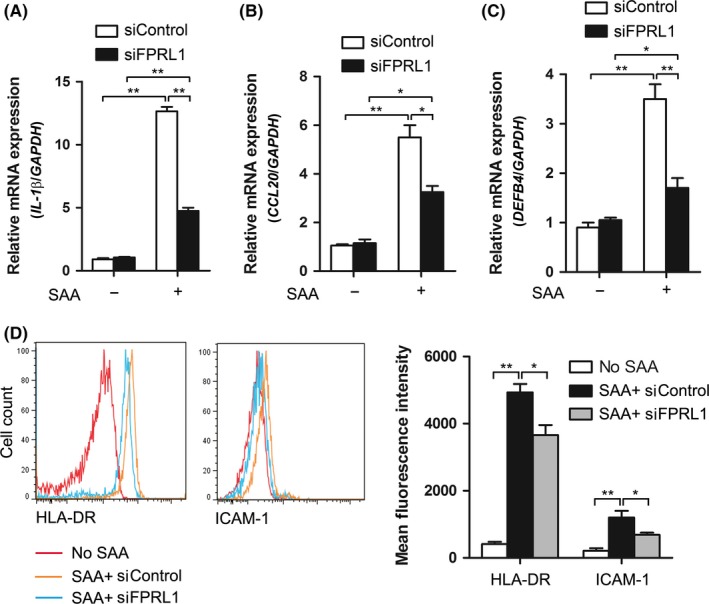
FPRL1 mediates the activation of keratinocytes induced by SAA. The siRNA‐transfected keratinocytes were treated with SAA for 24 h. (A) IL‐1β, (B) CCL20 and (C) DEFB4 gene expressions were then assessed by real‐time PCR and normalised against the amount of GAPDH mRNA. (D) Expression levels of HLA‐DR and ICAM‐1 were analysed by flow cytometry. Representative histograms are shown. The data from (A‐D) represent the mean ± SEM of three experiments (*P<.05, **P<.01, one‐way ANOVA)
3.6. SAA activates PI3K/Akt signalling pathway to regulate keratinocyte proliferation and differentiation
To further explore signalling pathways downstream of SAA/FPRL1, we analysed the activation of signalling molecules using Western blot. As shown in Figure 6A, phosphorylations of PI3K, Akt, Erk1/2 and NF‐κB p65 were detected in keratinocyte following treatment of SAA. To investigate the role of these signalling molecules in SAA‐induced activation of keratinocyte, we conducted blocking experiments using pharmacological inhibitors of the described signalling molecules. As shown in Figure 6B, pre‐treatment of keratinocytes with the PI3K/Akt inhibtors Wortmannin or LY294002 resulted in significant blockage of the proliferative activities of SAA. However, these effects were not influenced by Erk1/2 inhibitor PD98059 and NF‐κB inhibitor Bay 11‐7082 (data not shown). Although we have previously shown that SAA activated the NLRP3 inflammasome signalling pathway,15 NLRP3 inflammasome inhibitor Z‐YVAD‐fmk did not impact on the viability of keratinocytes (data not shown). To confirm that PI3K/Akt is the downstream pathway for FPRL1, we next examined the SAA‐induced Akt phosphorylation after FPRL1 was silenced. SAA markedly increased the phosphorylation of Akt and the increase was inhibited after FPRL1 was silenced (Figure 6D). All these data suggest that SAA activates FPRL1/PI3K/Akt signalling pathway to regulate keratinocyte proliferation and differentiation.
Figure 6.
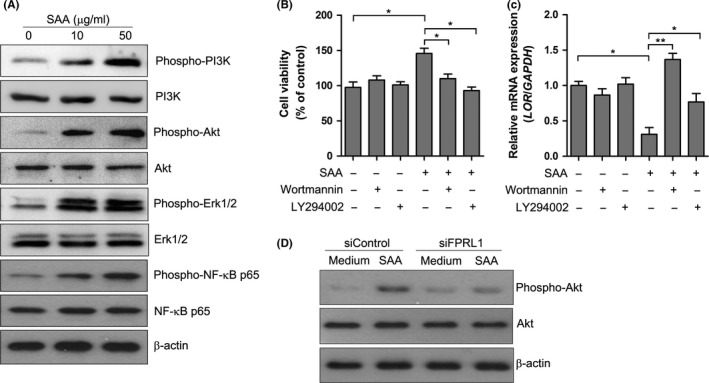
SAA activated PI3K/Akt signalling pathway. A, Keratinocytes were treated with different concentrations of SAA for 15 min, and the phosphorylation of PI3K, Akt, Erk1/2 and NF‐κB p65 was determined by Western blot. β‐actin was used for the verification of equal protein loading in each lane. Representative blots of three independent experiments are shown. B, Keratinocytes were pre‐treated with Wortmannin (100 nmol/L) or LY294002 (50 μmol/L) before the addition of SAA (20 μg/mL). After 72 h of incubation, the cell viability was evaluated with a CCK‐8 assay. C, The LOR mRNA level was measured by real‐time PCR. The data from (B and C) represent the mean ± SEM of three experiments with similar results (*P<.05, **P<.01, one‐way ANOVA). (D) The siRNA‐transfected keratinocytes were treated with SAA for 15 min, and the phosphorylation of Akt was analysed by Western blot. Representative blots of three independent experiments are shown
4. Discussion
In the present study, we found that SAA was upregulated in the lesional skin of IMQ‐induced psoriasis‐like mice. Neutralisation of SAA function with monoclonal antibody in vivo reduced the epidermal hyperplasia and inflammation associated with psoriasis. In vitro experiments revealed that SAA induced the proliferative and proinflammatory phenotype of keratinocytes. Furthermore, the activities of SAA were mediated by FPRL1/PI3K/Akt pathway. These results revealed that SAA is closely linked to psoriatic skin lesions by promoting keratinocyte proliferation and inflammation.
Serum amyloid A is a highly conserved, acute phase plasma protein, which is synthesised predominantly by the liver. During acute inflammation, the liver directs a significant proportion of its synthetic capacity into producing SAA.20 In addition to the liver, SAA is synthesised in various normal tissues such as kidney, breast and intestine, as well as in diseased tissues including atherosclerotic plaques, brain tissue of patients with Alzheimer' s disease and synovial tissue of patients with rheumatoid arthritis.21, 22, 23, 24 In our previous study, as well as Rooney et al. study showed that the expression of SAA was upregulated in psoriatic epidermis.15, 16 To further investigate the expression and function of SAA in a mouse model of psoriasis, we adopted the well‐described IMQ‐induced psoriasis‐like mice model. The observation of increased expression of SAA in the epidermis of IMQ‐treated mice was consistent with the results of human skin. The SAA expression is regulated by proinflammatory cytokines like TNF‐α, IL‐1β and IL‐6.25 The IL‐23/IL‐17A axis plays a crucial role in the pathogenesis of the psoriasis, as demonstrated by the detection of IL‐23‐producing dendritic cells and the expression of IL‐17A and IL‐22 by T cells and type 3 innate lymphoid cells in psoriatic lesions. We previously demonstrated that IL‐17A could stimulate the SAA expression in human keratinocytes,15 while Sano et al.26 reported that IL‐22 induced SAA expression in murine intestine epithelial cells. As both IL‐17A and IL‐22 are required in the IMQ‐induced psoriasis‐like mice model, it is likely that IL‐17A, IL‐22 and other proinflammatory cytokines might act synergistically in upregulating SAA in epidermis.
Proliferation and differentiation of keratinocytes is dysregulated in psoriasis. SAA has been reported to regulate the proliferation of human fibroblast‐like synoviocytes,14 but was not known to be involved in skin homeostasis. Here we found that treatment of SAA triggered cell proliferation in human keratinocytes, indicating the increased SAA expression in psoriatic epidermis may be involved in the pathogenesis of the disease. Indeed, psoriasis is characterised by excessive proliferation of keratinocytes. In addition, psoriatic keratinocytes show a number of proliferation markers, such as Ki67, which has been found to be induced by SAA treatment. The epidermal hyperplasia could also be partly due to an alteration of keratinocyte differentiation leading to a thickening of supra‐basal layers of epidermis. This is supported by a lowered expression of terminal differentiation genes (KRT10, LOR and FLG).
Most of the immune‐related functions of SAA are mediated by its interactions with various cell surface receptors.20 Previous studies have reported that FPRL1 is one of these receptors. FPRL1 is a G protein‐coupled receptor that possesses seven transmembrane domains and is expressed by a wide range of cell types.27, 28, 29 FPRL1 is activated by multiple ligands, including mitochondrial and bacterial peptides, lipoxin A4, chemokines and amyloidogenic proteins.30 SAA mediates human neutrophil production of IL‐8 and TNF‐α through FPRL127; in monocytes, it induces the secretion of chemokine CCL2 via FPRL119; the binding of SAA to FPRL1 also promoted the proliferation of synoviocytes.14 Our results showed that the expression of FPRL1 was high in keratinocytes, and the knockdown of FPRL1 expression resulted in impaired proliferation and activation of keratinocytes stimulated by SAA. Thus, we determined that FPRL1 was the receptor of SAA in human keratinocytes.
The upregulation of FPRL1 expression by SAA in our report suggests an increased sensitivity of keratinocytes to SAA, representing a self‐enhancing mechanism. However, the regulatory mechanism of FPRL1 expression in keratinocytes is not well understood. Upregulation of FPRL1 by proinflammatory TNF‐α and IL‐1β has previously been observed in synovial fibroblasts.28 Our data in keratinocytes extend the number of stimuli leading to FPRL1 upregulation by additionally identifying SAA as effective stimuli. As we previously reported that SAA induces the production of IL‐1β from keratinocytes,15 whether SAA upregulates FPRL1 expression in a direct or indirect way remains to be further investigated. A previous study suggested that Erk1/2 and NF‐κB activation mediates the IFN‐γ‐induced FPRL1 expression in microglial cells.31 As we showed that Erk1/2 and NF‐κB were activated by SAA in our data, we postulated that these signalling molecules might be involved in SAA‐induced FPRL1 upregulation.
Blockage of SAA‐induced signalling pathways revealed specific functions in regard to proliferation and differentiation. Our present analysis indicated that PI3K/Akt was essential for SAA‐induced keratinocyte proliferation. Although we previously demonstrated that NF‐κB and NLRP3 inflammasome activation were required for SAA‐mediated IL‐1β production, blocking NF‐κB or inflammasome pathways was not efficient in reducing SAA‐stimulated proliferation in the present study. It is very well known from different studies that PI3K/Akt signalling cascade is an important regulator of growth and proliferation of cells in hyperproliferative diseases like tumour and psoriasis.32, 33 Thus, our study provides evidence that SAA utilises well‐characterised pathways to induce proliferation and to inhibit differentiation in keratinocytes.
There are several potential mechanisms whereby SAA might exert positive effects on the pathogenesis of psoriasis. Firstly, as was suggested in this study, SAA, which is produced locally by keratinocytes, can exert a stimulatory effect on the proliferation of keratinocytes, while inhibiting cellular differentiation. Secondly, keratinocytes are now considered to play a key role as bona fide innate immune cells, capable of secreting cytokines, chemokines and antimicrobial peptides in response to various stimuli. In previous and present studies, we demonstrated that SAA could act as a DAMP by activating the proinflammatory cascades in keratinocytes.15 Thirdly, SAA can promote neovascularisation,16, 34, 35 a crucial hallmark of psoriasis pathogenesis. Consequently, activated keratinocytes might secrete increased quantities of SAA, which would then further stimulate the proliferation of keratinocytes in an autocrine manner, thereby forming a positive feedback loop. Considering that SAA production was induced by IL‐17A, the key cytokine in psoriasis, SAA acts as a link in keratinocyte activation and skin inflammation, promoting the formation of a feed‐forward and vicious cycle in psoriasis pathogenesis.
In conclusion, these findings support our discovery that SAA has a stimulatory role in keratinocyte proliferation and inflammation, leading to the formation of main pathological features of psoriasis. SAA was shown to induce the activation of keratinocytes, via its binding to its receptor, FPRL1. The effect of SAA is achieved via the stimulation of PI3K/Akt activity in the keratinocytes. Our findings suggest that the interaction between SAA and FPRL1 may be critical to the hyperplasia of keratinocytes, implicating the potential of SAA or FPRL1 as a therapeutic target in psoriasis.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81200675).
Yu N, Zhang S, Lu J, et al. Serum amyloid A, an acute phase protein, stimulates proliferative and proinflammatory responses of keratinocytes. Cell Prolif. 2017;50:e12320 10.1111/cpr.12320
References
- 1. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. [DOI] [PubMed] [Google Scholar]
- 2. Deng Y, Chang C, Lu Q. The inflammatory response in psoriasis: a comprehensive review. Clin Rev Allergy Immunol. 2016;50:377–389. [DOI] [PubMed] [Google Scholar]
- 3. Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol. 2012;7:385–422. [DOI] [PubMed] [Google Scholar]
- 4. Chiricozzi A, Saraceno R, Chimenti MS, Guttman‐Yassky E, Krueger JG. Role of IL‐23 in the pathogenesis of psoriasis: a novel potential therapeutic target? Expert Opin Ther Targets. 2014;18:513–525. [DOI] [PubMed] [Google Scholar]
- 5. Tonel G, Conrad C. Interplay between keratinocytes and immune cells–recent insights into psoriasis pathogenesis. Int J Biochem Cell Biol. 2009;41:963–968. [DOI] [PubMed] [Google Scholar]
- 6. Kim J, Krueger JG. The immunopathogenesis of psoriasis. Dermatol Clin. 2015;33:13–23. [DOI] [PubMed] [Google Scholar]
- 7. Sodin‐Semrl S, Zigon P, Cucnik S, et al. Serum amyloid A in autoimmune thrombosis. Autoimmun Rev. 2006;6:21–27. [DOI] [PubMed] [Google Scholar]
- 8. Migita K, Izumi Y, Jiuchi Y, et al. Effects of Janus kinase inhibitor tofacitinib on circulating serum amyloid A and interleukin‐6 during treatment for rheumatoid arthritis. Clin Exp Immunol. 2014;175:208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dogan S, Atakan N. Is serum amyloid A protein a better indicator of inflammation in severe psoriasis? Br J Dermatol. 2010;163:895–896. [DOI] [PubMed] [Google Scholar]
- 10. Connolly M, Marrelli A, Blades M, et al. Acute serum amyloid A induces migration, angiogenesis, and inflammation in synovial cells in vitro and in a human rheumatoid arthritis/SCID mouse chimera model. J Immunol. 2010;184:6427–6437. [DOI] [PubMed] [Google Scholar]
- 11. Meek RL, Urieli‐Shoval S, Benditt EP. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid A function. Proc Natl Acad Sci U S A. 1994;91:3186–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Siegmund SV, Schlosser M, Schildberg FA, et al. Serum amyloid A induces inflammation, proliferation and cell death in activated hepatic stellate cells. PLoS ONE. 2016;11:e0150893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tan SZ, Ooi DS, Shen HM, Heng CK. The atherogenic effects of serum amyloid A are potentially mediated via inflammation and apoptosis. J Atheroscler Thromb. 2014;21:854–867. [DOI] [PubMed] [Google Scholar]
- 14. Lee MS, Yoo SA, Cho CS, Suh PG, Kim WU, Ryu SH. Serum amyloid A binding to formyl peptide receptor‐like 1 induces synovial hyperplasia and angiogenesis. J Immunol. 2006;177:5585–5594. [DOI] [PubMed] [Google Scholar]
- 15. Yu N, Liu S, Yi X, Zhang S, Ding Y. Serum amyloid A induces interleukin‐1β secretion from keratinocytes via the NACHT, LRR and PYD domains‐containing protein 3 inflammasome. Clin Exp Immunol. 2015;179:344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rooney P, Connolly M, Gao W, et al. Notch‐1 mediates endothelial cell activation and invasion in psoriasis. Exp Dermatol. 2014;23:113–118. [DOI] [PubMed] [Google Scholar]
- 17. Van Belle AB, de Heusch M, Lemaire MM, et al. IL‐22 is required for imiquimod‐induced psoriasiform skin inflammation in mice. J Immunol. 2012;188:462–469. [DOI] [PubMed] [Google Scholar]
- 18. Sun Y, Zhang J, Zhou Z, et al. CCN1, a pro‐inflammatory factor, aggravates psoriasis skin lesions by promoting keratinocyte activation. J Invest Dermatol. 2015;135:2666–2675. [DOI] [PubMed] [Google Scholar]
- 19. Lee HY, Kim SD, Shim JW, et al. Serum amyloid A induces CCL2 production via formyl peptide receptor‐like 1‐mediated signaling in human monocytes. J Immunol. 2008;181:4332–4339. [DOI] [PubMed] [Google Scholar]
- 20. Eklund KK, Niemi K, Kovanen PT. Immune functions of serum amyloid A. Crit Rev Immunol. 2012;32:335–348. [DOI] [PubMed] [Google Scholar]
- 21. Urieli‐Shoval S, Cohen P, Eisenberg S, Matzner Y. Widespread expression of serum amyloid A in histologically normal human tissues. Predominant localization to the epithelium. J Histochem Cytochem. 1998;46:1377–1384. [DOI] [PubMed] [Google Scholar]
- 22. King VL, Thompson J, Tannock LR. Serum amyloid A in atherosclerosis. Curr Opin Lipidol. 2011;22:302–307. [DOI] [PubMed] [Google Scholar]
- 23. Chung TF, Sipe JD, McKee A, et al. Serum amyloid A in Alzheimer's disease brain is predominantly localized to myelin sheaths and axonal membrane. Amyloid. 2000;7:105–110. [DOI] [PubMed] [Google Scholar]
- 24. O'Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. Acute‐phase serum amyloid A production by rheumatoid arthritis synovial tissue. Arthritis Res. 2000;2:142–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uhlar CM, Grehan S, Steel DM, Steinkasserer A, Whitehead AS. Use of the acute phase serum amyloid A2 (SAA2) gene promoter in the analysis of pro‐ and anti‐inflammatory mediators: differential kinetics of SAA2 promoter induction by IL‐1 beta and TNF‐alpha compared to IL‐6. J Immunol Methods. 1997;203:123–130. [DOI] [PubMed] [Google Scholar]
- 26. Sano T, Huang W, Hall JA, et al. An IL‐23R/IL‐22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell. 2015;163:381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. He R, Sang H, Ye RD. Serum amyloid A induces IL‐8 secretion through a G protein‐coupled receptor, FPRL1/LXA4R. Blood. 2003;101:1572–1581. [DOI] [PubMed] [Google Scholar]
- 28. O'Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. Local expression of the serum amyloid A and formyl peptide receptor‐like 1 genes in synovial tissue is associated with matrix metalloproteinase production in patients with inflammatory arthritis. Arthritis Rheum. 2004;50:1788–1799. [DOI] [PubMed] [Google Scholar]
- 29. Su SB, Gong W, Gao JL, et al. A seven‐transmembrane, G protein‐coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med. 1999;189:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein‐coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006;17:501–519. [DOI] [PubMed] [Google Scholar]
- 31. Chen K, Iribarren P, Huang J, et al. Induction of the formyl peptide receptor 2 in microglia by IFN‐gamma and synergy with CD40 ligand. J Immunol. 2007;178:1759–1766. [DOI] [PubMed] [Google Scholar]
- 32. Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial‐mesenchymal transition. Cell Adh Migr. 2015;9:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chamcheu JC, Chaves‐Rodriquez MI, Adhami VM, et al. Upregulation of PI3K/AKT/mTOR, FABP5 and PPARβ/δ in human psoriasis and imiquimod‐induced murine psoriasiform dermatitis model. Acta Derm Venereol. 2016;96:854–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lv M, Xia YF, Li B, et al. Serum amyloid A stimulates vascular endothelial growth factor receptor 2 expression and angiogenesis. J Physiol Biochem. 2016;72:71–81. [DOI] [PubMed] [Google Scholar]
- 35. Connolly M, Rooney PR, McGarry T, et al. Acute serum amyloid A is an endogenous TLR2 ligand that mediates inflammatory and angiogenic mechanisms. Ann Rheum Dis. 2016;75:1392–1398. [DOI] [PubMed] [Google Scholar]