

Abstract
Objectives
Maternal gestational diabetes leads to an adverse in utero environment and increases the risk of malformations during embryo organogenesis. In the present study, we analysed the effects of maternal diabetes on tooth germ cell proliferation and apoptosis in offspring, and investigated their underlying mechanisms.
Materials and methods
A rat model of maternal diabetes was induced by intraperitoneal injection of streptozotocin and the pregnant rats were divided into three groups: controls, the diabetic group and diabetic group with insulin treatment. Offspring of the three groups were collected and cell proliferation and apoptosis in tooth germs were analysed. Primary dental papilla cells and dental epithelial stem cells were isolated and treated with high glucose in vitro, in an attempt to simulate maternal diabetes‐induced hyperglycaemia in vivo.
Results
Maternal diabetes significantly affected cell proliferation and apoptosis in offspring tooth germs. The TLR4/NF‐ĸB signalling pathway was activated in the tooth germs of offspring of diabetic dams. High glucose treatment activated the TLR4/NF‐ĸB signalling pathway in primary dental papilla cells and dental epithelial stem cells in vitro, resulting in suppression of cell proliferation and enhancement of apoptosis. TLR4 knockdown significantly reduced adverse effects induced by high glucose treatment.
Conclusions
Maternal gestational diabetes significantly impaired dental epithelial and mesenchymal cell proliferation and apoptosis in offspring, possibly by activation of the TLR4/NF‐ĸB signalling pathway.
Keywords: apoptosis, cell proliferation, maternal diabetes, TLR4/NF‐ĸB signalling, tooth development
1. Introduction
Due to the characteristics of modern lifestyle, such as changes in dietary habits and advanced maternal age at first pregnancy, gestational diabetes is becoming more and more prevalent throughout the world.1 An increasing number of studies have indicated that maternal gestational diabetes is the key risk factor for birth defects in the offspring of diabetic mothers. Both human and animal model studies have previously indicated that hyperglycaemia induced by diabetic pregnancy is responsible for the malformations during embryo organogenesis.2, 3, 4, 5 Exposure of offspring to intrauterine hyperglycaemia might result in developmental dysplasia, which can affect many organ systems of the body, including the central nervous system, craniofacial structures, cardiovascular system and immune system.6, 7, 8, 9, 10, 11 Maternal diabetes is also associated with altered offspring tooth development. Epidemiological and animal models have demonstrated that maternal diabetes affects the tooth developmental process by impairing tooth eruption and mineralization, and altering tooth dimensions.12, 13, 14, 15 However, the mechanisms by which maternal diabetes affects tooth development have not yet been well investigated.
Maternal diabetes is often accompanied by chronic systemic inflammation and an increased level of proinflammatory cytokines.16, 17 It has been profoundly studied that chronic inflammation induced by maternal diabetes is likely to be responsible for the adverse effects of maternal gestational diabetes on both mother and offspring.16, 17, 18 Toll‐like receptor 4/nuclear factor kappa B (TLR4/NF‐ĸB) signalling pathway is one of the important pathways activated by maternal diabetes, and its activation leads to the downstream release of inflammatory modulators, including interleukin (IL)‐1, IL‐6 and tumour necrosis factor‐alpha (TNF‐α).19, 20 Ligands binding to TLR4 cause conformational changes to recruit the adaptor proteins, MYD88 and TRAF6. This increases the phosphorylation and translocation of NF‐ĸB to the cell nucleus, where it subsequently binds to its target promoter region and activates transcription.21 Moreover, recent reports have demonstrated that the TLR4/NF‐ĸB signalling pathway is of critical importance in impairing embryo development.22, 23 TLR4 knockout has been shown to significantly attenuate the proinflammatory state induced by diabetes, indicating that TLR4 might play a key role in triggering intracellular signalling cascades leading to the activation of proinflammatory signalling pathways.24, 25 TLR4/NF‐ĸB signalling is also closely associated with the dental tissue‐derived stem cells.26, 27, 28 However, the role of TLR4/NF‐ĸB signalling activated by maternal diabetes during the offspring tooth development remains largely unknown.
2. Materials and methods
2.1. Animals and animal care
All animal experimental procedures used were approved by the Institutional Animal Care and Use Committee of Sichuan University (No.: WCCSIRB‐D‐2016‐048). All rats (body weight: 206±25 g) were housed under standard humidity and lighting conditions (12 hours light‐dark cycles) with free access to standard rat chow and water. Maternal diabetes rat model was induced by a single intraperitoneal injection of streptozotocin (STZ) at a dosage of 75 mg kg−1 body weight. STZ is a pancreatic beta‐cell toxin that is widely used to experimentally manipulate insulin levels. The rodent model of STZ‐induced diabetes has been accepted internationally in the research of diabetic embryopathy for decades.29, 30, 31, 32, 33, 34, 35 On E9.5 of pregnancy, when tooth development initiated,36 the pregnant rats were administered with STZ. Induction of diabetes was confirmed at E11.5 by measuring the blood glucose concentrations via the tail vein, and only those animals with plasma glucose levels higher than 15 mmol L−1 were included. For the insulin treatment group (Dia+Ins; n=18), dams at E11.5 were surgically implanted with insulin pellets subcutaneously. Control group (Con; n=18) and diabetic group (Dia; n=18) animals underwent sham surgery that did not involve insulin pellet implantation. The body weight and blood glucose levels of animals were monitored during pregnancy, and after delivery, they were monitored from the neonatal stage to 6 weeks of age (n=9 pups/group for each group; total three separate litters pups [3 pups/litter] were randomly collected). In this study, only male offspring were selected from each litter and used in subsequent experiments.
2.2. Sample collection
At E15.5, E17.5 and postnatal day 0.5 (P0.5), the offspring mandibular specimens were collected and placed in 4% paraformaldehyde and 0.1 mol L−1 phosphate‐buffered saline at pH 7.0 at 4°C for 24 hour, and then embedded in paraffin for histopathological detection. At P0.5, the newborn offspring from each of the dams were collected and the lower first molar was obtained for detection of gene or protein expression. Because of their small size, the first molars from the same litter were pooled and considered as an experimental unit. A total of three experimental units were used for gene or protein expression analysis (n=3).
2.3. Cell culture
For primary dental papilla cells (DPCs) cultures, the dental papilla was separated from the first mandibular molar germs of neonatal rats and digested in a solution containing 3 mg mL−1 type I collagenase and 4 mg mL−1 dispase for 10 minutes at 37°C. Single cell suspensions and dental papilla tissue were cultured in alpha‐minimum essential medium supplemented with 10% foetal bovine serum. DPCs in passage 5 were used for this study. For dental epithelial stem cells (DESCs) cultures,37 the cervical loops were dissected from neonatal rats and digested with collagenase to obtain single cell suspensions. Cells were cultured in DMEM/F12 medium supplemented with epidermal growth factor, basic fibroblast growth factor and 10% foetal bovine serum. All cells were grown in a humidified atmosphere at 37°C with 5% CO2, and the medium was changed every 3 days. For high glucose (HG) treatment, the cells were incubated in a medium containing 30 mmol L−1 d‐glucose, and the control cells were incubated in normal glucose medium (5.6 mmol L−1 d‐glucose) containing 24.4 mmol l −1‐mannitol to ensure equiosmolar control.
2.4. Real‐time PCR
Total RNA was extracted using RNAiso plus (TaKaRa Biotechnology, Shig, Japan) according to the manufacturer's protocol. Reverse transcription of the isolated RNA was performed using the Thermo Scientific RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Vilnius, Lithuania). Real‐time PCR was performed on a Mastercycler ep realplex (Eppendorf, Hamburg, Germany) with the SYBR Premix Ex Taq (TaKaRa Biotechnology, Shiga, Japan). PCR primer sequences are shown in Table S1. The relative gene expression levels were calculated using the comparative CT method (2−ΔΔCt) with β‐actin as an internal control. Experiments were performed independently for each sample and at least three technical replicates were run for each treated sample and controls.
2.5. Western blot and immunohistochemistry
Western blot and immunohistochemistry were conducted as previously described by us.38 For western blot analysis, the tissue samples or cell pellets were lysed with RIPA buffer containing complete protease inhibitor cocktail (Millipore Calbiochem, San Diego, USA). Proteins were quantified using the BCA Protein Assay (Bio‐Rad, Richmond, USA) and equal amounts (20 μg) of protein lysates from each sample were separated by SDS–PAGE. The proteins were then transferred onto PVDF membranes. The membranes were blocked with 5% skim milk and then incubated with primary antibodies, such as mouse anti‐TLR4 (1:1000; ab22048; Abcam,Cambridge, UK), rabbit anti‐MYD88 (1:1000; AB21009a; BBI, Sangon Biotech, Shanghai, China), rabbit anti‐TRAF6 (1:1000; AB21362a; BBI, Sangon Biotech, Shanghai, China), rabbit anti‐p‐P65 (1:1000; sc‐101751; Santa Cruz Biotechnology, Santa Cruz, USA), rabbit anti‐P65 (1:1000; ab7970; Abcam, Cambridge, UK), and mouse anti‐GAPDH (1:5000; 200306‐7E4; Zen Bioscience, Chengdu, China). The membranes were then thoroughly rinsed and incubated with species‐matched HRP‐conjugated secondary antibody. The protein bands were visualized using Amersham ECL Select Western blotting detection reagent (GE Healthcare Life Sciences, Piscataway, USA) in accordance with the manufacturer's protocol. Blot images were captured on ImageQuant LAS 4000 mini (GE Healthcare Life Sciences) and quantified by densitometric scanning (Image Quant TL; GE Healthcare Life Sciences). For immunohistochemical staining, tooth germ samples were fixed in 4% buffered paraformaldehyde and 6‐μm sections were made for staining. Immunohistochemical staining was performed with the ChemMateTM EnVisionTM Detection Kit (Gene Tech, Shanghai, China) according to the manufacturer's protocol. Primary antibodies against TLR4 (1:200; ab22048; Abcam), MYD88 (1:200; AB21009a; BBI), TRAF6 (1:200; AB21362a; BBI), p‐P65 (1:200; sc‐101751; Santa Cruz) and Ki67 (1:200; ab15580; Abcam) were used. All experiments were performed independently for each sample and at least three technical replicates were run for each treated sample and controls.
2.6. Cell proliferation and apoptosis detection
In the tooth germ of the mandibular first molar, cell apoptosis was detected using TUNEL assay kit (Roche Diagnostics, Indianapolis, USA) according to the supplier's instructions, and cell proliferation was detected via Ki67 immunostaining. TUNEL‐ or Ki67‐positive cells in the tooth germ area of each section were counted. At least five serial sections were counted in one animal and at least three animals were included in one group. Thus, the mean apoptotic or proliferative cell numbers were expressed as total TUNEL‐ or Ki67‐positive cells per tooth germ area per section, respectively. In the cultured cells, apoptosis was detected by flow cytometry using annexin V‐FITC and propidium iodide (PI) dye (BD Biosciences, San Jose, USA) according to the supplier's instructions.
2.7. Small interfering RNA (siRNA) treatment
The cells (5×106) were seeded into six‐well plates and were grown until 60–80% confluence. The cells were transiently transfected with 150 pM of TLR4‐specific siRNA or negative control (NC) siRNA (Ribo Bio, Guangzhou, China) using Lipofectamine ™ RNAiMAX (Invitrogen, Carlsbad, USA) transfection reagent according to the manufacturer's instructions. After 72 hour, TLR4 mRNA and protein levels were detected by quantitative real‐time PCR and western blotting.
2.8. Statistical analysis
All data are presented as the mean value±standard deviation (SD) of each group. Statistical significance between experimental groups was analysed initially by t tests or ANOVA, followed by the Bonferroni test, as appropriate. P<.05 was considered to be statistically significant.
3. Results
3.1. Characteristics of diabetic mothers and offspring
The blood glucose levels of diabetic dams were significantly higher than those of the control dams on pregnancy day 11.5 (Figure 1A). Diabetes induced a significant loss of body weight (Figure 1B). Hyperglycaemia and loss of body weight in dams were ameliorated via insulin administration (Figure 1A and B). In case of the offspring from diabetic dams, their body weights were significantly lower than those of control offspring until 6 weeks of age (Figure 1C), and insulin administration to the mothers significantly ameliorated the effect of maternal diabetes on the offspring body weight. However, there was no significant difference in the blood glucose levels of the three groups of offspring (Figure 1D).
Figure 1.
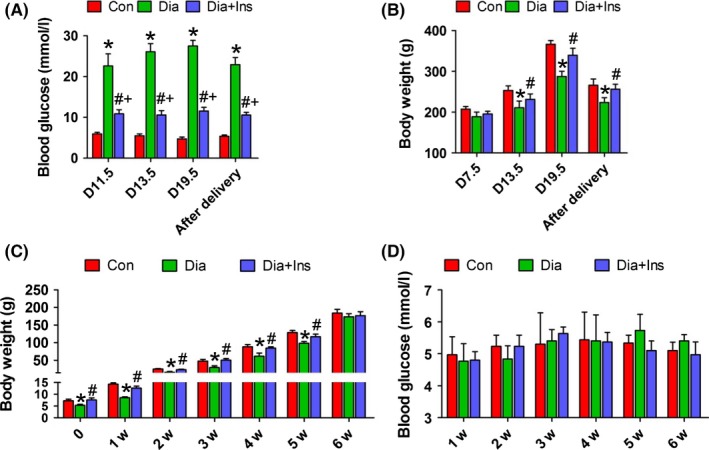
Characteristics of diabetic mothers and offspring. A, blood glucose levels of dams. B, body weight of dams. C, body weight of offspring from neonates to 6 weeks of age. D, blood glucose levels of offspring from neonates to 6 weeks of age. *P<.05 for diabetic vs control group, # P<.05 for diabetic+insulin vs diabetic group and + P<.05 for diabetic+insulin vs control group
3.2. Maternal diabetes or high glucose treatment affected dental mesenchymal and epithelial cell proliferation and apoptosis
To determine whether STZ toxicity could affect odontogenesis and cell proliferation and apoptosis of tooth germs in vivo, we first analysed the morphology, cell proliferation and apoptosis of the tooth germ of embryos or neonatal rats from mothers with or without STZ administration. STZ is unstable and its biological half‐life in solution is approximately 19 minutes (http://www.sigmaaldrich.com). Because STZ administration does not induce diabetes at all times, we examined the tooth germ morphology, proliferation and apoptosis of the embryos or neonates from STZ‐exposed rats with or without diabetes. The results showed that tooth germ cell proliferation (Figure 2A and E) and apoptosis (Figure 2B and F) seemed to depend on the level of maternal hyperglycaemia, but was independent of STZ administration (Figure 2C and D). Thus, data from our model suggested that it is unlikely that a small amount of STZ exerts toxicity in the foetus.
Figure 2.
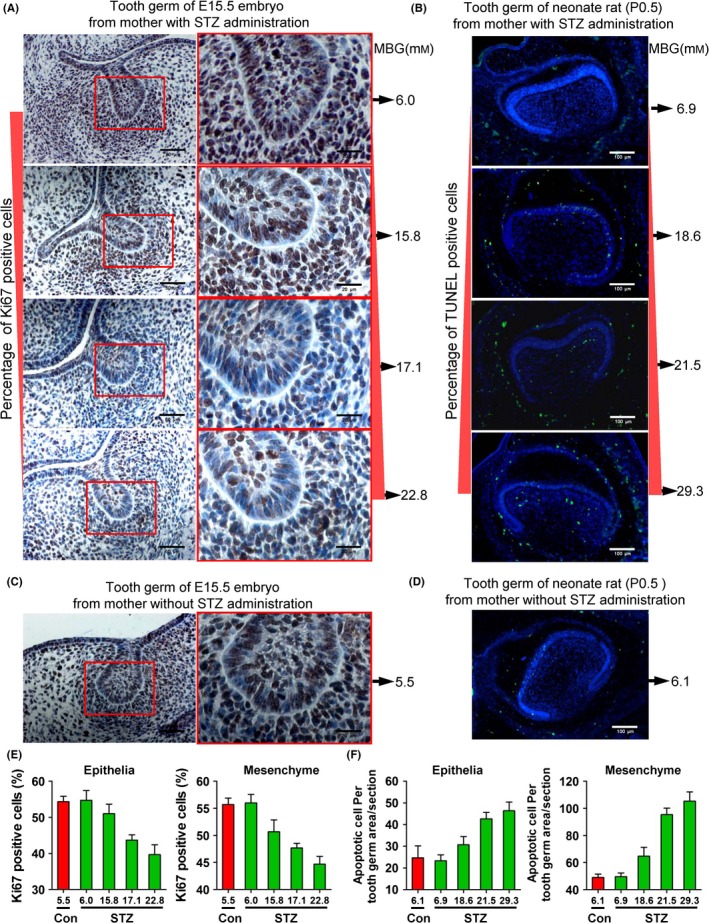
STZ toxicity analysis. A, Cell proliferation detection (Ki67 immunostaining) of E15.5 tooth germ of embryos from pregnant rats with four different maternal hyperglycaemic levels induced by STZ administration. B, Cell apoptosis (TUNEL) detection of P0.5 tooth germ of neonatal rats from pregnant dams with four different maternal hyperglycaemic levels induced by STZ administration. C, Cell proliferation detection (Ki67 immunostaining) of E15.5 tooth germ of embryos from pregnant rats in normal maternal glucose range without administration of STZ. D, Cell apoptosis (TUNEL) detection of P0.5 tooth germ of neonatal rat from pregnant dams in normal maternal glucose range without administration of STZ; MBG, maternal blood glucose. E and F, Quantitative analysis of Ki67 (E)‐ and TUNEL (F)‐positive cells of tooth epithelia and mesenchyme; the horizontal axis represents the blood glucose concentrations with or without (Con) administration of STZ
As shown in Figure 3, maternal diabetes significantly decreased the number of Ki67‐positive cells in the tooth germ epithelium and mesenchyme from E15.5 to P0.5, and a reversion of decreased cell proliferation was observed in the tooth germ of offspring from insulin‐treated diabetic dams (Figure 3A–C). As shown in Figure 4, TUNEL‐positive cells were observed in the E17.5 and P0.5 tooth germs, but not in the E15.5 tooth germs of all the three offspring groups; however, in the E17.5 tooth germ, TUNEL‐positive cells were only detected in the tooth mesenchyme (Figure 4A). Compared to the control offspring, the tooth germ of offspring of diabetic dams showed a higher number of TUNEL‐positive cells in the epithelium and mesenchyme in P0.5, and attenuation of increased apoptosis was also observed in the offspring of insulin‐treated dams (Figure 4B and C). The in vitro treatment of primary DPCs and DESCs with HG for 72 hour significantly suppressed cell growth (Figure 3D and E) and enhanced cell apoptosis (Figure 4D). In addition, treatment of primary DPCs or DESCs with mannitol did not affect cell growth (Fig. S1) or apoptosis (Figure 4D and Fig. S2). These results indicated that tooth germ cell proliferation and apoptosis is related to a HG milieu induced by maternal diabetes.
Figure 3.
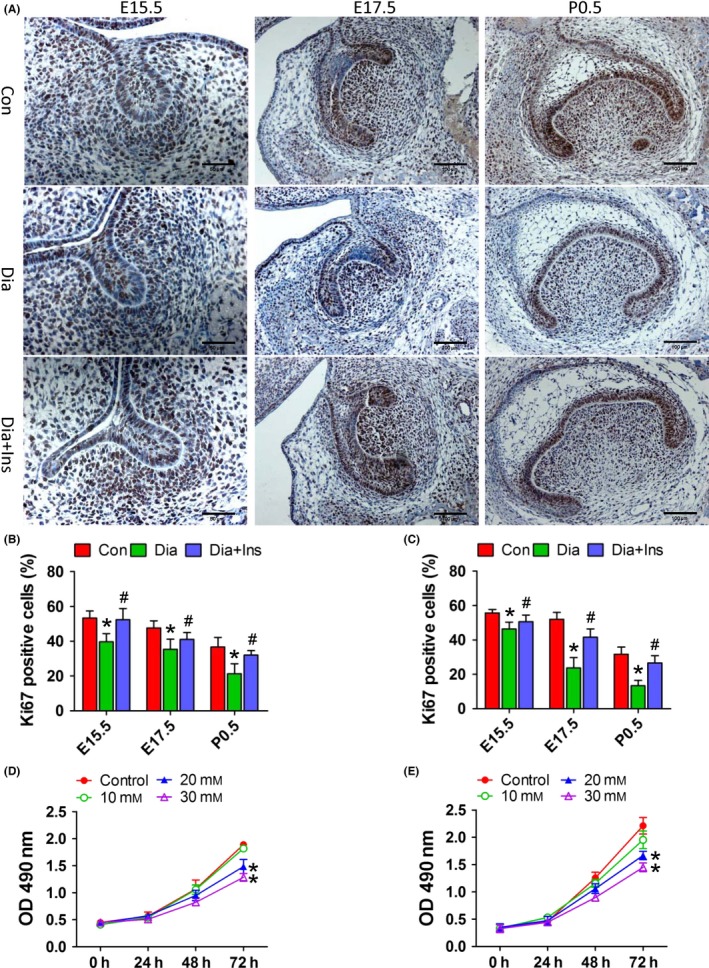
Maternal diabetes in vivo and high glucose in vitro suppressed dental epithelial and mesenchymal cell proliferation. A, Ki67 immunohistochemical analysis of molar germs from E15.5 to P0.5 in the offspring from control dams, diabetic dams and diabetic dams with insulin treatment. B and C, Quantitative analysis of Ki67‐positive cells of tooth epithelia (B) and mesenchyme (C). D and E, Cell growth curve of primary DESCs (D) and DPCs (E) under increasing glucose concentrations. *P<.05 for diabetic vs control group, # P<.05 for diabetic+insulin vs diabetic group
Figure 4.
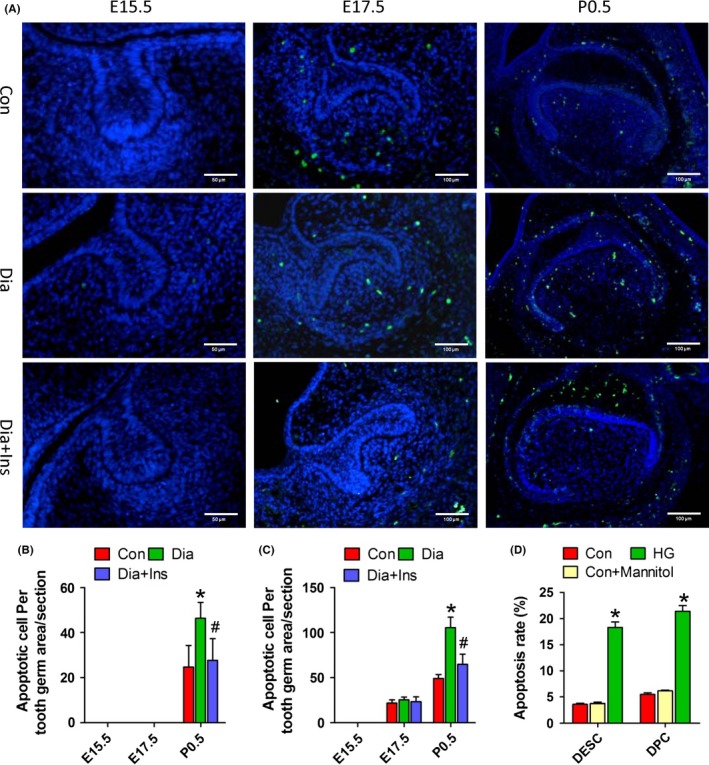
Maternal diabetes in vivo and high glucose in vitro enhanced dental epithelial and mesenchymal cell apoptosis. A, TUNEL assay of molar germs from E15.5 to P0.5 in the offspring from control dams, diabetic dams and diabetic dams with insulin treatment. B and C, Quantitative analysis of TUNEL‐positive cells of tooth epithelia (B) and mesenchyme (C). D, Quantitative assessment of apoptosis rate by annexin V/FITC‐PI labelling using flow cytometry. Primary DESCs or DPCs were treated with HG or mannitol for 72 hour. *P<.05 for diabetic vs control group, # P<.05 for diabetic+insulin vs diabetic group
3.3. Maternal diabetes activated TLR4/NF‐κB p65 signalling pathway in the molars of offspring
Immunohistochemical (Figure 5A) and western blot analysis (Figure 5B) revealed that the expression of TLR4, MYD88, TRAF6 and p‐P65 was significantly increased in the neonatal tooth germ of offspring of diabetic dams, and this increase was attenuated in the offspring of insulin‐treated diabetic dams. Moreover, the expression of proinflammatory cytokines, such as IL‐1α, IL‐1β, IL‐6 and TNF‐α was also increased in offspring of diabetic dams, but significantly decreased with insulin treatment (Fig. S3). These results indicated that maternal diabetes activates the TLR4/NF‐κB p65 signalling pathway during molar development.
Figure 5.
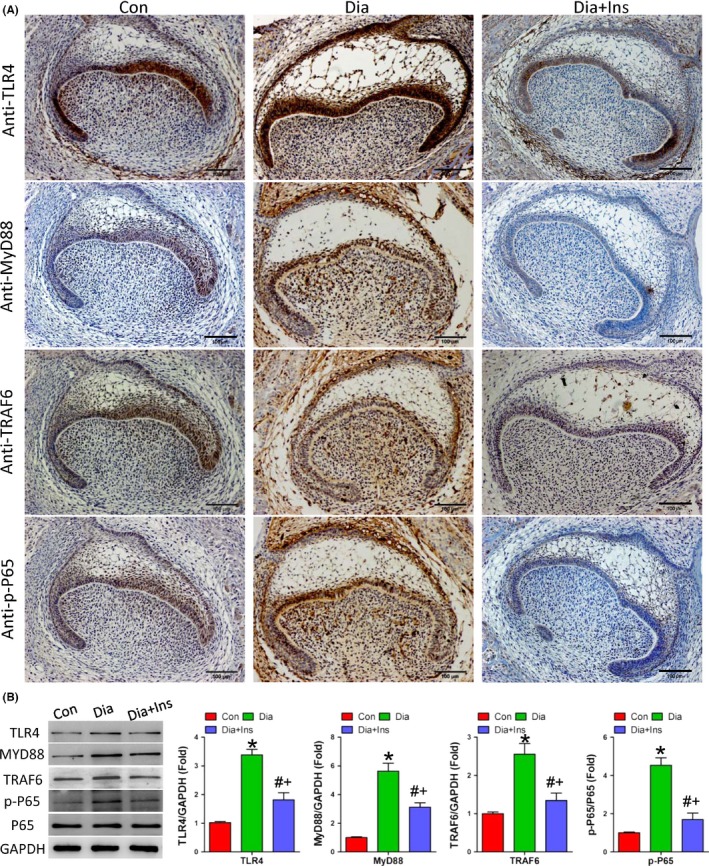
Maternal diabetes modulated TRL4/NF‐κB p65 signalling pathway in the offspring tooth germ. A, Immunohistochemical detection of TLR4, MYD88, TRAF6 and p‐P65 expression in offspring molar in P0.5 of each group. B, Western blot and quantitative analysis of TLR4, MYD88, TRAF6 and p‐P65 expression in each group offspring molar germ in P0.5. *P<.05 for diabetic vs control group, # P<.05 for diabetic+insulin vs diabetic group and + P<.05 for diabetic + insulin vs control group
3.4. High glucose treatment activated TLR4/NF‐κB p65 signalling which affected dental epithelial and mesenchymal cell proliferation and apoptosis
As shown in Figure 6, HG treatment activated the TLR4/NF‐κB p65 signalling pathway in both DPCs and DESCs in a dose‐dependent manner (Figure 6A and B). Transfection with TLR4‐specific siRNA significantly attenuated the activation of MYD88, TRAF6 and NF‐κB p65 induced by HG treatment, in both DPCs and DESCs (Figure 6C and D). The expression of proinflammatory mediators, such as IL‐1α, IL‐1β, IL‐6 and TNF‐α was also activated by high HG treatment, and attenuated by the TLR4‐specific siRNA transfection of DPCs and DESCs (Figure 6E and F).
Figure 6.
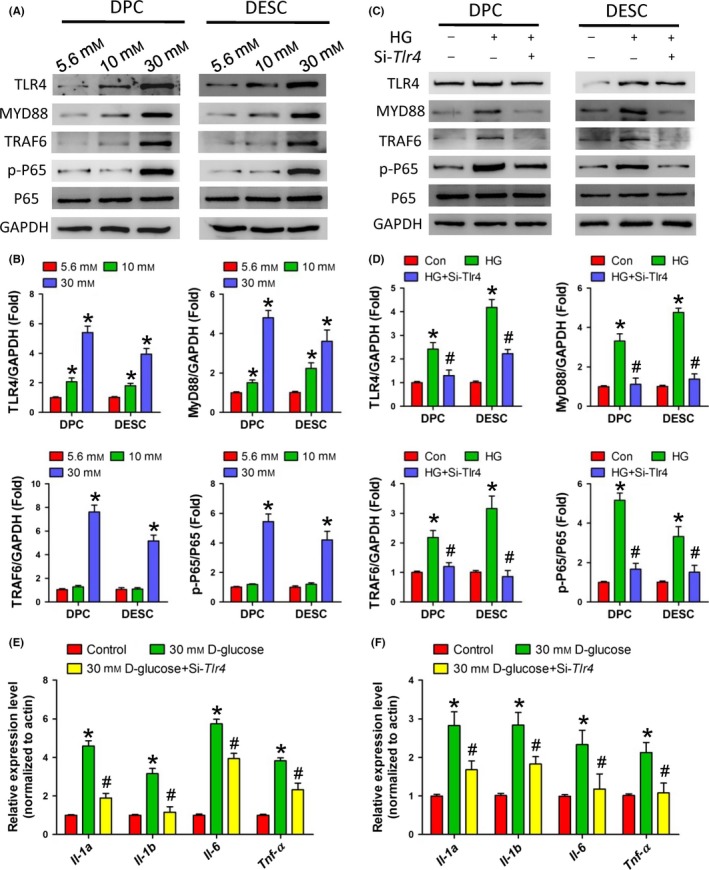
High glucose treatment in vitro activated the TRL4/NF‐κB p65 signalling pathway in dental epithelial and mesenchymal cells. A and B, Western blot (A) and quantitative analysis (B) showed the activation of TRL4/NF‐κB p65 signalling pathway in primary DPCs and DESCs induced by HG treatment. C and D, Western blot (C) and quantitative analysis (D) showed the activation of NF‐κB p65 in primary DPCs and DESCs induced by HG treatment via TLR4. E and F, Real‐time PCR detected the expression of the proinflammatory cytokines, IL‐1α, IL‐1β, IL‐6 and TNF‐α, induced by HG treatment via TLR4. *P<.05 vs control group, # P<.05 vs HG group
For cell proliferation, HG (30 mmol L−1) treatment significantly suppressed DPCs (Figure 7A) or DESCs (Figure 7B) growth in 48 hour or 72 hour, while transfection with a TLR4‐specific siRNA significantly reversed this effect (Figure 7A and B). Cell apoptosis detected by flow cytometry also showed that HG treatment enhanced apoptosis and this effect was significantly abolished by a transient transfection with TLR4‐specific siRNA (Figure 7C and D and Fig. S2). These results indicated that HG affects DPCs and DESCs proliferation and apoptosis via the activation of TLR4/NF‐κB p65 signalling pathway.
Figure 7.
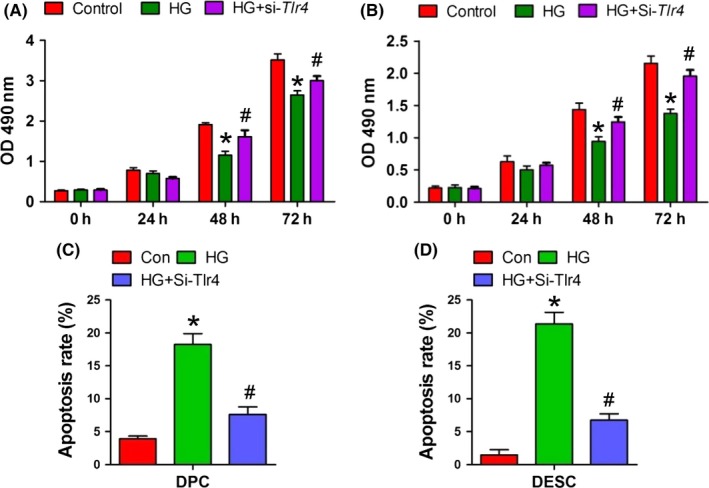
High glucose treatment in vitro affected dental mesenchymal and epithelial cell proliferation and apoptosis via TRL4/NF‐κB p65 signalling pathway. A and B, Cell growth analysis by CCK8 assay after treatment with HG alone or in combination with TLR4‐specific siRNA (A, DPCs; B, DESCs). C and D, Analysis of cell apoptosis by flow cytometry after treatment with HG alone or in combination with TLR4‐specific siRNA (C, DPCs; D, DESCs). *P<.05 vs control group, # P<.05 vs HG group
4. Discussion
In the present study, we aimed to demonstrate the functional role of maternal diabetes in modulating offspring dental development and to investigate its underlying mechanisms. Our data indicated that a hyperglycaemic environment in utero affects dental epithelial and mesenchymal cell proliferation and apoptosis. These changes were accompanied by the activation of TLR4/NF‐κB p65 signalling pathway. Insulin treatment of diabetic dams normalized these parameters in the teeth of their offspring. In vitro cell culture and knockdown of TLR4 significantly abolished the adverse effects of HG treatment on dental epithelial and mesenchymal cells, suggesting that TLR4/NF‐κB p65 signalling was responsible for the aberrant cell proliferation and apoptosis of tooth germs induced by maternal diabetes. To our knowledge, this is the first study reporting that maternal diabetes affects offspring tooth development via activation of TLR4/NF‐κB p65 signalling pathway.
Maternal diabetes presents an environmental challenge in utero and may fundamentally and dynamically impair the process of embryogenesis and organogenesis. Numerous studies have shown that maternal diabetes results in an increased risk of congenital malformations in the offspring.3, 4, 5, 39, 40 There are many organ systems in the offspring that can be affected by maternal diabetes, such as the nerve system, cardiovascular system and urinary system.6, 7, 8 Previous studies have also indicated that maternal diabetes affects offspring tooth development.12, 13, 14, 15 However, the pathological mechanisms by which it induces abnormal tooth development in the offspring are not fully understood. Studies have shown that diabetes is a proinflammatory state, and in vivo hyperglycaemia or in vitro HG level induces NF‐κB activity and inflammatory cytokine expression.39, 41 Maternal diabetes has also been reported to contribute to the proinflammatory state in the embryonic or postnatal stage of the offspring,3, 40 which modulates organogenesis of the neural tube, heart and kidney.41, 42 The present study also revealed that maternal diabetes elevates the expression of offspring tooth germ proinflammatory mediators, including TLR4/NF‐ĸB and its targets IL‐1α, IL‐1β, IL‐6 and TNF‐α (Figures 5 and S3). Studies performed in both diabetic and control rat embryos, cultured under hyperglycaemic conditions during organogenesis, have suggested that hyperglycaemia is the major teratogen in diabetic pregnancies.39, 40 During embryo organogenesis, maternal diabetes leads to the production of a wide range of proinflammatory agents, which increase embryo resorption and malformation rates, placental dysfunction and foetal alterations that lead to increased neonatal morbidity and mortality rates.16 Together with these data, we speculated that maternal diabetes‐induced activation of TLR4/NF‐ĸB in the tooth germ might alter the offspring tooth development.
Toll‐like receptor family, belonging to innate immune receptors, plays a critical role in pathogen recognition and activation of innate immunity. Toll‐like receptor signalling induces immune responses through the adaptor molecules MYD88 and TRAF6, leading to the activation of NF‐κB, and subsequently elevating the expression of proinflammatory cytokines and chemokines.21 Recently, studies have shown that the TLR4/NF‐ĸB signalling pathway is involved in various diseases, including rheumatoid arthritis, cardiovascular diseases and nervous system diseases.21 In the present study, we found that maternal diabetes activated the TLR4/NF‐κB signalling pathway in the offspring tooth germ. To investigate the mechanism underlying this activation, we performed an in vitro knockdown of TLR4 in DPCs and DESCs using siRNA. Results showed that HG treatment significantly activated the TLR4/NF‐κB signalling pathway and upregulated the expression of the proinflammatory cytokines, IL‐1α, IL‐1β, IL‐6 and TNF‐α. However, knockdown of TLR4 significantly inhibited the activation of NF‐κB and proinflammatory cytokine expression, which was induced by HG treatment. Previous mice studies have also reported that knockout of TLR4 attenuates the proinflammatory state of diabetes.24 More importantly, in our study the knockdown of TLR4 expression significantly reversed the proliferation‐inhibition, and attenuated cell apoptosis under HG conditions. These data were consistent with the results from our animal model, suggesting that cell proliferation and apoptosis play an important role in normal tooth development.43 The aberrant cell proliferation, together with apoptosis, might induce odontodysplasia. All these data indicated that maternal diabetes affects the offspring tooth germ cell proliferation and apoptosis by activating the TLR4/NF‐κB signalling pathway.
In summary, we demonstrated that maternal diabetes leads to an abnormal development of offspring molar and affects offspring dental mesenchymal and epithelial cell proliferation and apoptosis via activation of TLR4/NF‐κB signalling pathway.
Supporting information
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81271119), the China Postdoctoral Science Foundation (2012M511934), and the Basic Research Program of Sichuan Province of China (2013JY0019).
Chen G, Sun W, Liang Y, Chen T, Guo W , Tian W. Maternal diabetes modulates offspring cell proliferation and apoptosis during odontogenesis via the TLR4/NF‐κB signalling pathway. Cell Prolif. 2017;50:e12324 10.1111/cpr.12324
Contributor Information
Weihua Guo, Email: guoweihua943019@163.com.
Weidong Tian, Email: drtwd@sina.com.
References
- 1. Ferrara A. Increasing prevalence of gestational diabetes mellitus: a public health perspective. Diabetes Care. 2007;30:S141–S146. [DOI] [PubMed] [Google Scholar]
- 2. Eriksson UJ, Cederberg J, Wentzel P. Congenital malformations in offspring of diabetic mothers–animal and human studies. Rev Endocr Metab Disord. 2003;4:79–93. [DOI] [PubMed] [Google Scholar]
- 3. Gäreskog M, Cederberg J, Eriksson UJ, et al. Maternal diabetes in vivo and high glucose concentration in vitro increases apoptosis in rat embryos. Reprod Toxicol. 2007;23:63–74. [DOI] [PubMed] [Google Scholar]
- 4. Akazawa S. Diabetic embryopathy: studies using a rat embryo culture system and an animal model. Congenit Anom (Kyoto). 2005;45:73–79. [DOI] [PubMed] [Google Scholar]
- 5. Zhao Z, Reece EA. New concepts in diabetic embryopathy. Clin Lab Med. 2013;33:207–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hokke SN, Armitage JA, Puelles VG, et al. Altered ureteric branching morphogenesis and nephron endowment in offspring of diabetic and insulin‐treated pregnancy. PLoS ONE. 2013;8:e58243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vijaya M, Manikandan J, Parakalan R, et al. Differential gene expression profiles during embryonic heart development in diabetic mice pregnancy. Gene. 2013;516:218–227. [DOI] [PubMed] [Google Scholar]
- 8. Wei D, Loeken MR. Increased DNA methyltransferase 3b (Dnmt3b)‐mediated CpG island methylation stimulated by oxidative stress inhibits expression of a gene required for neural tube and neural crest development in diabetic pregnancy. Diabetes. 2014;63:3512–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Badr G, Mohany M. Maternal perinatal undernutrition attenuates T‐cell function in adult male rat offspring. Cell Physiol Biochem. 2011;27:381–390. [DOI] [PubMed] [Google Scholar]
- 10. Badr G, Alwasel S, Ebaid H, et al. Perinatal supplementation with thymoquinone improves diabetic complications and T cell immune responses inrat offspring. Cell Immunol. 2011;267:133–140. [DOI] [PubMed] [Google Scholar]
- 11. Badr G, Mahmoud MH, Farhat K, et al. Maternal supplementation of diabetic mice with thymoquinone protects their offspring from abnormal obesityand diabetes by modulating their lipid profile and free radical production and restoring lymphocyte proliferationvia PI3K/AKT signaling. Lipids Health Dis. 2013;12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Villarino ME, Goya JA, De Lucca RC, et al. Alterations of tooth eruption and growth in pups suckling from diabetic dams. Pediatr Res. 2005;58:695–699. [DOI] [PubMed] [Google Scholar]
- 13. Silva‐Sousa YT, Peres LC, Foss MC. Enamel hypoplasia in a litter of rats with alloxan‐induced diabetes mellitus. Braz Dent J. 2003;14:87–93. [DOI] [PubMed] [Google Scholar]
- 14. Bennett KA, Cheverud JM, Booth SN. Deciduous tooth dimensions in fetal rhesus monkeys from mothers with induced diabetes. Am J Phys Anthropol. 1981;55:411–417. [DOI] [PubMed] [Google Scholar]
- 15. Lal S, Cheng B, Kaplan S, et al. Accelerated tooth eruption in children with diabetes mellitus. Pediatrics. 2008;121:e1139–e1143. [DOI] [PubMed] [Google Scholar]
- 16. Jawerbaum A, González E. Diabetic pregnancies: the challenge of developing in a pro‐inflammatory environment. Curr Med Chem. 2006;13:2127–2138. [DOI] [PubMed] [Google Scholar]
- 17. Jawerbaum A, Capobianco E. Review: effects of PPAR activation in the placenta and the fetus: implications in maternal diabetes. Placenta. 2011;32:S212–S217. [DOI] [PubMed] [Google Scholar]
- 18. Segovia SA, Vickers MH, Gray C, et al. Maternal obesity, inflammation, and developmental programming. Biomed Res Int. 2014;2014:418975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mrizak I, Grissa O, Henault B, et al. Placental infiltration of inflammatory markers in gestational diabetic women. Gen Physiol Biophys. 2014;33:169–176. [DOI] [PubMed] [Google Scholar]
- 20. Xie BG, Jin S, Zhu WJ. Expression of toll‐like receptor 4 in maternal monocytes of patients with gestational diabetes mellitus. Exp Ther Med. 2014;7:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol. 2010;11:373–384. [DOI] [PubMed] [Google Scholar]
- 22. Kramer BW, Kallapur SG, Moss TJ, et al. Intra‐amniotic LPS modulation of TLR signaling in lung and blood monocytes of fetal sheep. Innate Immun. 2009;15:101–107. [DOI] [PubMed] [Google Scholar]
- 23. Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8. [DOI] [PubMed] [Google Scholar]
- 24. Devaraj S, Tobias P, Jialal I. Knockout of toll‐like receptor‐4 attenuates the pro‐inflammatory state of diabetes. Cytokine. 2011;55:441–445. [DOI] [PubMed] [Google Scholar]
- 25. Lin M, Yiu WH, Wu HJ, et al. Toll‐like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol. 2012;23:86–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu Y, Gao Y, Zhan X, et al. TLR4 activation by lipopolysaccharide and Streptococcus mutans induces differential regulation of proliferation and migration in human dental pulp stem cells. J Endod. 2014;40:1375–1381. [DOI] [PubMed] [Google Scholar]
- 27. Lisboa RA, Andrade MV, Cunha‐Melo JR. Toll‐like receptor activation and mechanical force stimulation promote the secretion of matrix metalloproteinases 1, 3 and 10 of human periodontal fibroblasts via p38, JNK and NF‐kB. Arch Oral Biol. 2013;58:731–739. [DOI] [PubMed] [Google Scholar]
- 28. Park JH, Kwon SM, Yoon HE, et al. Lipopolysaccharide promotes adhesion and migration of murine dental papilla‐derived MDPC‐23 cells via TLR4. Int J Mol Med. 2011;27:277–281. [DOI] [PubMed] [Google Scholar]
- 29. Salbaum JM, Kappen C. Neural tube defect genes and maternal diabetes during pregnancy. Birth Defects Res A Clin Mol Teratol. 2010;88:601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hagay ZJ, Weiss Y, Zusman I, et al. Prevention of diabetes‐associated embryopathy by overexpression of the free radical scavenger copper zinc superoxide dismutase in transgenic mouse embryos. Am J Obstet Gynecol. 1995;173:1036–1041. [DOI] [PubMed] [Google Scholar]
- 31. Yang P, Reece EA. Role of HIF‐1α in maternal hyperglycemia‐induced embryonic vasculopathy. Am J Obstet Gynecol 2011;204:332.e1–332.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eriksson UJ, Dahlström E, Hellerström C. Diabetes in pregnancy. Skeletal malformations in the offspring of diabetic rats after intermittent withdrawal of insulin in early gestation. Diabetes. 1983;32:1141–1145. [DOI] [PubMed] [Google Scholar]
- 33. Li X, Xu C, Yang P. c‐Jun NH2‐terminal kinase 1/2 and endoplasmic reticulum stress as interdependent and reciprocal causation in diabetic embryopathy. Diabetes. 2013;62:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li X, Weng H, Xu C, et al. Oxidative stress‐induced JNK1/2 activation triggers proapoptotic signaling and apoptosis that leads to diabetic embryopathy. Diabetes. 2012;61:2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jawerbaum A, White V. Animal models in diabetes and pregnancy. Endocr Rev. 2010;31:680–701. [DOI] [PubMed] [Google Scholar]
- 36. Tucker A, Sharpe P. The cutting‐edge of mammalian development; how the embryo makes teeth. Nat Rev Genet. 2004;5:499–508. [DOI] [PubMed] [Google Scholar]
- 37. Chavez MG, Yu W, Biehs B, et al. Characterization of dental epithelial stem cells from the mouse incisor with two‐dimensional and three‐dimensional platforms. Tissue Eng Part C Methods. 2013;19:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen J, Chen G, Yan Z, et al. TGF‐β1 and FGF2 stimulate the epithelial‐mesenchymal transition of HERS cells through a MEK‐dependent mechanism. J Cell Physiol. 2014;229:1647–1659. [DOI] [PubMed] [Google Scholar]
- 39. Dasu MR, Devaraj S, Zhao L, et al. High glucose induces toll‐like receptor expression in human monocytes: mechanism of activation. Diabetes. 2008;57:3090–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loeken MR. Advances in understanding the molecular causes of diabetes‐induced birth defects. J Soc Gynecol Investig. 2006;13:2–10. [DOI] [PubMed] [Google Scholar]
- 41. Jawerbaum A, Higa R, White V, et al. Peroxynitrites and impaired modulation of nitric oxide concentrations in embryos from diabetic rats during early organogenesis. Reproduction. 2005;130:695–703. [DOI] [PubMed] [Google Scholar]
- 42. Tran S, Chen YW, Chenier I, et al. Maternal diabetes modulates renal morphogenesis in offspring. J Am Soc Nephrol. 2008;19:943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Setkova J, Lesot H, Matalova E, et al. Proliferation and apoptosis in early molar morphogenesis – voles as models in odontogenesis. Int J Dev Biol. 2006;50:481–489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials