

Abstract
Enzyme-mediated cascade reactions are widespread in biosynthesis. To facilitate comparison with the mechanistic categorizations of cascade reactions by synthetic chemists and delineate the common underlying chemistry, we discuss four types of enzymatic cascade reactions: those involving nucleophilic, electrophilic, pericyclic, and radical reactions. Two subtypes of enzymes that generate radical cascades exist at opposite ends of the oxygen abundance spectrum. Iron-based enzymes use O2 to generate high valent iron-oxo species to homolyze unactivated C–H bonds in substrates to initiate skeletal rearrangements. At anaerobic end, enzymes reversibly cleave S-adenosylmethionine (SAM) to generate the 5’-deoxyadenosyl radical as a powerful oxidant to initiate C–H bond homolysis in bound substrates. The latter enzymes are termed radical SAM enzymes. We categorize the former as “thwarted oxygenases”.
Keywords: electrophilic cascades, natural products, nucleophilic cascades, pericyclic cascades, radical cascades
Graphical Abstract


Fantastic four: Generally, enzymes are highly selective catalysts for single reactions. However, some enzymes instead control a series of reactions in a cascade-like fashion. This Review highlights four types of enzymatic cascade strategies, mediated by nucleophilic, electrophilic, pericyclic, and radical-based reactions, observed in the biosynthesis of complex natural products
1. Introduction to Cascade Reactions in Total Synthesis
Cascade reactions have attracted special attention both historically and among current organic chemists for both intellectual and practical challenges (e.g. atom economy[1] and synthetic ideality[2]) in the generation of molecular scaffold complexity.[3] Cascade reactions, both planned and somewhat serendipitous, have advanced mechanistic understanding and molecular reactivity, for example, Robinson s tropinone synthesis in 1917,[4] the polyolefin cyclization of Johnston and colleagues in the synthesis of progesterone in 1971,[5] and many dozens over the past five decades.[6]
Nicolaou and colleagues[3a] authored an extensive review on Cascade Reactions in Total Synthesis in 2006 that collected many examples of strategies for the assembly of several natural product scaffolds in which cascade reactions of different types created dramatic increases in framework complexity. They often represent the apotheosis of both skill and elegance as organic chemists use synthetic campaigns as proving grounds for understanding and predicting reactivity rules.
Cascade reactions transform reactants into intermediates/products in multiple distinct steps. Thus, the definition of where a cascade stops may be the practical stage of a one-pot reaction. This is a different endpoint from the enzymatic cases discussed in this Review, where release of a product from the active-site microenvironment is the reaction endpoint.
Nicolaou and colleagues[3a] also noted that the nature of multistep cascade transformations can complicate classification but they divided their analyses into five mechanistic categories: nucleophilic, electrophilic, radical-based, pericyclic, and transition-metal-based mechanisms for the framework rearrangements (Scheme 1).
Scheme 1.

Examples of natural product chemical syntheses exemplifying the five mechanistic categories of cascade reactions. A) A nucleophilic cyclization cascade in the total synthesis of tetronasin by Ley and colleagues.[7] B) An electrophilic cascade involving an epoxy-olefin cyclization in the total synthesis of hemibrevetoxin B by Holton and colleagues.[8] C) A radical cyclization cascade in the total synthesis of morphine by Parker and Fokas.[9] D) A pericyclic cascades involving Diels-Alder and [3+2] cycloadditions in the total synthesis of vindorosine by Boger and colleagues.[10] E) Transition-metal-catalyzed cascades involving multiple ring-opening/ring-closing olefin-metatheses in the total synthesis of cyanthiwigin U by Pfeiffer and Phillips.[11] AIBN =2,2’-azobisisobutyronitrile, KHMDS =potassium bis(trimethylsilyl)amide, MOM =methoxymethyl, N-PSP =N-(phenylseleno)phthalimide, TIPB =triisopropylbenzene, Ts =p-tolylsulphonyl.
Readers are referred to that review and related articles.[6] Many of the end products of enzymatic cascades noted here have attracted the attention of natural product chemists who have completed total syntheses. Some readers may wish to go back and compare nature s actual cascade strategies with those of synthetic chemists, who proceeded, with or without a preconceived notion of biomimicry, often before the relevant enzymes were even identified and characterized.
This rubric seems a useful starting point as well for the enzyme-mediated cascades, since both enzymatic and non-enzymatic synthetic transformations follow the same rules of organic chemistry, although there are no known equivalents of olefin metathesis reactions (yet) in nature. Thus, we present four categories of nucleophilic, electrophilic, pericyclic, and radical-based cascades in enzymatic catalysis.
2. Enzymatic Cascades
For more than a century, it has been clear that proteins with catalytic activity, enzymes, carry out essentially all of the controlled chemical transformations in living organisms. In an RNA-world scenario, they were preceded by catalytic RNAs, remnants of which exist today, for example, in the ribonucleoprotein ribosomes that carry out protein biosynthesis.[12] There are some cascade reactions in primary metabolism, the interconversion of small-molecule metabolites into product-(s) carried out by all cells. For example, the tandem action of thiolase and hydroxymethylglutaryl-CoA synthase converts three acetyl-CoA molecules to the C6 half thioester HMG-CoA.[13] However, the major enzymatic complexity-generating events occur in conditional pathways, known collectively as secondary metabolism.
This is the realm of natural product biosynthesis, that is, biosynthesis of the isoprenoids, alkaloids, phenylpropanoids, polyketides, and nonribosomal peptides that account for most of the approximately 500,000 known natural products.[13] These also include the bioactive molecular frameworks that have been the targets of hundreds of natural product synthetic studies by chemists over the past 80 years.
Cascade reaction planning, implementation, and analysis have been core activities of synthetic chemists in the context of biomimetic or “bioinspired” routes.[3a, 6] Many of these synthetic campaigns were undertaken before the enzymes of a particular pathway were identified, purified, and examined for mechanism and specificity. Nonetheless, they often had predictive power, given that chemical mechanisms play out in biology. Indeed, a recent review by Liu and co-workers has catalogued examples of dozens of organic named reactions in enzymatic catalysis,[14] many of which are key steps in both nonenzymatic and enzymatic synthetic cascades.
In this Review, we delve into enzymatic examples of cascade reactions to illustrate how barriers for multistep transformations are lowered in specific enzyme active sites. These are part and parcel of the ability of enzymes involved in natural product biosynthesis to build complexity from simple primary metabolites. The coverage is not meant to be encyclopedic but rather to illustrate how some of the organic named reactions are put to use in cascades within a given enzyme active site, nature s equivalent of the one-pot synthetic reaction. We will mention a few tandem reactions, involving the consecutive action of two enzymes to promulgate the cascades. We exclude multienzyme participations in full biosynthetic pathway reconstitutions, although we have noted elsewhere the remarkable efficiency of enzymes to function in short pathways that build remarkable scaffold complexity.[13, 15]
While one could divide the presentation according to specific natural product categories, we think it more useful to categorize enzymatic cascade reactions by mechanism. To that end we use the Nicolaou[3a] approach of nucleophilic, electrophilic, radical-based, and pericyclic cascades. There is no enzymatic analogue of nonenzymatic olefin metathesis cascades and no indication that palladium or platinum are involved in biology. Molybdenum is used in a small number of enzymes[16] but not detectably for olefin metatheses.
3. Enzymatic Nucleophilic Cascades on Assembly Lines
Cascade reactions initiated or carried out by nucleophiles are common in the enzymatic assembly lines that build many thousands of polyketide, nonribosomal peptide, and hybrid nonribosomal peptide/polyketide natural product frameworks. A large subset of polyketide synthases (PKSs) and essentially all of the nonribosomal peptide synthetases (NRPSs) hold onto the growing intermediate chains as covalent thioesters and thus qualify as cascade catalysts.[13]
Carbanion equivalents as nucleophiles.
The celebrated deoxyerythronolide B (DEB) synthase, for example, takes seven molecules of 2S-methylmalonyl-CoA and elongates them to the 14-carbon linear thioester and then carries out regiospecific cyclization to release the 14-member DEB macrocyclic lactone as only one of 1024 possible diastereomers[17] (Scheme 2 A). Analogously, in the biosynthesis of tetracycline, the enzyme trio of OxyABC builds a tethered 19-carbon nonaketidyl thioester chain (Scheme 2 B). Tailoring enzymes then carry out transannular aldol and Claisen condensation reactions to release preteramid as the first soluble intermediate.[18]
Scheme 2.

Two polyketides generated by enzymatic cascades. A) The 14-member deoxyerythronolide B (DEB) lactone is released from the three-subunit DEB synthase assembly line after incorporation of seven methylmalonyl units. B) Oxytetracycline biosynthesis involves a chain elongation cascade to a nineteen carbon nonaketonyl thioester that is converted into the tetracyclic pretetramide product.
To build these nucleophilic chain-elongating cascade reactions, polyketide synthases need carbon nucleophiles to generate the C–C bonds. These are typically the CoA thioester enolates of malonyl and methylmalonyl acids (all seven monomers incorporated in DEB above). The thioester grouping activates C1 as the electrophile and adjacent C2 as the nucleophile for iterative thioclaisen condensations (see Ref. [13] for a review).
Amines as nucleophiles.
In contrast to polyketide cascades, where C–C bond formations are the chain elongation steps, NRPSs make amide bonds and use the amino nitrogen atoms of tethered amino acid monomers as the chain-elongating nucleophiles.[19] In the active sites of the condensation/chain elongation catalytic domains of NRPS assembly lines, the predominant -NH3+ ionization states must be converted into nucleophilic NH2 groups (Scheme 3 A). Concomitantly, in hybrid NRPS/PKS assembly lines, amines and thioester enolates are the requisite nucleophiles, for example, in rapamycin (Scheme 3 B) or FK506 assembly.[20]
Scheme 3.

Nonribosomal peptide synthetase assembly lines carry out chain-elongation cascades to the heptapeptide vancomycin (A) and the hybrid NRPS/PKS product rapamycin (B), in which amine nucleophiles are utilized for the insertion of amino acids.
An example of an unleashed cascade reaction occurs in the action of the fungal trimodular NRPS that makes fumiquinazoline F, in which anthranilate, tryptophan, and alanine building blocks are combined into a fused tricyclic quinazoline core.[21] The assembly line enzyme builds a tethered linear anthranilyl-Trp-Ala-thioester, which undergoes intramolecular capture by the anthranilyl amine, releasing a presumptive 6,10-macrocycle. This is never detected. Instead, one observes only the transannular, cyclodehydrated quinazoline product (Scheme 4).
Scheme 4.

The fumiquinazoline F NRPS assembly line carries out a cascade employing the amino group of the anthranilyl-1 residue for cyclizing release, followed by transannular capture of the transient 6,0-macrocycle to yield the tricyclic quinazolinone framework.
We note in the section on radical-driven cascades below that the tethered heptapeptidyl chain in the vancomycin synthetase assembly line is acted on successively by three “thwarted oxygenases” that make side-chain radicals that form cross links rather than undergo hydroxylations, as emblematic of tailoring reactions that occur on NRPS assembly lines. There are many examples of tailoring reaction on PKS assembly lines as well, including Michael additions by side chain -OH groups on conjugated enoyl thioesters to form cyclic ethers (Scheme 5).[22]
Scheme 5.

Oxa-1,4-cojugate additions create pyran rings on the PKS assembly lines during pederin (A), ambruticin S (B), and salinomycin (C) polyketide assembly.
3.1. Nucleophilic Enzymatic Cascades Enabled by Redox Steps
There are a number of cases where enzymes that carry out an oxidative or reductive change on a substrate/nascent product uncover reactions that can be categorized as cascade reactions. One simple case, extending the above discussion of NRPS assembly lines, is the recent discovery of the pathway to the cyclic peptide lugdunin, produced by Staphylococcus lugdunensis, which competes with pathogenic Staphylococcus aureus strains in human oral cavities.[23] The lugdunin NRPS assembly line generates a tethered heptapeptidyl-enzyme adduct and then reductively releases it through hydride transfer from the co-substrate NADH to yield the nascent linear peptide aldehyde (Scheme 6). This can be cyclized through attack of the N-terminal Cys1 amino group on the Val7 aldehyde to give the cyclic imine. The equilibrium in favor of cyclization is further driven by addition of the Cys1 side-chain -SH onto the imine to yield the cyclic thiazolidine. This is the accumulating form of lugdunin, which acts as an antibiotic against S. aureus strains in the oral cavity.
Scheme 6.

The antibiotic lugdunin arises from an NRPS assembly line cascade. The release step involves reduction of tethered peptidyl thioester by hydride transfer from NADH catalyzed by the LugC terminal reductase domain. The released aldehyde can circularize as the cyclic imine, which is further driven to accumulate as the cyclic thiazolidine from addition of the cysteine thiolate to the imine.
Another cascade reaction set in motion by a redox step with a nicotinamide co-substrate is catalyzed by the Sadenosylhomocysteine (SAH) lyase, the key enzyme in returning homocysteine carbons to the cellular metabolic pool after SAM has been used for millions of methyl transfers in every cell cycle (Scheme 7). The enzyme is erroneously termed a hydrolase because the thioether linkage in SAH is converted into free homocysteine and adenosine. The thioether is stable to hydrolysis. Instead the SAH lyase first oxidizes C-3 of the ribose ring to the ketone while generating NADH, which is kept tightly bound in the active site.[24] The value of the alcohol to ketone oxidation is in acidification of the adjacent C4–OH since the resultant carbanion is now stabilizable as the enolate anion. This easily accessible carbanion can be used to eliminate the homocysteine moiety with C5–S cleavage to yield the conjugated enone with a 4,5-exomethylene. This conjugate enone is the electrophile for water addition to yield the 3’-ketoadenosine. Then back transfer of the hydride from bound NADH gives the observed product adenosine.
Scheme 7.

The enzyme SAH lyase starts a cascade leading to thioether cleavage by initial hydride-mediated oxidation of the ribose-3-OH to the ketone. This lowers energy for C4 carbanion as the internal nucleophile required for C5–S cleavage.
Initiation of nucleophilic cascades can occur through an initial transfer of a hydride from NAD(P)H to an olefin in a co-substrate, thereby creating a transient carbon nucleophile to drive subsequent carbocycle formation. Examples of this enzymatic strategy include the formation of plant iridoid monoterpene scaffolds[25] and also the tricyclic core of bacterial polycyclic tetramate macrolactams.[26] Iridoid scaffolds, such as in nepetalactone and the secologanin-derived moiety in the oncology agent vincristine, arise from geranyl-diphosphate (GPP) that is processed to 8-oxogeranial (1,8-geranyl dialdehyde). Hydride conjugate addition from NADPH by iridoid synthase sets up the enolate that reacts intramolecularly through a Michael cyclization to form the irodial nucleus nepetalactol (Scheme 8 A).[25b] In analogy, the ikarugamycin biosynthetic enzyme IkaB similarly transfers a hydride to the terminal olefin of the nascent product from the IkaA polyketide synthase (Scheme 8 B). A proposed 4+2 cyclization yields the substrate for IkaC and another hydride-initiated cyclization to yield the fused 5–6-5 core of the bacterial metabolite ikarugamycin.[26b]
Scheme 8.

Reductive strategies to carbocycle formation in plant iridoid scaffolds (A) and bacterial polycyclic tetramate macrolactams (B).
In addition to these NAD/NADH-dependent redox enzymes that set off cascade reactions, there are several flavin-dependent enzymes that conduct redox catalysis that uncover cryptic reactivity in nascent products. Three examples are presented. The first is the FAD-enzyme Sol5 that converts the exocyclic alcohol group in prosolanapyrone II into the pyrone aldehyde in prosolanapyrone III in a fungus that causes potato blight (Scheme 9).[27] The aldehyde is now in conjugation with the adjacent olefin and that apparently lowers the energy barrier so that it can act as a dienophile towards the terminal diene. The result is an apparent Diels-Alder [4+2] cyclization to the decalin bicycle in solanapyrone.
Scheme 9.

The redox step catalyzed by the FAD-containing solanapyrone synthase brings the aldehyde into conjugation with the exocyclic olefin to increase its reactivity as a dienophile for the ensuing Diels-Alder cyclization.
The second enzyme is an FAD-containing monooxygenase that converts the tetracyclic core of glycosylated premithramycin B in to the tricyclic ring in mithramycin DK (Scheme 10). The enzyme MtmOIV is a Baeyer-Villiger catalyst, which delivers a nucleophilic oxygen to the d-ring enone with ring expansion to the lactone. The lactone as a cyclic ester is labile to water-catalyzed ring opening as shown. Collapse of the tetrahedral adduct generates an acid attached to the C-ring and an initial enediolate from what had been the D-ring. Ketonization yields the hydroxyketone grouping in the product mithramycin, with net oxygenative/ hydrolytic fragmentation of the d-ring cyclohexenone.[28]
Scheme 10.

The FAD-enzyme that catalyzes a Baeyer-Villiger oxygenation on the d-ring of tetracyclic premithramycin converts the cylohexenone D-ring into the ring-expanded lactone. This is now labile to water-mediated hydrolytic opening. The released enediolate isomerizes to the tricyclic ketone mithramycin product.
The third FAD enzyme, EncM, catalyzes oxidation of C4 in the side chain of the advanced polyketide chain tethered to the type II PKS EncC protein in enterocin biosynthesis.[29] A notable aspect of the EncM catalytic mechanism is the discovery that it uses a newly identified flavin-N5-oxide as its oxygen atom transfer catalyst to the 1,3-diketone methylene[30] (Scheme 11). This creates a transient 1,2,3-triketo moiety and sets in motion a Favorskii-type rearrangement, presumably generating a hydroxycyclopropanone intermediate. That highly strained electrophile can be captured by an intramolecular OH group acting as nucleophile, as shown, to create an internal lactone and altered framework connectivity. Lactonizing release from the acyl carrier protein EncC produces the observed deoxyenterocin product. The complete pathway has been reconstituted from pure enterocin biosynthesis enzymes in vitro to generate the natural product and designed unnatural products.[31]
Scheme 11.

The EncM flavoenzyme doubly oxygenates a 1,3-diketo moiety of the enzyme-bound poly-β-keto intermediate to a 1,2,3-triketo nascent product via the newly discovered flavin-N5-oxide cofactor. This product intermediate is subject to a Favorskii-type nucleophilic rearrangement cascade with formation of a transient hydroxycyclopropanone.
4. Enzymatic Electrophilic Cascades
Two categories of electrophilic cascades are discussed in this section. The most extensive occurs in terpene biosynthetic pathways in which carbocation chemistry dominates enzymatic reaction sequences. In the second category, we describe the use of epoxides as disappearing electrophiles, particular in the context of cyclic ether formations. Both of these strategies have been previously reviewed in the context of biosynthetic cascades.[32]
4.1. Carbocation-Driven Cascade Rearrangements of Terpene Scaffolds
The cyclization of terpene substrates is synonymous with cascade biochemistry. The enzymology of monoterpene (C10), sesquiterpene (C15), diterpene (C20), and triterpene (C30) scaffold diversification is replete with cascade reactions that are mediated by carbocation rearrangements and quenched in given enzyme sites at specific loci and at specific points in the reaction manifold.[13, 33]
At the C15 level, farnesyl-diphosphate (FPP) is converted into the tricyclic-5,5,5-alkene in the active site of pentalenene synthase via at least four cation rearrangement intermediates during the assembly of the sesquiterpene antibiotic pentalenolactone (Scheme 12).[34] Catalysis is initiated as usual by early dissociation of the C1–OPP carbon-oxygen bond to the delocalized allyl cation. Capture of the C1 cation by the C11 double-bond terminus yields the E,Z-humulyl cation, which can then proceed to a tricyclic 5,6,4-cation before rearrange-ment to the 5,5,5-tricyclic cation. Proton loss creates the double bond in the pentalenene product framework as shown.[35]
Scheme 12.

A sequiterpene cation rearrangement cascade converts the acyclic C15 farnesyl-diphosphate into the tricyclic-5,5,5-framework of pentalenene during the biosynthesis of pentalenolactone.
At the C20 (diterpene) level, the classic building block is geranylgeranyl-diphosphate (GGPP). It is the direct precursor of the pentacyclic framework of taxadiene, the 6,8,6-tricyclic diene that is the product of taxadiene synthase (Scheme 13).[36] In turn, taxadiene is subsequently oxygenated to introduce eight oxygen atoms to increase solubility and yield the end product taxol,[37] a potent ligand for tubulin and an antimitotic antitumor agent of wide clinical utility. The conversion of linear tetraenoic GGPP to tricyclic taxadiene is clearly a cascade process, with the first step converting a bound conformer of GGPP into the C1 allyl cation with capture by the terminal C17 olefinic carbon. That initial macrocyclic cation undergoes three further rearrangements, leading to two new C–C bonds on the way to releasing taxadiene.
Scheme 13.

A diterpene rearrangement cascade converts acyclic C20 geranylgeranyl-diphosphate into tricyclic taxadiene.
The most famous of the polyolefin cyclization cascades occur in many variants of triterpene cyclases, with the acyclic C30-hexaene squalene as the starting point. The most straight-forward may be the bacterial squalene-hopene cyclase.[38] The 2,3-double bond is protonated in the enzyme active site to the hopanyl cation to start the multistep capture of that cation by the remaining five olefins. This creates a pentacyclic tertiary cation that can be quenched in one of two ways. Proton abstraction yields hopene (Scheme 14 A), while water addition gives hopanol.
Scheme 14.

Triterpene cyclization cascades. A) Squalene-hopene cyclase initiates the electrocyclic cascade by protonation of the 2,3-terminal double bond of the acyclic C30 hexaene squalene. B) Cyclization of squalene to lanosterol via a cascade of cation, hydride, and methyl group migrations after oxidosqualene formation. C) Enzymatic cyclization cascade from 2,3-epoxysqualene to the pentacyclic β-amyrin product is the predominant mode in plant metabolism.
4.2. Enzymatic Methylation as Initiator of a Cyclization Cascade
A variant of controlled induction of an electrophilic cyclization occurs in the late stages of assembly of the prenylated indole teleocidin B, an inhibitor of protein kinase C (Scheme 15).[39] The precursor of teleocidin B is the indolactam lyngbyatoxin A, itself the causative agent of seaweed dermatitis.[40] The biosynthesis of lyngbyatoxin A is understood to involve a two-module NRPS and then a P450 enzyme (thwarted oxygenase; see the section below on radical cascades), with a mechanism involving radical coupling of the valyl nitrogen and C4 on the indole moiety.[41] To go from lyngbyatoxin A to the teleocidin B stereoisomers requires a new C–C bond at C6 of the indole ring. Studies by Abe and colleagues[39] revealed that the methyltransferase TleD is the enzyme that sets the appropriate chemical cascade in motion. S-adenosylmethionine donates a [CH3+] equivalent to the terminal olefin at C25, creating the initial C26 tertiary carbocation. Through deuterium labeling, Abe and colleagues established that the C25–H migrates to C26 as a hydride, creating a transient 28 carbocation at C25. Capture of this cation by C7 of the indole would yield the indicated 5,6-ring spiro cycle with an exocyclic iminium ion. Ring expansion through single-bond migration would quench the charge on the indole lactam exocyclic nitrogen and give teleocidin B, thus showing that the capture of the C25 carbanion had been a re face attack. This cascade expands the enzymatic repertoire of electrophilic initiation of cyclizations to double-bond protonation, C-methylation, and chlorination and epoxidation (examined in the next two subsections).
Scheme 15.

Enzymatic C-methylation at an olefin initiates an electrophilic cascade in teleocidin B biosynthesis.
4.3. Enzymatic Chlorination to Initiate an Electrophilic Cascade
The merochlorin meroterpenoid antibiotics contain a 1,3,6,8-tetrahydroxynaphthalene core (from a type III polyketide synthase) connected to a rearranged sesquiterpene chain arising from prenyltransferase activity.[42] The premerochlorin scaffold is then the substrate for a vanadium-dependent chloroperoxidase that delivers chloronium ions to the naphthalene ring and also sets in motion an oxidative dearomatization/terpene cyclization cascade via carbocation intermediates that lead to the unusual frameworks of merochlorins A-D (Scheme 16).[42]
Scheme 16.

Enzymatic double chlorination of a naphthol ring by chloronium ion equivalents sets a carbocation cascade in motion in merochlorin A and B formation. Merochlorin C formation rather involves a Cl+-mediated α-hydroxyketone rearrangement and a third chlorination event.
The vanadium-dependent chloroperoxidase Mcl24 delivers a “Cl+” equivalent, most likely from a V OCl species via an enzyme-bound chloramine intermediate, to C2 of the tetrahydroxynaphthol nucleus as shown in Scheme 16. Transfer of a second chloronium ion equivalent to the C1–OH would give the naphthyl hypochlorite transiently, followed by elimination of that Cl as a chloride ion by intramolecular participation of the aromatic double bonds, leading to the dearomatized cation. Now cyclization by regioselective capture of that cation by the middle double bond of the side chain triene yields the new tertiary cation that can be quenched by either of two routes. Route a) yields the product merochlorin A with fused chloro-cyclopentanone, while route b) gives the fused tetrahydrofuran ring of merochlorin B. More recently, Miles et al have shown that merochlorin X, the putative precursor to merochlorins C and D, can come from similar chemical logic in the active site of the Mcl24 chloroperoxidase, in this case via route c) through a chloroniumion assisted α-hydroxyketone rearrangement (Scheme 16).[43] Thus Mcl24 single-handedly provides the three merochlorin natural product skeletons from the same pre-merochlorin precursor.
Total syntheses of both merochlorins A and B have been achieved.[42b, 44] The four-enzyme pathway to merochlorins in a single test tube has also been achieved,[45] as well as a unifying hypothesis for naphthoquinone-containing meroterpenoid scaffolds.[43, 46] Comparison of the strategic use of “Cl+” in this case versus “CH3+” in the teleocidin case above shows the different tools available in enzymatic sites to initiate electrophilic cascade chemistry.
We will describe a different halogenation strategy in Section 8, in which chlorine radical equivalents are employed in an enzymatic cascade to cyclindrocyclophane, and compare it to nonenzymatic metathesis synthetic strategy.
4.4. Tandem Epoxide Formation and Opening in Enzymatic Electrophilic Cascades
The more common modes of squalene enzymatic cyclization involve prior epoxidation of the 2,3-double bond to the β-epoxide by a vitamin B2-dependent squalene epoxidase. This epoxide is then the electrophilic substrate for oxidosqualene cyclase, with participation of the π electrons from four of the olefins (not five as in squalene-hopene cyclase, presumably because of folded conformer differences in the active sites) to the protosterol cation (Scheme 14 B, see Ref. [13] for a summary).[47] A cascade of two hydride and two methyl migrations occurs before cation quenching and release of tetracyclic lanosterol. It has been argued that the squalene substrate in the hopene cyclase is an all pre-chair foldamer but is folded as a pre-chair-boat-chair conformer in oxidosqualene cyclase.[47] Correspondingly, a chair-chair-chair foldamer for squalene epoxide would give the 6,6,6,6,6-pentacyclic scaffold of β-amyrin, the most common steroid scaffold in plant metabolism. Conversion of the initial dammarenyl cation via a lupanyl cation into the oleanyl cation is followed by quenching through proton abstraction by a specifically placed enzymatic base (Scheme 14 C).[48]
While many of the steroid cyclases terminate the cation-driven cascade rearrangements at one specific stage and region by proton abstraction or water addition, there are family members that show some promiscuity. The baruol synthase from the Arabidopsis plant generates baruol at nearly 90 % of the flux, with the remaining material distributed over 22 minor products, varying in amounts from 0.022−2.7 % (Scheme 17).[49] This product range proves that a cascade of multiple rearranged cationic species exist and can be quenched, perhaps as 22 minor mistakes of timing and placement.
Scheme 17.

Baruol synthase leaks a set of minor products reflecting capture of intermediates at different points in the cationic cascade process. Bold arrows show the primary route to baruol with representative pathway byproducts shown to other cyclic triterpenes with relative percentage product distribution.
Several examples of disappearing-epoxide strategies occur at the interface of indole alkaloid and prenyl transfer enzymology. One such example occurs during assembly of the paxilline scaffold. Indole capture of the C20 GGPP yields 3-geranylgeranyl indole (Scheme 18 A). This is the substrate for PaxM, which bis-epoxidizes the terminal two of four side-chain olefins. The bis-epoxide is now subject to enzyme-mediated capture by π electrons of the remaining two double bonds by the PaxB enzyme to create the hexacyclic frame-work of paspaline,[50] which upon subsequent double oxygenation serves as a precursor to the end product paxillin. A related strategy is used in assembly of indolosesquiterpenes, represented by the pentacyclic family of xiamycin.[51] The conversion of farnesylindole into xiamyicin involves three kinds of enzymatic oxygenations (Scheme 18 B). The first is a prototypic epoxidation of the terminal olefin of farnesyl indole and electrophilic closure of two carbacyclic rings to form preindosespene. The second round of three P450-mediated oxygenations converts an exocyclic methyl into the carboxylate via an intermediate alcohol and aldehyde, consuming three molecules of O2. C–C bond formation to yield the xiamycin scaffold occurs by flavoenzyme action through a cryptic indole ring hydroxylation. The resultant indoline intermediate then undergoes capture at C2 of the indole ring by the neighboring olefin, loss of water, and oxidative aromatization.[51d]
Scheme 18.

Disappearing-epoxide cascade reactions in the enzymatic conversion of 3-prenylindole substrates into paspaline (A) and xiamycin (B).
The actions of PaxM and PaxB illustrate a widely used cascade strategy of tandem enzymes that make and then open epoxides as “disappearing electrophiles” in cascade reactions.[52] The squalene epoxidase and oxidocyclase pair above are such examples. So are Lsd18 and Lsd19 in lasalocid biosynthesis, but in that case the nucleophiles initiating the cascades are not ? electrons of olefins but instead the alkoxide forms of internal -OH groups.[53] As shown in Scheme 19, Lsd19 acts on the bis-epoxide generated by Lsd18 with regio- and stereospecific control to generate two sizes of cyclic ethers: the five-member dihydrofuran and the sixmember dihydropyran. Although the enzymes remain to be discovered for the dinoflagellate polyether toxins, such as the 11-fused ether scaffold of brevetoxin B and 13-cyclic ethers in ciguatoxins, it is anticipated that polyepoxide precursors will be converted in comparable cascades (Scheme 20).[54] Analogously, Leadlay and colleagues have presented evidence for MonC involvement as an epoxidase to create a triepoxide that, on opening by MonB, yields the five cyclic ethers in the polyether antibiotic monensin.[55] The sequence of polyolefin to polyepoxide to fused pyran and furan ring scaffolds in cyclic polyethers was predicted by Cane, Celmer, and Westley,[53b] and further refined by Spencer and colleagues[55b] for monensin.
Scheme 19.

Tandem action of the epoxidase Lsd18 and “epoxide hydrolase” Lsd19 convert a bis-olefin by way of bis epoxide into the tetrahydrofuran and tetrahydropyran rings of the ionophore lasalocid A.
Scheme 20.

It is proposed that 11 double bonds are converted into 13 epoxides as precursors to the fused cyclic ethers in the potent marine ciguatoxin.
One final small-molecule example of the strategy of deploying epoxides as intermediate electrophiles in enzymatic cascades is the assembly of the 2,6-dioxabicyclo-[3.2.1]octane scaffold of the ATP synthase inhibitor aurovertin E (Scheme 21).[56] The E,E,E-terminal triene of the polyketide precursor is enzymatically isomerized to the E,E,Z geometric isomer that is now a substrate for the FAD-containing monooxygenase AurC, which epoxidizes two of the three double bonds. AurD is the paired epoxide hydrolase that catalyzes intermolecular regioselective attack of water on one of the epoxides to create the incipient oxyanion to open the second epoxide and yield the dihydroxyfuran species. This has one double bond and is again a substrate for the epoxidase AurC. That mono-epoxide is once again tandemly acted on by AurD, now acting as an intramolecular “epoxide hydrolase”, in a 6-endo-tet opening to create the bicyclo-octane scaffold with fused terahydrofuran and tetrahydropyran rings, reflecting their epoxide heritages. The use of epoxides as disappearing electrophiles to initiate cascade reactions is well established as part of nature s biosynthetic tool set.
Scheme 21.

Three disappearing epoxide intermediates in assembly of the bicycloctane scaffold of the ATP synthase inhibitor aurovertin.
5. Pericyclases
Pericyclic reactions have a central place in many of the multistep syntheses of complex natural product scaffolds. Nicolaou et al.[3a] and Baran[6] list a variety of 2+2 and 4+2 cyclizations, and even a 5+2 cyclization, as well as several variants of 3,3-Claisen and Cope, azaCope, oxyCope rearrangements in a multitude of synthetic campaigns. Hetero-4+2 rearrangements have also been used to in biochemical pathways for complexity generation in several contexts.
For some decades, there has been debate as to whether comparable pericyclic reactions occur in metabolism, particularly in the secondary metabolic pathways where many complex scaffolds and frameworks are generated. The debate has often centered on whether transformations were actually concerted or stepwise. We note 3,3-rearrangements of the Claisen and Cope types below. Over the past decade, a number of purified enzymes have also been assigned as 4+2 cyclization catalysts, although it remains to be proven that any one of the purported 4+2 reactions proceed by a concerted mechanism (either synchronous or partially asynchronous).[57] Certainly, the 4+2 conversions dramatically alter framework connectivity in the enzymatic products.[58] Some of them, we would put in the cascade category.
In connection with this, we highlight the tandem action of the enzymes PyrE3 and PyrE4 in the pathway to pyrroindomycin B (Scheme 22).[59] It is not so much that either enzyme by itself is a cascade catalyst but rather that tandem action leads to the generation of the pentacyclic core of the natural product. Furthermore, the two enzymes illustrate the two variants of the kinds of 4+2 outcomes that have been catalogued to date in enzyme systems. PyrE3 is a “typical” decalin-forming [4+2] catalyst. The next enzyme, PyrE4, takes this dialkyl decalin product and makes a spirotetronate ring system as the pentacyclic scaffold is constructed through the second type of apparent [4+2] cyclization.
Scheme 22.

Tandem action of PyrE3 and PyrE4 as catalysts of two types of Diels-Alder cyclization in the biosynthetic pathway to pyrroindomycin
With some degree of analogy, the insecticide molecule spinosyn A has a 5,6,5-tricyclic core that is also of pericyclic origin. SpnF has been purified to homogeneity and shown to accelerate a slow non-enzymatic cyclization of the macrocyclic substrate to the central cyclohexene and the right-hand cyclopentane rings. Following enzymatic glycosylation by SpnG, SpnL then cyclizes the remaining diene to the cyclopentene ring in what may be a Rauhut-Currier-type of C?C bond formation.[60] The two enzymes SpnF and SpnL thus build the constrained 5,6,5-tricyclic core of spinosyn A (Scheme 23). Energetic calculations for the SpnF transition states support a [4+2] reaction but also suggest a possible [6+4] route that would need to be followed by a 3,3-Cope rearrangement to give the observed product.[60]
Scheme 23.

SpnF and SpnL build the fused 5,6,5-tricyclic core of the insecticidal agent spinosyn through Diels-Alder and Rauhut-Currier reactions.
The Diels-Alder [4+2] cyclization was originally described in 1928.[61] An oxo-Diels-Alder reaction to give dihydropyran derivatives was reported in 1949.[62] The corresponding intramolecular aza-Diels-Alder reaction on an imino alkyne was reported in 2009.[63] The biosyntheses of around 80 members of a peptide antibiotic class that has been morphed into products with a central 2,4,6-trithiazolylpyridine embedded in a peptide macrocycle have recently been formulated to involve an aza-Diels-Alder-type reaction catalyzed by the purified enzyme ThiM (Scheme 24).[64] The total synthesis of thiocillin and several analogues was reported more than a decade ago, and aza-Diels-Alder and aza-Mannich chemistry was at the heart of the approaches, which means that they in fact resemble biomimetic strategies (see Ref. [3a] and references therein). The ThiM catalysis would be a cascade reaction in that the initial hetero-Diels- Alder product would be the dihydropyridine still containing the upstream leader peptide. Aromatizing elimination would free the upstream leader peptidylamide and yield the trithiazolyl pyridine. This aza-Diels-Alder reaction also closes the macrocycle at the same time and imposes a nonplanar geometry on the heteroaromatic trithiazolylpyridine core (Scheme 24), which is essential to set the three-dimensional conformation of the active antibiotic at the binding site of the 50S bacterial ribosome subunit.[65] Structural insights of pyridine synthases involved in the biosynthesis of the related thiopeptides thiomuracin and GE2270A recently revealed the molecular logic in folding the peptide precursor and catalyzing formation of the unusual aza-cyclic product.[66]
Scheme 24.

Biosynthesis of the trithiazolylpyridine core of thiocillin-type antibiotics. Proposed aza-Diels-Alder cyclization in the reaction catalyzed by ThiM during thiocillin assembly. Creation of the pyridine ring at the core of the trithiazolylpyridine array also closes the 26-membered macrocyclic ring in thiocillin.
An example of an oxo-Diels-Alder enzymatic reaction with an additional novel twist has recently been reported for the fungal enzyme LepI, which generates the insecticidal leporin C product (Scheme 25).[67] Because leporin C has a fused dihydropyran ring system, its biosynthetic gene cluster was examined to evaluate whether a hetero-Diels- Alder enzyme might be involved. Indeed, the purified LepI catalyzed loss of water from the indicated substrate, presumably to yield the exocyclic trienyl ketone. A hetero-Diels-Alder reaction would produce the observed leporin C product directly. On the other hand, a conventional [4+2]-catalyzed pathway would yield the indicated spirodecalin. Indeed, this was observed as a co-product with leporin C at early time points of substrate conversion. Later, the enzyme utilized this co-product and converted it all into leporin C. This is most readily formulated as a retro-Claisen rearrangement (Scheme 25). This direction for a 3,3 conversion is the first seen in an enzyme-catalyzed reaction. In summary, LepI appears to be able to conduct both a conventional [4+2] and a hetero-[4+2] cyclization reaction competitively (ambivalent transition state), and then retrieve the stranded nascent Diels-Alder product through a retro-Claisen reaction in a remarkable cascade of pericyclic transformations. A novel feature of the LepI enzyme is that it requires the cofactor SAM, not for its methylating capacity, but rather to use the positive charge of SAM to direct the pericyclic reactions.
Scheme 25.

The enzyme LepI carries out both a conventional [4+2] cyclization and an oxa-[4+2] cyclization competitively. The latter reaction yields the main product leporin C. The spiro product from the conventional [4+2] pathway is then subjected to an enzymatic [3,3] retro-Claisen reaction to rescue the stranded material and convert it into leporin C.
The forward 3,3-Claisen rearrangement of ally vinyl ethers has been known for decades to be the mechanism of the key enzymatic transformation in aromatic amino acid biosynthesis in microbes and plants (Scheme 26). Chorismate mutase converts the metabolite chorismate into prephenate.[68] In turn prephenate is the substrate for two types of aromatizing decarboxylation enzymes. One mediates loss of CO2 with elimination of the -OH substituent as the cyclohexadiene ring aromatizes. The resultant phenylpyruvate is then a precursor to l-phenylalanine upon reductive amination. A competing decarboxylase enzyme ejects not the -OH but the -H substituent with its electron pair, as a hydride equivalent to NAD, generating NADH and the para-hydroxyphenylpyruvate precursor to the proteinogenic amino acid Ltyrosine. These are not cascade reactions but do establish that catalyzed pericyclic reactions occur in nature. The other common 3,3-rearrangement, the Cope rearrangement of 1,5-dienes, has been less clear in enzymatic systems until very recently. Although the Cope rearrangement pathway has been suggested as one alternative for the common formation of 4-prenyltryptophan in the large class of prenylated indole alkaloids,[69] it has been difficult to prove that mechanistic alternative.
Scheme 26.

Chorismate mutase catalyzes the only known [3,3]-rear-rangement in primary metabolism of microbes and plants.
There is strong presumptive evidence for a Cope rearrangement in the biosynthesis of C11 cycloalkene pheromones by female gametes of marine brown algae. Among the plethora of related algal cycloalkenes is ectocarpene.[70] This C11 cycloheptadiene is thought to arise from oxygenative fragmentation of the Δ5,8,11,14,17 penta-unsaturated C20 fatty acid primary metabolite by radical-based peroxide chemistry to the indicated two fragments. Isotope labeling studies are consistent with this proposed mechanism but there are no reports on the enzymatic details (Scheme 27). The 3,3-rearrangement step would occur from the indicated trienyl cyclopropane pheromone, detected as an unstable intermediate, and that may be a nonenzymatic thermal reaction at room temperature. It certainly qualifies as a biosynthetic cascade reaction.
Scheme 27.

Proposed Cope rearrangement in the biosynthesis of the brown algal feeding deterrent ectocarpene from a transient cyclo-propane pheromone, which in turn arises from peroxidative fragmentation of a polyunsaturated fatty acyl peroxide.
There now appears to be compelling evidence for an enzymatic Cope reaction in the biosynthetic route to hapalindole and fischerindole cyanobacterial natural product scaffolds (Scheme 28).[71] The first step is prenylation of 3-cis-isocyano-indole by geranyl-diphosphate. The resultant adduct with two carbon chains at C3 of the indole is proposed to undergo a Cope rearrangement, not to C4 of the indole, but as shown onto the isocyano side-chain carbon via a boat transition state. The indoline is a good electron sink for an aza-Prins cyclization to the indicated carbocation. Three modes for quenching the carbenium ion can lead to three products. Loss of a proton yields the terminal olefin in 12-epi hapalindole C. Capture by C4 of the indole ring yields the distinct tetracyclic framework of 12-epi hapalindole U. Alternatively, capture by C2 yields the alternate tetracyclic connectivity of 12-epi fischerindole U, all known products in this series.[72] An X-ray structure of the enzyme forming 12-epi hapalindole U has recently been reported.[73]
Scheme 28.

Hapalindole and fischerindole biosynthesis arise via a [3,3]-Cope rearrangement followed by an aza Prins reaction.
6. Radical-Based Cascades
Two general enzymatic routes exist to convert unactivated C–H bonds into carbon-centered radicals that can then participate in radical-based cascades before release of the products (Scheme 29). The two alternatives operate at the opposite ends of the spectrum of O2 concentrations. Iron-based enzymes that reductively activate co-substrate O2 to generate highvalent oxo-iron species as proximal oxidizing reagents represent one evolutionary variant.[74]
Scheme 29.

Two enzymatic strategies to homolyze unactivated C–H bonds in substrates and create transient carbon-centered radicals involving oxygenative and nonoxygenative paths. A) Two variants of iron-based oxygenases, namely heme-containing cytochrome P450s and mononuclear nonheme iron enzymes that require co-substrate a-ketoglutarate, generate high-valent FeV=O and FeIV=O oxidants, respectively, for co-substrate C H bond homolysis. B) The strategy for homolytic cleavage of S-adenosylmethionine (SAM) in radical SAM enzymes by one-electron transfer from a 4Fe/4S cluster to generate the 5’-deoxyadenosyl radical (dA•) as initiator of substrate C–H bond homolysis.
At the other end of aerobic/anaerobic range are enzymes that are typically inactivated by ambient O2. These are the (predicted) hundreds of thousands of open reading frames in the radical S-adenosylmethionine (SAM) family.[75] This enzyme class typically has a tightly bound air-sensitive 4Fe/ 4S cluster as a one-electron reductant for bound SAM molecules.
The SAM cofactor usually participates in methyl transfers of [CH3+] equivalents in SN2 type transfers to O, N, S, and even nucleophilic C atoms in a wide variety of small molecule and macromolecular substrates. An alternate mode of reactivity is evinced in the radical SAM enzyme family.[76] As the name suggests, one-electron transfer from the 4Fe/4S center to coordinated SAM, yields coordinated methionine and the 5’-adenosyl radical (dAC). This is the proximal abstractor of HC from C–H bonds in bound substrates in active sites of this enzyme family to then create carbon-centered radicals in bound co-substrates.
We present the oxygenase families before the radical SAM enzymes. The fate of the carbon-centered radicals in these two distinct enzyme classes depends on the substrate framework s functional groups and alternative modes and timing of radical quenching. In accord with the multiple turnover roles of catalysts, both classes of enzymes, the ironoxygenases and the radical SAM enzymes, must be returned to starting oxidation states at the end of each catalytic cycle.[77]
6.1. Oxygenative Paths
There are two kinds of iron-based enzymes with monooxygenase activity, distinguished by the coordination sites around iron, which control the redox potentials and kinetic reactivities: heme proteins of the cytochrome P450 (Cyt P430) superfamily and nonheme mononuclear iron of the two histidine, one carboxylate ligand family (Scheme 29 A). On reductive fragmentation of O2, Cyt P450s reach an FeV=O oxidation state while the nonheme enzymes reach an FeIV=O oxidation state. Both high-valent oxo-iron species can cleave unactivated C–H bonds in bound substrates homolytically to yield the substrate radical and Fe(IV/III) OH. For a prototypic monoxygenase outcome, transfer of OH• to substrate• yields hydroxylated product and regenerates either the FeIII-heme or FeII nonheme resting state for the next catalytic cycle. There have been many dozens of examples of such hydroxylases reported.[78] (See Ref. [13] for examples in natural product biosynthesis). Some of these oxygenases act iteratively, for example, on substrate methyl groups to carry out three steps of two-electron oxidations each, to go from a methyl group to an alcohol to an aldehyde to a carboxylic acid. By some criteria, these could be considered cascade reactions, but they are not the subject of this discussion.
Rather, a significant subset of catalysts in these enzyme superfamilies never complete the OH• rebound step.[13, 79] An alternate fate of the carbon-centered radical intervenes, typically in a rearrangement cascade. The released product has not incorporated an oxygen atom. Both atoms from the co-substrate O2 end up as in H2O molecules. These enzymes, we term “thwarted oxygenases”: O2 is reductively activated and fragmented, high-valent iron-oxo species are formed as strong oxidants, substrate C–H bonds are cleaved homolytically, but the oxygen transfer step (OH• rebound) is not completed.
The most famous of these rerouted oxygenases are perhaps isopenicillin N synthase (IPNS) and its immediate downstream partner desacetoxycephalosporin-C synthase (DAOCS) in the formation of these major antibiotic classes (Scheme 30).[78, 80] These O2-reducing nonheme iron oxygenases dramatically alter their substrates in cascade reactions. IPNS converts the acyclic tripeptide aminoadipoyl-L-cysteinyl-D-valine into the fused 4,5-warhead of isopenicillin. The L-amino group of the aminoadipoyl side chain is next epimerized by an enzyme to penicillin N. Deep dissections of the mechanism of both IPNS and DAOCS mechanisms have validated the radical-based cascades.[81]
Scheme 30.

Isopenicillin N synthase (IPNS) is a “thwarted oxygenase” that converts the acyclic tripeptide ACV into the fused 4,5-ring system of the penicillin family of β-lactam antibiotics through a cascade of radicals.
For IPNS, the β-lactam ring is formed first and then the fused five-member thiane ring is constructed, without loss of the monocyclic lactam intermediate. Carbon-based and sulfur-based odd-electron intermediates have been proposed. DAOCS in turn uses a high-valent FeIV=O intermediate to cleave a C–H bond of penicillin N homolytically at one of the two prochiral CH3 substituents in the 5-member thiane ring. One can formulate the set of rearrangements as proceeding through the 3-member episulfide radical that opens to give the ring expansion, followed by transfer of HC back to FeIII to regenerate starting FeII. The final expanded product has a 6-membered sulfur-containing ring with a C=C double bond fused to the beta lactam-the hallmark of the cephalosporin warheads.
Many examples of “thwarted oxygenase outcomes” are found that reflect cascade intramolecular reactions of bound substrate radicals that effectively outcompete the intermolecular transfer of OH•. Among them are transformation of the acyclic heptapeptide backbones of vancomycin and congeneric nonribosomal peptides with triple crosslinks connecting side chains 2–4, 4–6, and 5–7 to generate the cup-shaped architecture of this cell-wall-targeting antibacterial agent (Scheme 31). Three O2-consuming P450 enzymes, oxyABC, act while the heptapeptide is still tethered on the vancomycin assembly line, at its last carrier protein domain, to build two aryl-ether bonds (residues 2–4, 4–6) and one direct C–C bond (residues 5–7) through radical chemistry on the electron-rich tyrosyl (residues 2 and 6) and hydroxyphenylglycyl (residues 4, 5 and 7) moieties.[82] This is tandem peptide cascade chemistry, first to build the tethered heptapeptide backbone (Scheme 3 A), and then tandem radical chemistry three times by the tailoring “pseudo-oxygenases”. It is clear that in these cases, the oxygen insertion is epiphenomenal to the generation of the substrate side-chain carbon radicals.
Scheme 31.

The vancomycin heptapeptide is tailored on the NRPS assembly line by three dedicated P450 enzymes. Each acts as a thwarted oxygenase, generating carbon-based radicals in the electron-rich aromatic side chains of the heptapeptide and ultimately producing two aryl ether crosslinks (C-O-D and D-O-E aryl ether bonds) and one direct C–C link (A-B biaryl linkage).
Moving from peptide scaffolds to polyketide scaffolds, one can likewise see evidence of enzymatic cascades from thwarted oxygenases. Notable is the enzymatic conversion of griseophenone B into the 5,6-spirocyclic system in the antifungal drug griseofulvin (Scheme 32) by the P450 enzyme GsfF.[83]
Scheme 32.

The P450-containing GsfF enzyme catalyzes a radical-based reaction pathway to create the spirocyclic ring system of the fungal metabolite griseofulvin in a “thwarted oxygenase” mode.
Alkaloid biosynthesis also presents a number of such cases of intramolecular routing of substrate radicals generated by high-valent iron-oxo species in biosynthetic enzyme active sites (Scheme 33). These include the conversion of R-reticuline into salutaridine in the morphine biosynthetic pathway,[84] and the apparent coupling of two indolyl radicals to form the indolecarbazole core in staurosporine and rebeccamycin by the StaP and RebP heme proteins, respectively.[85] In the prenylated indole alkaloid family of fumitremorgins, tryprostatin A is converted into fumitremorgin C by the P450 enzyme FtmE. O2 is reduced but not incorporated into the product framework during formation of the new C–N bond.[86]
Scheme 33.

Additional enzyme-mediated radical redirection reactions in the biosynthesis of the alkaloid salutaridine (A), the indolecarbazoles staurosporine and rebeccamycin (B), and the diketopiperazine fumitremorgin C (C).
Among the most spectacular of the cascade reactions with a thwarted oxygenase P450 enzyme is the generation of the core of the communesin natural products by CsnC (Scheme 34).[87] Tryptophan is the precursor primary metabolite to tryptamine by decarboxylation and to aurantioclavine by prenylation at C4 and cyclization of the allyl cation.[88] The FeV=O oxidant in the CsnC heme group is proposed to cleave the indole N H bond of tryptamine homolytically and generate a bound tryptamine C3 radical. If a comparable C3 aurantioclavine radical could also be generated, and the two radicals couple, a 3–3’-dimeric adduct would result as nascent product. Imine exchange and double aminal condensation would yield the communesin core. Subsequent N-methylation, epoxidation, and addition of a polyketide hexadienyl acyl group yields communesin B. Because of communesin s complex core structure, many syntheses have been established, including a recent biomimetic enantioselective approach.[89]
Scheme 34.

The biosynthesis of communesin B involves the thwarted P450 oxygenase CnsC, which mediates coupling between the tryptamine and aurantioclavine radical partners.
6.1.1. Ring Contraction and Formation by Iron-Enzyme Cascades
The complexity generation reactions that carbon radicals, formed in enzyme active sites by high valent oxo-iron species, participate in include framework rearrangements that can result in ring contraction and carbon extrusion as well as ring formation. Two recent examples are presented here. One is the ring contraction of ent-kaurenoic acid by Cyp88A on the way to gibberellic acid 12-aldehyde, an intermediate in the biosynthesis of the gibberellin diterpenoid phytohormone.[90] The second are a set of maturations of the anditomin scaffold by AntA and AntF to create alternate ring connectivity within the starting pentacyclic framework.[91]
The tetracyclic ent-kaurenoic acid (Scheme 35) undergoes one hydroxylation at C7 to the 7-α-OH by the plant enzyme Cyp88A. In the process of a second hydroxylation to the gemdiol and thereby the aldehyde, a rearrangement intervenes, initiated by C6–H cleavage to yield the C6 radical.[90] This intermediate undergoes ring contraction of the six-membered B ring to a five-member ring with an extruded -C6H•–OH. This is the rearranged radical that catches the OH• equivalent on rebound to form the gem diol. Loss of water gives the ring-contracted gibberellic acid-12-aldehyde. This is not a thwarted oxygenase but one that pauses between carbon radical formation and delivery of the OH• equivalent, leaving time for framework rearrangement. A convergent pathway to gibberellin has also been characterized in fungi and bacteria.[92] The fungal gibberellin pathway also employs a single P450 enzyme (P450–1) to catalyze the conversion of ent-kaurenoic acid into gibberellic acid-12-aldehyde and the more oxidized gibberellic acid-7 lactone,[92b] while bacteria employ two P450s and a dehydrogenase to catalyze the conversion to gibberellic acid-12.[92c]
Scheme 35.

P450 CYP88A-mediated conversion of ent-kaurenoic acid into giberellic acid-12-aldehyde via a reaction sequence onvloving a radical-based ring contraction and aldehyde carbon -C6H•(OH) extrusion concomitant with oxygen transfer.
The anditomin meroterpenoids are highly oxygenated molecular structures with some novel bridging rings in the complex scaffolds, including a highly congested bicycle-[2.2.2]octane component (Scheme 36). The building blocks of 3,5-dimethylorsellinic acid and farnesyl-diphosphate are condensed, epoxidized, and enzymatically cyclized by AndB to the pentacyclic preandiloid framework. The iron-based oxygenase AndA then sets off a radical-based framework rearrangement that alters connectivity, and sets up a fused γ-lactone and a new carbon bridge on the way to the bicyclooctane unit.[91] The cascade is set in motion by homolytic cleavage of one of the methyl C12–H bonds by the FeIV=O active site oxidant. That primary radical is converted into an exomethylene group as the neighboring C8–O ether bond is cleaved to generate a 5’ carbon radical and adjacent ketone. Reaction of the C12 olefin terminus with that 5’ carbon radical creates the new CH2 bridge with unpaired electron density at C8. Another olefin-radical coupling ensues to set the final hexacyclic connectivity before H• transfer by reducing agent generates product andiconin, with an embedded bicyclo[2.2.2]octane moiety. No oxygenation of the scaffold has occurred. The X-ray crystal structure of AndA in complex with its substrates and computational calculations have recently provided further insight in how AndA initiates this cascade reaction.[91b] The enzyme AndF, which occurs further along in the pathway after the action of a Baeyer-Villigerase, is likewise a mononuclear nonheme oxygenase family member that also enables radical-based nonoxygenative rearrangement outcomes to get to anditomin at the end of 12 enzymatic steps in this pathway (Scheme 36).[91a]
Scheme 36.

Enzyme mediated O2-dependent radical cascades in the late stages of the biosynthesis of anditomin meroterpenoids. AndA creates a bicyclooctane ring embedded in the meroterpenoid scaffold, while AndF introduces the final nonoxygenative radical rearrangements in the 12-enzyme pathway to anditomin.
6.1.2. Okaramine: One Satisfied and Two Thwarted Oxygenases in a Pathway That Builds an Octacyclic Scaffold in Five Steps
Penicillium molds are famous for their biosynthetic capacity to build heterocyclic scaffolds, among them of course the penicillins and cephalosporins. Recently the pathway to the octacyclic framework of the fungal metabolite okaramine E, an insect ion-channel blocker, has been shown to occur in a short efficient five-enzyme pathway from the primary metabolites l-tryptophan and ∆2-isopentenyl diphosphate (Scheme 37).[93] The first enzyme is a two-module NRPS assembly line, which condenses two molecules of L-tryptophan and mediates intramolecular capture by the amine of the second tryptophan residue to release the cyclic diketopiperazine. A prenyltransferase then asymmetrically prenylates one indole moiety at N1 and the other at C2 with capture of the allyl cations at their C3 rather than C1 centers to give “reversed” regiochemistry of prenylation, which is important for subsequent cascades with the last two enzymes in the pathway to okaramine E. These two steps set up the action of three oxygenases that represent the three known monoxoygenase types: a flavoenzyme, a P450 heme ironcontaining enzyme, and a mononuclear nonheme iron enzyme.
Scheme 37.

Enzymatic assembly of the octacyclic framework of the insect ion channel blocker okaramine E in a short, efficient pathway involving two thwarted oxygenases, the P450 OkaD and the nonheme iron enzyme OkaE, that catalyze radical cascade reactions to generate the eight-member azocine ring and the four-member azetidine ring, respectively.
The latter two are the “thwarted oxygenases” while the FAD enzyme, OkaB, actually acts to epoxidize the indole moiety that is prenylated at N1. This reaction sets off a short cascade in which the adjacent amide NH of the diketopiperazine, weak nucleophile that it is, opens the epoxide regio-and stereospecifically to create a new cyclopentane ring interposed between the indole and the diketopiperazine in a fused tetracyclic array in okaramine C.
This is the substrate for OkaD, the P450 enzyme. It uses the FeV=O oxidant to cleave the remaining diketopiperazine N–H bond homolytically (Scheme 37). Rather than completing a net N-hydroxylation event, the nitrogen radical reacts with the nearby terminal olefin to form a new C N bond. This olefin-radical coupling is analogous to the step noted for AndA above. In this case, it creates an eight-membered heterocycle, an azocine ring radical that can be reduced by one electron/H• transfer to the azocine.
Now the third “oxygenase”, OkaE, goes to work (Scheme 37). It acts on the other end of the substrate, cleaving the C H bond between the two five-member pyrroline rings homolytically to generate the indicated carbon radical. As with the previous enzyme, this carbon-centered radical can react with the neighboring prenyl-groupderived olefin, in this case to form a four-member azetidine ring radical. One electron/H• reduction completes the five-step formation of the octacyclic scaffold of okaramine E, with new 4-ring, 5-ring and 8-ring fused heterocycles from three cascades. Oxygen delivery to substrate is kinetically competent in only the first of the three oxygenases and emphasizes the different roles for flavoenzymes versus iron enzyme oxygenases.
6.1.3. Phenylpropanoid Thwarted Oxygenases
A final two examples, of many that could be cited (see Ref. [13], Chapter 7), come from plant phenylpropanoid natural product pathways. One is the dimerization of coniferyl alcohol to a set of three distinct regioisomeric dimers, with dramatically different 8–8’ connectivities (Scheme 38 A).[94] Pinoresinol is the precursor to a variety of downstream natural products in plants,[95] including sesamin and podophyllotoxin. A second classic case is the enzymatic conversion of flavonoid to isoflavonoid scaffolds in plant secondary metabolism, as exemplified by the O2-and P450-mediated conversion of naringenin into genistein,[96] itself a precursor to many plant defense phytoalexins (Scheme 38 B). The reaction catalyzed by isoflavone synthase features as a short cascade with a 1,2-aryl group shift among carbon radical intermediates.
Scheme 38.

Phenylpropanoid metabolite O2-dependent diverted radical cascades. A) Oxidative dimerization of coniferyl alcohol to different lignan frameworks. B) Enzymatic flux of flavonoids to isoflavonoids by 1,2-aryl migration in a radical intermediate.
6.2. Nonoxygenative Paths: The 5’-Deoxyadenosyl Radical as Initial Oxidant of Bound Substrates
A second and independent route to generate substrate radicals is presumed to have evolved early in anaerobes and retained for use in anaerobic/microaerophilic microenvironments in aerobic organisms. In lieu of high-valent oxo-iron species as powerful oxidants in these enzyme active sites, the common coenzyme S-adenosylmethionine (SAM) undergoes radical fragmentation on receipt of one electron from a conserved 4Fe/4S cluster in the active site (Scheme 29 B). The methionine fragment remains coordinated to the ironsulfur cluster for subsequent religation, while the 5’-deoxyadenosyl radical (5’dA•) is most often the initiator of subsequent chemistry on a bound substrate molecule. As with the high-valent oxo-iron states in the above class of monooxygenases, the 5’dA• is a strong enough oxidant to cleave a nearby C H bond, even when unactivated, to create the methyl group of 5’-deoxyadenosine (5’-dA) and the resultant substrate radical.[97]
Depending on the substrate, a variety of chemical outcomes are possible via radical cascades. These include isomerization of L-(2S)-lysine to beta-lysine,[98] insertion of a sulfur atom into desthiobiotin,[99] and double sulfur insertion into octanoyl thioesters to yield the 7,8-dithioctanoyl product that is the coenzyme lipoic acid.[100] The fate of the SAM substrate can be regeneration at the end of a catalytic cycle or net fragmentation to methionine and 5’-deoxyadenosine.
Vitamin K, a menaquinone, has a naphthoquinone nucleus that functions as a one-electron redox carrier in bacterial electron transport and plant photosynthetic electron transport chains as well as mammalian blood coagulation. In addition to the long known aerobic biosynthetic pathway from chorismate via isochorismate and ortho-succinylbenzoate, an anaerobic pathway involving radical chemistry has recently been discovered.[101] Two radical SAM enzymes, MqnE and MqnC, process the same starting chorismate first to aminofutalosine,[102] then to cyclic dehypoxanthine futalosine[103] on the way to 1,4-dihydroxy-6-naphthoate (Scheme 39), a remarkable tandem set of radical-based chemical transformations in cofactor biosynthesis.[104]
Scheme 39.

Two radical SAM enzymes in the anaerobic aminofutalosine pathway to menaquinone (vitamin K).
In the conversion of the inactive apo forms of microbial [FeFe]-hydrogenases to the active forms containing both carbon monoxide and cyanide ion as ligands to active-site iron-sulfur clusters, the common amino acid tyrosine is fragmented to para-cresol and the two desired one-carbon cyanide ion and carbon monoxide molecules (Scheme 40).[105] This is a fascinating radical SAM enzyme cascade. About a third of the more than 100 000 open reading frames predicted to be radical SAM enzymes in protein data bases are also thought to contain vitamin B12, also a potential radical generator as well as a methyl donor, thus indicating much new radical cascade chemistry to be elucidated.
Scheme 40.
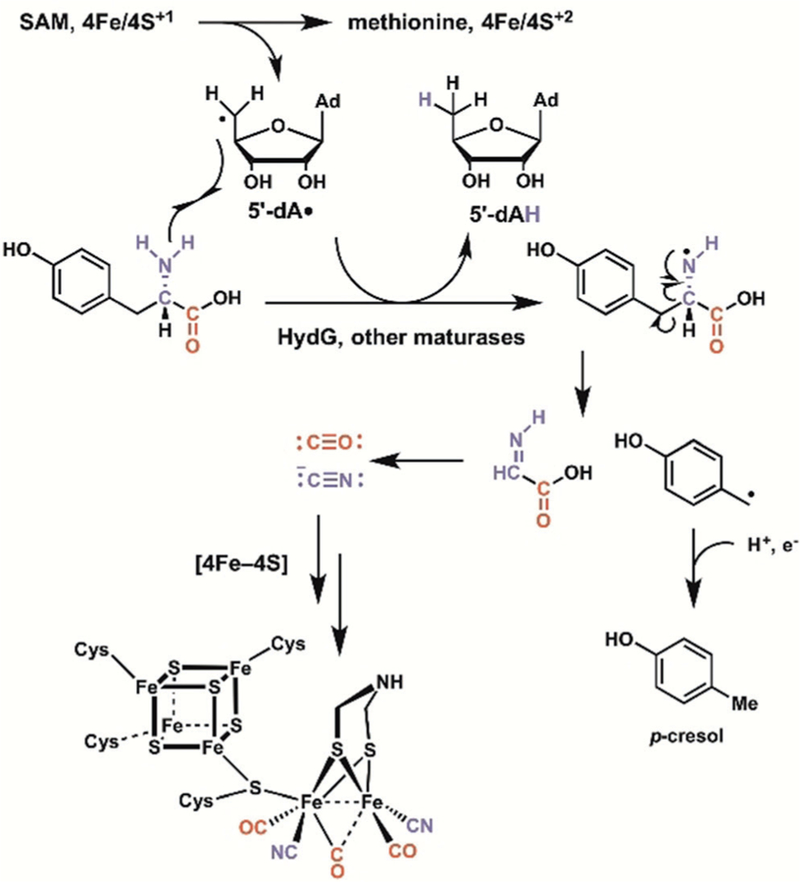
In hydrogenase activation, a radical SAM enzyme supplies CO and CN as ligands for the active-site iron through radical fragmentation of substrate L-tyrosine.
7. Cylindrocyclophane: Nonenzymatic Metathesis Versus Apparent Friedel-Crafts Enzymatic Bis-alkylation
One of the mainstays of synthetic chemical strategies to complex natural product scaffolds has been the use of transition-metal-catalyzed olefin metathesis -to stitch frameworks together. A prominent example noted by Nicolaou et al.[3a] was the approach by Smith and colleagues[106] to employ olefin metathesis dimerization -to form the 7,7-para-cyclophane core of the natural product cylindrocyclophane F (Scheme 41 A). Although this molecule had been synthesized in a stepwise manner previously, Smith s group built the indicated resorcinol monomer and exposed it to a Schrock molybdenum carbene reagent to effect two metathesis steps, via the presumed intermediate shown. The E,E-diene-7,7-cyclophane product is a reduction and deprotection step away from cylindrocyclophane F. Mechanistic analysis suggested that the dimerization-macrocyclization product was the thermodynamically favored isomer, arising from a cascade of reversible olefin metatheses that settle at the thermodynamic minimum.
Scheme 41.

Synthesis and biosynthesis of the 7,7-para-cyclophane cylindrocyclophane F by different cascade strategies. A) Synthesis by a double metathesis cascade. B) Biosynthesis by an apparent Friedel Crafts type bis alkylation cascade to create the para-cyclophane macrocycle.
To date, no known biosynthetic analogues of metalcoordinated olefin metathesis equivalents are known in enzymatic catalysis. On the other hand, cyanobacteria do produce cylindrocyclophane F,[107] albeit by a quite different cascade logic.[108] Balskus and co-workers established that the primary metabolite decanoic acid is the starting substrate for tethering by thioester linkage to a 10 kDa acyl carrier protein.[108] Then a novel member of the diiron-dicarboxylate enzyme family CylC carries out a regio-and stereospecific chlorination of the unactivated methylene group at C6 of the 10-carbon acyl chain to yield the 6R-chloro-decanoyl-S-acyl carrier protein. This is the substrate for a PKS assembly line that builds and then releases the S-methyl-R-chloroalkyl resorcinol (Scheme 41 B). The proximal chlorination reagent is likely to be a chlorine atom [Cl•] rather than the [Cl+] equivalent noted earlier in the merochlorin pathway. The diiron reagent in the active site likely homolyzes one of the two prochiral C–H bonds at C6 of the decanoyl chain, with [Cl•] rebound. Now, this saturated chloroalkyl chain, not a metathesis substrate even if such a bioreagent were present, undergoes stepwise condensation by the Ca2+-dependent enzyme CylK, which acts as a double Friedel-Crafts alkylating agent (Scheme 41 B). The exact mechanism of bond formation is yet to be identified but tandem Friedel-Crafts alkylations to set the para-cyclophane macrocycle is the default proposal. This cascade is a novel reaction sequence that has not previously been reported.
8. Summary and Outlook
Cascade reactions in synthetic chemistry, particularly in assembly of the complex frameworks of many classes of bioactive natural products, have garnered attention and admiration for elegant control of developing molecular architecture. Many synthetic campaigns have cited biomimetic strategies as inspiration, based on known building blocks and chemical reactivity considerations, although the constituent enzymes most often had not been identified.
The renaissance in natural product biosynthesis, driven by two decades of genome sequencing, has now dramatically changed our understanding of biocatalysts. Genome mining for biosynthetic gene clusters, transfer of the genes to surrogate hosts for expression of encoded proteins, and sensitive mass spectrometry methods have allowed the investigation of dozens of enzymes that in fact carry out sets of cascade reactions as substrates are converted into products with remarkable complexity generation.
In this Review, we have used the same categories of cascade reactions-nucleophilic, electrophilic, pericyclic, and radical-that have been advanced for total synthesis to categorize the related enzymatic transformations. The efficiency of enzymatic complexity generation through short metabolic pathways is illustrated in a number of cascade examples, particularly in carbacyclic and heterocyclic framework constructions and rearrangements. Enzymes have access to a limited array of accessible carbon nucleophilic (e.g., thioester enolates) and carbon electrophile building blocks (e.g., allyl cations), but make the most of them in polyketide, nonribosomal peptide, and isoprenoid cascade reactions. Examples of pericyclases have emerged over the past two decades as more natural product biosynthetic chemistry has come into view.
Perhaps the richest recent advances have been in understanding the two strategies that enzymes use, namely O2-dependent and O2-shunning strategies, for the generation of carbon-centered radicals in substrate frameworks. Dozens of iron-based oxygenases are now known to generate radicals that escape [OH•] rebound and react in intramolecular rearrangements without oxygen incorporation. On the radical SAM enzyme side, bioinformatics indicates hundreds of thousands of open reading frames that carry sequence signatures unique to this enzyme family, thus suggesting the existence for novel radical cascades have yet to be found and characterized.
Of course, synthetic chemists have a number of abiotic reagents, protecting groups, forcing reaction conditions at their disposal, compared to the neutral aqueous solutions and room-temperature parameters of physiological conditions. In this context, there is as yet no indication that there are enzymatic cascade reactions that mimic the transition-metalmediated olefin metatheses that are the backbone of many contemporary synthetic transformations. The final example of the Review, the biosynthesis of cylindrocyclophane F, shows the two parallel strategies, namely double metatheses in the synthetic realm versus double Friedel-Crafts-type alkylations in the enzymatic realm, that can provide the 7,7-para-cyclophane macrocycle. We anticipate that as we delve into the biosynthetic capacity of understudied organisms, something made possible through new genome mining methods, we are bound to uncover new cascade strategies in nature.
Acknowledgements
We thank Yi Tang (UCLA) for assistance with the preparation of the images and Stefan Diethelm (UCSD) for insightful discussions that led to the inception of this Review article. B.S.M. acknowledges support of the NIH through grant R01-AI047818 for supporting his work on biosynthetic cascade reactions to bioactive natural products.
Biographies

Christopher Walsh is a consulting professor to the Stanford University department of chemistry and an advisor to the Stanford CheM-H Institute. He was Chemistry Department Head at MIT and the Hamilton Kuhn Professor of Biological Chemistry and Molecular Pharmacology at Harvard Medi-cal School from 1987–2013. His research has explored the chemical mechanisms used in nature to build complexity into natural product scaffolds.

Bradley Moore is Professor of Marine Chemical Biology at the Scripps Institution of Oceanography and Chair of Pharmaceutical Chemistry at the Skaggs School of Pharmacy and Pharmaceutical Sciences at UC San Diego. He received his B.S. in chemistry from the University of Hawaii, his PhD from the University of Washington, and was a postdoc at the University of Zurich. His research focuses on understanding how microbes and marine organisms construct complex bioactive molecules with an eye toward biomedicine and biotechnology.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Christopher T. Walsh, Stanford University Chemistry, Engineering, and Medicine for Human Health (CheM-H), Stanford University Stanford, CA 94305 (USA)
Bradley S. Moore, Center for Marine Biotechnology and Biomedicine Scripps Institution of Oceanography University of California, San Diego, La Jolla, CA 92093 (USA) and Skaggs School of Pharmacy and Pharmaceutical Sciences University of California, San Diego, La Jolla, CA 92093 (USA).
References
- [1].a) Trost BM, Science 1991, 254, 1471–1477 [DOI] [PubMed] [Google Scholar]; b) Trost BM, Angew. Chem. Int. Ed. Engl 1995, 34, 259–281; Angew. Chem. 1995, 107, 285 – 307. [Google Scholar]
- [2].a) Wender PA, Miller BL, Nature 2009, 460, 197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gaich T, Baran PS, J. Org. Chem 2010, 75, 4657–4673. [DOI] [PubMed] [Google Scholar]
- [3].a) Nicolaou KC, Edmonds DJ, Bulger PG, Angew. Chem. Int. Ed 2006, 45, 7134–7186; Angew. Chem. 2006, 118, 7292 – 7344 [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Chen JS, Chem. Soc. Rev 2009, 38, 2993–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Robinson R, J. Chem. Soc. Trans 1917, 111, 762–768 [Google Scholar]; b) Medley JW, Movassaghi M, Chem. Commun 2013, 49, 10775–10777. [DOI] [PubMed] [Google Scholar]
- [5].Johnson WS, Gravestock MB, McCarry BE, J. Am. Chem. Soc 1971, 93, 4332–4334. [DOI] [PubMed] [Google Scholar]
- [6].Baran P, J. Am. Chem. Soc 2018, 140, 4751–4755. [DOI] [PubMed] [Google Scholar]
- [7].Ley SV, Brown DS, Clase JA, Fairbanks AJ, Lennon IC, Osborn HMI, Stokese ESE (nee Owen), Wadsworth DJ, J. Chem. Soc. Perkin Trans 1 1998, 2259–2276. [Google Scholar]
- [8].Zakarian A, Batch A, Holton RA, J. Am. Chem. Soc 2003, 125, 7822–7824. [DOI] [PubMed] [Google Scholar]
- [9].Parker KA, Fokas D, J. Org. Chem 2006, 71, 449–455. [DOI] [PubMed] [Google Scholar]
- [10].Elliott GI, Velcicky J, Ishikawa H, Li Y, Boger DL, Angew. Chem. Int. Ed 2006, 45, 620–622; Angew. Chem. 2006, 118, 636 – 638. [DOI] [PubMed] [Google Scholar]
- [11].Pfeiffer MW, Phillips AJ, J. Am. Chem. Soc 2005, 127, 5334–5335. [DOI] [PubMed] [Google Scholar]
- [12].Gleitsman KR, Sengupta RN, Herschlag D, RNA 2017, 23, 1745–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Walsh CT, Tang Y, Natural Product Biosynthesis: Chemical Logic and Enzymatic Machinery, Royal Society of Chemistry, London, 2017. [Google Scholar]
- [14].Lin GM, McCarty RM, Liu HW, Angew. Chem. Int. Ed 2017, 56, 3446–3489; Angew. Chem. 2017, 129, 3498 – 3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Walsh CT, Fischbach MA, J. Am. Chem. Soc 2010, 132, 2469–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mendel RR, J. Biol. Chem 2013, 288, 13165–13172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cane DE, J. Biol. Chem 2010, 285, 27517–27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pickens LB, Tang Y, J. Biol. Chem 2010, 285, 27509–27515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Finking R, Marahiel MA, Annu. Rev. Microbiol 2004, 58, 453–488. [DOI] [PubMed] [Google Scholar]
- [20].a) Schwecke T, Aparacio JF, MolnQr I, Knig A, Khaw LE, Haydock SF, Oliynyk M, Caffrey P, Cortes J, Lester JB, Proc. Natl. Acad. Sci. USA 1995, 92, 7839–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Aparicio JF, Molnar I, Schwecke T, Konig A, Haydoc SF, Khaw LE, Staunton J, Leadlay PF, Gene 1996, 169, 9–16 [DOI] [PubMed] [Google Scholar]; c) Gatto GJ, Mcloughlin SM, Kelleher NL, Walsh CT, Biochemistry 2005, 44, 5993–6002. [DOI] [PubMed] [Google Scholar]
- [21].Walsh CT, Haynes SW, Ames BD, Gao X, Tang Y, ACS Chem. Biol 2013, 8, 1366–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Pöplau P, Frank S, Morinaka BI, Piel J, Angew. Chem. Int. Ed 2013, 52, 13215–13218; Angew. Chem. 2013, 125, 13457 – 13460 [DOI] [PubMed] [Google Scholar]; b) Berkhan G, Hahn F, Angew. Chem. Int. Ed 2014, 53, 14240–14244; Angew. Chem. 2014, 126, 14464 – 14468 [DOI] [PubMed] [Google Scholar]; c) Luhavaya H, Dias MV, Williams SR, Hong H, de Oliveira LG, Leadlay PF, Angew. Chem. Int. Ed 2015, 54, 13622–13625; Angew. Chem. 2015, 127, 13826 – 13829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zipperer MA, Konnerth MC, Laux C, Berscheid A, Janek D, Marschal M, Burian M, Schilling NA, Slavetinsky C, Weidenmaier C, Willman M, Kahlbacher H, Schittek B, Brotz-Osterhelt H, Grond S, Peschel A, Krismer B, Nature 2016, 535, 511–516. [DOI] [PubMed] [Google Scholar]
- [24].Palmer JL, Abeles RH, J. Biol. Chem 1979, 254, 1217–1226. [PubMed] [Google Scholar]
- [25].a) Geu-Flores F, Sherden N, Courdavault V, Burlat V, Glenn W, Wu C, Nims E, Cui Y, O’Connor S, Nature 2012, 492, 138–142 [DOI] [PubMed] [Google Scholar]; b) Kries H, Caputi L, Stevenson C, Kamileen M, Sherden N, Geu-Flores F, Lawson D, O’Connor S, Nat. Chem. Biol 2016, 12, 6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Antosch J, Schaefers F, Gulder T, Angew. Chem. Int. Ed 2014, 53, 3011–3014; Angew. Chem. 2014, 126, 3055 – 3058 [DOI] [PubMed] [Google Scholar]; b) Zhang G, Zhang W, Zhang Q, Shi T, Ma L, Zhu Y, Li S, Zhang H, Zhao Y, Shi R, Zhang C, Angew. Chem. Int. Ed 2014, 53, 4840–4844; Angew. Chem. 2014, 126, 4940 – 4944. [DOI] [PubMed] [Google Scholar]
- [27].a) Kasahara K, Miyamoto T, Fujimoto T, Oguri H, Tokiwano T, Oikawa H, Ebizuka Y, Fujii I, ChemBioChem 2010, 11, 1245–1252 [DOI] [PubMed] [Google Scholar]; b) Kim W, Park CM, Park JJ, Akamatsu HO, Peever TL, Xian M, Gang DR, Vandemark G, Chen W, Mol. Plant-Microbe Interact 2015, 28, 482–496. [DOI] [PubMed] [Google Scholar]
- [28].Beam MP, Bosserman MA, Noinaj N, Whenkel M, Rohr J, Biochemistry 2009, 48, 4476–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Xiang L, Kalaitzis JA, Moore BS, Proc. Natl. Acad. Sci. USA 2004, 101, 15609–15614 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Teufel R, Miyanaga A, Michaudel Q, Stull F, Louie G, Noel JP, Baran PS, Palfey B, Moore BS, Nature 2013, 503, 552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a) Teufel R, Stull F, Meehan MJ, Michaudel Q, Dorrestein PC, Palfrey B, Moore BS, J. Am. Chem. Soc 2015, 137, 8078–8085 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Saleem-Batcha R, Stull F, Sanders JN, Moore BS, Palfey BA, Houk KN, Teufel R, Proc. Natl. Acad. Sci. USA 2018, 115, 4909–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].a) Cheng Q, Xiang L, Izumikawa M, Meluzzi D, Moore BS, Nat. Chem. Biol 2007, 3, 557–558 [DOI] [PubMed] [Google Scholar]; b) Kalaitzis JA, Cheng Q, Thomas PM, Kelleher NL, Moore BS, J. Nat. Prod 2009, 72, 469–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ueberbacher BT, Hall M, Faber K, Nat. Prod. Rep 2012, 29, 337–350. [DOI] [PubMed] [Google Scholar]
- [33].Christianson DW, Chem. Rev 2017, 117, 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lesburg CA, Zhai G, Cane DE, Christianson DW, Science 1997, 277, 1820–1824. [DOI] [PubMed] [Google Scholar]
- [35].Tetzlaff CN, You Z, Cane DE, Takamtsu S, Omura S, Ikeda H, Biochemistry 2006, 45, 6179–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jin KMY, Coates RM, Croteau R, Christianson DW, Nature 2011, 469, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guerra-Bubb J, Croteau R, Williams RM, Nat. Prod. Rep 2012, 29, 683–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].a) Siedenburg G, Jendrossek D, Appl. Environ. Microbiol 2011, 77, 3905–3915 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wendt KU, Poralla K, Schulz GE, Science 1997, 277, 1811–1815. [DOI] [PubMed] [Google Scholar]
- [39].Awakawa T, Zhang L, Wakimoto T, Hoshino S, Mori T, Ito T, Ishikawa J, Tanner M, Abe I, J. Am. Chem. Soc 2014, 136, 9910–9913. [DOI] [PubMed] [Google Scholar]
- [40].Cardellina JH, Marner FJ, Moore RE, Science 1979, 204, 193–195. [DOI] [PubMed] [Google Scholar]
- [41].Ongley SE, Bian X, Zhang Y, Chau R, Gerwick WH, Muller R, Neilan BA, ACS Chem. Biol 2013, 8, 1888–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].a) Kaysser L, Bernhardt P, Nam SJ, Loesgen S, Ruby JG, Skewes-Cox P, Jensen PR, Fenical W, Moore BS, J. Am. Chem. Soc 2012, 134, 11988–11991 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Diethelm S, Teufel R, Kaysser L, Moore BS, Angew. Chem. Int. Ed 2014, 53, 11023–11026; Angew. Chem. 2014, 126, 11203 – 11206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Miles ZD, Diethelm S, Pepper HP, Huang DM, George JH, Moore BS, Nat. Chem 2017, 9, 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].a) Pepper HP, George JH, Angew. Chem. Int. Ed 2013, 52, 12170–12173; Angew. Chem. 2013, 125, 12392 – 12395 [DOI] [PubMed] [Google Scholar]; b) Meier R, Strych S, Trauner D, J. Org. Chem 2014, 16, 2634–2637. [DOI] [PubMed] [Google Scholar]
- [45].Teufel R, Kaysser L, Villaume MT, Diethelm S, Carbullido MK, Baran PS, Moore BS, Angew. Chem. Int. Ed 2014, 53, 11019–11022; Angew. Chem. 2014, 126, 11199 – 11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Moore BS, Synlett 2017, 29, 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wendt KU, Angew. Chem. Int. Ed 2005, 44, 3966–3971; Angew. Chem. 2005, 117, 4032 – 4037. [DOI] [PubMed] [Google Scholar]
- [48].a) Thimmappa R, Geisler K, Louveau T, O’Maille P, Osbourn A, Annu. Rev. Plant Biol 2014, 65, 225–257 [DOI] [PubMed] [Google Scholar]; b) Hoshino T, Org. Biomol. Chem 2017, 15, 2869–2891. [DOI] [PubMed] [Google Scholar]
- [49].Lodeiro S, Xiong Q, Wilson WK, Kolesnikova MD, Onak CS, Matsuda SP, J. Am. Chem. Soc 2007, 129, 11213–11222. [DOI] [PubMed] [Google Scholar]
- [50].Tagami K, Liu C, Minami A, Noike M, Isaka T, Fueki S, Shichijo Y, Toshima H, Gomi K, Dairi T, Oikawa H, J. Am. Chem. Soc 2013, 135, 1260–1263. [DOI] [PubMed] [Google Scholar]
- [51].a) Li H, Zhang Q, Li S, Zhu Y, Zhang G, Zhang H, Tian X, Zhang S, Ju J, Zhang C, J. Am. Chem. Soc 2012, 134, 8996–9005 [DOI] [PubMed] [Google Scholar]; b) Xu Z, Baunach M, Ding L, Hertweck C, Angew. Chem. Int. Ed 2012, 51, 10293–10297; Angew. Chem. 2012, 124, 10439 – 10443 [DOI] [PubMed] [Google Scholar]; c) Li H, Sun Y, Zhang Q, Zhu Y, Li SM, Zhang C, Org. Lett 2015, 17, 306–309 [DOI] [PubMed] [Google Scholar]; d) Kugel S, Baunach M, Baer P, Ishida-Ito M, Sundaram S, Xu Z, Groll M, Hertweck C, Nat. Commun 2017, 8, 15804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].a) Tang MC, Zhou Y, Watanabe K, Walsh CT, Tang Y, Chem. Rev 2017, 117, 5226–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Walsh CT, Tang Y, Biochemistry 2018, 57, 3087–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].a) Minami A, Shimaya M, Suzuki G, Migita A, Shinde S, Sato K, Watanabe K, Tamura T, Ogura H, Oikawa H, J. Am. Chem. Soc 2012, 134, 7246–7249 [DOI] [PubMed] [Google Scholar]; b) Cane DE, Celmer WD, Westley JW, J. Am. Chem. Soc 1983, 105, 3594–3600 [Google Scholar]; c) Liu T, Cane DE, Deng Z, Methods Enzymol 2009, 459, 187–214 [DOI] [PubMed] [Google Scholar]; d) Gallimore AR, Nat. Prod. Rep 2009, 26, 266–280. [DOI] [PubMed] [Google Scholar]
- [54].Kellmann R, Stuken A, Orr RJ, Svendsen HM, Jakobsen KS, Mar. Drugs 2010, 8, 1011–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].a) Oliynyk M, Stark CB, Bhatt A, Jones MA, Hughes-Thomas ZA, Wilkinson C, Oliynyk Z, Demydchuk Y, Staunton J, Leadlay PF, Mol. Microbiol 2003, 49, 1179–1190 [DOI] [PubMed] [Google Scholar]; b) Gallimore AR, Stark CBW, Bhatt A, Harvey BM, Demydchuk Y, Bolanos-Garcia V, Fowler DJ, Staunton JF, Leadlay PF, Spencer JB, Chem. Biol 2006, 13, 453–460. [DOI] [PubMed] [Google Scholar]
- [56].Mao XM, Zhan ZJ, Grayson MN, Tang MC, Xu W, Li YQ, Yin WB, Lin HC, Chooi YH, Houk KN, Tang Y, J. Am. Chem. Soc 2015, 137, 11904–11907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jeon BS, Wang SA, Ruszczcky M, Liu HW, Chem. Rev 2017, 117, 5367–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Minami A, Oikawa H, J. Antibiot 2016, 69, 500–506. [DOI] [PubMed] [Google Scholar]
- [59].Tian Z, Sun P, Zan Y, Wu Z, Zheng Q, Zhou S, Zhang H, Yu F, Jia X, Chen D, Mandi A, Kurtan T, Liu W, Nat. Chem. Biol 2015, 11, 259–265. [DOI] [PubMed] [Google Scholar]
- [60].Jeon BS, Ruszczcky MW, Russell WK, Lin GM, Kim N, Choi SH, Wang SA, Liu YN, Russell DH, Houk KN, Liu HW, Proc. Natl. Acad. Sci. USA 2017, 114, 10408–10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Diels O, Alder K, Liebigs Ann. Chem 1928, 460, 98–122. [Google Scholar]
- [62].Gresham TL, Steadman TR, J. Am. Chem. Soc 1949, 71, 737–738. [Google Scholar]
- [63].Desrat S, van de Wege P, J. Org. Chem 2009, 74, 6728–6734. [DOI] [PubMed] [Google Scholar]
- [64].Wever WJ, Bogart JW, Baccile JA, Chan AN, Schroeder FC, Bowers AA, J. Am. Chem. Soc 2015, 137, 3494–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Walsh CT, Acker MG, Bowers AA, J. Biol. Chem 2010, 285, 27525–27531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Cogan DP, Hudson GA, Zhang Z, Pogorelov TV, van der Donk WA, Mitchell DA, Nair SK, Proc. Natl. Acad. Sci. USA 2017, 114, 12928–12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ohashi M, Liu F, Hai Y, Chen M, Tang MC, Yang Z, Sato M, Watanabe K, Houk KN, Tang Y, Nature 2017, 549, 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lee AY, Stewart JD, Clardy J, Ganem B, Chem. Biol 1995, 2, 195–203. [DOI] [PubMed] [Google Scholar]
- [69].Luk LYP, Qian Q, Tanner ME, J. Am. Chem. Soc 2011, 133, 12342–12345. [DOI] [PubMed] [Google Scholar]
- [70].Boland W, Proc. Natl. Acad. Sci. USA 1995, 92, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].a) Li S, Lowell AN, Yu F, Taveh A, Newmister SA, Baiir N, Schaub JM, Williams RM, Sherman DH, J. Am. Chem. Soc 2015, 137, 15366–15369 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li S, Lowell AN, Newmister SA, Yu F, Williams RM, Sherman DH, Nat. Chem. Biol 2017, 13, 467–469 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhu Q, Liu X, Chem. Commun 2017, 53, 2826–2829 [DOI] [PubMed] [Google Scholar]; d) Zhu Q, Liu X, Angew. Chem. Int. Ed 2017, 56, 9062–9066; Angew. Chem. 2017, 129, 9190 – 9194. [DOI] [PubMed] [Google Scholar]
- [72].Moore RE, Cheuk C, Qiang X, Patterson GML, Bonjouklian R, Smitka TA, Mynderse JS, Foster RF, Jones ND, Swartzendruber JK, Deeter JB, J. Org. Chem 1987, 52, 1036–1043. [Google Scholar]
- [73].Newmister SA, Li S, Garcia-Borras M, Sanders JN, Yang S, Lowell AN, Yu F, Smith JL, Williams RM, Houk KN, Sherman DH, Nat. Chem. Biol 2018, 14, 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].a) Krebs C, Galonic Fujimori D, Walsh CT, Bollinger JMJ, Acc. Chem. Res 2007, 40, 484–492 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nakamura H, Matsuda Y, Abe I, Nat. Prod. Rep 2018, 35, 633–645. [DOI] [PubMed] [Google Scholar]
- [75].Landgraf BJ, McCarthy EL, Booker SJ, Annu. Rev. Biochem 2016, 85, 485–514. [DOI] [PubMed] [Google Scholar]
- [76].a) Zhang Q, van der Donk WA, Liu W, Acc. Chem. Res 2012, 45, 555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yokoyama K, Lilla EA, Nat. Prod. Rep 2018, 35, 660–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ruszczycky MW, Zhong A, Liu HW, Nat. Prod. Rep 2018, 35, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Schofield CJ, Zhang Z, Curr. Opin. Struct. Biol 1999, 9, 722–731. [DOI] [PubMed] [Google Scholar]
- [79].Mizutani M, Sato F, Arch. Biochem. Biophys 2011, 507, 194–203. [DOI] [PubMed] [Google Scholar]
- [80].Hamed RB, Gomez-Castellanos JR, Ducho HL, McDonough MA, Schofield CJ, Nat. Prod. Rep 2013, 30, 21–107. [DOI] [PubMed] [Google Scholar]
- [81].a) Roach PL, Clifton IJ, Hensgens CM, Shibata N, Schofield CJ, Hajdu J, Baldwin JE, Nature 1997, 387, 827–830 [DOI] [PubMed] [Google Scholar]; b) Valeg rd K, van Scheltinga AC, Lloyd MD, Hara T, Ramaswamy S, Perrakis A, Thompson A, Lee HJ, Baldwin JE, Schofield CJ, Hajdu J, Andersson I, Nature 1998, 394, 805–809. [DOI] [PubMed] [Google Scholar]
- [82].a) Pylypenko O, Vitali F, Zerbe K, Robinson JA, Schlichting I, J. Biol. Chem 2003, 278, 46727–46733 [DOI] [PubMed] [Google Scholar]; b) Zerbe K, Woithe K, Li DB, Vitali F, Bigler L, Robinson JA, Angew. Chem. Int. Ed 2004, 43, 6709–6713; Angew. Chem. 2004, 116, 6877 – 6881 [DOI] [PubMed] [Google Scholar]; c) Woithe K, Geib N, Zerbe K, Li DB, Heck M, Fournier-Rousset S, Meyer O, Vitali F, Matoba N, Abou-Hadeed K, Robinson JA, J. Am. Chem. Soc 2007, 129, 6887–6895 [DOI] [PubMed] [Google Scholar]; d) Haslinger K, Peschke M, Brieke C, Maximowitsch E, Cryle MJ, Nature 2015, 521, 105–109 [DOI] [PubMed] [Google Scholar]; e) Forneris CC, Seyedsayamdost MR, Angew. Chem. Int. Ed 2018, 57, 8048–8052; Angew. Chem. 2018, 130, 8180 – 8184. [DOI] [PubMed] [Google Scholar]
- [83].Grandner JM, Cacho RA, Tang Y, Houk KN, ACS Catal 2016, 6, 4506–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].a) Gesell A, Rolf M, Ziegler J, Diaz Chavez ML, Huang FC, Kutchan TM, J. Biol. Chem 2009, 284, 24432–24442 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Galanie S, Thodey K, Trenchard IJ, Filsinger Inter-rante M, Smolke CD, Science 2015, 349, 1095–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].a) Howard-Jones AR, Walsh CT, J. Am. Chem. Soc 2007, 129, 11016–11017 [DOI] [PubMed] [Google Scholar]; b) Makino M, Sugimoto H, Shiro Y, Asamizu S, Onaka H, Nagano S, Proc. Natl. Acad. Sci. USA 2007, 104, 11591–11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kato N, Suzuki H, Takagi H, Asami Y, Kakeya H, Uramoto M, Usui T, Takahashi S, Sugimoto Y, Osada H, ChemBioChem 2009, 10, 920–928. [DOI] [PubMed] [Google Scholar]
- [87].Lin HC, McMahon T, Patl A, Corsello M, Simon A, Xu W, Zhao M, Houk KN, Garg NK, Tang Y, J. Am. Chem. Soc 2016, 138, 4002–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lin HC, Chiou G, Chooi YH, McMahon TC, Xu W, Garg NK, Tang Y, Angew. Chem. Int. Ed 2015, 54, 3004–3007; Angew. Chem. 2015, 127, 3047 – 3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lathrop SP, Pompeo M, Chang WT, Movassaghi M, J. Am. Chem. Soc 2016, 138, 7763–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Helliwell CA, Chandler PM, Poole A, Dennis ES, Peacock WJ, Proc. Natl. Acad. Sci. USA 2001, 98, 2065–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].a) Matsuda Y, Wakimoto T, Mori T, Awakawa T, Abe I, J. Am. Chem. Soc 2014, 136, 15326–15336 [DOI] [PubMed] [Google Scholar]; b) Nakashima Y, Mitsihashi T, Matsuda Y, Senda M, Sato H, Yamazaki M, Uchiyama M, Senda T, Abe I, J. Am. Chem. Soc 2018, 140, 9743–9750. [DOI] [PubMed] [Google Scholar]
- [92].a) Salazar-Cerezo S, Martinez-Montiel N, Garcia-Sanchez J, Perez-Y-Terron R, Martinez-Contreras RD, Microbiol. Res 2018, 208, 85–98 [DOI] [PubMed] [Google Scholar]; b) Tudzynski B, Appl. Microbiol. Biotechnol 2005, 66, 597–611 [DOI] [PubMed] [Google Scholar]; c) Nett RS, Montanares M, Marcassa A, Lu X, Nagel R, Charles TC, Hedden P, Rojas MC, Peters RJ, Nat. Chem. Biol 2017, 13, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Lai CY, Lo IW, Hewage RT, Chen YC, Chen CT, Lee CF, Lin S, Tang MC, Lin HC, Angew. Chem. Int. Ed 2017, 56, 9478–9482; Angew. Chem. 2017, 129, 9606 – 9610. [DOI] [PubMed] [Google Scholar]
- [94].a) Davin LB, Wang HB, Crowell AL, Bedgar DL, Martin DM, Sarkanen S, Lewis NG, Science 1997, 275, 362–366 [DOI] [PubMed] [Google Scholar]; b) Pickel B, Schaller A, Appl. Microbiol. Biotechnol 2013, 97, 8427–8438. [DOI] [PubMed] [Google Scholar]
- [95].Teponno RB, Kusari S, Spiteller M, Nat. Prod. Rep 2016, 33, 1044–1092. [DOI] [PubMed] [Google Scholar]
- [96].Jung W, Yu O, Lau SM, O’Keefe DP, Odell J, Fader G, McGonigle B, Nat. Biotechnol 2000, 18, 208–212. [DOI] [PubMed] [Google Scholar]
- [97].Broderick J, Duffus BR, Duschene KD, Shepard EM, Chem. Rev 2014, 114, 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lepore BW, Ruzicka FJ, Frey PA, Ringe D, Proc. Natl. Acad. Sci. USA 2005, 102, 13819–13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Fugate CJ, Jarrett JT, Biochim. Biophys. Acta Proteins Proteomics 2012, 1824, 1213–1222. [DOI] [PubMed] [Google Scholar]
- [100].McLaughlin MI, Lanz ND, Goldman PJ, Lee KH, Booker SJ, Drennan CL, Proc. Natl. Acad. Sci. USA 2016, 113, 9446–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Hiratsuka T, Furihata K, Ishikawa J, Yamashita H, Itoh N, Seto H, Dairi T, Science 2008, 321, 1670–1673. [DOI] [PubMed] [Google Scholar]
- [102].a) Mahanta N, Fedoseyenko D, Dairi D, Begley TP, J. Am. Chem. Soc 2013, 135, 15318–15321 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Joshi S, Mahanta N, Fedoseyenko D, Williams H, Begley TP, J. Am. Chem. Soc 2017, 139, 10952–10955. [DOI] [PubMed] [Google Scholar]
- [103].Cooper LE, Fedoseyenko D, Abdelwahed SH, Kim SH, Dairi T, Begley TP, Biochemistry 2013, 52, 4592–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Mehta AP, Abdelwahed SH, Mahanta N, Fedoseyenko D, Philmus B, Cooper LE, Liu Y, Jhulki I, Ealick SE, Begley TP, J. Biol. Chem 2015, 290, 3980–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].a) Muldera DW, Boyd ES, Sarma R, Lange RK, Endrizzi JA, Broderick JB, Peters JW, Nature 2010, 465, 248–251 [DOI] [PubMed] [Google Scholar]; b) Byer AS, Shepard EM, Peters JW, Broderick JB, J. Biol. Chem 2015, 290, 3987–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Suess DL, Kuchenreuther JM, De La Paz L, Swartz JR, Britt RD, Inorg. Chem 2016, 55, 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].a) Smith AB, Kozmin SA, Adams CM, Paone DV, J. Am. Chem. Soc 2000, 122, 4984–4985 [Google Scholar]; b) Smith AB, Adams CM, Kozmin SA, Paone DV, J. Am. Chem. Soc 2001, 123, 5357–5359. [DOI] [PubMed] [Google Scholar]
- [107].Moore BS, Chen JL, Patterson GML, Moore RE, Tetrahedron 1992, 48, 3001–3006. [Google Scholar]
- [108].a) Nakamura H, Hamer HA, Sirasani G, Balskus EP, J. Am. Chem. Soc 2012, 134, 18518–18521 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nakamura H, Schultz EE, Balskus EP, Nat. Chem. Biol 2017, 13, 916 –921 [DOI] [PubMed] [Google Scholar]