

Abstract
Objectives
Adipose tissue plays a fundamental role in glucose homeostasis. For example, fat removal (lipectomy, LipX) in lean mice, resulting in a compensatory 50% increase in total fat mass, is associated with significant improvement in glucose tolerance. This study was designed to further examine the link between fat removal, adipose tissue compensation and glucose homeostasis using a peroxisome proliferator‐activated receptor γ (PPAR γ; activator of adipogenesis) knockout mouse.
Material and methods
The study involved PPARγ knockout (FKOγ) or control mice (CON), subdivided into groups that received LipX or Sham surgery. We reasoned that as the ability of adipose tissue to expand in response to LipX would be compromised in FKOγ mice, so would improvements in glucose homeostasis.
Results
In CON mice, LipX increased total adipose depot mass (~60%), adipocyte number (~45%) and changed adipocyte distribution to smaller cells. Glucose tolerance was improved (~30%) in LipX CON mice compared to Shams. In FKOγ mice, LipX did not result in any significant changes in adipose depot mass, adipocyte number or distribution. LipX FKOγ mice were also characterized by reduction of glucose tolerance (~30%) compared to shams.
Conclusions
Inhibition of adipose tissue PPARγ prevented LipX‐induced increases in adipocyte expansion and produced a glucose‐intolerant phenotype. These data support the notion that adipose tissue expansion is critical to maintain and/or improvement in glucose homeostasis.
Keywords: adipocytes, adipokines, compensation, lipids, PPAR gamma, proliferation, visceral obesity
1. Introduction
The metabolic consequences of obesity are closely aligned with how lipids are distributed among adipose tissue depots. Dyslipidaemia, type 2 diabetes and insulin resistance are more likely to occur in obese individuals characterized by central/abdominal and upper body lipid storage.1, 2, 3, 4, 5 In contrast, these comorbidities are less likely in individuals with6, 7, 8 lower body subcutaneous adipose tissue (SAT) expansion.9 Indeed, lower body SAT expansion is a primary characteristic of the metabolically healthy obese individual.10 Therefore, accumulation of lipids in this adipose tissue region has been suggested to be protective from typical obesity‐mediated metabolic impairments.
Adipose tissue depots appear to have distinct, inherent characteristics that may explain different metabolic outcomes. For example, whereas upper body SAT expansion appears to involve hypertrophy which results in large, typically insulin resistance adipocytes, lower body SAT expansion involves increases in smaller adipocytes that retain insulin sensitivity.11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 Hence, some studies suggests that depots characterized by smaller adipocytes impart protection from obesity‐related metabolic impairments.22, 23 However, not all studies support this view.24, 25
One common element that has been linked to metabolic dysregulation associated with obesity involves the recruitment of new fats cells and regulation of differentiation. In this context, impairments in the differentiation and proliferation of new fat cells can lead to spill over and accumulation of lipids and lipid intermediates in non‐adipose tissue cells.26 Non‐adipose tissue or ectopic lipid accumulation has been associated with insulin resistance and increased risk for a number of obesity‐related comorbidities. Adipogenesis and maintenance of mature adipocytes is driven by the activation of peroxisome proliferator‐activated receptor γ (PPARγ), a nuclear receptor.27, 28 PPARγ activation in adipocytes lowers circulating insulin levels, improves whole‐body insulin sensitivity and adipokine profiles, and reduces serum lipids.29 Adipose PPARγ activation also suppresses inflammation induced by high‐fat diet.29 It is proposed that these improvements occur, in part, through sequestration of excess fatty acids and triglycerides by adipocyte hyperplasia.30 We have therefore elected to manipulate PPARγ in the context of lipectomy (LipX) or sham surgery to further investigate the relationship between adipose tissue expansion and glucose homeostasis.
PPARγ knockout mice are characterized by reduced adipocyte maturation after initial adipogenic events, reduced hyperplasia leading to hypertrophic mature adipocytes and elevated plasma fatty acids.31 We have previously demonstrated that removal of discrete adipose tissue depots (LipX) improves glucose tolerance and increases sequestration of fatty acids in non‐excised adipose tissue of lean young and old rodent models.32, 33 We and others have also demonstrated that compensation in non‐excised depots was characterized by increased adipocyte number and adipose tissue mass.34, 35, 36 In the current study, we have employed LipX to induce adipose tissue depot growth in CON and FKOγ mice. We hypothesized that LipX‐induced increases in adipose tissue growth would be impaired in PPARγ knockout mice and this impairment would also lead to unfavourable outcomes in glucose homeostasis.
2. Materials and methods
2.1. Mice and housing
2.1.1. Breeding colony
Twelve transgenic adult mice were purchased from The Jackson Laboratory (Bar Harbor, Maine), six males and six females (~24 g), for tissue‐specific Cre/Lox inducible knockouts. Half of the mice from each sex were homozygous for PPARγ flanked with LoxP sites, targeted for its deletion. The other half were hemizygous for the expression of Cre recombinase enzyme under control of FABP4, fatty acid‐binding protein‐4, an adipocyte lipid transport protein. A second‐generation breeding scheme was used to obtain knockouts (KO) and control littermates used in this study. First, homozygous PPARγ loxP‐flanked mice were mated with Cre recombinase FABP4 mice. The first generation of offspring heterozygous for the loxP allele and hemizygous/heterozygous for the cre transgene were then mated with the original homozygous loxP‐flanked mice. Second‐generation offspring, homozygous for the loxP‐flanked allele and hemizygous/heterozygous for the cre transgene were designated FKOγ KO mice (n=13). Homozygous LoxP offspring were designated as Control C (CON) mice (n=15).
2.1.2. Experimental mice
A total of 13 male adipose tissue‐specific PPARγ knockout transgenic mice (FKOγ) and 15 male homozygous LoxP CON mice were produced from a colony of ~175 mice. An a priori power analysis using previous data from other breeding colonies to determine the expected variance was used to estimate the number of animals necessary for this experiment. Sample sizes were determined by providing a 90% chance of finding a significant effect of 25% or greater for the between‐ and within‐subject variables. It was predicted 8–10 mice were needed to provide adequate statistical power. Due to the limitation of breeding numbers, we did not reach the predicted number of mice; however, the animal numbers acquired were adequate to observe a number of statistically significant differences.
Offspring of interest were weaned 21 days post‐birth and transferred to individual housing under controlled conditions (12:12 light‐dark cycle, 50–60% humidity and 25°C). Experiments (surgery) started at 3 months of age before metabolic dysregulation was exacerbated in FKOγ mice. For experiment duration, mice were given ad libitum access to a standard chow diet (Harlan Teklad 7002, Madison, WI, USA) and unlimited water. Weekly body mass and food intake were monitored and recorded. Procedures were reviewed and approved by the Colorado State University Institutional Animal Care and Use Committee.
2.2. Surgical procedures
Surgeries were performed while mice were anesthetized with isoflurane. Half of the experimental mice received sham surgery (mid‐ventral abdominal incision through skin and muscle without adipose tissue removed), while the other half received bi‐lateral excision of the intra‐abdominal epididymal depot (~550 mg; connected to testes). Both groups received abdominal muscle suture closure and skin was closed with wound clips. A subcutaneous injection of meloxicam analgesic (0.025 mg/10 g body weight) was given immediately after surgery was completed. The four experimental groups consist of FKOγ or CON with or without surgery (FKO surgery n=6, FKO sham n=7, CON surgery n=7 and CON sham n=8).
2.3. Glucose tolerance test
Pre‐surgery (1 week prior to adipose tissue removal surgery) and terminal (1 week prior to termination, 12 weeks post‐surgery) glucose tolerance tests (GTTs) were performed on mice. Mice were fasted, but allowed water, for 6 hours after lights on. Blood was collected from the tail vein and glucose concentration was determined using a Freestyle Lite Glucometer (Abbott, Abbott Park, IL, USA). After fasting blood glucose was collected (time point 0), mice received a 1.5 g kg−1 dextrose injection in the intraperitoneal cavity and blood glucose was measured from tail vein blood samples at 15, 30, 45, 60 and 120 minutes post‐injection.
2.4. Termination
Termination occurred 13 weeks post‐surgery. Final body weights were collected before mice were fasted for 4 hours for terminal collection. First, following isoflurane anesthetization, systemic blood was collected via decapitation and serum was separated and stored at −80°C. Femoral muscle and liver were removed and snap‐frozen in liquid nitrogen and stored at −80°C. Inguinal (IWAT), epididymal (EWAT), perirenal (PWAT), dorsal (DWAT), and visceral (VWAT) white adipose tissue, as well as inter‐scapular brown adipose tissue (BAT), were collected and weighed, snap‐frozen and stored at −80°C. IWAT, VWAT and PWAT depots were halved and also fixed in osmium for cell size distribution (see below). Subcutaneous lymph nodes were removed from IWAT and visceral lymph nodes were removed from VWAT prior to being frozen.
2.5. RNA isolation and cDNA synthesis
Lipid‐specific RNAeasy mini‐kit columns were used to isolate RNA from adipose tissue using QIAzol Lysis Reagent (QIAGEN, Valencia, CA, USA). Aggregate RNA was converted to complementary‐DNA using iScript (Bio‐Rad, Hercules, CA, USA) with a normalized quantity of RNA totalling 0.25 μg.
2.6. Quantitative real‐time PCR
Sequences of primers for adipose tissue and liver are shown in Figure S1. Primers were optimized as previously described.37 Samples were run in triplicate using an iCycler (Bio‐Rad) and iQ SYBR Green Supermix (Bio‐Rad). Expression patterns of genes of interest were normalized to constitutively expressed β2 microglobulin (B2M) and relative expression was quantified as previously described.37 Genes were selected for their known associations with adipose tissue metabolism and adipogenesis. Genes of interest are reported as relative change compared with control.
2.7. Plasma and tissue measurements
Systemic plasma at termination was analysed for insulin, leptin and resistin using a commercial kit (EMD Millipore Corporation, Billerica, MA, USA) and analysed on a Luminex instrument (LX200; Millipore, Austin, TX, USA). Skeletal muscle and liver lipids were extracted using the procedure of Bligh and Dyer.38 Muscle and liver triglyceride concentration (Sigma Chemical Co, St. Louis, MO, USA) and plasma non‐esterified fatty acids (Wako, Richmond, VA, USA) were determined enzymatically using commercially available kits.
2.8. Adipocyte distribution
Approximately half of the collected total IWAT, VWAT and PWAT depot was fixed in osmium tetroxide according to the method of Hirsch and Gallain.39 Fixation was completed in a warm water bath for at least 24 hours. Cell number and size distribution were determined by Coulter Counter analysis (Beckman Coulter, Fullerton, CA, USA), as suspended particles were passed through an aperture in the counter to provide a histogram in per unit volume of suspension. Distribution is presented as a per cent in cell size bin, which represents a range of 10 μm; hence, 25 μm bin contains cell sizes 25 μm through 34 μm.
2.9. Statistical analysis
Data are expressed as mean±standard error of the mean (SEM). Statistics of multiple groups were completed using two‐way between‐subject analysis of variance (ANOVA) (IBM SPSS for Windows, release 22; Chicago, IL, USA) with group (CON and FKO) and surgery (Sham and LipX) as factors. This was utilized for the following dependent variables: body and adipose mass, lipids, gene expression, adipocyte mean size and adipokine concentration. Adipocyte cells size distribution and glucose tolerance test were analysed using two‐way repeated measures ANOVA with adipocyte size as a within‐subject variable. Per cent change in insulin and area under the curve (AUC) as well as total adipocyte number within the visceral, inguinal and perineal depots were analysed by ANOVA. Post hoc test of individual groups were accomplished with Tukey's test. Differences among groups was considered significantly different if P≤.05. Exact probabilities are shown when applicable.
3. Results
3.1. Food intake, body and adipose tissue mass and lipids
Thirteen weeks post‐surgery, cumulative food intake (CON Sham; 334.5 g±3.8, CON LipX; 341.4 g±1.8, FKO Sham; 320.2 g±9.1 and FKO LipX 330.4 g±7.2) and terminal body mass (Table 1) were not different among groups. Both CON and FKO LipX groups had ~550 mg of epididymal adipose tissue removed, but only the FKO LipX mice had significantly smaller epididymal depot mass at termination compared with respective Sham (Table 1; P≤.05). Despite similar body mass among groups, adipose tissue mass of FKO mice was significantly less than CON. In particular, IWAT (Group main effect, P=.011), PWAT (Group main effect P=.017), EWAT (Group main effect P=.006) and total depot mass (summed IWAT, VWAT, PWAT and DWAT, Group main effect P=.011) were significantly reduced in FKOγ compared to CON mice. VWAT was also less in FKOγ, though not significant. In CON mice, LipX was associated with significant increases in non‐excised adipose tissue depot mass including a ~65% increase in IWAT, ~110% increase in VWAT, ~50% increase in both PWAT and DWAT depots (Table 1; P≤.05) compared to CON Sham. This resulted in an overall ~65% increase in total adipose depot mass in CON LipX mice when compared to CON Sham. No increase in adipose tissue mass was observed in FKOγ LipX mice when compared to FKOγ Sham. Circulating and muscle lipids were not significantly different among groups; however, FKOγ liver triglycerides tended to be higher than CON (Table 1; P=.063).
Table 1.
Terminal body mass, total and individual (inguinal, visceral, perirenal and dorsal white adipose tissue and inter‐scapular brown adipose tissue) adipose tissue mass, adipocyte mean size, systemic circulating adipokines, free fatty acid (FFA) and tissue triglyceride (TG: muscle and liver) concentration 13 weeks post‐surgery. Values are reported as mean±SEM
| CON Sham | CON LipX | FKO Sham | FKO LipX | P value | |||
|---|---|---|---|---|---|---|---|
| Model | Group | Sur | |||||
| Final body mass (g) | 34.4±2.4 | 37.4±2.5 | 32.7±0.5 | 32.2±1.6 | — | — | — |
| Adipose tissue mass (mg) | — | — | — | ||||
| EWAT | 0.97±0.18 | 0.66±0.13 | 0.99±0.10 | 0.26±0.05a | 0.001 | — | 0.0009 |
| IWAT | 0.63±0.09 | 1.04±0.23a | 0.40±0.05 | 0.42±0.10 | 0.034 | 0.011 | — |
| VWAT | 0.37±0.04 | 0.79±0.16a | 0.45±0.05 | 0.43±0.06 | 0.041 | 0.075 | 0.026 |
| PWAT | 0.51±0.06 | 0.80±0.13a | 0.36±0.05 | 0.40±0.07 | 0.046 | 0.017 | — |
| DWAT | 0.54±0.05 | 0.81±0.21 | 0.30±0.05 | 0.36±0.06 | 0.018 | 0.006 | — |
| Total | 2.33±0.25 | 3.70±0.07a | 1.70±0.12 | 1.81±0.30 | 0.029 | 0.011 | — |
| BAT | 0.28±0.03 | 0.25±0.05 | 0.20±0.01 | 0.20±0.04 | — | — | — |
| Adipocyte mean size (μm) | |||||||
| IWAT | 50.4±1.72 | 49.3±0.76 | 53.95±2.56 | 52.23±1.68 | — | — | — |
| VWAT | 58.18±1.49 | 54.50±2.38 | 58.18±1.24 | 60.25±2.23 | — | — | — |
| PWAT | 53.82±1.90 | 54.40±2.50 | 58.94±2.59 | 56.93±2.59 | — | — | — |
| Adipokine (μg mL−1) | |||||||
| Leptin | 4918±621 | 2728±846a | 3269±491 | 1484±430a | 0.041 | 0.040 | 0.019 |
| Resistin | 1516±147 | 826±109a | 725.5±195 | 767±287 | 0.011 | 0.02 | — |
| Systemic FFA (mmol L−1) | 0.55±0.04 | 0.51±0.02 | 0.57±0.04 | 0.45±0.03 | — | — | — |
| Muscle TG (mg g−1 tissue) | 25.6±4.9 | 19.7±3.8 | 23.2±4.9 | 28.1±11.2 | — | — | — |
| Liver TG (mg g−1 tissue) | 31.5±6.2 | 28.3±2.7 | 43.8±12.5 | 52.3±12.0 | — | 0.063 | — |
CON, Control; LipX, Lipectomy; Sur, Surgery; μm, micrometre.
Chart contains P values of model and main effect of group and surgery (— = not significant).
P ≤ 0.05, compared with respective control.
3.2. Glucose tolerance test and insulin concentration
One week before termination (12 weeks post‐surgery) glucose concentration following a 6‐hour fast was not different among groups (Data not shown). Glucose tolerance test results are reported as per cent difference between LipX and Sham at each time point (0, 15, 30, 60 and 120 minutes) for CON and FKOγ mice. LipX decreased the glucose response by ~30% in CON mice, whereas LipX increased the glucose response by ~30% in the FKOγ group, these two groups were significantly different than one another (Figure 1A and B; P≤.05). Systemic insulin concentration was significantly higher, ~2‐fold, in FKOγ mice (CON Sham=161±20, CON LipX=169±18, FKO Sham=339±58, FKO LipX=375±75; P=.033, FKO Sham vs CON Sham and P=.01, FKO LipX vs CON Sham). LipX did not result in significant effects on insulin concentration (Figure 1C). Overall, these data demonstrate that LipX resulted in improvements in glucose tolerance in CON mice and impairments in glucose tolerance in FKOγ mice.
Figure 1.
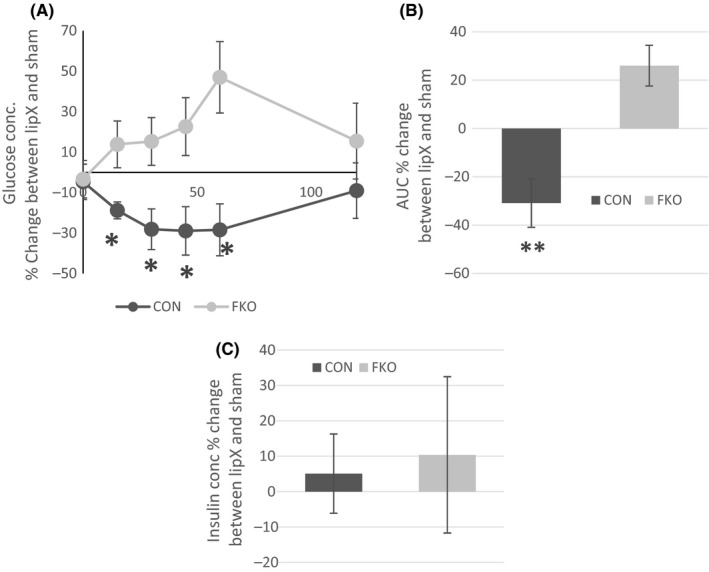
Glucose Tolerance Test (GTT) and Insulin Concentration – A, GTT – Per cent change in glucose concentration between the LipX or Sham group of CON and FKOγ mice. LipX is associated with decreased glucose concentration in FKOγ mice, whereas it is associated with an increase in glucose concentration in the CON (*P≤.05). B, Per cent change in area under the curve between the LipX or Sham group was decreased in control mice demonstrating improved glucose tolerance, whereas it was increased in FKOγ mice which is indicative decreased glucose tolerance (**P=.002). C, Insulin concentration expressed as per cent change between the LipX or Sham surgery was not different between CON and FKO mice
3.3. Adipocyte number and cell size distribution
Adipose tissue removal provokes compensation in non‐excised depots via differentiation/proliferation, which is typically assessed by the measurement of adipocyte number and adipocyte size distribution.35, 40 Therefore, we evaluated adipocyte number and size distribution in a subcutaneous depot, inguinal (IWAT) and two intra‐abdominal depots, the visceral depot (VWAT) that releases effluent to the portal circulation and the perirenal depot (PWAT) that releases effluent to the systemic circulation. LipX resulted in increased total adipocyte number in CON but not FKOγ mice. In particular, the number of adipocytes in the inguinal and perirenal depots of CON mice was significantly increased by ~45% following LipX (Figure 2 A and E inset; P≤.05). LipX also induced a significant shift in adipocyte distribution in the inguinal (cell size/surgery interaction: Figure 2A; P=.001) and visceral (cell size/surgery interaction: Figure 2C; P=.04) depot. In CON mice, adipocytes in these depots shifted towards a higher percent of small cells in CON, but not FKOγ mice. In the inguinal depot of CON mice, LipX caused a significant increase in the per cent of 25–44 μm (Figure 2A; P≤.05) adipocytes and a significant decrease in the percent of 45–84 μm (Figure 2A; P≤.05). Similarly, in the visceral depot, LipX increased the percent of 25–34 μm adipocytes and significantly decreased 45–64 μm size adipocytes (Figure 2B; P≤.05). LipX did not affect perirenal depot adipocyte distribution in either group. These changes in adipocyte number and distribution were prevented in FKOγ mice. These data demonstrate that LipX‐induced adipose depot compensation was prevented with the inhibition of PPARγ in adipose tissue.
Figure 2.

Adipocyte Number and Cell Size Distribution of Inguinal, Visceral and Perirenal Adipose Tissue – Inset of A and E.) LipX in CON mice, but not FKOγ, resulted in a significant increase in adipocyte number in the inguinal and perirenal adipose depots (*P≤.05, compared with Sham). In CON mice, LipX also shifted the (A) inguinal (Cell size/surgery interaction: Figure 2A; P<.05) and C, visceral (Cell size/surgery interaction: Figure 2C; P≤.05) depot adipocyte distribution towards smaller cell size. In the inguinal depot of CON mice, LipX resulted in a significant increase in the percent of 25–44 μm (**P≤.001, *P≤.05, compared with Sham) adipocytes and significant decrease in the percent of 45–84 μm (**P≤.000, *P≤.05, compared with Sham). For the visceral depot, LipX increased the percent of 25–34 μm adipocytes and significantly decreased 45–64 μm (*P≤.05, compared with Sham). LipX did not affect inguinal (B), visceral (D) or perirenal (F) depot adipocyte distribution or change distribution of cell size in FKOγ mice
3.4. Adipose tissue gene expression
Given the dramatic changes in adipose tissue mass, adipocyte number and size distribution observed in CON but not FKOγ mice we examined gene markers related to adipose tissue growth/expansion and maturation in both the inguinal and visceral depot. These two depots were further evaluated for gene expression because of their differential associations with metabolic risk. We focused on gene markers of cellular differentiation (peroxisome proliferator‐activated receptor γ; PPARγ and CCAAT enhancer‐binding protein α (C/EBPα)), fatty acid uptake and transport (fatty acid‐binding protein 4; FABP4 and fatty acid transport protein 4; FATP4) and genes encoding proteins secreted from adipose tissue (adiponectin; adipoQ). Overall, all of the adipose gene markers were significantly reduced in FKOγ mice compared with CON in both the inguinal and visceral depot. For the inguinal depot PPARγ, C/EBPα, FABP4, and adipoQ were significantly lower in FKOγ mice compared with CON (Figure 3A,C,E and I, P≤.007). Similarly, these genes along with FATP4 were significantly lower in the visceral depot of FKOγ mice (Figure 3B,D,F,H and J, P≤.025). LipX induced alterations in inguinal and visceral adipose tissue gene expression, but only in CON mice. In the inguinal depot, LipX caused a significant ~5‐fold decrease in gene expression of PPARγ (Figure 3A, interaction P=.009), C/EBPα (Figure 3C, interaction P=.005) and FABT4 (Figure 3G, interaction P=.000), whereas in the visceral depot, LipX resulted in a ~2‐fold increase in adipoQ (Figure 3J, approaching interaction P=.079). Therefore, as predicted, LipX‐induced alterations in adipose tissue gene expression were specific to CON mice and did not occur in FKOγ. The direction of most changes, however, was in opposition of what was anticipated.
Figure 3.
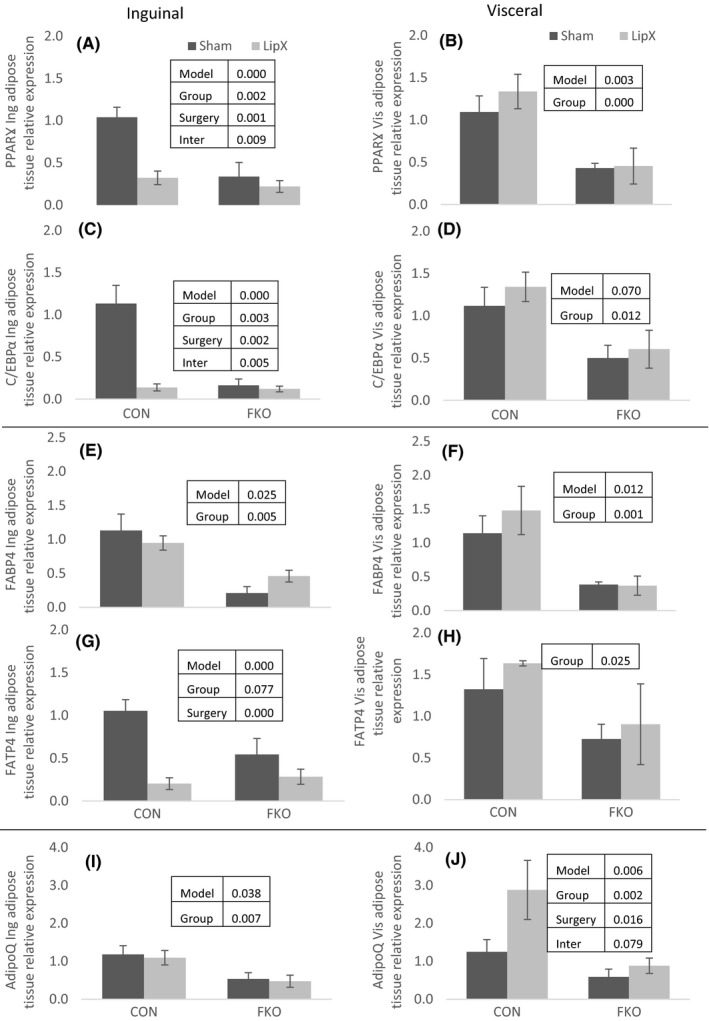
Adipose Tissue Gene Expression – Gene markers of cellular differentiation (peroxisome proliferator‐activated receptor γ; PPARγ and CCAAT enhancer‐binding protein α; C/EBPα), fatty acid uptake and transport (fatty acid binding protein 4; FABP4 and fatty acid transport protein 4; FATP4) and genes encoding proteins secreted from adipose tissue (adiponectin; adipoQ). (A–J) All of the adipose gene markers were significantly reduced in FKOγ compared to CON mice. (A) PPAR, (C) CEBP and (G) FATP4 were significantly reduced in inguinal but not visceral adipose tissue from LipX vs Sham CON mice. LipX did not result in any significant changes in FKOγ mice. Insets within figure include P values for significance of model, main effects of group (CON vs FKO) and surgery (LipX and Sham) and interactions when applicable
3.5. Circulating adipokines
Compared with CON mice, FKOγ mice had significantly lower leptin (Table 1, Group main effect P=.04) and resistin (Table 1, Group main effect P=.02) concentrations in systemic plasma. LipX significantly decreased leptin concentrations in both CON and FKOγ mice (Table 1, Surgery main effect P=.019); however, resistin was only reduced in CON LipX mice (~45%, Table 1, P≤.05; compared with sham control).
3.6. Liver gene expression
FKOγ mice were characterized by a ~2‐fold increase expression of selected gene markers of inflammation, caspase1 (CASP1) (Figure 4A, P=.000) and interleukin 1 α (Figure 4B, P=.007) and β (Figure 4C, P=.004) (IL1α and ILβ) (Figure 4A–C, P≤.05). None of these genes were affected by LipX.
Figure 4.
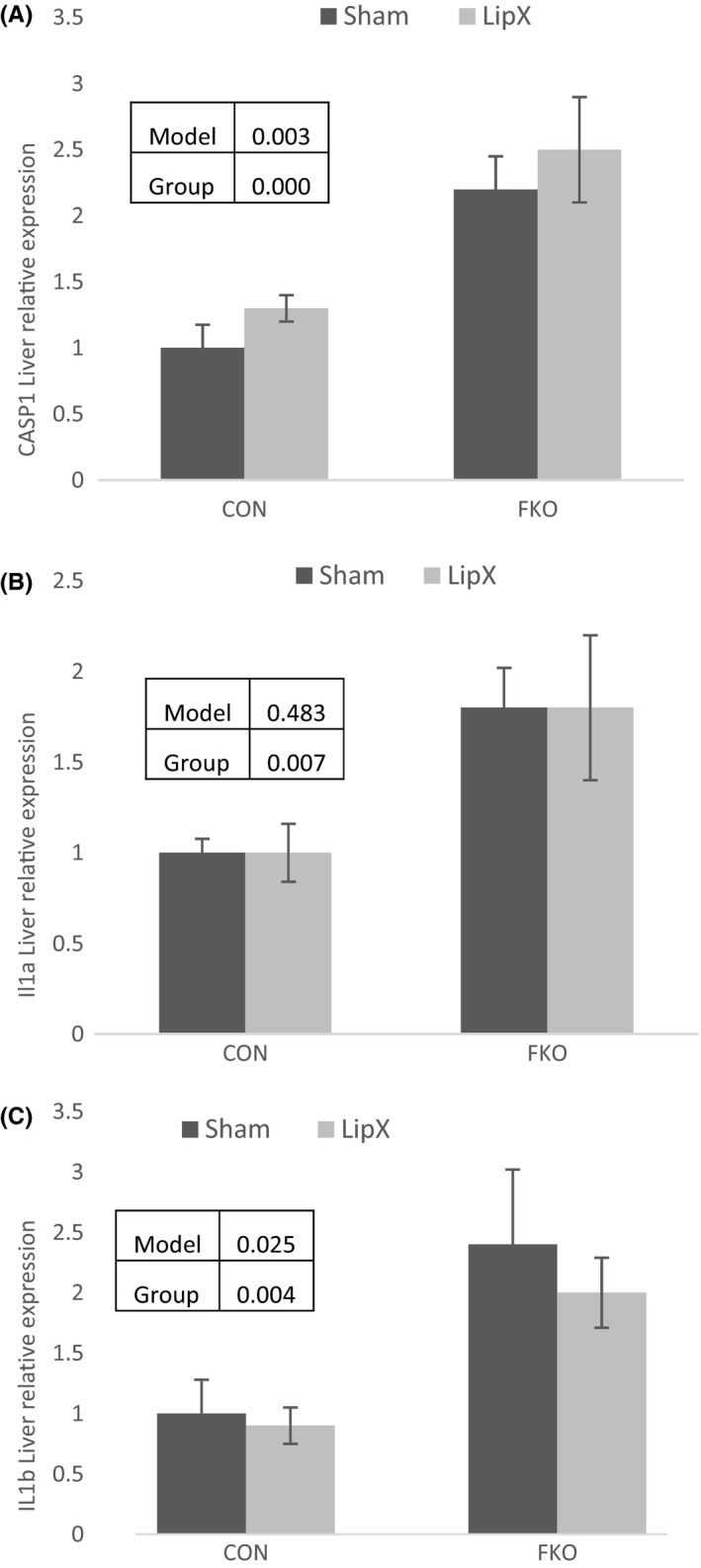
Liver Gene Expression – FKOγ mice were characterized by a significant increase in expression of selected gene markers of inflammation, A) caspase1 (CASP1) and interleukin (B) 1 α and (C) β (IL1α and Ilβ). None of these genes were significantly affected by LipX. Insets within figure include p values for significance of model, main effects of group (CON vs FKO) and surgery (LipX and Sham) and interactions when applicable.
4. Discussion
The present study was designed to examine the link between fat removal, adipose tissue compensation and glucose homeostasis. We employed lipectomy to induce compensatory adipose tissue depot growth in Control and FKOγ mice. We hypothesized that lipectomy‐induced increases in adipose tissue compensation would be impaired in FKOγ knockout mice and this impairment would also be associated with unfavourable outcomes in glucose homeostasis. Our results support the notion that adipose tissue expansion, via PPARγ‐mediated mechanisms, plays a role in glucose homeostasis.
The removal of gonadal adipose tissue (epididymal) in CON mice increased total fat mass, adipocyte number in inguinal and perirenal adipose tissue depots, and shifted cell distributions towards smaller adipocytes in the inguinal and visceral adipose tissue depots. This is consistent with other rodent studies where gonadal LipX increases the mass of intra‐abdominal and subcutaneous adipose depots.41, 42, 43 In contrast, the removal of gonadal adipose tissue in FKOγ mice did not increase total fat mass, adipocyte number or shift cell size distribution. These data suggest that LipX‐induced adipose tissue hyperplasia is mediated, at least in part, by PPARγ. As LipX did not result in compensatory increases in adipose tissue mass, adipocyte number or decreases in adipocyte size in FKOγ mice, we speculate that limitations placed on adipogenesis and adipocyte proliferation by the absence of PPARγ restrict sequestration of lipids in adipose tissue. Given the simultaneous improvement in glucose homeostasis, our results are consistent with the notion that smaller adipocytes are associated with metabolic protection.
Adipose tissue expandability is often associated with favourable metabolic outcomes and appears to play a fundamental role in the regulation of glucose homeostasis. Indeed, studies demonstrate that the hyperplastic potential of white adipose tissue is directly associated with insulin sensitivity44 and improved glucose tolerance.45 Consistent with this, intra‐abdominal LipX resulted in both adipose tissue expansion and improved glucose tolerance in CON mice. Adipose tissue expansion and improvements in glucose tolerance, however, did not occur in FKOγ mice. Rather, LipX in FKOγ mice reduced glucose tolerance. It should be emphasized that LipX‐induced improvements are not due to fat removal alone, but rather the consequential compensation. Therefore, we propose that LipX‐mediated improvements in glucose homeostasis are linked to the ability of adipose tissue to expand via mechanisms that include increases in cell number and shift in size distribution.
A restriction or impairment of adipose tissue expansion and continued sequestration of lipids can lead to lipid deposition in non‐adipose tissues, such as liver and muscle (for review see: 46). In the present study, FKOγ sham mice were characterized by a 40% increase in liver triglycerides compared to CON sham mice. Although this increase was not statistically significant (approaching P=.063), the data suggest that the lack of PPARγ and the resultant limitations placed on adipose tissue expansion resulted in ectopic lipid sequestration in the liver. In addition, muscle and liver triglycerides tended to be higher (~25%) in FKOγ mice who received lipectomy compared to their sham counterparts. These data, while not definitive, support the notion that adipose tissue expansion is linked to the degree of ectopic lipid accumulation.
We also analysed genes involved in adipocyte differentiation and growth to gain insight into the FKOγ phenotype and the effects of lipectomy. Consistent with previous studies,31 we observed that genes involved in cellular differentiation and fatty acid uptake, transport and storage, as well as genes that encode secreted proteins were lower in the white adipose tissue of FKOγ mice compared with CON. These differences were consistent across the inguinal and visceral adipose tissue depots. In CON mice, LipX altered genes involved in adipocyte differentiation and fatty acid uptake in the inguinal, but not visceral depot. In opposition to our prediction of enhanced gene expression of adipose tissue growth/compensation markers in LipX CON mice, gene expression of PPARγ, C/EBPα and FATP4 were decreased in the inguinal depot of CON LipX mice compared with Sham. Though it is possible that the downregulation of gene expression is due to decreased rate of expansion of the inguinal depot, it is important to acknowledge that this is a single time point and perhaps not indicative of alterations occurring at an early time point. Previous studies in rodents have consistently demonstrated that body fat loss following lipectomy was normalized ~3 months post‐surgery42, 43, 47; hence, gene markers of compensation should be decreased at the 13‐week time point that we chose for our study. LipX did not alter expression of these genes in the inguinal or visceral adipose tissue depot of FKOγ mice.
It is proposed that dysregulated visceral adipose tissue contributes to the development of hepatic steatosis and insulin resistance48 because of its proximity to and release of lipids and cytokines to the liver.49 However, in the current study, LipX induced visceral adipose tissue compensation in CON mice and was associated with improved glucose tolerance despite an increase in visceral adiposity which is proposed to be detrimental. We propose that events following lipectomy enhance the inherent ability of visceral adipocytes to expand and sequester triglycerides, thus decreasing lipid effluent to the liver. At 13 weeks, however, circulating free fatty acids, liver triglycerides and liver markers of inflammation were not lower in mice with fat removed relative to their sham counterparts. It is important to note all mice received standard rodent chow and remained lean throughout the experiment, thus some liver measures may already be at a minimum and therefore less flexible towards change.
Despite considerable adipose tissue compensation, lipectomy decreased both leptin and resistin in CON mice. The reduction in leptin was not expected given this adipokine is a lipostatic signal and LipX mice were characterized by increased adiposity. However, leptin can also inhibit cell proliferation, therefore high levels may be counterproductive during periods that involve adipose tissue compensation.50 LipX in CON mice also decreased resistin, an adipokine highly associated with obesity 51 and impaired insulin action in the liver.52, 53, 54 Healthy, insulin‐sensitive adipocytes produce less resistin,55 thus a depot with a higher distribution of small proliferating cells, as observed in lipectomy‐induced compensation, would be expected to produce less resistin, and potentially play a role in improvements in glucose regulation. In general, FKOγ mice had lower circulating concentrations of adipokines compared with CONs. This is consistent with previous studies 31 and suggests that PPARγ is also associated with normal adipocyte function and hormone release related to cell maturation.
In summary, in the present study, we have elected to manipulate PPARγ (present or absent in adipose tissue) in the context of lipectomy or sham surgery to further investigate the relationship between adipose tissue expansion and glucose homeostasis. We hypothesized that lipectomy‐induced increases in adipose tissue growth would be impaired in FKOγ knockout mice and this impairment would lead to unfavourable outcomes in glucose homeostasis. Our results support the notion that adipose tissue expansion, via PPARγ‐mediated mechanisms, plays a role in glucose homeostasis. More specifically data from the present study suggest that increased adipocyte number and changes in size distribution to smaller adipocytes is closely linked to improvements in glucose homeostasis observed in response to lipectomy.
Supporting information
Acknowledgements
We thank Hamlin Barnes who completed RNA extraction for qPCR procedure. This study was funded by NIH, grant K01DK087816.
Booth AD, Magnuson AM, Cox‐York KA, Wei Y, Wang D, Pagliassotti MJ, and Foster MT. Inhibition of adipose tissue PPARγ prevents increased adipocyte expansion after lipectomy and exacerbates a glucose‐intolerant phenotype. Cell Prolif. 2017;50:e12325. 10.1111/cpr.12325
References
- 1. Bjorntorp P. Metabolic implications of body fat distribution. Diabetes Care. 1991;14:1132–1143. [DOI] [PubMed] [Google Scholar]
- 2. Kissebah AH, Krakower GR. Regional adiposity and morbidity. Physiol Rev. 1994;74:761–811. [DOI] [PubMed] [Google Scholar]
- 3. Vague J. The degree of masculine differentiation of obesities: A factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. Am J Clin Nutr. 1956;4:20–34. [DOI] [PubMed] [Google Scholar]
- 4. Bjorntorp P. “Portal” adipose tissue as a generator of risk factors for cardiovascular disease and diabetes. Arteriosclerosis. 1990;10:493–496. [PubMed] [Google Scholar]
- 5. Ross R, Aru J, Freeman J, Hudson R, Janssen I. Abdominal adiposity and insulin resistance in obese men. Am J Physiol Endocrinol Metab. 2002;282:E657–E663. [DOI] [PubMed] [Google Scholar]
- 6. Ohlson LO, Larsson B, Svardsudd K, et al. The influence of body fat distribution on the incidence of diabetes mellitus. 13.5 years of follow‐up of the participants in the study of men born in 1913. Diabetes. 1985;34:1055–1058. [DOI] [PubMed] [Google Scholar]
- 7. Goodpaster BH, Thaete FL, Simoneau JA, Kelley DE. Subcutaneous abdominal fat and thigh muscle composition predict insulin sensitivity independently of visceral fat. Diabetes. 1997;46:1579–1585. [DOI] [PubMed] [Google Scholar]
- 8. Lapidus L, Bengtsson C, Larsson B, Pennert K, Rybo E, Sjostrom L. Distribution of adipose tissue and risk of cardiovascular disease and death: A 12 year follow up of participants in the population study of women in Gothenburg, Sweden. Br Med J (Clin Res Ed). 1984;289:1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McLaughlin T, Lamendola C, Liu A, Abbasi F. Preferential fat deposition in subcutaneous versus visceral depots is associated with insulin sensitivity. J Clin Endocrinol Metab. 2011;96:E1756–E1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arsenault BJ, Beaumont EP, Despres JP, Larose E. Mapping body fat distribution: A key step towards the identification of the vulnerable patient? Ann Med. 2012;44:758–772. [DOI] [PubMed] [Google Scholar]
- 11. Laurencikiene J, Skurk T, Kulyte A, et al. Regulation of lipolysis in small and large fat cells of the same subject. J Clin Endocrinol Metab. 2011;96:E2045–E2049. [DOI] [PubMed] [Google Scholar]
- 12. Krotkiewski M, Bjorntorp P, Sjostrom L, Smith U. Impact of obesity on metabolism in men and women. Importance of regional adipose tissue distribution. J Clin Invest. 1983;72:1150–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Patel P, Abate N. Body fat distribution and insulin resistance. Nutrients. 2013;5:2019–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Acosta JR, Douagi I, Andersson DP, et al. Increased fat cell size: A major phenotype of subcutaneous white adipose tissue in non‐obese individuals with type 2 diabetes. Diabetologia. 2016;59:560–570. [DOI] [PubMed] [Google Scholar]
- 15. Joe AW, Yi L, Even Y, Vogl AW, Rossi FM. Depot‐specific differences in adipogenic progenitor abundance and proliferative response to high‐fat diet. Stem Cells. 2009;27:2563–2570. [DOI] [PubMed] [Google Scholar]
- 16. Weyer C, Foley JE, Bogardus C, Tataranni PA, Pratley RE. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia. 2000;43:1498–1506. [DOI] [PubMed] [Google Scholar]
- 17. Eriksson JW, Smith U, Waagstein F, Wysocki M, Jansson PA. Glucose turnover and adipose tissue lipolysis are insulin‐resistant in healthy relatives of type 2 diabetes patients: Is cellular insulin resistance a secondary phenomenon? Diabetes. 1999;48:1572–1578. [DOI] [PubMed] [Google Scholar]
- 18. Paolisso G, Tataranni PA, Foley JE, Bogardus C, Howard BV, Ravussin E. A high concentration of fasting plasma non‐esterified fatty acids is a risk factor for the development of NIDDM. Diabetologia. 1995;38:1213–1217. [DOI] [PubMed] [Google Scholar]
- 19. Foley JE, Lillioja S, Zawadzki J, Reaven G. Comparison of glucose metabolism in adipocytes from Pima Indians and Caucasians. Metabolism. 1986;35:193–195. [DOI] [PubMed] [Google Scholar]
- 20. Kursawe R, Eszlinger M, Narayan D, et al. Cellularity and adipogenic profile of the abdominal subcutaneous adipose tissue from obese adolescents: Association with insulin resistance and hepatic steatosis. Diabetes. 2010;59:2288–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brook CG, Lloyd JK. Adipose cell size and glucose tolerance in obese children and effects of diet. Arch Dis Child. 1973;48:301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Manolopoulos KN, Karpe F, Frayn KN. Gluteofemoral body fat as a determinant of metabolic health. Int J Obes (Lond). 2010;34:949–959. [DOI] [PubMed] [Google Scholar]
- 23. Heilbronn L, Smith SR, Ravussin E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int J Obes Relat Metab Disord. 2004;28(Suppl 4):S12–S21. [DOI] [PubMed] [Google Scholar]
- 24. McLaughlin T, Sherman A, Tsao P, et al. Enhanced proportion of small adipose cells in insulin‐resistant vs insulin‐sensitive obese individuals implicates impaired adipogenesis. Diabetologia. 2007;50:1707–1715. [DOI] [PubMed] [Google Scholar]
- 25. Fang L, Guo F, Zhou L, Stahl R, Grams J. The cell size and distribution of adipocytes from subcutaneous and visceral fat is associated with type 2 diabetes mellitus in humans. Adipocyte. 2015;4:273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Danforth E Jr. Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000;26:13. [DOI] [PubMed] [Google Scholar]
- 27. Tontonoz P, Spiegelman BM. Fat and beyond: The diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. [DOI] [PubMed] [Google Scholar]
- 28. Imai T, Takakuwa R, Marchand S, et al. Peroxisome proliferator‐activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc Natl Acad Sci USA. 2004;101:4543–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sugii S, Olson P, Sears DD, et al. PPARgamma activation in adipocytes is sufficient for systemic insulin sensitization. Proc Natl Acad Sci USA. 2009;106:22504–22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rader DJ. Effect of insulin resistance, dyslipidemia, and intra‐abdominal adiposity on the development of cardiovascular disease and diabetes mellitus. Am J Med. 2007;120(3 Suppl 1):S12–S18. [DOI] [PubMed] [Google Scholar]
- 31. He W, Barak Y, Hevener A, et al. Adipose‐specific peroxisome proliferator‐activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci USA. 2003;100:15712–15717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Foster MT, Shi H, Seeley RJ, Woods SC. Removal of intra‐abdominal visceral adipose tissue improves glucose tolerance in rats: Role of hepatic triglyceride storage. Physiol Behav. 2011;104:845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Foster MT, Shi H, Seeley RJ, Woods SC. Transplantation or removal of intra‐abdominal adipose tissue prevents age‐induced glucose insensitivity. Physiol Behav. 2010;101:282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mauer MM, Bartness TJ. Fat pad‐specific compensatory mass increases after varying degrees of lipectomy in Siberian hamsters. Am J Physiol. 1997;273:R2117–R2123. [DOI] [PubMed] [Google Scholar]
- 35. Shi H, Song CK, Giordano A, Cinti S, Bartness TJ. Sensory or sympathetic white adipose tissue denervation differentially affects depot growth and cellularity. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1028–R1037. [DOI] [PubMed] [Google Scholar]
- 36. Hausman DB, Lu J, Ryan DH, Flatt WP, Harris RB. Compensatory growth of adipose tissue after partial lipectomy: Involvement of serum factors. Exp Biol Med (Maywood). 2004;229:512–520. [DOI] [PubMed] [Google Scholar]
- 37. Kim DH, Sandoval D, Reed JA, et al. The role of GM‐CSF in adipose tissue inflammation. Am J Physiol Endocrinol Metab. 2008;295:E1038–E1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. [DOI] [PubMed] [Google Scholar]
- 39. Hirsch J, Gallian E. Methods for the determination of adipose cell size in man and animals. J Lipid Res. 1968;9:110–119. [PubMed] [Google Scholar]
- 40. Bowers RR, Festuccia WT, Song CK, Shi H, Migliorini RH, Bartness TJ. Sympathetic innervation of white adipose tissue and its regulation of fat cell number. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1167–R1175. [DOI] [PubMed] [Google Scholar]
- 41. Mauer MM, Bartness TJ. Temporal changes in fat pad mass and cellularity after lipectomy in Siberian hamsters. Physiol Behav. 1997;62:1029–1036. [DOI] [PubMed] [Google Scholar]
- 42. Mauer MM, Bartness TJ. Body fat regulation after partial lipectomy in Siberian hamsters is photoperiod dependent and fat pad specific. Am J Physiol. 1994;266:R870–R878. [DOI] [PubMed] [Google Scholar]
- 43. Mauer MM, Bartness TJ. Photoperiod‐dependent fat pad mass and cellularity changes after partial lipectomy in Siberian hamsters. Am J Physiol. 1996;270:R383–R392. [DOI] [PubMed] [Google Scholar]
- 44. Kim SM, Lun M, Wang M, et al. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014;20:1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lu Q, Li M, Zou Y, Cao T. Induction of adipocyte hyperplasia in subcutaneous fat depot alleviated type 2 diabetes symptoms in obese mice. Obesity (Silver Spring). 2014;22:1623–1631. [DOI] [PubMed] [Google Scholar]
- 46. Mittendorfer B. Origins of metabolic complications in obesity: Adipose tissue and free fatty acid trafficking. Curr Opin Clin Nutr Metab Care. 2011;14:535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mauer MM, Bartness TJ. Short‐day‐like body weight changes do not prevent fat pad compensation after lipectomy in Siberian hamsters. Am J Physiol. 1997;272:R68–R77. [DOI] [PubMed] [Google Scholar]
- 48. Koda M, Kawakami M, Murawaki Y, Senda M. The impact of visceral fat in nonalcoholic fatty liver disease: Cross‐sectional and longitudinal studies. J Gastroenterol. 2007;42:897–903. [DOI] [PubMed] [Google Scholar]
- 49. van der Poorten D, Milner KL, Hui J, et al. Visceral fat: A key mediator of steatohepatitis in metabolic liver disease. Hepatology. 2008;48:449–457. [DOI] [PubMed] [Google Scholar]
- 50. Wagoner B, Hausman DB, Harris RB. Direct and indirect effects of leptin on preadipocyte proliferation and differentiation. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1557–R1564. [DOI] [PubMed] [Google Scholar]
- 51. Kusminski CM, McTernan PG, Kumar S. Role of resistin in obesity, insulin resistance and Type II diabetes. Clin Sci (Lond). 2005;109:243–256. [DOI] [PubMed] [Google Scholar]
- 52. Qi YNZ, Lee YS, Singhal NS, Scherer PE, Lazar MA, Ahima RS. Loss of resistin improved glucose homeostasis in leptin deficiency. Diabetes. 2006;55:3083–3090. [DOI] [PubMed] [Google Scholar]
- 53. Steppan CMBS, Bhat S, Brown EJ, et al. The hormones resistin links obestity to diabetes. Nature. 2001;409:307–312. [DOI] [PubMed] [Google Scholar]
- 54. Costandi J, Melone M, Zhao A, Rashid S. Human resistin stimulates hepatic overproduction of atherogenic ApoB‐containing lipoprotein particles by enhancing ApoB stability and impairing intracellular insulin signaling. Circ Res. 2011;108:727–742. [DOI] [PubMed] [Google Scholar]
- 55. Shojima N, Sakoda H, Ogihara T, et al. Humoral regulation of resistin expression in 3T3‐L1 and mouse adipose cells. Diabetes. 2002;51:1737–1744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials