

Abstract
Objectives
Ubiquitin specific protease 32 (USP32) is a highly conserved but uncharacterized gene, which has been reported to be associated with growth of breast cancer cells. However, the role of USP32 in human small cell lung cancer (SCLC) has not been uncovered. The aim of this study was to investigate and evaluate the clinical significance of USP32 in patients with SCLC.
Materials and methods
Expression of USP32 was firstly investigated using public online data sets and then determined in SCLC tissues and cell lines using quantitative real‐time PCR, Western blotting and immunohistochemical staining. SCLC cells were transfected with a small‐interfering RNA targeting USP32 mRNA and analysed for cell viability, proliferation ability, cell cycle distribution, apoptosis and invasion.
Results
USP32 was found to be overexpressed in SCLC tissues compared with normal tissues. High USP32 expression was significantly correlated with disease stage and invasion. In vitro experiments demonstrated that silencing of USP32 caused a significant decrease in the proliferation and migration rate of cells. Furthermore, USP32 silencing arrested cell cycle progression at G0/G1 phase via decreasing CDK4/Cyclin D1 complex and elevating p21. In addition, downregulation of USP32 significantly induced cell apoptosis by activating cleaved caspase‐3 and cleaved PARP, as well as inhibiting cell invasiveness via altering epithelial mesenchymal transition expression.
Conclusions
Our results suggest for the first time that USP32 is important for SCLC progression and might be a potential target for molecular therapy of SCLC.
Keywords: cell apoptosis, cell cycle, cell proliferation, invasion, small cell lung cancer, USP32
1. INTRODUCTION
Lung cancer is thought as one of the most common causes of cancer‐related death worldwide with higher incidence, mortality and lower survival rate.1 As a histological subtype of lung cancer, small cell lung cancer (SCLC) accounts for 13%‐15% of lung cancers and considered as the most malignant of all lung cancers, because of its rapid growth and early metastasis.2, 3 According to the anatomical extent, patients with SCLC are staged as limited disease (LD) or extensive disease (ED).4 Compared with LD patients, ED patients have shorter survival rate <5%.5 At present, combined chemotherapy is the standard treatment for SCLC.6 Despite great progress has been made in the chemotherapeutic treatment, higher rate of rapidly growing drug‐resistant metastases is frequent. Therefore, it is uttermost importance to have a deeper understanding of the molecular mechanism underlying SCLC tumorigenesis and seek new therapeutic strategies for SCLC.
Deubiquitinating enzymes (DUBs) play an important role in regulating cellular ubiquitination by catalysing the removal of ubiquitin from substrates and disassembling ubiquitin chains.7 It is reported there are five related classes, including 100 DUBs were encoded by the mammalian genome.8 As the largest and most diverse subclass, ubiquitin‐specific proteases (USPs) contain two highly conserved short motifs termed the Cys and His boxes and are believed to target specific protein substrates, which have been shown to be involved in cell cycle progression, apoptosis, gene silencing and immune signalling.9 Recently, more studies have indicated that USPs participate in the pathogenesis of many human diseases, including inflammation and cancer.10 USP10, as a novel regulator of p53, is constitutively downregulated in renal clear cell carcinoma11 and also involves in the Ras signal transduction pathway.12 USP18 is identified as a key regulator of interferon‐induced pancreatic beta cell apoptosis. USP22 is a tumour promoter in oral squamous cell carcinoma patients13 and papillary thyroid carcinoma.14 In addition to as oncogenes, USP46 has been found to be a tumour suppressor in colon cancer by regulating PHLPP‐dependent attenuation of Akt signalling.15
USP32 localizes on chromosome 17q23 and belongs to the USPs family of cysteine proteases. As an ancient and highly conserved gene, USP32 has more than 90% sequence identity to proto‐oncogene USP6.16 Interestingly, USP6 has been reported to be an oncogene as the first DUB to activate tumour promoter NF‐κB in aneurysmal bone cyst.17 In the study of predicting breast cancer metastasis, USP32 is shown to have increased copy number in oestrogen receptor‐positive tumours.18 Moreover, the mRNA levels of USP32 were also reported to be upregulated in malignant breast epithelium.19 Shiva et al.20 have reported that USP32 is overexpressed in breast cancer, and involved in the proliferation of tumour cells. However, there has been no definitive data showing the expression and biological function of USP32 in SCLC.
Thus, the objective of this study was to investigate whether USP32 is overexpressed by SCLC tissues analysis and online data‐mining, and whether USP32 plays a crucial role in regulating tumour cell biological function, in an attempt to gain novel insights into tumorigenesis of SCLC.
2. MATERIALS AND METHODS
2.1. Gene expression data sets analysis
To investigate the expression of USP32 in lung cancer, USP32 gene expression was analysed using microarray gene expression data sets derived from the Oncomine database (http://www.oncomine.org). Briefly, the cancer type was defined as lung cancer, data type was mRNA and analysis type was cancer vs normal analysis. In addition, five sets of USP32 mRNA expression data were downloaded from the Cancer Genome Atlas project (TCGA data set, https://gdc-portal.nci.nih.gov/) database, including lung adenocarcinoma (LUAD), gastric adenocarcinoma (STAD), breast cancer (BRCA), oesophageal squamous carcinoma (ESCC), and stomach and oesophageal carcinoma (STES). The expression of USP32 was regarded to be significantly differentially expressed using cut‐off criteria of P‐value <.05 by t‐test method.
2.2. SCLC tissue specimens and cell lines
Total 10 fresh SCLC tissue samples and corresponding paired non‐cancerous lung tissue samples, as well as 83 paraffin‐embedded SCLC samples were obtained from the Department of Oncology of Shandong Cancer Hospital from December 2015 to January 2016. All participants voluntarily signed informed consent in this study, who were diagnosed SCLC by pathological biopsy samples and did not undergo any chemotherapy and radiotherapy. Clinical and pathological data of the 83 patients with SCLC were collected and summarized in Table 1. This study was approved by the Medical Ethics Committee of Shandong Cancer Hospital, China.
Table 1.
Association between USP32 expression and clinicopathologic parameters in patients with small cell lung cancer (n=83)
| Clinical pathologic parameters | Total (n=83) | Expression of USP32 | P‐value (chi‐square test) | |
|---|---|---|---|---|
| Low (45.4%) | High (54.6%) | |||
| Sex | ||||
| Male | 72 | 35 (48.61) | 37 (51.39) | .953 |
| Female | 11 | 6 (54.55) | 5 (45.45) | |
| Age | ||||
| <70 | 75 | 55 (73.33) | 20 (26.67) | .235 |
| ≥70 | 8 | 5 (62.5) | 3 (37.5) | |
| Stage | ||||
| LD | 47 | 18 (38.30) | 29 (61.70) | .004 |
| ED | 36 | 22 (61.11) | 14 (38.89) | |
| Invasion | ||||
| Vascular invasion | 30 | 9 (30.0) | 21 (70.0) | .005 |
| Lymphatic invasion | 53 | 32 (60.38) | 21 (39.62) | |
LD, limited disease; ED, extensive disease.
The bold of p value means significant difference.
Human SCLC cell lines, including H1688, DMS79, H446, DMS114, SBC3 and GLC4, were purchased from the cell bank of the Chinese Academy of Sciences (Shanghai, China). H1688, H446, SBC3, DMS79, and GLC4 cells were cultivated in RPMI‐1640 medium containing 10% foetal bovine serum (FBS) and penicillin/streptomycin. DMS114 cells were maintained Waymouth's media with 10% FBS and penicillin/streptomycin.
2.3. RNA extraction and quantitative real‐time PCR
Total RNA was extracted from fresh tissue samples and cell lines using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) following the manufacturer's instructions. The cDNA was synthesized from 2 μg RNA using SuperScript II RT 200 U/mL (Invitrogen) and used as a template for PCR amplification. The following primers were used: USP32 (Forward): 5′‐GGCTGCTCGTGATATGCTGTTC‐3′ and USP32 (Reverse): 5′‐GTTTCTGGGCTGACACCTTGC‐3′; β‐actin (Forward): 5′‐GTGGACATCCGCAAAGAC‐3′ and β‐actin (Reverse): 5′‐AAAGGGTGTAACGCAACTA‐3′. The 2−ΔΔCt method was used to calculate the relative mRNA expression of USP32. Each experiment was performed in triplicate.
2.4. Western blot analysis
Total cell lysates were isolated from tissues samples and cell lines using RIPA lysis buffer containing protease inhibitors (Sigma, St. Louis, MO, USA). Then cell lysates were centrifuged at 12 000 g for 10 minutes at 4°C, and cell supernatants were collected. BCA assay was used to determine the protein concentration. Approximately, 15 μg proteins was separated on 10% sodium dodecyl sulphate‐polyacrylamide gel and transferred to a PVDF membrane using Bio‐Rad semidry transfer system. Then the membrane was blocked in 5% non‐fat dry milk dissolved in TBST (Tris‐buffered saline, 0.1% Tween‐20) for 1 hour at room temperature, and then incubated with the corresponding primary antibodies, including anti‐USP32 (#2745; Cell signaling, Danvers, MA, USA), CDK4 (11026‐2‐AP; Proteintech, Chicago, IL, USA), Cyclin D1 (60186‐1‐1 g; Proteintech, Chicago, IL, USA), P21 (#2947; Cell signaling, Danvers, MA, USA), caspase‐3 (25546‐1‐AP; Proteintech, Chicago, IL, USA), PARP (#9542; Cell signaling, Danvers, MA, USA), P53 (sc‐126; Santa Cruz Biotechnology), E‐cadherin (#3195; Cell signaling, Danvers, MA, USA), N‐cadherin (#4016; Cell signaling, Danvers, MA, USA) and β‐actin (#3120; Cell signaling, Danvers, MA, USA) overnight. β‐actin was used as an internal control. Subsequently, the membrane was incubated with appropriated horseradish peroxidase‐conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 2 hours at room temperature. Bands were monitored with super ECL detection reagent (Amersham Pharmacia Biotech, Shanghai, China).
2.5. Immunohistochemical analysis
The paraffin‐embedded sections were dewaxed with xylenes, and rehydrated in gradient alcohol. Endogenous peroxidase was removed by incubation in 3% H2O2 for 30 minutes, and antigen retrieval was done by heating sections with EDTA antigenic retrieval buffer (0.05 mol/L glycine‐HCl buffer, pH 3.6, containing 0.01% (w/v) EDTA) in a microwave oven. The sections were then blocked with 10% goat serum at room temperature for 30 minutes, then incubated with primary antibody anti‐USP32 (1:50; Santa Cruz Biotechnology, Dallas, TX, USA) overnight at 4°C. After incubation with HRP‐conjugated secondary antibody for 30 minutes at 37°C, the sections were stained using Histostain‐Plus 3rd Gen IHC Detection Kit (Invitrogen) according to the manufacturer's directions. Two pathologists independently defined the score and intensity of the percentage of positive tumour cells according to the previous report.21 Finally, the expression of USP32 was classified into two groups, including low (<3) and high (>3) based on the sum of intensity and extent score scaling from 0 to 6.
2.6. Cell transfection
Small‐interfering RNAs (siRNAs) targeting USP32 (5′‐GGGAUAGCUAGAGGUUAGTTA‐3′) and a scramble siRNA (5′‐UUCUCCGAACGUGUCACGUTT‐3′) were synthesized and modified by Santa Cruz (Santa Cruz Biotechnology, Carlsbad, CA, USA). For transfection, H1688 and GLC4 cells were cultured in six‐well plates. After the cell density was about 50%, cells were transfected with the above siRNAs using Lipofectamine 2000 transfection reagent kit according to the manufacturer's instructions (Invitrogen). After 48 hours transfection, the interference efficiency was evaluated by Western blotting and quantitative real‐time PCR analysis.
2.7. CCK‐8 assay
The effects of USP32 on SCLC cell viability were detected by CCK‐8 assay. Briefly, cells from different treatments were collected and seeded into 96‐well plates at a density of 2000 cells per well. After incubation in 10% CCK‐8 solution for 2 hours at 37°C, the absorbance of each well was measured at 450 nm under a microplate reader. Each experiment was performed in triplicate.
2.8. Colony formation assay
Small cell lung cancer cells from different treatments were seeded in a six‐well plate in complete medium until the colonies were visible. Then the plates were washed twice in phosphate‐buffered saline (PBS) after removing complete medium. The colonies were fixed in 95% ethanol for 10 minutes, dried and then stained with 0.1% crystal violet solution for 10 minutes. The number of colonies (contained >50 cells) was counted. The experiment was performed three times.
2.9. Flow cytometry analysis
Small cell lung cancer cells from different treatments underwent cell cycle analysis using nuclear stain PI and apoptosis analysis using Annexin V‐APC/7‐AAD apoptosis detection KIT (Invitrogen) according to the manufacturer's instructions. Briefly, the cells were fixed in ice‐cold 70% (v/v) ethanol overnight and resuspended in 500 μL PBS containing 50 mg/mL RNase (Sigma‐Aldrich), then incubated for 30 minutes at 37°C. After staining with 50 mg/mL PI (Sigma‐Aldrich) for 30 minutes at 4°C in the dark, cell cycle distribution was analysed using FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) with appropriate software (ModFit LT; BD, Topsham, ME, USA). For cell apoptosis analysis, cells were harvested by trypsinization and resuspended in 500 μL binding buffer, and then stained with an Annexin V‐APC and 7‐AAD solution for 30 minutes at room temperature in the dark. The samples were analysed immediately using the FACSCalibur flow cytometer (BD Biosciences). The Annexin V‐positive and 7‐AAD‐negative cells represent early apoptotic populations. Annexin V‐positive and 7‐AAD‐positive cells represent late apoptotic proportions. Each sample was run in triplicate.
2.10. Cell matrigel invasion assay
To further investigate the role of USP32 in SCLC cell metastasis, a matrigel invasion assay was performed using the BiocoatMatrigel Invasion Chamber (BD) in SCLC cells transfected with different recombinants following the manufacturer's protocols. Briefly, cells were seeded into the upper chamber at 4000 cells per well in 100 μL serum‐free medium. The lower chamber was added into a chemoattractant. After incubation for 24 hours, the viable cells in lower surface of the filter were fixed in 4% paraformaldehyde and stained with crystal violet. The cells on the dissected stained membrane were counted in five random fields (100× magnification) under a light microscope. Each matrigel invasion assay was performed three times.
2.11. Statistical analysis
Data were analysed using spss software, version 13.0 and expressed as the mean ± standard deviation (SD) of more than three samples. The Student's t‐test (two‐tailed) was used to analyse the differences between two groups and P‐value <.05 was considered as a statistical significant difference.
3. RESULTS
3.1. USP32 expression is upregulated in lung cancer tissues
To investigate the expression of USP32 in lung cancer, we performed data‐mining to compare the gene expression profiles of USP32 between normal and cancer tissues. The results from Oncomine data sets analysis showed USP32 mRNA expression was significantly elevated in human LUAD tissues compared with that in normal tissues using Su Lung data set22 (Figure 1A, P=9.45 × 10−4, fold change=1.279), Landi Lung data set23 (Figure 1B, P=2.068 × 10−8, fold change=1.184), Selamat Lung data set24 (Figure 1C, P=3.65 × 10−6, fold change=1.171) and Okayama Lung data set25 (Figure 1D, P=3.84 × 10−5, fold change=1.199). Consistently, the expression of USP32 mRNA level was also significantly elevated in LUAD tissues in TCGA data set (Figure 1E, P=7.1 × 10−8). In addition, USP32 mRNA expression was significantly elevated in other four types of tumours, including TCGA BRCA data set (Figure 1F, P=9.1 × 10−3), TCGA ESCC (Figure 1G, P=1.4 × 10−3), TCGA STES data set (Figure 1H, P=1.08 × 10−8) and TCGA STAD data set (Figure 1I, P=3.18 × 10−3). These observations highlighted a deregulated expression of USP32 in cancers, especially in lung cancer.
Figure 1.
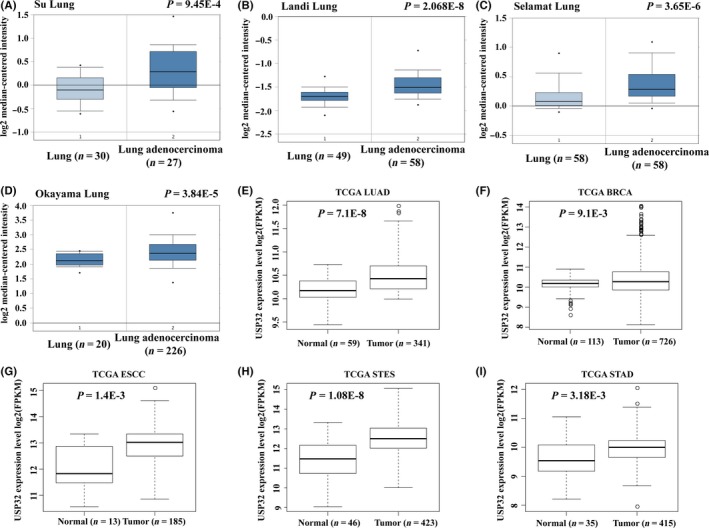
USP32 expression is upregulated in lung cancer tissues by publicly available database. The mRNA expression of USP32 in Oncomine data sets including Su Lung data set (A), Landi Lung data set (B), Selamat Lung data set (C) and Okayama Lung data set (D); the mRNA expression of USP32 in TCGA database including lung adenocarcinoma (LUAD) data set (E), TCGA BRCA data set (F), TCGA ESCC (G), TCGA STES data set (H) and TCGA STAD data set (I)
We then examined the mRNA and proteins expression of USP32 in 10 fresh SCLC tumours and their paired adjacent normal tissues. As shown in Figure 2A,B, both the mRNA and proteins expression of USP32 were commonly overexpressed in SCLC tissues, which is consistent with the results of the online data sets analysis. Furthermore, we detected the expression of USP32 in 83 paraffin‐embedded SCLC samples and 20 normal lung samples by immunohistochemical analysis. Representative photomicrographs of different degrees of USP32 expression intensity are shown in Figure 2C. As shown in Figure 2D, about 45.4% of paraffin‐embedded SCLC tissues showed weak staining of USP32 protein, while 37.5% SCLC tissues showed moderate USP32 staining, and 17.1% showed strong staining. In normal tissues, there was existed relatively less moderate and strong staining area compared with that in SCLC tissues.
Figure 2.
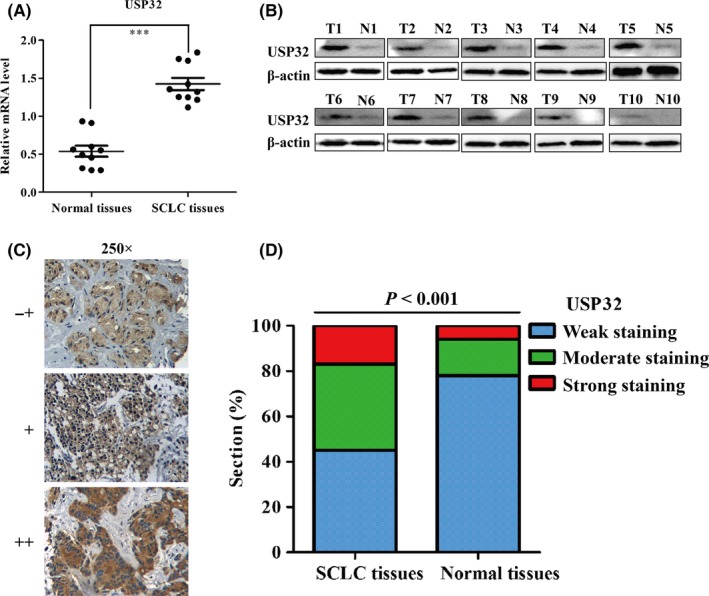
USP32 is frequently upregulated in small cell lung cancer (SCLC) tissues. The mRNA (A) and proteins (B) expression of USP32 in 10 pairs of paracancerous and cancer tissues from SCLC patients; N, paracarcinoma (normal) lung tissues. T, SCLC tissues; (C) representative images of immunohistochemistry staining of USP32 in SCLC tissues (−+, weak staining, + moderate staining, ++ strong staining); (D) Analysis of USP32 protein in tissues by immunohistochemistry. USP32 expression was significantly increased in SCLC tissues, when compared with normal lung tissues, P<.001
3.2. Correlation of USP32 expression with clinicopathological features
To evaluate the association between USP32 expression and the clinicopathological outcomes, the 83 patients with SCLC were divided into two groups: low expression (n=38, 45.4%) and high expression (n=45, 54.6%) according to the positive staining intensity as described in Methods. As described in Table 1, USP32 expression was significantly correlated with disease stage (LD vs ED, P=.004) and microscopic invasion (P=.005). There was no significant correlation between USP32 expression and age or gender.
3.3. USP32 expression was efficiently suppressed in SCLC cells
Subsequently, the protein level of USP32 was detected in six SCLC cell lines using Western blot analysis. As shown in Figure 3A,B, the protein level of USP32 was more highly expressed in H1688 and GLC4 cells compared with the other four cell lines. Therefore, H1688 and GLC4 were chosen to explore the role of USP32 in proliferation and metastasis in SCLC. To assess the functional role of USP32 on SCLC cells, both H1688 and GLC4 cells were transfected with siUSP32 or scrambled siRNA. As shown in Figure 3C, the mRNA and protein levels of USP32 were significantly downregulated in H1688 cells (P<.001). Similar results were also found in GLC4 cells after transfection with siUSP32 (Figure 3D, P<.001), suggesting a satisfactory knockdown efficiency.
Figure 3.
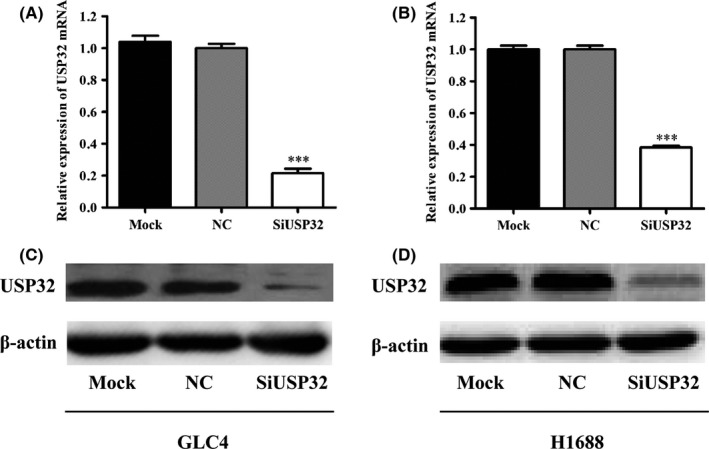
The expression of USP32 is efficiently silenced in two small cell lung cancer (SCLC) cell lines. (A) USP32 expression in six SCLC cell lines using Western blot analysis; (B) statistical analysis of the ratio of USP32/ β‐actin; SCLC, small cell lung cancer; ***P<.001; β‐actin was used as an internal control. The mRNA and proteins expression of USP32 was significantly reduced when USP32 was silenced by specific siRNA targeting USP32 (SiUSP32) compared with untreated (Mock) and scrambled siRNA (NC) in GLC4 (C) and H1688 (D) cells. SCLC, small cell lung cancer; all the results are represented as mean ± SD from three independent experiments. ***P<.001 as compared with NC cells; β‐actin was used as an internal control
3.4. Silencing of USP32 significantly inhibited cell proliferation
As USP32 has been shown to be overexpressed in SCLC, the present study aimed to determine whether knockdown of USP32 may inhibit uncontrolled cell growth in H1688 and GLC4 cells. The results from the CCK‐8 assay demonstrated that both GLC4 (Figure 4A) and H1688 (Figure 4B) cells transfected with siUSP32 exhibited significantly reduced cell viability compared with cells transfected with scrambled siRNA (P<.001). Subsequently, we evaluated the colony formation ability in both H1688 and GLC4 cells. As illustrated in Figure 4C,D, the smaller and fewer colonies were observed in siUSP32 groups than control cells in H1688 and GLC4 cells (P<.001). Collectively, silencing of USP32 could significantly suppress the cell viability and colony formation ability of SCLC cells.
Figure 4.
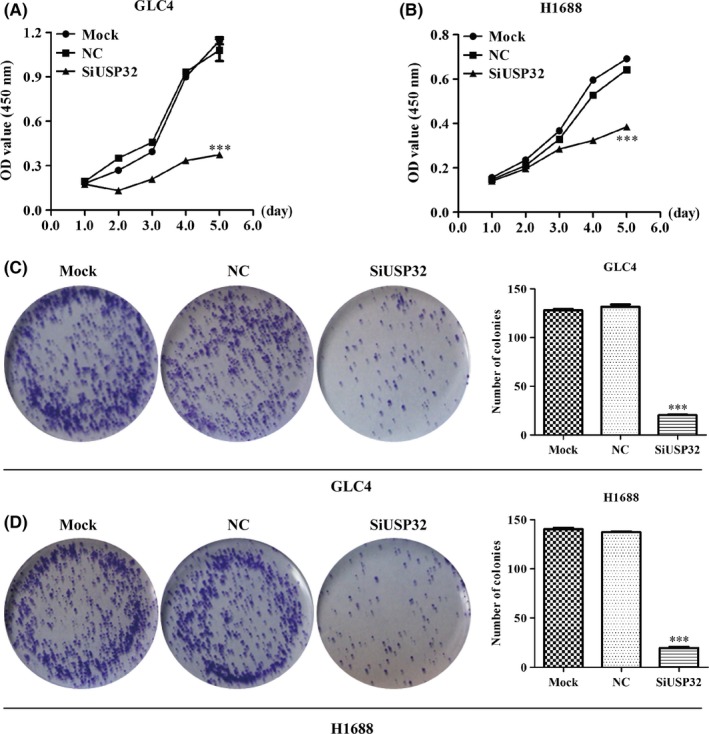
USP32 silencing significantly suppressed small cell lung cancer (SCLC) cell proliferation. (A, B) CCK‐8 assay indicated that the cell viability of GLC4 and H1688 was decreased after USP32 silencing. (C, D) The number of colonies in GLC and H1688 cells was significantly suppressed after USP32 knockdown. SCLC, small cell lung cancer; Mock: untreated cells; NC: cells were treated with scrambled siRNA; SiUSP32: cells were treated with siRNA targeting USP32; all the results are represented as mean ± SD from three independent experiments. ***P<.001 as compared with NC cells
3.5. Silencing of USP32 induced cell cycle arrested at G0/G1 phase and apoptosis
To uncover the inhibitory effects of USP32 silencing on cellular proliferation, flow cytometry was performed to analyse the cell cycle distribution and apoptosis in GLC4 cells from different treatments. As shown in Figure 5A, representative images indicated that knockdown of USP32 altered cell cycle distribution profiles in GLC4 cells compared with the negative controls (Mock or NC). Quantitative analysis further confirmed the percentages of cells in G0/G1 phase were significantly increased, whereas the number of cells in S phase was significantly decreased in GLC4 cells infected with siUSP32 (Figure 5B, P<.001). In addition, USP32 silencing decreased the expression levels of CDK4 and Cyclin D1 and slightly elevated the expression level of p21, suggesting that CDK4, Cyclin D1 and p21 play important roles in USP32‐induced cell cycle arrest (Figure 5C). Interestingly, in the absence of USP32, more cells were remarkably accumulated in the sub‐G1 phase, representing apoptotic cells (Figure 5D, P<.001).
Figure 5.

USP32 silencing remarkably induced G0/G1 arrest in GLC4 cells. A, Representative images showed knockdown of USP32 altered cell cycle distribution profiles in GLC4 cells. B, The cell cycle was significantly arrested in G0/G1 phase in SiUSP32 group. C, Western blot analysis of proteins associated with cell cycle regulation in GLC4 cells after USP32 knockdown. D, The percentage of cells in sub‐G1 phase was significantly elevated in SiUSP32 group. Mock: untreated cells; NC: cells were treated with scrambled siRNA; SiUSP32: cells were treated with siRNA targeting USP32; all the results are represented as mean ± SD from three independent experiments. **P<.01, ***P<.001 as compared with NC cells; β‐actin was used as an internal control
As illustrated in Figure 6A, Annexin V‐APC vs 7‐AAD plots from the gated cells showed the population corresponding to viable and non‐apoptotic (Annexin V‐/7‐AAD−), early (Annexin V+/7‐AAD−) and late apoptotic (Annexin V+/7‐AAD+) cells. Representative images indicated more cells presented Annexin V+/7‐AAD− and Annexin V+/7‐AAD+ signals in siUSP32 groups than that in mock or NC groups in GLC4 cells. Statistical analysis further demonstrated knockdown of USP32 significantly promoted cell early and late apoptosis in GLC4 cells after USP32 silencing (Figure 6B, P<.001). Further investigation demonstrated that USP32 silencing upregulated the expression of pro‐apoptotic proteins, including cleaved caspase‐3, cleaved PARP and P53 (Figure 6C). Collectively, knockdown of USP32 significantly suppressed cell cycle progression and induced cell apoptosis, which might be a good explanation for cell growth inhibition caused by USP32 silencing.
Figure 6.
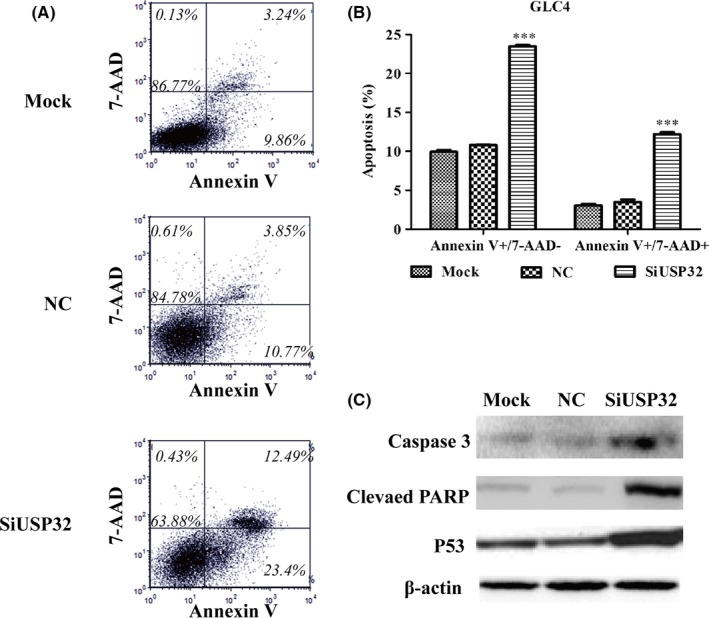
USP32 silencing remarkably promoted cell apoptosis, including early apoptosis and late apoptosis in GLC4 cells. A, GLC4 cells stained with Annexin V and 7‐AAD analysed using flow cytometry analysis. B, Quantification of the percentage of early apoptotic cells (Annexin V+/7‐AAD−) and late apoptotic cells (Annexin V+/7‐AAD+). C, Western blot analysis of proteins associated with cell apoptosis in GLC4 cells after USP32 knockdown. Mock: untreated cells; NC: cells were treated with scrambled siRNA; SiUSP32: cells were treated with siRNA targeting USP32; all the results were represented as mean ± SD from three independent experiments. ***P<.001 as compared with NC cells; β‐actin was used as an internal control
3.6. Silencing of USP32 by siRNA leading to decreased invasiveness
In the invasion assay, our results showed siUSP32 successfully reduced matrix invasiveness as compared with controls (Mock or NC) in H1688 cells (Figure 7A,B, P<.001). To determine the mechanism by which USP32 regulates the invasiveness in H1688 cells, we examined the expression levels of E‐cadherin and N‐cadherin, as markers of epithelial‐mesenchymal transition (EMT). As shown in Figure 7C, USP32 silencing significantly enhanced the expression of E‐cadherin, whereas decreased the expression of N‐cadherin.
Figure 7.
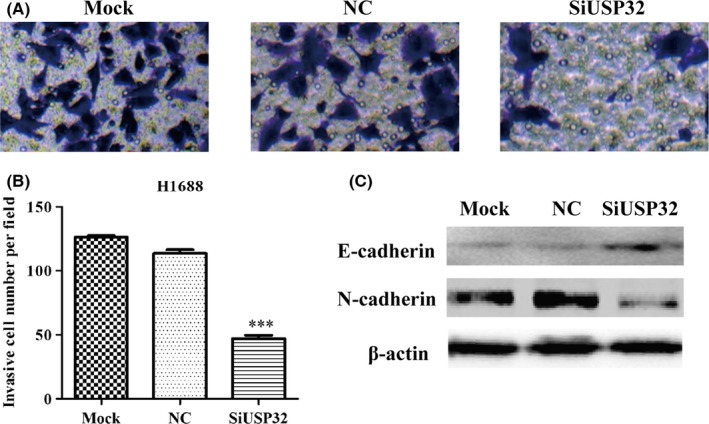
Knockdown of USP32 impaired the invasive potential of H1688 cells. A, Crystal violet staining images of invasive H1688 infected with siUSP32 or scrambled siRNA are shown. B, Quantification of the number of invaded cells are shown as mean ± SD. C, Western blot analysis of proteins associated with cell mobility in H1688 cells after USP32 knockdown. Mock: untreated cells; NC: cells were treated with scrambled siRNA; SiUSP32: cells were treated with siRNA targeting USP32; ***P<.001 as compared with NC cells; β‐actin was used as an internal control
4. DISCUSSION
It has long been known that USPs with more than 60 members comprise the largest class of DUBs, which have attracted great attentions from more and more researches for their function, substrates and important roles in the pathogenesis of many diseases, including cancer. USP32 is an active, membrane‐bound ubiquitin protease and has high sequence similarity to oncogene USP6. It has been reported that USP32 is overexpressed in breast cancer and its stably silencing causes a significant decrease in the proliferation and migration rate of cells;20 thus, its role in SCLC remains elusive. Here, we explored the effect of USP32 in SCLC in vitro. First, we showed USP32 was increased approximately 1.2‐fold in lung cancer by Oncomine and TCGA data‐mining. To further confirm its role in SCLC, we determined the expression of USP32 protein level in 10 pairs of fresh‐frozen SCLC tissue samples and found it was elevated nearly 2.5‐fold. This notably difference might be ascribed to small size of tissue specimens. Furthermore, we found that 54.6% paraffin‐embedded SCLC tissues showed strong and moderate USP32 staining, while the non‐cancerous tissues presented mainly negative expression of USP32. These findings strongly proved the higher expression of USP32 might be associated with tumorigenesis of SCLC.
Additionally, we found USP32 expression was correlated with disease stage and invasion, indicating high level of USP32 expression might be positively associated with worse tumour biological features. To further investigate the relationships between USP32 and malignant biological behaviour of SCLC, we determined the effects of USP32 silencing on SCLC cell growth and colony formation, two important indexes for tumorigenesis. Our studies indicated knockdown of USP32 significantly impaired SCLC cell growth and colony formation ability, which is consistent with the report of USP32 in breast cancer cells.20
Previous studies have shown that overexpression of USPs could cause the increase level of deubiquitinated variants of its corresponding substrates,26 which has also been described as an oncogene due to its resistance to apoptosis.27 Cyclin D1 is the best characterized substrate of USP,28 which has been demonstrated to be downregulated by small molecule inhibition of the USP2 leading to cell cycle arrest in colon cancer.29 These evidences suggest USP32, as USP might affect cell cycle progression and apoptosis in SCLC. In our results, inhibition of USP32 induced cell cycle arrest at G0/G1 phase by altering the expression of CDK4, Cyclin D1 and p21, which suggests that USP32 could positively regulate cell cycle progression in SCLC. Beside the induction of the cell cycle arrest in G0/G1 phase, we also observed USP32 silencing activated apoptosis, accompanied by increased cleaved caspase‐3, cleaved PARP and p53 as similarly reported in other USPs in cancer, including USP8,30, 31 USP1432 and USP22.33. These results were also in agreement with the role of USP39, and also belongs to the USPs family in tumour progression, such as hepatocellular carcinoma34 and prostate cancer.35 However, further investigation was still needed to confirm whether this well‐characterized DUB activity of USP32 knockdown altered cell cycle and apoptosis regulatory‐related proteins via direct transcriptional or post‐translational regulation in the cell proliferation machinery of SCLC cells.
In addition, some DUBs are now emerging as potential therapeutic targets that control cell migration and metastasis of multiple types of malignancies. For example, enforced expression of USP2a caused enhanced invasion and migration in bladder cancer.36 USP22 is positively associated with invasion and metastasis of thyroid cancer by altering EMT expression.33 EMT‐inducing transcriptional factors are extremely labile proteins and tightly regulated by the ubiquitin–proteasome system, which indicates the important role of USPs in controlling EMT.37 In this study, we showed that USP32 depletion significantly decreased the migration ability of SCLC cells accompanied by increased E‐cadherin and decreased N‐cadherin expression. These findings suggest the possibility of USP32 knockdown as an approach to control cancer cell metastasis therapy.
5. CONCLUSIONS
In summary, we first demonstrated that USP32 was upregulated in SCLC tissues compared with normal tissues. Functionally, USP32 depletion reduced the growth and migration of SCLC cells by regulating the expression of a series of pro‐tumorigenesis molecules. However, further investigation was still needed to elucidate whether USP32 is involved in other signalling pathways required for the progression of SCLC, which will help to better understanding tumour progression of SCLC and guide the development of therapeutic targets for SCLC.
AUTHORS’ CONTRIBUTIONS
WH and HW mainly performed experiments, interpreting data and drafting the manuscript. KL and PL were responsible for collecting tissue specimens and relevant clinicopathological data. JL participated in the interpretation of data and revised the manuscript. RF designed, supervised this study and revised the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ETHICAL APPROVAL
This article was approved by Medical Ethics Committee of Shandong Cancer Hospital, China.
INFORMED CONSENT
Informed consent was obtained from all individual participants included in the study.
Hu W, Wei H, Li K, Li P, Lin J, Feng R. Downregulation of USP32 inhibits cell proliferation, migration and invasion in human small cell lung cancer. Cell Prolif. 2017;50:e12343 10.1111/cpr.12343
REFERENCES
- 1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐86. [DOI] [PubMed] [Google Scholar]
- 2. Bordi P, Tiseo M, Barbieri F, et al. Gene mutations in small‐cell lung cancer (SCLC): results of a panel of 6 genes in a cohort of Italian patients. Lung Cancer. 2014;86:324‐8. [DOI] [PubMed] [Google Scholar]
- 3. Bunn PA Jr. Worldwide overview of the current status of lung cancer diagnosis and treatment. Arch Pathol Lab Med. 2012;136:1478‐81. [DOI] [PubMed] [Google Scholar]
- 4. Lally BE, Urbanic JJ, Blackstock AW, Miller AA, Perry MC. Small cell lung cancer: have we made any progress over the last 25 years? Oncologist. 2007;12:1096‐104. [DOI] [PubMed] [Google Scholar]
- 5. Gelsomino F, Rossi G, Tiseo M. MET and small‐cell lung cancer. Cancers. 2014;6:2100‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chan BA, Coward JI. Chemotherapy advances in small‐cell lung cancer. J Thorac Dis. 2013;5(suppl 5):S565‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wing SS. Deubiquitinating enzymes–the importance of driving in reverse along the ubiquitin‐proteasome pathway. Int J Biochem Cell Biol. 2003;35:590‐605. [DOI] [PubMed] [Google Scholar]
- 8. Nijman SM, Luna‐Vargas MP, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773‐86. [DOI] [PubMed] [Google Scholar]
- 9. Shen C, Ye Y, Robertson SE, Lau AW, Mak DO, Chou MM. Calcium/calmodulin regulates ubiquitination of the ubiquitin‐specific protease TRE17/USP6. J Biol Chem. 2005;280:35967‐73. [DOI] [PubMed] [Google Scholar]
- 10. Daviet L, Colland F. Targeting ubiquitin specific proteases for drug discovery. Biochimie. 2008;90:270‐83. [DOI] [PubMed] [Google Scholar]
- 11. Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010;140:384‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soncini C, Berdo I, Draetta G. Ras–GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene. 2001;20:3869‐79. [DOI] [PubMed] [Google Scholar]
- 13. Songlin P, Yanlong L, Jing H, et al. USP22 is useful as a novel molecular marker for predicting disease progression and patient prognosis of oral squamous cell carcinoma. PLoS ONE. 2012;7:e42540‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang H, Li YP, Chen JH, et al. Prognostic significance of USP22 as an oncogene in papillary thyroid carcinoma. Tumour Biol. 2013;34:1635‐9. [DOI] [PubMed] [Google Scholar]
- 15. Li X, Stevens PD, Yang H, et al. The deubiquitination enzyme USP46 functions as a tumor suppressor by controlling PHLPP‐dependent attenuation of Akt signaling in colon cancer. Oncogene. 2013;32:471‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paulding CA, Maryellen R, Haber DA. The Tre2 (USP6) oncogene is a hominoid‐specific gene. Proc Natl Acad Sci USA. 2003;100:2507‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pringle LM, Young R, Quick L, et al. Atypical mechanism of NF‐kappaB activation by TRE17/ubiquitin‐specific protease 6 (USP6) oncogene and its requirement in tumorigenesis. Oncogene. 2012;31:3525‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y, Martens JW, Yu JX, et al. Copy number alterations that predict metastatic capability of human breast cancer. Cancer Res. 2009;69:3795‐801. [DOI] [PubMed] [Google Scholar]
- 19. Grigoriadis A, Mackay A, Reis‐Filho JS, et al. Establishment of the epithelial‐specific transcriptome of normal and malignant human breast cells based on MPSS and array expression data. Breast Cancer Res. 2006;8:R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Akhavantabasi S, Akman HB, Sapmaz A, Keller J, Petty EM, Erson AE. USP32 is an active, membrane‐bound ubiquitin protease overexpressed in breast cancers. Mamm Genome. 2010;21:388‐97. [DOI] [PubMed] [Google Scholar]
- 21. Yoshikawa D, Ojima H, Iwasaki M, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer. 2008;98:418‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Su LJ, Chang CW, Wu YC, et al. Selection of DDX5 as a novel internal control for Q‐RT‐PCR from microarray data using a block bootstrap re‐sampling scheme. BMC Genom. 2007;8:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landi MT, Dracheva T, Rotunno M, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE. 2008;3:e1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Selamat SA, Chung BS, Girard L, et al. Genome‐scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012;22:1197‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Okayama H, Kohno T, Ishii Y, et al. Identification of genes upregulated in ALK‐positive and EGFR/KRAS/ALK‐negative lung adenocarcinomas. Cancer Res. 2012;72:100‐11. [DOI] [PubMed] [Google Scholar]
- 26. Graner E, Tang D, Rossi S, et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell. 2004;5:253‐61. [DOI] [PubMed] [Google Scholar]
- 27. Priolo C, Tang D, Brahamandan M, et al. The isopeptidase USP2a protects human prostate cancer from apoptosis. Cancer Res. 2006;66:8625‐32. [DOI] [PubMed] [Google Scholar]
- 28. Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Davis MI, Pragani R, Fox JT, et al. Small molecule inhibition of the ubiquitin‐specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. J Biol Chems. 2016;291:24628‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jeong CH. Inhibition of ubiquitin‐specific peptidase 8 suppresses growth of gefitinib‐resistant non‐small cell lung cancer cells by inducing apoptosis. J Cancer Prev. 2015;20:57‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jian FF, Li YF, Chen YF, et al. Inhibition of ubiquitin‐specific peptidase 8 suppresses adrenocorticotropic hormone production and tumorous corticotroph cell growth in AtT20 cells. Chin Med J. 2016;129:2102‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Y, Wang J, Zhong J, et al. Ubiquitin‐specific protease 14 (USP14) regulates cellular proliferation and apoptosis in epithelial ovarian cancer. Med Oncol. 2015;32:379. [DOI] [PubMed] [Google Scholar]
- 33. Zhao HD, Tang HL, Liu NN, et al. Targeting ubiquitin‐specific protease 22 suppresses growth and metastasis of anaplastic thyroid carcinoma. Oncotarget. 2016;7:31191‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pan Z, Pan H, Zhang J, et al. Lentivirus mediated silencing of ubiquitin specific peptidase 39 inhibits cell proliferation of human hepatocellular carcinoma cells in vitro. Biol Res. 2015;48:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang Y, Pan XW, Li L, et al. Overexpression of USP39 predicts poor prognosis and promotes tumorigenesis of prostate cancer via promoting EGFR mRNA maturation and transcription elongation. Oncotarget. 2016;7:22016‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim J, Kim WJ, Liu Z, Loda M, Freeman MR. The ubiquitin‐specific protease USP2a enhances tumor progression by targeting cyclin A1 in bladder cancer. Cell Cycle. 2012;11:1123‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inoue Y, Itoh Y, Sato K, et al. Regulation of epithelial‐mesenchymal transition by E3 ubiquitin ligases and deubiquitinase in cancer. Curr Cancer Drug Targets. 2016;16:110‐8. [DOI] [PubMed] [Google Scholar]