

Abstract
Voltage-dependent sodium (NaV) 1.8 channels regulate action potential generation in nociceptive neurons, identifying them as putative analgesic targets. Here, we show that NaV1.8 channel plasma membrane localization, retention, and stability occur through a direct interaction with the postsynaptic density-95/discs large/zonula occludens-1–and WW domain–containing scaffold protein called membrane-associated guanylate kinase with inverted orientation (Magi)-1. The neurophysiological roles of Magi-1 are largely unknown, but we found that dorsal root ganglion (DRG)–specific knockdown of Magi-1 attenuated thermal nociception and acute inflammatory pain and produced deficits in NaV1.8 protein expression. A competing cell-penetrating peptide mimetic derived from the NaV1.8 WW binding motif decreased sodium currents, reduced NaV1.8 protein expression, and produced hypoexcitability. Remarkably, a phosphorylated variant of the very same peptide caused an opposing increase in NaV1.8 surface expression and repetitive firing. Likewise, in vivo, the peptides produced diverging effects on nocifensive behavior. Additionally, we found that Magi-1 bound to sequence like a calcium-activated potassium channel sodium-activated (Slack) potassium channels, demonstrating macrocomplexing with NaV1.8 channels. Taken together, these findings emphasize Magi-1 as an essential scaffold for ion transport in DRG neurons and a central player in pain.—Pryce, K. D., Powell, R., Agwa, D., Evely, K. M., Sheehan, G. D., Nip, A., Tomasello, D. L., Gururaj, S., Bhattacharjee, A. Magi-1 scaffolds NaV1.8 and Slack KNa channels in dorsal root ganglion neurons regulating excitability and pain.
Keywords: sodium channels, potassium channels, trafficking, nociception, peptidomimetics
Nociceptive neurons are endowed with a specific subset of voltage-dependent sodium (NaV) channels, allowing neurons to uniquely respond to noxious and inflammatory stimuli. Therefore, a current strategy for new analgesic development relies on targeting nociceptor-specific sodium channels. For example, the NaV1.8 channel sodium voltage-gated channel alpha subunit 10 (SCN10A) has distinctive biophysical properties (1), permitting nociceptive neurons to repetitively fire action potentials (APs) under compromised conditions associated with tissue damage (2). This has led to the clinical testing of NaV1.8 channel–specific blockers for pain relief (3). Because channels also traffic to the membrane during inflammatory signaling (4, 5), an alternative approach to affect NaV1.8 channel functioning would be to perturb their trafficking. However, the precise molecular mechanisms controlling NaV1.8 channel trafficking are not completely understood (6). NaV1.8 channels and many potassium channels, including sequence like a calcium-activated potassium channel (Slack) sodium-activated potassium (KNa) channels [potassium channel subfamily T (Kcnt) 1], contain putative postsynaptic density-95/discs large/zonula occludens-1 (PDZ) binding motifs (7, 8), and there is evidence that ion channels can associate with PDZ-containing scaffolds to boost membrane expression (9). Additionally, membrane regulation of NaV1.8 channels was shown to be dependent upon ubiquitin ligases (ULs) (10, 11). ULs use WW binding domains to bind to recognition sites in proteins for subsequent ubiquitination and degradation (12). WW domains are 40-aa protein modules that mediate protein-protein interactions through recognition of proline-rich peptide motifs and phosphorylated serine–threonine-proline sites (13). Group 1 WW domains bind the motif PPXY, where X can be any amino acid. Interestingly, most of the NaV pore-forming subunits, including NaV1.8, contain highly conserved PPXY motifs in their respective C termini (10), serving as sites for WW domain interaction. An effective scaffold protein should be able to anchor NaV1.8 channels at the membrane and shield channels against ULs.
Repetitive firing during inflammatory signaling can also be facilitated by a decrease in potassium channel activity. For example, PKA-mediated internalization of the Slack KNa channel in dorsal root ganglion (DRG) neurons resulted in a loss of firing accommodation and hyperexcitability (14). This channel internalization process was shown to be dependent upon adaptor protein 2–clathrin-mediated endocytosis (AP2-CME) (15). Inhibiting AP2-CME with a cell-penetrating peptide prevented PKA-induced repetitive firing of DRG neurons (15). Importantly, immunohistochemistry studies found overlapping Slack KNa immunolabeling with NaV1.8 channels in DRG neurons (16), and electrophysiological studies suggested that KNa channels reside in close proximity and possibly are functionally coupled to NaV channels (17). The scaffolding mechanism anchoring Slack KNa channels at the DRG neuronal membrane is unknown, but Slack and the other KNa channel subunit called sequence like an intermediate conductance potassium channel (Slick) (Kcnt2) contain identical class I PDZ binding motifs at the ends of their respective C termini (18). Using web-based bioinformatics tools (19) and inputting the Slack channel sequence, we determined that membrane-associated guanylate kinase with inverted orientation (Magi)-1 (20), a protein with multiple PDZ domains, was a potential binding partner to both Slack and Slick (Kcnt2) KNa channels.
In addition to containing PDZ domains, the Magi family of scaffold proteins is unique among all other PDZ scaffolding proteins because it also contains WW domains (21). Sequence alignment demonstrated that Magi-1 WW domains are highly homologous to neural precursor cell expressed developmentally down-regulated protein 4-2 (Nedd4-2) WW domains (22), and the WW domains of Magi-1 have an absolute requirement for a PPXY motif for protein interaction compared with other WW domain–containing proteins (23). Although there are many PDZ binding proteins known to associate with Magi-1, it remains unclear as to why Magi-1 contains WW domains. Indeed, there are only a few WW domain–containing proteins that do not have UL activity, but their function may be to protect other proteins against UL-dependent degradation.
The neurophysiological roles of Magi-1 are largely unknown; however, human genetic studies have revealed that MAGI-1 mutations are associated with psychiatric disease, particularly schizophrenia (24–26). Notably, the extensive and diverse literature of clinical and experimental reports suggests that many individuals with schizophrenia are less sensitive to pain than normal individuals (27). Moreover, this appears not to be dependent upon the psychotic state but is rather trait dependent because family members of schizophrenic patients also exhibit pain insensitivity (28). Here, we studied the consequences of Magi-1 deficiency on pain sensitivity. We demonstrated that membrane targeting of NaV1.8 and KNa channels is dependent upon Magi-1. We characterized the expression and distribution of Magi-1 in DRG neurons and found that knockdown of Magi-1 caused a reduction in inward sodium currents (INa) and inward potassium currents (IK)and diminished excitability in neurons. We also determined that NaV1.8 and Slack KNa channels are complexed together. In vivo knockdown of Magi-1 suppressed pain behaviors and produced a significant loss of NaV1.8 channel protein expression. Finally, using WW motif cell-penetrating peptidomimetics, we show that NaV1.8 channel trafficking can be pharmacologically manipulated.
MATERIALS AND METHODS
Animals
All animals used in the present study were housed at the University at Buffalo Laboratory Animal Facility on a 12-h light/dark cycle with free access to food and water. All experimental procedures were in accordance with the guidelines in Guide for the Care and Use of Laboratory Animals from the National Institutes of Health (NIH; Bethesda, MD, USA) and were approved by the University at Buffalo Institutional Animal Care and Use Committee.
Primary DRG neuronal culture
Timed-pregnant Sprague-Dawley rats (Harlan, Indianapolis, IN, USA) were used for culturing neurons. On the day of the dissection, rats were euthanized by CO2 asphyxiation and embryonic d 15 embryos were extracted. DRG neurons were dissected from the embryos and enzymatically digested with trypsin (2.5 mg/ml) at 37°C for 45 min, followed by dissociation and plating. DRG neurons were plated onto coverslips coated with poly-d-lysine (100 μg/ml; MilliporeSigma, Burlington, MA, USA) and laminin (3 μg/ml; Thermo Fisher Scientific, Waltham, MA, USA). Neurons were maintained at 37°C in a 7% CO2 humidified incubator in serum-free medium composed of the trophic factors N2 NeuroPlex serum-free medium (1%; Gemini Bio-Products, West Sacramento, CA, USA), l-glutamine (200 μg/ml; Thermo Fisher Scientific), and nerve growth factor (100 ng/ml; Harlan), which is essential for embryonic neuronal survival, in 50% DMEM and 50% F-12 (14). The reliance of embryonic DRG neurons on nerve growth factor selects for the small-diameter population that is thought to underlie nociception and thermoception (29) Two successive days after DRG dissection, DRG neurons were cultured in C2 medium containing the antimitotic agent cytosine β-d-arabinofuranoside hydrochloride (3 μM; MilliporeSigma). After 2 days of recovery, neurons received regular serum-free medium before being used for experiments. All subsequent experiments using embryonic cultures were performed on d 5–10 of neuronal culture.
Cell culture
Chinese hamster ovary (CHO) cells were cultured at 37°C in 5% CO2 in Iscove’s modified Dulbecco’s medium supplemented with 10% fetal bovine serum, 1% hypoxanthine-thymidine supplement (Thermo Fisher Scientific), and 1% penicillin-streptomycin. CHO cells were plated on 12-mm coverslips for immunolabeling experiments, in 35-mm dishes for all electrophysiology experiments, and in 6-well culture plates for biochemical experiments. Cells were cotransfected with either 0.5 μg of Slick (pTRACER) or 0.5 μg of Slack (pTRACER) plus 0.5 μg of Magi-1 (pcDNA3.1; Addgene, Watertown, MA, USA) or 0.5 μg of empty vector using Lipofectamine (Thermo Fisher Scientific) as per the manufacturer’s guidelines. The Magi-1 clone was mutated to include a Kozak sequence at the 5′ end to boost protein expression.
DRG neuron small interfering RNA transfection
Small interfering RNA (siRNA) directed against Magi-1 was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). A negative control siRNA composed of a scrambled sequence was obtained from the same vendor. For each experiment, neurons were transfected with Magi-1 siRNA or a nontargeting control siRNA. Cultured DRG neurons (described above) were transfected using Lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer’s protocol. Briefly, we diluted 1.5 μl of Lipofectamine 2000 in 50 μl of Opti-mem medium and allowed the mixture to sit at room temperature for 5 min. After 5 min, this mixture was combined with 40 pmol of scrambled or Magi-1 siRNA (3 different siRNA duplexes pooled) in 50 μl of Opti-mem; the mixture was allowed to incubate at room temperature for 30 min before being added to cells plated on 12-mm coverslips in 24-well plates. The siRNA mixture added to the DRG culture medium was allowed to incubate with DRG neurons for 48–72 h before being used for electrophysiological recordings. For Western blotting, 300 pmol of siRNA was used in each 6-well plate. siRNA-transfected DRG neurons were used in experiments 48–72 h post-transfection. For electrophysiological experiments, DRG neurons were cotransfected with the GFP-containing plasmid pTRACER and siRNA duplexes for positive indication of transfection. Imunofluorescence procedures on cultured neurons were performed as previously described (15). The investigator was blinded to the transfection condition.
Electrophysiology
All data were acquired using the Axopatch 200B amplifier (Molecular Devices, San Jose, CA, USA) and Multiclamp 700B (Molecular Devices), digitized and filtered at 5 kHz. Data acquisition was monitored and controlled using pClamp 10 (Molecular Devices). Whole-cell patch-clamp recordings were performed on cultured DRG neurons, and CHO cells were transiently transfected with plasmid constructs containing wild-type (WT) or mutated Slack cDNA and Magi-1 cDNA. Glass electrodes were pulled using a vertical pipette puller (Narishige International USA, Amityville, NY, USA) and fire polished to be of 5–8 MΩ resistance. Pipettes were filled with solution containing (in millimolars) 124 K-gluconate, 2 MgCl2, 13.2 NaCl, 1 EGTA, 10 HEPES, 4 Mg-ATP, and 0.3 Na-GTP at pH 7.2 for neuronal recordings and with solution containing (in mM) 32.5 KCl, 97.5 potassium gluconate, 5 EGTA, and 10 HEPES (pH 7.2) (7) for CHO cell Slack and Slick recordings. The bath solution for all cells contained (in mM) 140 NaCl, 5.4 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose (pH 7.4). Identical bath and pipette solutions were used in both the voltage clamp and current clamp modes. In voltage clamp mode, macroscopic currents were recorded at voltages ranging from −120 to +120. Cells were clamped at −70 mV, and voltage steps of 20 mV were applied for 200-ms durations. The cell capacitance recorded under these conditions ranged from 10 to 15 pF for CHO cells and 20 to 25 pF for cultured DRG neurons. A current clamp protocol consisting of depolarizing steps in increments of 10 pA from 10 to 200 pA (20-ms duration) was used to examine AP firing. Firing frequency of individual neurons was assessed by measurement of repetitive discharge upon injection of a suprathreshold stimulus of 400 pA for 1000 ms. For INa recordings, the pipette solution contained (in millimolars) 130 CsCl, 13 CsF, 10 tetraethylammonium chloride, 1 MgCl2, 1 EGTA, 2.5 Na2-ATP, and 10 HEPES, and pH was adjusted to 7.2 with CsOH. The bath solution contained (in millimolars) 140 NaCl, 5.4 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose (pH 7.4). For tetrodotoxin (TTX)–resistant INa, DRG neurons were recorded in a bath solution containing 250 nM TTX. DRG neurons were recorded at voltages ranging from −60 to +60 mV. Cells were held at −70 mV, and INa was evoked by incremental 10-mV depolarizing steps for a duration of 50 ms.
Immunohistochemistry
Sciatic nerves (SNs), lumbar spinal cords (SCs), lumbar and thoracic DRGs were isolated from adult mice. Briefly, animals were anesthetized with Fetal Plus (Thermo Fisher Scientific) and perfused transcardially with ice-cold PBS containing heparin (50 μg/ml) and sodium nitrite (5 mg/ml) followed by ice-cold 4% paraformaldehyde. DRGs, SNs, and SCs were subsequently removed, cleaned of surrounding tissue, and postfixed in 4% paraformaldehyde overnight at 4°C. The following day, SNs and DRGs were cryoprotected in 20% sucrose (Cryoprotect). After 2 d, DRGs, SNs, and SCs were removed from sucrose and embedded in freezing media and stored at −80°C for future use. Using a cryostat, 16-μm sections of DRGs, 10-μm sections of SNs, and 20-μm sections of SCs were made. Slices were permeabilized with a PBS solution containing 0.4% Triton X-100. Sections were then blocked for 2 h at room temperature with PBS containing 5% bovine serum albumin (BSA). Then, sections were incubated with a mixture of primary antibodies in PBS containing 5% BSA overnight at 4°C. All primary antibodies were previously validated and included the following: mouse anti-NaV1.8 antibody (1:250; NeuroMab, Davis, CA, USA) (11, 30), rabbit anti–Magi-1 antibody (1:100; Abcam, Cambridge, MA, USA) (31), mouse anti–Magi-1 antibody (1:100; Novus Biologicals, Centennial, CO, USA) (32), rabbit anti–contactin associated protein like (Caspr; 1:250; Abcam) (33), chicken anti-Slack antibody (1:750) (34), and chicken anti-Slick antibody (35). The specificity of the rabbit anti–Magi-1 antibody was further verifed in Magi-1 heterolgous expression experiments Supplemental Fig. S1). After several rinses, secondary antibodies Alexa Fluor 633 goat anti-mouse, Alexa Fluor 488 goat anti-rabbit, and Alexa Fluor 546 goat anti-chicken were added (1:1000) overnight. Coverslips were then mounted on slides using Prolong Gold antifade reagent (Thermo Fisher Scientific) with 4′,6′-diamidino-2-phenylindole dihydrochloride. Cell size characterization of Magi-1 immunolabeling was analyzed using the MetaMorph software (Molecular Devices). Magi-1 expression was verified using the Allen Mouse Spinal Cord Atlas (http://mousespinal.brain-map.org/imageseries/showref.html) and BioGPS (http://biogps.org/#goto=welcome). Phosphorylation sites were determined using Scansite (Massachusetts Institute of Technology, Cambridge, MA, USA) and PhosphoSitePlus (https://www.phosphosite.org/homeAction.action).
Western blot analysis
Total proteins were collected from transfected CHO cells, SCs, and DRGs. Tissue was homogenized in RIPA buffer supplemented with protease inhibitor cocktail (MilliporeSigma). Immunoblotting was performed as previously described (14). Briefly, proteins were separated on 4–15% Mini-Protean TGX Precast Gel (Bio-Rad, Hercules, CA, USA) and transferred to a 0.45-μm nitrocellulose membrane (Bio-Rad). Membranes were probed overnight at 4°C with antibodies against Slack anti-mouse (1:500; NeuroMab) (36), rabbit anti–β-actin (1:500; MilliporeSigma), rabbit anti–Magi-1 (1:100; Abcam), mouse anti–Magi-1 (1:100; Novus Biologicals), mouse anti-NaV1.8 (1:200; NeuroMab), mouse anti-NaV1.7 (1:200; NeuroMab) (11), and mouse anti-Flag (1:500; MilliporeSigma) in 5% milk prepared in Tris-buffered saline with 0.05% Tween-20. On the following day, the membrane was washed 3 times for 5 min in 1-time Tris-buffered saline Tween before being incubated for 1 h at room temperature in anti-mouse or anti-rabbit horseradish peroxidase conjugate (1:5000; Promega, Madison, WI, USA) and 0.1% BSA prepared in 1-time PBS. The membrane was again washed 3 times for 5 min before being developed and imaged. Bands were visualized with ECL (Thermo Fisher Scientific) and quantified with ImageJ Software (NIH). Each experiment was repeated at least 3 times.
Coimmunoprecipitation
CHO cells in 6-well plates were transiently transfected with WT or mutated Slack with or without Magi-1 plasmids, respectively. Cells were then lysed with ice-cold RIPA buffer supplemented with protease inhibitor cocktail (100 μl per well; MilliporeSigma). Sixty microliters per well of protein G–linked Sepharose bead slurry (GE Healthcare, Chicago, IL, USA) was washed 3 times with ice-cold lysis buffer and incubated on a rotator overnight at 4°C with 4 μg of rabbit anti-Magi1 antibody (Abcam) or mouse anti-Slack antibody (NeuroMab) in PBS with 0.1% Tween 20 and cell lysate (3 wells per sample). On the following day, samples were centrifuged and supernatants were stored separately. Pellets were washed 3 times with cold lysis buffer, and bound protein was eluted via boiling 3 times at 9°C for 8 min. each. Samples were centrifuged to separate proteins into the supernatant, which was then denatured with sodium dodecyl sulfate (SDS) and loaded onto a Ready Gel (Bio-Rad) (4–15% Tris-HCl) as the immunoprecipitate. The supernatants were collected, and whole-cell lysates (total input) were also denatured with SDS and run as controls. Samples were probed for Slack or NaV1.8 and actin protein by Western blot, as described above.
Surface protein biotinylation
Plasma membrane protein expression was detected using a protein biotinylation assay. Briefly, CHO cells or DRG neurons in 6-well plates were used at 48 h after transient transfection of plasmid constructs or Magi-1–targeting siRNA, respectively. For peptide incubation, neurons were incubated for 24 h. A total of 160 μl of 10 mM Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific) was added to each well and incubated at room temperature for 45 min. The biotinylation reaction was terminated using 10 mM glycine (quenching solution; 89881, Thermo Fisher Scientific). Cells were harvested, washed in Tris-buffered saline, and lysed on ice for 30 min in a lysis buffer–protease inhibitor cocktail. Lysates were collected and incubated with 500 μl of NeutrAvidin Agarose (Thermo Fisher Scientific) for 60 min at room temperature with rotation. Following incubation, the column underwent centrifugation to collect the unbiotinylated protein. To elute the biotinylated proteins, SDS and DTT were added to the column membrane and incubated with rotation for 1 h at room temperature. Biotinylated and unbiotinylated samples were probed for Slack or NaV1.8 and actin protein by Western blot, as described above.
Nociception testing
Baseline thermal nociceptive behavior was measured using the automated Hargreaves apparatus by Ugo Basile (Varese, Italy). Naive C57Bl/6 mice (8–10 wk old) (Envigo, Huntingdon, United Kingdom) underwent 2 d of habituation followed by 3 d of measurements. On d 1 and 2 (habituation), mice spent 30 min in homecages adjusting to the testing room and were then transferred to testing chambers for 1 h. On d 3 through 5, mice underwent testing. An infrared stimulus (IR 40) was delivered through the plexiglass floor to the plantar surface of the hind paw, and the latency to withdrawal was measured automatically. For each subject, 3–6 measurements per hind paw were taken and used to calculate the mean latency to withdrawal (seconds). A maximum IR exposure time of 15 s was established to ensure no tissue damage occurred, and at least 5 min was allowed between measurements taken from the same mouse.
In vivo transfection with jetPEI–Magi-1 short hairpin RNA plasmid DNA polyplexes
The spinal nerve injection protocol was adopted from Chang et al. (37) and optimized for spinal nerve injection in mice. Three days after baseline thermal behavior was established, mice were anesthetized using isoflurane (induction: 4%; maintenance: 2%) and placed in a prone position. A 3-cm posterior longitudinal skin incision was made at the lumbar segment of the spine. The ipsilateral paraspinal muscles were carefully separated, using a pair of sterile toothpicks, from their attachments at the L4 to S1 (Lumbar vertebrae 4 to Sacral vertebrae 1) levels of the vertebral column. A total of 1.5 μl of polyethylenimine–short hairpin RNA (shRNA) plasmid DNA polyplexes at an N/P (nitrogen to phosphate) ratio of 6 was slowly injected directly in the spinal nerve of the right hind paw using a syringe connected to a 26-gauge needle (Hamilton 80030; Hamilton, Reno, NV, USA). Magi-1 shRNAs and control shRNAs were purchased from Santa Cruz Biotechnology and were identical to the siRNA sequences described above. In vivo-JetPEI transfection reagents were purchased from Polyplus Transfection. After injection, the needle was held at the spinal nerve for 1 min to prevent leakage. Complete hemostasis was confirmed, and the wound was sutured with wound clips. Mice were allowed to recover for 7 d before thermal nociceptive behavior was tested again.
Formalin test
The formalin assay for acute inflammatory pain was used to assess the impact of DRG Magi-1 knockdown on nocifensive behaviors. Mice were habituated in the behavior room for 15 min and then 30 min in the formalin chamber before formalin injection. Twenty microliters of 5% formalin (in sterile saline) was injected intraplantarly into the right hind paw, and mice were placed back in the chamber for video recording. The total time spent lifting paw, the total number of licks, and the number of paw flinches were recorded during a 1-min period at every 5-min interval for 60 min. The measurements from 2 observers blinded to the experimental condition for each video recording were averaged to obtain final measurements at each time point.
RNA extraction and cDNA synthesis
An RNeasy Micro Kit (Qiagen, Germantown, MD, USA) was used for total RNA extraction from mouse lumbar DRG neurons. RNA was reverse transcribed with SuperScript III Reverse Transcriptase (Thermo Fisher Scientific). A PCR was performed using this cDNA as the template with previously validated primers against Magi-1 (32) and NaV1.8 (38). Transcriptional abundance was measured by a thermocycler using SYBR Green PCR Master Mix. For quantification, a 50-cycle, 2-step denaturing and annealing protocol was used, with a 15-s absorbance reading on a Bio-Rad iQ5 cycler. Each sample was performed in triplicate.
Peptides
We designed the N-terminal myristoylated PDZ peptide peptidomimetic NPETRDETQL based upon the C-terminal sequence of the rat Slack channel. This peptide and the scrambled variant peptide, QPNTRLDETE, were synthesized by GenScript (Piscataway, NJ, USA). Similarly, the proline tyrosine motif (PY) peptide SATSFPPSYDSVTRG and the phosphorylated-PY (phospho-PY) peptide SATSFPPSYDSV(pT)RG were designed based upon the WW binding domain in rat NaV1.8 channels. These peptides and the scrambled peptide SDRPVTSYSFSAPG were also synthesized by GenScript. Peptides were initially dissolved in DMSO and diluted to a final working concentration in saline. A peptide concentration of 10 μM was used as previously described (15), and the final DMSO concentration was 0.05%. Intraplantar dosing was chosen based on a prior study by Weng et al. (39) demonstrating the analgesic effects of hind paw intradermal injections of a different myristoylated peptide.
Statistics
Clampfit (Molecular Devices) and Origin 8.0 (OriginLab, Northampton, MA, USA) software were used for all electrophysiology data analysis. Densitometry analyses of Western blots were done using ImageJ software. Statistical analysis was done using GraphPad Prism 4 (GraphPad, La Jolla, CA, USA). Single between-group comparisons were made using Student’s t test. Multiple comparisons were investigated using 1-way or 2-way ANOVA followed by Bonferroni’s test to detect pairwise between-group differences. Data are presented as means ± sem.
RESULTS
KNa channel expression is affected by the PDZ binding motif
Slack and Slick channels contain a type 1 PDZ binding motif at their respective distal C termini (Fig. 1A). Using the PDZ protein interactive predictor (PDZPepInt; University of Freiburg, Freiburg im Breisgau, Germany) and inputting the Slack amino acid sequence, we identified Magi-1 as a Slack channel interactor, specifically the second and fifth PDZ domain of Magi-1 Supplemental Table S1) (19, 40). Heterologous coexpression of Magi-1 and the Slack-B subunit (41) increased Slack current density (Fig. 1B); however, coexpression of Magi-1 with a mutated Slack construct with a truncated PDZ motif did not affect Slack current density (Fig. 1B). We confirmed that Magi-1 interacted with Slack channels in CHO cells and in DRG neurons using coimmunoprecipitation (Co-IP) assays (Fig. 1C, G). Double immunolabeling studies depicted colocalization between Magi-1 and Slack KNa channels in CHO cells in cultured and intact DRG neurons (Fig. 1F, H). We confirmed by Co-IP that Slack KNa channels interacted with Magi-1 via its C-terminal PDZ motif (ETQL) (Fig. 1D). To verify the role of Magi-1 on Slack channel membrane expression, we performed a surface biotinylation assay and confirmed that coexpression of Slack with Magi-1 increased Slack channel surface expression (Fig. 1E). Slick, the other member of the KNa family of channels, which shares ∼74% sequence homology to Slack (42), has the same evolutionarily conserved class 1 PDZ binding motif (ETQL) (Fig. 1A). We assessed whether Magi-1 also modulated Slick current activity in CHO cells by coexpressing Magi-1 and Slick. Patch-clamp recordings revealed that Magi-1 similarly potentiated Slick current density (Fig. 2A), but differing with Slack-B, Western blot analysis surprisingly showed an increased total Slick protein expression (8-fold) (Fig. 2B). Magi-1 was also found to colocalize with Slick channels when heterologously expressed in CHO cells (Fig. 2C). Therefore, Magi-1 influenced KNa currents by increasing Slack channel membrane expression. With respect to Slick channels, Magi-1 seemed to serve an additional protein stabilizing function because Magi-1 expression resulted in increased total Slick channel protein expression.
Figure 1.

PDZ binding motif regulates KNa channel expression. A) Amino acid alignment of the distal C termini from orthologous Slack subunits (Xenopus, chicken, rat, and human Slack) and the rat Slick subunit. The final 4 evolutionarily conserved amino acids (ETQL) (red) represent a consensus type 1 PDZ motif (X–S/T–X–V/L/I). Green, AP-2 binding site; magenta, putative PKA phosphorylation site; blue, putative PKC phosphorylation site. B) Representative current traces of Slack and mutated Slack channels (Mut) where the PDZ motif was truncated and recombinantly expressed in CHO cells with or without Magi-1 (top). Current density analysis for each experimental condition (bottom). For each experimental condition, currents from 20 to 25 cells were analyzed. Recordings were performed 48 h after transfection. Values are expressed as ± sem. *P < 0.05 vs. respective controls. C) Representative immunoblots from Co-IP between Magi-1 and Slack when recombinantly expressed in CHO cells. D) Co-IP assay of Magi-1 with WT and a mutant Slack variant with a truncated PDZ motif. Truncating the Slack PDZ motif prevented Co-IP with Magi-1. E) Representative immunoblot of surface biotinylation assay from CHO cells coexpressing Magi-1 with Slack or Slack alone (left). Quantification of surface Slack expression is shown on the right. Data was normalized to input to account for transfection efficiency. t6 = 4.276, n = 4 per group, 2-tailed t test. *P < 0.0129. F) Double immunolabeling experiments showing overlapping expression between Magi-1 (green) (Flag antibody) and Slack (red) (top) and Magi-1 (green) (pAb) and Slack (red) (bottom) when coexpressed in CHO cells. Original magnification value, ×20. G) Representative immunoblots of Co-IP assay between Magi-1 and Slack from intact DRG neurons from adult mice. H) Double immunolabeling experiments depicting colocalization between Magi-1 (green) (pAb) and Slack (red) in cultured DRG neurons. Scale bars, 50 μm. IP, Co-Immunoprecipitation; WB, Western Blot.
Figure 2.
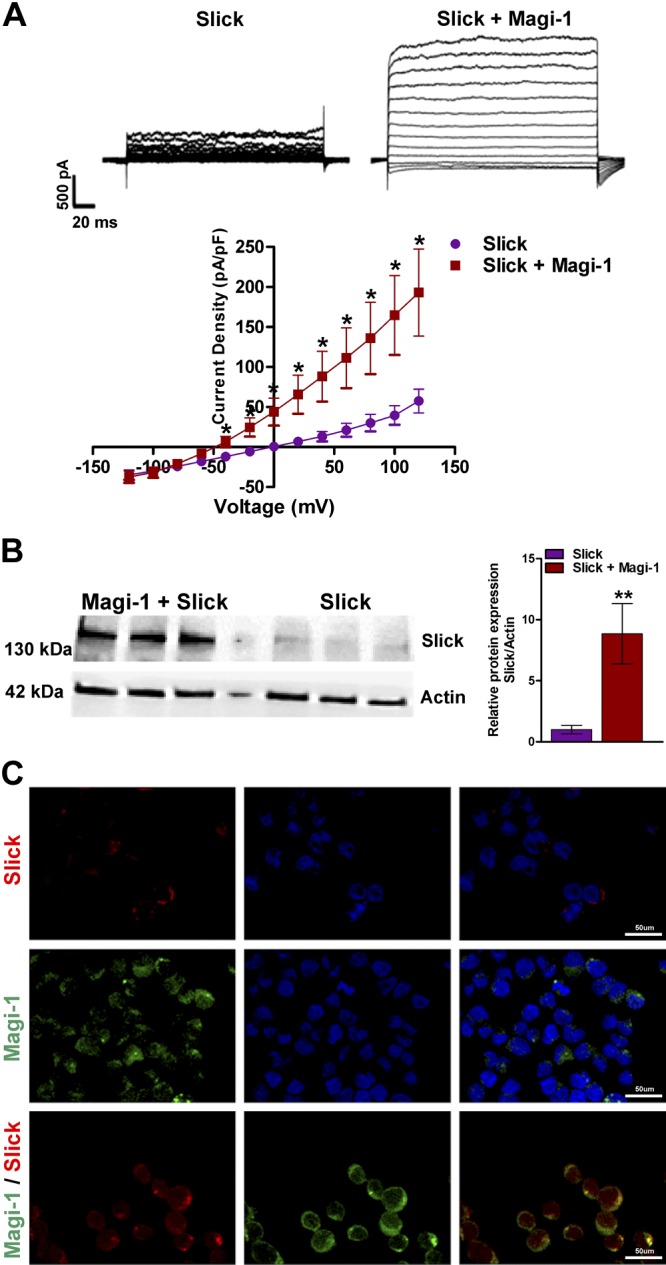
Magi-1 regulates Slick channels in CHO cells. A) Representative current traces of Slick currents recombinantly expressed with or without Magi-1 in CHO cells (top). Current density analysis of Slick currents for each condition (bottom). A total of 25 cells were analyzed, and values are expressed as ± sem. *P < 0.05 vs. respective controls. B) Immunoblot depicting total increased Slick protein expression during coexpression with Magi-1. Results were taken from 3 independent cultures, and values are expressed as means ± sem (t4 = 6.152, n = 3 cultures per group, 2-tailed t test). **P < 0.0021. C) Immunolabeling of recombinant Slick channels (red) and Magi-1 (green) when expressed alone or in combination in CHO cells. Scale bars, 50 μm.
Magi-1 knockdown suppressed IK but produced hypoexcitability in cultured DRG neurons
We examined the neurophysiological function of Magi-1 by using knockdown strategies with previously validated siRNAs (31, 43) in cultured DRG neurons. We verified Magi-1 knockdown by immunolabeling and Western blot analyses (Fig. 3A, B) using a previously validated polyclonal Magi-1 antibody (31). Substantial Magi-1 knockdown was achieved: we observed ∼70–75% reduction in Magi-1 protein when compared with a noncoding scrambled control siRNA 72 h after transfection (Fig. 3A, B). Additional immunofluorescence images depicting knockdown can be found in Supplemental Fig. S2. We examined the consequences of Magi-1 knockdown on KNa Slack channel surface expression. Membrane biotinylation assays revealed a significant decrease in membrane Slack channel expression (∼70% compared with controls) (Fig. 3C). Voltage clamp recordings also showed a significant reduction in outward IK density after Magi-1 knockdown, although the transient IK was still present (Fig. 3D). To our surprise, Magi-1 knockdown resulted in DRG hypoexcitability, with neurons failing to fire APs (Fig. 3E and Supplemental Fig. S3A). We expected the decrease in surface Slack KNa channels to result in repetitive firing (14, 15); however, the observed severely stunted APs suggested that Magi-1 deficiency was also affecting sodium channel functioning.
Figure 3.

Magi-1 knockdown decreases ionic currents and excitability in DRG neurons. A) Representative Magi-1 immunolabeling from cultured DRG neurons 3 d after transfection with Magi-1–targeting siRNA and nontargeting scrambled siRNA (left) using a previously validated polyclonal Magi-1 antibody. Quantification of Magi-1 immunoreactivity is shown on the right. The integrated fluorescence intensity was calculated as the product of the area and the mean pixel intensity using Metamorph software. Values from 4 independent DRG neuronal cultures per experimental condition were analyzed. Values are expressed as means ± sem [ANOVA, F(2,11) = 32.25]. Scale bar, 50 μm. ***P < 0.001 vs. respective controls. B) Representative immunoblots depicting Magi-1 expression after siRNA-mediated Magi-1 knockdown. Magi-1 antibodies normally detect multiple splice variants as indicated by the multiple bands observed on Western blot. Quantification of Magi-1 knockdown in DRG neurons (right). Three different cultures per experimental condition were analyzed. Values expressed as means ± sem [ANOVA, F(2,6) = 42.94]. ***P < 0.001 vs. respective controls. C) Representative immunoblots of surface biotinylation from DRG neurons after Magi-1 knockdown (left). Quantification of Slack channel surface expression is shown on the right. Three independent cultures were analyzed, and values are expressed as means ± sem [ANOVA, F(2,6) = 10.84]. **P < 0.01 vs. respective controls. D) Representative current traces of IK in DRG neurons after Magi-1 knockdown (top). A total of 11–12 neurons per experimental condition were analyzed, and values are expressed as means ± sem. *P ≤ 0.05. E) Representative Action Potential (AP) firing from neurons after siRNA-mediated Magi-1 knockdown during suprathreshold current stimulation (400 pA) for 1000 ms, untransduced (10 out of 10), scrambled DRG neurons 12 out of 12 fire 1 AP, whereas 12 out of 18 neurons transfected with Magi-1 siRNA failed to fire a single AP. A.u., arbitrary unit.
Magi-1 knockdown also decreased INa and NaV1.8 plasma membrane expression in DRG neurons
We consequently examined the effects of Magi-1 knockdown on the INa in DRG neurons using whole-cell voltage clamp recordings. Magi-1 knockdown produced a significant reduction in total INa (Fig. 4A–C). The peak TTX-sensitive and the TTX-resistant components of INa were both significantly reduced compared with neurons treated with control siRNA, accounting for the hypoexcitability phenotype seen in DRG neurons during Magi-1 knockdown despite decreased membrane Slack expression. Notably, the culture conditions of these DRG neurons favor tropomyosin receptor kinase A–positive, nociceptive DRG neurons (see Materials and Methods) that express high levels of NaV1.8 channels. In most mature nociceptive DRG neurons, NaV1.8 channels account for up to 90% of the upstroke of the AP (44). Because Magi-1 knockdown reduced the TTX-resistant component of the INa, of which NaV1.8 is a significant contributor, we concentrated our investigation on the surface expression of NaV1.8. Using surface biotinylation assays, we found decreased membrane expression of NaV1.8 (∼50%) after Magi-1 knockdown (Fig. 4D). Together, these results suggested that Magi-1 is an essential scaffold for the membrane localization of both NaV1.8 and Slack channels in DRG neurons.
Figure 4.
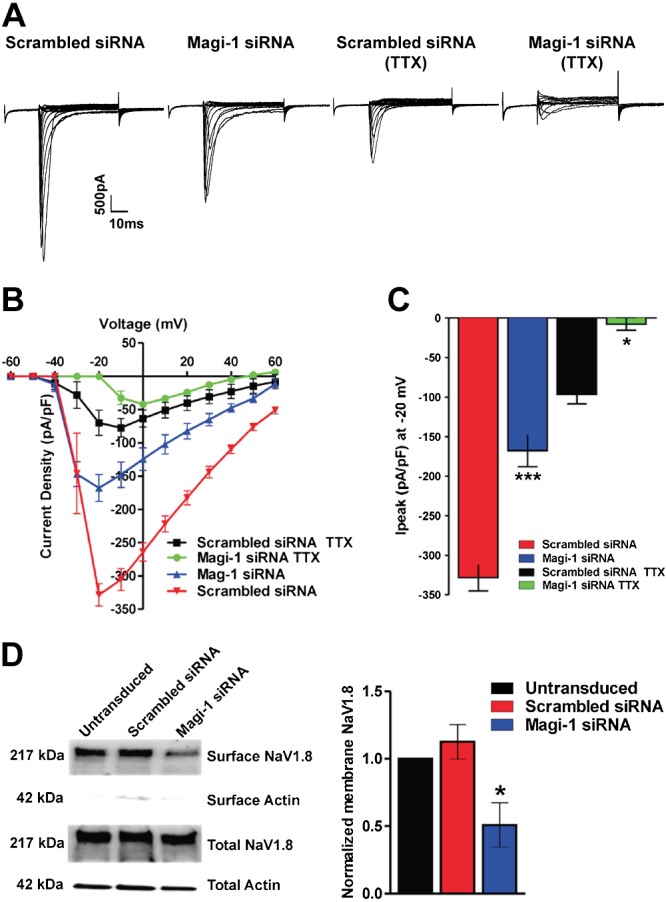
Magi-1 knockdown decreases NaV1.8 plasma membrane expression. A) Representative whole-cell voltage clamp current traces of total INa and TTX-resistant INa in cultured DRG neurons 3 d after transfection with Magi-1–targeting siRNA or nontargeting scrambled siRNA. B) Current density analysis of INa with different conditions. Sodium currents in neurons were recorded in either the presence or absence of 250 nM TTX. The total and TTX-resistant INa was significantly reduced after siRNA-mediated Magi-1 knockdown in cultured DRG neurons. A total of 9–12 cells per experimental group were analyzed, and values are expressed as means ± sem. C) Quantification of peak INa and TTX-resistant peak INa (at voltage step −20 mV) after Magi-1 knockdown. A total of 9–12 cells per experimental group were analyzed, and values are expressed as means ± sem [ANOVA, F(3,26) = 66.24]. *P < 0.0106, ***P < 0.001 vs. respective controls (scrambled siRNA with or without TTX). D) Representative immunoblots from surface biotinylation experiments of DRG neurons depicting reduced NaV1.8 surface expression after Magi-1 knockdown (left). Quantification of NaV1.8 surface expression is shown on the right. For quantification, 4 independent DRG cultures per experimental condition were analyzed, and values are expressed as ± sem [ANOVA, F(2,6) = 7.319]. *P < 0.05 vs. respective controls.
Magi-1 is expressed in small- and medium-sized DRG neurons, in their axonal tracts, and at some nodes of Ranvier
We verified the expression of Magi-1 in intact DRG neurons for indication of its broader physiologic function. The Allen Mouse Spinal Cord Atlas and BioGPS support high Magi-1 message within DRG neurons. According to BioGPS, DRG tissue has the second highest Magi-1 mRNA tissue expression profile, with the hypothalamus accounting for the highest expression. Moreover, the Allen Spinal Cord Atlas depicts differential Magi-1 expression in small- and medium-sized, presumably nociceptive DRG neurons. Magi-1 immunolabeling was also previously shown in the growth cones of cultured DRG neurons and within the dorsal root entry zone of embryonic SCs (45). We confirmed Magi-1 expression in adult mouse DRG neurons and SC tissue by Western blot (Fig. 5A) and immunohistochemistry analyses (Fig. 5B). Immunohistochemistry was performed using a previously validated monoclonal anti–Magi-1 antibody (32). Histologic examination of the SN showed high Magi-1 immunoreactivity along axonal fibers and at some nodes of Ranvier using the paranodal marker Caspr (Fig. 5C). Cell size analysis indicated that the highest distribution of Magi-1 was in small- and medium-sized DRG neurons (<600 μm2) (Fig. 5D), similar to the data that can be found in the Allen Mouse Spinal Cord Atlas. The preferential tissue expression profile of Magi-1 to small- and medium-sized DRG neurons and the dorsal horn of the SC indicated a potential function for Magi-1 in pain signaling.
Figure 5.

Magi-1 is expressed in DRG neurons, the SC, the SN, and at nodes of Ranvier. A) Representative immunoblots depicting Magi-1 expression from intact DRG (left) and SC (right). Untransfected CHO cell lysates were used as the control lane. CHO cells do not endogenously express Magi-1 Supplemental Fig. S1). B) Immunolabeling images showing Magi-1 (green) expression in cultured DRG neurons (panel 1), DRG sections (panel 2 and 3), and the SC (panels 4 and 5) using a previously validated monoclonal antibody. Panels 2 and 4 depict control immunolabeling, stained with secondary antibody only. DAPI (blue) labels all nuclei of cells. Scale bars, 50 μm for DRGs and 200 μm for SCs. C) Double immunolabeling depicting Magi-1 (red) and the paranodal marker Caspr (green) in SN sections (top). Arrows indicate Magi-1 labeling at nodes of Ranvier. Insets represents high-magnification images of Magi-1 immunoreactivity at nodes (bottom). Scale bars, 20 μm (top) and 10 μm (bottom). D) Frequency distribution of Magi-1 in intact DRG neurons of varying cell body size. A total of 735 neurons from 4 mice were analyzed. Neurons larger than 800 μm2 did not show high levels of Magi-1 expression.
Magi-1 mediated coupling between slack KNa channels and NaV1.8 in DRG neurons
Previous reports showed that neuronal KNa channel activity decreased when sodium entry pathways were blocked, suggesting that NaV channels reside in close proximity to KNa channels (17, 46). Moreover, a coimmunolocalization was previously observed between the Slack KNa channel and NaV1.8 in DRG neurons (16). Our data also suggested possible coupling as indicated by decreased membrane localization of NaV1.8 and Slack channels after Magi-1 knockdown. We therefore examined the possibility of Magi-1 interacting with NaV1.8 in DRG neurons and whether Magi-1 facilitated a coupling of NaV1.8 with Slack KNa channels. In Co-IP assays, we confirmed that Magi-1 interacted with NaV1.8 channels (Fig. 6A). Double immunolabeling studies also depicted a colocalization between Magi-1 and NaV1.8 in cultured and intact DRG neurons and within the SC (Fig. 6B). Furthermore, we performed Co-IP experiments using Slack- and NaV1.8-specific antibodies from intact DRG lysates and successfully coimmunoprecipitated Slack with NaV1.8 (Fig. 6C), indicating that NaV1.8 and Slack KNa channels are complexed together in DRG neurons. These findings implicated the scaffolding of Slack and NaV1.8 in DRG neurons by Magi-1. However, Magi-1 might not be the only scaffold responsible for clustering these ion channels together; potentially, there are other yet-undiscovered proteins that can cause ion channel clustering.
Figure 6.
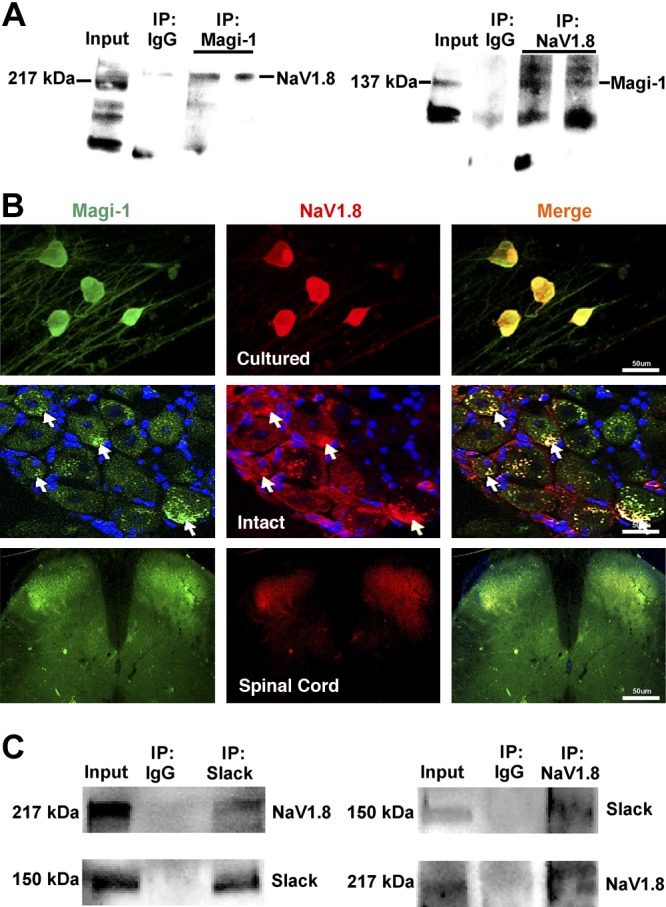
Magi-1 complexes NaV1.8 channels with Slack KNa channels in DRG neurons. A) Representative whole immunoblots from Co-IP assays demonstrating binding between Magi-1 and NaV1.8 using intact adult DRG tissue. IP product samples were run in duplicate. The polyclonal Magi-1 antibody also recognized a 50-kDa band during blotting thought to be a degradation product (as per manufacturer’s description). B) Double immunolabeling experiments demonstrate similar localization between Magi-1 (green) and NaV1.8 (red) in cultured DRG neurons (panel 1), intact DRG sections (panel 2), and the spinal cord (panel 3). Scale bars, 50 μm. C) Representative immunoblots of Co-IP between Slack and NaV1.8 from intact adult DRG neurons.
In vivo Magi-1 knockdown in DRG neurons reduced pain sensitivity
Because Magi-1 was essential for ion channel scaffolding in DRG neurons, we hypothesized that in vivo knockdown of Magi-1 would attenuate pain behavior. To test this hypothesis, we used a novel spinal nerve injection technique of nonviral vectors (37) containing shRNA sequences. We are the first to adapt this in vivo transfection method for mice, and the technique allows for shRNA plasmid uptake by DRG sensory neurons via axonal retrograde transport but did not require invasive paraspinal muscle dissection that is necessary for rats (37). The schematic representation of our experimental outline is depicted in Fig. 7A. Intraspinal nerve injection of Magi-1 shRNAs in naive male and female mice induced a marked and persistent reduction in thermal nociception compared with control shRNA (Fig. 7B, C). To assess the intra-animal differences with respect to withdrawal latency, we subtracted the contralateral (uninjected) paw withdrawal latency (PWL) from the ipsilateral (injected) PWL. With respect to individual animals, there was a significant ∼3-s increase in PWL in mice injected with Magi-1 shRNA compared with mice injected with nontargeting shRNA. We next examined the effects of Magi-1 knockdown in an acute inflammatory pain model (formalin assay) (47). Intraplantar injection of 5% formalin induced the typical biphasic inflammatory pain responses associated with this acute inflammatory pain model. Briefly, the formalin assay can be divided into 2 phases (phase I and phase II) based on behavioral and electrophysiological responses. Phase I is characterized by a brief behavioral response thought to be due to direct activation of nociceptors by formalin (48), and phase II is a prolonged response resulting from both peripheral and central sensitization, the latter of which is due to increased and ongoing nociceptive input to the SC (49). To this extent, 15 d after Magi-1 shRNA in vivo transfection, phase I flinching behavior and phase II licking, lifting, and flinching behaviors were all significantly reduced (Fig. 7D).
Figure 7.

In vivo Magi-1 knockdown attenuates thermal nociception and acute inflammatory pain behavior. A) Experimental timeline before and after Magi-1 knockdown in vivo. B) Hargreaves test for thermal nociception showed increased PWL in ipsilateral paw injected with Magi-1–targeting shRNA when compared with the contralateral paw. No significant difference was seen in PWL between paws in mice injected with nontargeting shRNA. Behavior was taken from 9 different animals (3 females and 6 males) per experimental condition and analyzed (9). Values are expressed as means ± sem. ****P < 0.001 vs. respective controls. C) Difference score analysis determined a ∼3-s difference in withdrawal latency between ipsilateral and contralateral paw after Magi-1 shRNA in vivo transfection (d 7, 11, and 15). Values are expressed as means ± sem. *P < 0.05 vs. control. D) Formalin-induced Phase II inflammatory pain, as measured by 3 nocifensive behaviors [paw licking (left), lifting (middle), and whole-body flinches (right)] in each interval of 5 min, is reduced in mice injected with Magi-1–targeting shRNA after 15 d as compared with controls. Behavior from 9 different animals (n = 9) per experimental condition was analyzed, and values are expressed as means ± sem [ANOVA, licking: F(1,16) = 7.545; lifting: F(1,16) = 11.67; flinching: F(1,16) = 5.007]. *P < 0.05, **P < 0.01, ***P < 0.001 vs. respective controls. E) Representative Magi-1 immunolabeling in DRG sections obtained from 1 mouse injected with Magi-1–targeting shRNA (bottom left) compared with 1 mouse injected with nontargeting scrambled shRNA (top left). Magi-1 immunoreactivity was significantly reduced in ipsilateral paw from mice injected with Magi-1 shRNA as compared with contralateral paw (right). No significant change in immunoreactivity was observed in mice injected with nontargeting scrambled shRNA. DRGs from 3 different animals were analyzed, and values are expressed as means ± sem [ANOVA, F(3,20) = 9.872]. Scale bars, 50 µm. **P < 0.01 vs. respective controls. F) Western blot analysis confirmed Magi-1 knockdown in DRGs 15 d after in vivo transfection of Magi-1–targeting shRNA (left). Quantification of Western blot is shown on the right. Intact DRGs from 3 different animals were analyzed, and values are expressed as means ± sem. [ANOVA, F(3,8) = 5.161]. *P < 0.05 vs. respective controls. A.u., arbitrary unit; contra, contralateral; ipsi, ipsilateral.
Next, we confirmed in vivo Magi-1 silencing 15 d after transfection within the DRG and SN of shRNA-injected mice by immunohistochemical and biochemical analyses. We observed a significant loss of Magi-1 immunoreactivity in the ipsilateral DRG and SNs from mice injected with Magi-1 shRNA when compared with the contralateral DRG from the same mouse and mice injected with control shRNA (Fig. 7E). We also verified Magi-1 transcript knockdown using RT-PCR Supplemental Fig. S4B). Magi-1 protein knockdown was confirmed by immunoblotting (∼70–75%) (Fig. 7F) and was comparable with the knockdown achieved in vitro (Fig. 3A, B). Together, these results suggest that Magi-1 regulates nociception and acute inflammatory pain.
NaV1.8 expression decreased after Magi-1 knockdown in vivo
Immunohistochemical analyses of the SN and at the nodes of Ranvier also revealed an unexpected but significant reduction of NaV1.8 immunoreactivity after Magi-1 shRNA treatment when compared with noncoding scrambled shRNA control (Fig. 8A). This finding was corroborated by an observed 75% decrease in NaV1.8 protein expression in DRG neurons after Magi-1 in vivo knockdown (Fig. 8B), as determined by Western blot analysis. These data revealed that in addition to scaffolding channels at the membrane, Magi-1 is required for NaV1.8 protein stability. A recent study by Shirata et al. (50) has demonstrated a protective role for Magi-2 in preventing the dendrin from Nedd4-2–mediated protein degradation via a WW-mediated interaction. Furthermore, the loss of NaV1.8 protein and the concomitant reduction in phase II inflammatory pain behavior is consistent with the reduced phase II behavior seen in NaV1.8 knockout mice (51). Using RT-PCR, we confirmed that NaV1.8 message remained unchanged during Magi-1 knockdown, reinforcing the notion that Magi-1 regulates NaV1.8 protein stability Supplemental Fig. S4C). These results suggested that Magi-1 plays a critical role in regulating ion channel protein stability.
Figure 8.

NaV1.8 expression decreases after Magi-1 knockdown in vivo. A) Representative immunolabeling of SN depicting NaV1.8 (red) expression in paw injected with nontargeting shRNA after 15 d (top); expression of NaV1.8 at nodes of Ranvier was detected using the paranodal marker Caspr (green). Boxed areas shown are a high-magnification image of NaV1.8 and Caspr immunoreactivity (original magnification value, ×63). NaV1.8 immunoreactivity was absent in SN and at nodes in paw injected with Magi-1–targeting shRNA after 15 d (bottom). Scale bars, 20 µm. B) Representative immunoblots of NaV1.8 expression from ipsilateral and contralateral DRG lysates of mice injected in the SN with nontargeting Magi-1 shRNA (scrambled) or Magi-1–targeting shRNA. Representative blot shown for each condition is taken from the same mice. C) Quantification of NaV1.8 expression is shown on the right. Lumbar DRGs from 3 different animals were analyzed, and values are expressed as ± sem. *P < 0.05 vs. representative controls. Contra, contralateral; ipsi, ipsilateral.
PY motif–mimicking peptides regulate NaV1.8 trafficking, DRG neuronal excitability, and pain behavior
We have demonstrated that a PDZ-mediated interaction was an absolute requirement for the Slack–Magi-1 interaction (Fig. 1D). NaV1.8 channels were reported to contain multiple internal putative PDZ binding motifs and bind to PDZ domain-containing protein 2 (8). However, PDZ domain-containing protein 2 knockout mice failed to show any alterations in pain behavior (8). On the other hand, NaV1.8 channels also contain PY motifs (PPXY) (10) Supplemental Fig. S5) at their distal C termini, and this motif was postulated to regulate the interaction with Nedd4-2 UL for targeted protein degradation (52). Interestingly, the WW domain of Nedd4-2 shares high sequence homology with the WW domains in Magi-1. Furthermore, Magi proteins have been shown to protect Nedd4-2 target proteins from degradation via WW interaction (50). Therefore, we opted to compete off WW domain binding of NaV1.8 using cell-penetrating PY motif peptide mimetics. We engineered 2 peptides of identical sequence based on the NaV1.8 WW binding motif; however, 1 of the peptides was phosphorylated (representing Thr1926 within the channel). This was done because scanning NaV1.8 with the PhosphoSitePlus post-translational modification resource tool revealed that Thr1926, 4 aa adjacent to the PPXY domain (4), is putatively phosphorylated Supplemental Fig. S5). Primary DRG neurons were then treated with 10 μM unphosphorylated peptide (PY, myristoyl-SATSFPPSYDSVTRG) or phosphorylated peptide [phospho-PY myristoyl-SATSFPPSYDSV(pT)RG] to outcompete NaV1.8 channel WW domain binding. Neurons exposed to the PY peptide for 24 h resulted in an almost complete loss of total INa, whereas in contrast, the phospho-PY peptide strongly increased peak INa. The PY and phospho-PY peptide showed time-dependent decreases and increases in INa, respectively (6 and 24 h) (Fig. 9A, B). PY peptide treatment (24 h) almost completely abolished AP firing (10 of 11 neurons), whereas the phospho-PY produced contrasting repetitive AP firing (7 of 12) (Fig. 9B). We assessed surface expression of NaV1.8 channels after treatment with the PY peptide and found a substantial decrease of NaV1.8 channels at the plasma membrane, whereas there was a significant increase of NaV1.8 membrane expression with the phospho-PY peptide (Fig. 9C). Moreover, we observed a substantial decrease in total NaV1.8 protein after incubation with the PY peptide, suggesting that NaV1.8 protein stability is dependent upon this WW binding motif. These results indicate that the phosphorylation status of our competitor NaV1.8 PY motif peptide was essential for the stabilization of NaV1.8 channels. Furthermore, our data imply that other sodium channels are potentially regulated by PY motif interactions (Fig. 9A, B).
Figure 9.

Cell-penetrating WW motif peptidomimetics alter neuronal excitability and affect pain behavior. A) Representative voltage clamp recordings depicting decreased INa (arrow) in cultured DRG neurons after 24 h of pretreatment with the peptide mimetic designated PY peptide, whereas the phospho-PY peptide increasd INa (top). Representative AP traces from cultured DRG neurons pretreated with PY peptide or phospho-PY peptide for 24 h during suprathreshold stimulation (400 pA) for 1000 ms (bottom). B) Peak INa (at voltage step −20 mV) with different peptide treatments in DRG neurons. Neurons were treated for 6 or 24 h with PY peptide or phospho-PY peptide. A total of 10–12 DRG neurons per experimental condition were analyzed, and values are expressed as means ± sem [ANOVA, F(4,35) = 19.11]. *P < 0.05, ***P < 0.001 vs. respective controls. C) NaV1.8 protein expression was altered after peptidomimetic treatment. Representative Western blot of total and surface NaV1.8 membrane expression after DRG neurons were treated with PY peptide, phospho-PY peptide, or a scrambled peptide (left) for 24 h. Quantification of Western blots shown to the right. Treatment with the PY peptide produced a significant reduction of both total and surface NaV1.8 expression when compared with scrambled peptide. The phospho-PY peptide increased surface expression of NaV1.8 when compared with scrambled peptide. Data from 3 independent cultures were analyzed, and values are expressed as means ± sem. *P < 0.05, **P < 0.01 vs. control. #P < 0.01 vs. phospho-PY peptide. D) Phase II formalin inflammatory pain was measured by nocifensive behaviors [paw licking (left), lifting (middle), and whole-body flinches (right)] in each interval of 5 min, is reduced by intraplantar pretreatment (24 h) with 100 μM (20 μl) of PY peptide, whereas phospho-PY peptide increased nocifensive behavioral responses compared with scrambled peptide control. Peptides were administered 24 h before the formalin injection (5%, 25 μl). Behavior from 6 different animals per experimental condition was analyzed, and values are expressed as means ± sem. *P < 0.05, **P < 0.01 vs. controls. #P < 0.05, ##P < 0.01 vs. phospho-PY peptide. E) Magi-1 constitutes the sodium signalosome in DRG neurons. Slack KNa channels were previously shown to internalize by AP2-CME. AP-2, adaptor complex; CL, clathrin.
To assess the potential analgesic effects of disrupting sodium channel membrane localization on pain behavior, we investigated the impact of our competing peptides in the formalin model of inflammatory pain. Mice were given a single intraplantar injection of either PY, phospho-PY, or a scrambled PY peptide [100 μM concentration (20 μl injection volume)] to the right hind paw 24 h before injection with 5% formalin into the same paw. Pretreatment with the PY peptide significantly reduced phase II acute inflammatory pain, and the phospho-PY peptide–pretreated mice exhibited a contrasting increase in phase II response when compared with scrambled peptide–treated mice (Fig. 9D). These data corroborate what was observed during in vitro experiments and demonstrate a capability of routing NaV1.8 channels in and out of the neuronal membrane using PY motif–based peptidomimetics. Additionally, these data suggest that the phosphorylation state of Thr1926 determines NaV1.8 channel trafficking, neuronal excitability, and acute pain behavior.
DISCUSSION
Here, we report that Magi-1 is expressed in the pain pathway: high Magi-1 expression was observed in the cell bodies and axons of nociceptive DRG neurons and within the superficial dorsal horn of the SC. Additionally, we demonstrated that Magi-1 was a critical scaffold for the membrane localization of NaV1.8 and Slack KNa channels in DRG neurons. Both NaV1.8 and Slack KNa channels have been previously implicated in rodent models of inflammatory and neuropathic pain (16, 51, 53), and we showed that Magi-1 interacted with both Slack and NaV1.8 channels. Furthermore, we found that Magi-1 silencing decreased membrane expression of both types of ion channels, resulting in net deficits in DRG neuronal excitability, suggesting that Magi-1 is a critical regulator of ion channel function in neurons. To assess the importance of Magi-1 in pain processing, we in vivo transfected silencing Magi-1 shRNAs into DRG neurons of naive mice using spinal nerve injection (37). This is a novel and rapid technique to manipulate gene functioning in the DRG neurons of naive rodents, in particular mice. It allows for internal control testing of ipsilaterally modified DRG neurons vs. unaltered contralateral DRG neurons within the same mouse. Using this in vivo transfection method, we found Magi-1 knockdown resulted in significant deficits in thermal nociception and acute inflammatory pain behavior.
In addition to localizing ion channels to the neuronal membrane, our findings are the first to indicate that Magi-1 is also crucial for ion channel protein stability. Indeed, unlike other scaffolding proteins, the Magi family of proteins may serve a broader function of protecting proteins from degradation. For example, the non-UL Yes-associated protein 1 was shown to protect against Nedd4-2–mediated protein degradation through its WW domain (54). Subsequently, Magi-2 was then reported to also protect the protein dendrin from Nedd4-2–mediated ubiquitination via a WW domain interaction with a conserved PY motif in dendrin (50). Like dendrin, NaV1.8 has an evolutionarily conserved PY motif that has been demonstrated to be the binding site for Nedd4-2, targeting NaV1.8 for subsequent proteasomal degradation (52). During prolonged in vivo Magi-1 knockdown, we observed substantial and statistically significant decreases in NaV1.8 immunolabeling and protein expression as determined by immunoblotting (Fig. 8). Likewise, after 24 h of PY peptide incubation in DRG neurons, we observed an almost complete loss of NaV1.8 expression. Indeed, INa density decreased by 50% after just 6 h, suggesting both TTX-resistant and TTX-sensitive NaV channel membrane expression is dependent upon the PY motif. However, subsequent immunoblotting analysis of NaV1.7 protein showed that although some protein reduction was observed during long-term in vivo Magi-1 shRNA knockdown, surface NaV1.7 protein levels were unchanged after peptidomimetic treatment Supplemental Fig. S8). It should be noted that embryonic DRG neurons also express NaV1.3 channels (55), which contain WW binding domains Supplemental Fig. S5). So, some of the in vitro effects we observed on TTX-sensitive channels might have been attributed to this channel. Nonetheless, our data suggest that there are differential sensitivities for NaV isoforms to scaffolding and protein stability by Magi-1. Furthermore, we observed that when recombinantly expressed with Magi-1, the Slick KNa subunit—which has a putative PY motif in its N terminus at amino acids 12–15 (-Proline-Proline-Arginine-Tyrosine-) (42)—produced ∼5-fold larger currents with concomitant increases in Slick channel protein levels than when the subunit was expressed alone (Fig. 2A, B). This contrasted with the Slack-B subunit, for which there is an absence of this PY motif in its N terminus (41); membrane currents increased to a lesser extent (2-fold), and no increase in Slack-B protein expression was observed. Indeed, the difficulty in expressing Slick channels compared with Slack channels in heterologous expression systems (42) may be due to this WW binding motif and susceptibility to UL-dependent degradation. Therefore, our results suggest that in addition to membrane targeting, Magi-1 also protects ion channels from degradation pathways, and indeed, targeting Magi-1 represents a novel pharmacological approach to affect ion channel levels and function.
Although both Magi-1 knockdown and the PY peptide caused a reduction in INa, NaV1.8 channel stability, and decreased pain behavior, the opposite effects produced by the phospho-PY peptide, specifically the increase in INa, repetitive firing, and exacerbation of nocifensive responses, were unexpected. Phosphoproteomic data available online from PhosphoSitePlus determined that Thr1926 in NaV1.8 channels is putatively phosphorylated. We used this information to design the second peptide because Thr1926 is 4 aa downstream of the PY motif Supplemental Fig. S5). Using the results from Scansite, Thr1926 is predicted to be either a casein kinase II or a glycogen synthase kinase–3β kinase consensus phosphorylation site. Both kinases are constitutively active kinases, suggesting that Thr1926 is likely basally phosphorylated. We speculate that the phospho-PY peptide competed with phosphorylated NaV1.8 channels at ULs, preventing a significant proportion of NaV1.8 channels from being ubiquitinylated and retained within the cytosol (Fig. 9E). In addition, we did not observe a statistically significant increase in input levels of NaV1.8 protein over 24 h, indicating that at least during this time window, the majority of internally localized channels are likely in a monoubiquitinylated state (56, 57). Similarly, we speculate that dephosphorylated Thr1926 has a higher affinity for Magi-1, which would explain why the competing PY peptide caused a loss of NaV1.8 channel membrane expression. In this case, we observed a substantial decrease in total NaV1.8 channel protein levels, suggesting that within 24 h, the final fate of the channels was degradation. How this process occurs remains a topic for future investigation, including determining whether Thr1926 is phosphorylated and identifying which kinases and phosphatases might regulate phosphorylation at Thr1926. Nevertheless, our results strongly suggest that the PY motif is a primary determinant for NaV1.8 channel trafficking and that Magi-1 is a critical component of the sodium signalosome in DRG neurons (Fig. 9E).
Although 5% formalin is typically used to study formalin behavioral responses in acute pain models, some studies have suggested that a formalin concentration above 0.5% has off-target effects on Aδ and Aβ fibers and that phase II formalin behavior (high formalin dose) is a result of continued afferent input to the spinal dorsal horn (58). Notwithstanding, these studies are not contradicted by our electrophysiological data, in which a loss of Magi-1 (particularly in Aδ fibers) and concomitant decreased neuronal firing would be expected to reduce pain sensitivity, as was observed after in vivo Magi-1 knockdown in the formalin assay.
Prior studies have shown that when preinjected into the hind paw of mice (10 min prior to formalin), lidocaine caused a signficant reduction in acute inflammatory pain (59). Here, we demonstrated that a single intradermal injection of the PY peptidomimetic 24 h prior to formalin administration produced significant analgesia (Fig. 9D). The PY peptide acted as a local, long-lasting analgesic and could have therapeutic value for invasive procedures that require long-lasting analgesia and/or could reduce the need for postsurgical opioids. In contrast, the phospho-PY peptide drove NaV1.8 channels to the DRG neuronal membrane and exacerbated nocifensive behavior and therefore has potential value for pain insensitivity–related diseases. Myristoylation allows the peptide to partition through the membrane, possibly by a flip-flop mechanism (60), but keeps most of the peptide tethered to the inner surface of the membrane (61, 62). This membrane-delimited feature may enhance the ability of peptides to exert their mimetic effects, especially for membrane-associated proteins (15, 62–64). In addition to causing cell permeability and anchoring peptides within the inner phospholipid bilayer, myristoylation of peptides and their inherent hydrophobic nature likely ensure that peptides are localized to the site of injection. Moreover, the metabolism of these peptides requires phospholipid membrane turnover, possibly contributing to their long-lasting effects in vivo. Prior studies using a similar intradermal injection approach of a peptidomimetic for transmembrane protein 100, important for the transient receptor potential cation channel subfamily A, member 1–transient receptor potential cation channel subfamily V, member 1 complex, showed an analgesic effect during paclitaxel-induced chronic pain (39). Therefore, the use of myristoylated cell-penetrating peptides offers a potential therapeutic approach to manipulate nerve-ending activity.
In addition to the high expression of Magi-1 in peripheral DRG neurons, Magi-1 is also robustly expressed within the CNS (45). Therefore, scaffolding and membrane stabilization of NaV1.8 and Slack KNa channels by Magi-1 in DRG neurons might similarly occur with TTX-sensitive, WW binding motif–containing NaV channels within central neurons. Indeed, our data showed that Magi-1 knockdown in cultured DRG neurons also resulted in a significant reduction in the TTX-sensitive INa, and this might be expected because multiple NaV channel isoforms contain WW binding domains Supplemental Fig. S5). NaV channel loss-of-function mutations are linked with mental health disorders (65). Our findings may then help resolve why Magi-1 deficiency is also associated with multiple psychiatric syndromes (24–26, 66, 67) because Magi-1 deficiency causes diminished sodium transport and hypoexcitability. Hence, we propose that the phospho-PY peptide could serve as a novel therapeutic platform to increase excitability in neurologic diseases associated with hypoexcitability.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank undergraduate students Kimberly Nguyen (State University of New York at Buffalo) and Morgan Paladino (University of Notre Dame, Notre Dame, ID, USA) for scoring the formalin behavior. The authors thank Dr. Wade Sigurdson (Jacobs School of Medicine and Biomedical Sciences, State University of New York at Buffalo) for assistance with microscopy for the immunofluorescence experiments. The authors thank Dr. Elsa Daurignac (Jacobs School of Medicine and Biomedical Research, State University of New York at Buffalo) for critical reading of this manuscript. D.A., K.M.E., D.L.T., and A.B. are also affiliated with the Program for Neuroscience at The State University of New York at Buffalo. This work was supported by the U.S. National Institutes of Health (NIH), National Institute of Neurological Disorders and Stroke Grant NS078184 (to A.B.). The authors declare no conflicts of interest.
Glossary
- AP2-CME
adaptor protein 2–clathrin-mediated endocytosis
- AP
action potential
- BSA
bovine serum albumin
- Caspr
contactin associated protein like
- CHO
Chinese hamster ovary
- Co-IP
coimmunoprecipitation
- DRG
dorsal root ganglion
- IK
inward potassium current
- INa
inward sodium current
- Kcnt
potassium channel subfamily T
- KNa
sodium-activated potassium
- Magi
membrane-associated guanylate kinase with inverted orientation
- NaV
voltage-dependent sodium
- PDZ
postsynaptic density-95/discs large/zonula occludens-1
- PWL
paw withdrawal latency
- PY
proline tyrosine motif
- SC
spinal cord
- SDS
sodium dodecyl sulfate
- shRNA
short hairpin RNA
- Slack
sequence like a calcium-activated potassium channel
- Slick
sequence like an intermediate conductance potassium channel
- SN
sciatic nerve
- siRNA
small interfering RNA
- TTX
tetrodotoxin
- UL
ubiquitin ligase
- WT
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
K. D. Pryce performed all the immunohistochemical and biochemical analyses of Magi-1 in neurons, all the Co-IP and membrane biotinylation assays, all in vitro and in vivo Magi-1 knockdown assays (including the electrophysiological recordings), the PY and phospho-PY peptide experiments, conducted all the nociception and most of the nocifensive behaviors, and performed all the data analysis; R. Powell conducted the other nocifensive behavior testing and performed the NaV1.7 immunoblotting assays; D. Agwa piloted the Magi1–Slack interaction experiments; K. M. Evely piloted the initial spinal nerve injection experiments; G. D. Sheehan initially set up the formalin behavior assay; A. Nip conducted the Magi-1–Slick coexpression electrophysiology studies in CHO cells; D. L. Tomasello engineered the Kozak sequence in the Magi-1 cDNA clone; S. Gururaj set up the initial cultured DRG neuronal system; A. Bhattacharjee conceived the idea for the project and designed the experiments; K. D. Pryce and A. Bhattacharjee wrote the manuscript; and all authors critically revised and gave final approval to this manuscript.
REFERENCES
- 1.Akopian A. N., Souslova V., England S., Okuse K., Ogata N., Ure J., Smith A., Kerr B. J., McMahon S. B., Boyce S., Hill R., Stanfa L. C., Dickenson A. H., Wood J. N. (1999) The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 2, 541–548 [DOI] [PubMed] [Google Scholar]
- 2.Cummins T. R., Sheets P. L., Waxman S. G. (2007) The roles of sodium channels in nociception: implications for mechanisms of pain. Pain 131, 243–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tibbs G. R., Posson D. J., Goldstein P. A. (2016) Voltage-gated ion channels in the PNS: novel therapies for neuropathic pain? Trends Pharmacol. Sci. 37, 522–542 [DOI] [PubMed] [Google Scholar]
- 4.Vijayaragavan K., Boutjdir M., Chahine M. (2004) Modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by protein kinase A and protein kinase C. J. Neurophysiol. 91, 1556–1569 [DOI] [PubMed] [Google Scholar]
- 5.Liu C., Li Q., Su Y., Bao L. (2010) Prostaglandin E2 promotes Na1.8 trafficking via its intracellular RRR motif through the protein kinase A pathway. Traffic 11, 405–417 [DOI] [PubMed] [Google Scholar]
- 6.Bao L. (2015) Trafficking regulates the subcellular distribution of voltage-gated sodium channels in primary sensory neurons. Mol. Pain 11, 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joiner W. J., Tang M. D., Wang L. Y., Dworetzky S. I., Boissard C. G., Gan L., Gribkoff V. K., Kaczmarek L. K. (1998) Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits. Nat. Neurosci. 1, 462–469 [DOI] [PubMed] [Google Scholar]
- 8.Shao D., Baker M. D., Abrahamsen B., Rugiero F., Malik-Hall M., Poon W. Y., Cheah K. S., Yao K. M., Wood J. N., Okuse K. (2009) A multi PDZ-domain protein Pdzd2 contributes to functional expression of sensory neuron-specific sodium channel Na(V)1.8. Mol. Cell. Neurosci. 42, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uchino S., Wada H., Honda S., Hirasawa T., Yanai S., Nakamura Y., Ondo Y., Kohsaka S. (2003) Slo2 sodium-activated K+ channels bind to the PDZ domain of PSD-95. Biochem. Biophys. Res. Commun. 310, 1140–1147 [DOI] [PubMed] [Google Scholar]
- 10.Fotia A. B., Ekberg J., Adams D. J., Cook D. I., Poronnik P., Kumar S. (2004) Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J. Biol. Chem. 279, 28930–28935 [DOI] [PubMed] [Google Scholar]
- 11.Laedermann C. J., Cachemaille M., Kirschmann G., Pertin M., Gosselin R. D., Chang I., Albesa M., Towne C., Schneider B. L., Kellenberger S., Abriel H., Decosterd I. (2013) Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J. Clin. Invest. 123, 3002–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ingham R. J., Gish G., Pawson T. (2004) The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 23, 1972–1984 [DOI] [PubMed] [Google Scholar]
- 13.Ingham R. J., Colwill K., Howard C., Dettwiler S., Lim C. S., Yu J., Hersi K., Raaijmakers J., Gish G., Mbamalu G., Taylor L., Yeung B., Vassilovski G., Amin M., Chen F., Matskova L., Winberg G., Ernberg I., Linding R., O’donnell P., Starostine A., Keller W., Metalnikov P., Stark C., Pawson T. (2005) WW domains provide a platform for the assembly of multiprotein networks. Mol. Cell. Biol. 25, 7092–7106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nuwer M. O., Picchione K. E., Bhattacharjee A. (2010) PKA-induced internalization of slack KNa channels produces dorsal root ganglion neuron hyperexcitability. J. Neurosci. 30, 14165–14172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gururaj S., Evely K. M., Pryce K. D., Li J., Qu J., Bhattacharjee A. (2017) Protein kinase A-induced internalization of Slack channels from the neuronal membrane occurs by adaptor protein-2/clathrin-mediated endocytosis. J. Biol. Chem. 292, 19304–19314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu R., Bausch A. E., Kallenborn-Gerhardt W., Stoetzer C., Debruin N., Ruth P., Geisslinger G., Leffler A., Lukowski R., Schmidtko A. (2015) Slack channels expressed in sensory neurons control neuropathic pain in mice. J. Neurosci. 35, 1125–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hage T. A., Salkoff L. (2012) Sodium-activated potassium channels are functionally coupled to persistent sodium currents. J. Neurosci. 32, 2714–2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhattacharjee A., Kaczmarek L. K. (2005) For K+ channels, Na+ is the new Ca2+. Trends Neurosci. 28, 422–428 [DOI] [PubMed] [Google Scholar]
- 19.Kundu K., Backofen R. (2014) Cluster based prediction of PDZ-peptide interactions. BMC Genomics 15(Suppl 1), S5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobrosotskaya I., Guy R. K., James G. L. (1997) MAGI-1, a membrane-associated guanylate kinase with a unique arrangement of protein-protein interaction domains. J. Biol. Chem. 272, 31589–31597 [DOI] [PubMed] [Google Scholar]
- 21.Zheng C. Y., Seabold G. K., Horak M., Petralia R. S. (2011) MAGUKs, synaptic development, and synaptic plasticity. Neuroscientist 17, 493–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Rodriguez S., Bacarizo J., Luque I., Camara-Artigas A. (2015) Crystal structure of the first WW domain of human YAP2 isoform. J. Struct. Biol. 191, 381–387 [DOI] [PubMed] [Google Scholar]
- 23.Yazicioglu M. N., Monaldini L., Chu K., Khazi F. R., Murphy S. L., Huang H., Margaritis P., High K. A. (2013) Cellular localization and characterization of cytosolic binding partners for Gla domain-containing proteins PRRG4 and PRRG2. J. Biol. Chem. 288, 25908–25914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlsson R., Graae L., Lekman M., Wang D., Favis R., Axelsson T., Galter D., Belin A. C., Paddock S. (2012) MAGI1 copy number variation in bipolar affective disorder and schizophrenia. Biol. Psychiatry 71, 922–930 [DOI] [PubMed] [Google Scholar]
- 25.Li J., Yoshikawa A., Brennan M. D., Ramsey T. L., Meltzer H. Y. (2018) Genetic predictors of antipsychotic response to lurasidone identified in a genome wide association study and by schizophrenia risk genes. Schizophr. Res. 192, 194–204 [DOI] [PubMed] [Google Scholar]
- 26.Piluso G., Monteleone P., Galderisi S., Giugliano T., Bertolino A., Rocca P., Rossi A., Mucci A., Aguglia E., Andriola I., Bellomo A., Comparelli A., Gambi F., Fagiolini A., Marchesi C., Roncone R., Sacchetti E., Santonastaso P., Siracusano A., Stratta P., Tortorella A., Steardo L., Jr., Bucci P., Nigro V., Maj M.; Italian Network for Research on Psychoses (2017) Assessment of de novo copy-number variations in Italian patients with schizophrenia: detection of putative mutations involving regulatory enhancer elements. World J. Biol. Psychiatry 1–11 [DOI] [PubMed] [Google Scholar]
- 27.Dworkin R. H. (1994) Pain insensitivity in schizophrenia: a neglected phenomenon and some implications. Schizophr. Bull. 20, 235–248 [DOI] [PubMed] [Google Scholar]
- 28.Hooley J. M., Delgado M. L. (2001) Pain insensitivity in the relatives of schizophrenia patients. Schizophr. Res. 47, 265–273 [DOI] [PubMed] [Google Scholar]
- 29.Ruit K. G., Elliott J. L., Osborne P. A., Yan Q., Snider W. D. (1992) Selective dependence of mammalian dorsal root ganglion neurons on nerve growth factor during embryonic development. Neuron 8, 573–587 [DOI] [PubMed] [Google Scholar]
- 30.Ritter D. M., Zemel B. M., Hala T. J., O’Leary M. E., Lepore A. C., Covarrubias M. (2015) Dysregulation of Kv3.4 channels in dorsal root ganglia following spinal cord injury. J. Neurosci. 35, 1260–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridgway L. D., Kim E. Y., Dryer S. E. (2009) MAGI-1 interacts with Slo1 channel proteins and suppresses Slo1 expression on the cell surface. Am. J. Physiol. Cell Physiol. 297, C55–C65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Z., Peng A. W., Oshima K., Heller S. (2008) MAGI-1, a candidate stereociliary scaffolding protein, associates with the tip-link component cadherin 23. J. Neurosci. 28, 11269–11276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.González C., Cánovas J., Fresno J., Couve E., Court F. A., Couve A. (2016) Axons provide the secretory machinery for trafficking of voltage-gated sodium channels in peripheral nerve. Proc. Natl. Acad. Sci. USA 113, 1823–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhattacharjee A., Gan L., Kaczmarek L. K. (2002) Localization of the Slack potassium channel in the rat central nervous system. J. Comp. Neurol. 454, 241–254 [DOI] [PubMed] [Google Scholar]
- 35.Bhattacharjee A., von Hehn C. A., Mei X., Kaczmarek L. K. (2005) Localization of the Na+-activated K+ channel slick in the rat central nervous system. J. Comp. Neurol. 484, 80–92 [DOI] [PubMed] [Google Scholar]
- 36.Gururaj S., Fleites J., Bhattacharjee A. (2016) Slack sodium-activated potassium channel membrane expression requires p38 mitogen-activated protein kinase phosphorylation. Neuropharmacology 103, 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang M. F., Hsieh J. H., Chiang H., Kan H. W., Huang C. M., Chellis L., Lin B. S., Miaw S. C., Pan C. L., Chao C. C., Hsieh S. T. (2016) Effective gene expression in the rat dorsal root ganglia with a non-viral vector delivered via spinal nerve injection. Sci. Rep. 6, 35612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerr B. J., Souslova V., McMahon S. B., Wood J. N. (2001) A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport 12, 3077–3080 [DOI] [PubMed] [Google Scholar]
- 39.Weng H. J., Patel K. N., Jeske N. A., Bierbower S. M., Zou W., Tiwari V., Zheng Q., Tang Z., Mo G. C., Wang Y., Geng Y., Zhang J., Guan Y., Akopian A. N., Dong X. (2015) Tmem100 is a regulator of TRPA1-TRPV1 complex and contributes to persistent pain. Neuron 85, 833–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kundu K., Mann M., Costa F., Backofen R. (2014) MoDPepInt: an interactive web server for prediction of modular domain-peptide interactions. Bioinformatics 30, 2668–2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown M. R., Kronengold J., Gazula V. R., Spilianakis C. G., Flavell R. A., von Hehn C. A., Bhattacharjee A., Kaczmarek L. K. (2008) Amino-termini isoforms of the Slack K+ channel, regulated by alternative promoters, differentially modulate rhythmic firing and adaptation. J. Physiol. 586, 5161–5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhattacharjee A., Joiner W. J., Wu M., Yang Y., Sigworth F. J., Kaczmarek L. K. (2003) Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J. Neurosci. 23, 11681–11691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou S., Pita-Almenar J. D., Eskin A. (2011) Regulation of glutamate transporter GLT-1 by MAGI-1. J. Neurochem. 117, 833–840 [DOI] [PubMed] [Google Scholar]
- 44.Renganathan M., Cummins T. R., Waxman S. G. (2001) Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol. 86, 629–640 [DOI] [PubMed] [Google Scholar]
- 45.Ito H., Morishita R., Sudo K., Nishimura Y. V., Inaguma Y., Iwamoto I., Nagata K. (2012) Biochemical and morphological characterization of MAGI-1 in neuronal tissue. J. Neurosci. Res. 90, 1776–1781 [DOI] [PubMed] [Google Scholar]
- 46.Takahashi I., Yoshino M. (2015) Functional coupling between sodium-activated potassium channels and voltage-dependent persistent sodium currents in cricket Kenyon cells. J. Neurophysiol. 114, 2450–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hunskaar S., Hole K. (1987) The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain 30, 103–114 [DOI] [PubMed] [Google Scholar]
- 48.McNamara C. R., Mandel-Brehm J., Bautista D. M., Siemens J., Deranian K. L., Zhao M., Hayward N. J., Chong J. A., Julius D., Moran M. M., Fanger C. M. (2007) TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. USA 104, 13525–13530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor B. K., Peterson M. A., Basbaum A. I. (1995) Persistent cardiovascular and behavioral nociceptive responses to subcutaneous formalin require peripheral nerve input. J. Neurosci. 15, 7575–7584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shirata N., Ihara K. I., Yamamoto-Nonaka K., Seki T., Makino S. I., Oliva Trejo J. A., Miyake T., Yamada H., Campbell K. N., Nakagawa T., Mori K., Yanagita M., Mundel P., Nishimori K., Asanuma K. (2017) Glomerulosclerosis induced by deficiency of membrane-associated guanylate kinase inverted 2 in kidney podocytes. J. Am. Soc. Nephrol. 28, 2654–2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abrahamsen B., Zhao J., Asante C. O., Cendan C. M., Marsh S., Martinez-Barbera J. P., Nassar M. A., Dickenson A. H., Wood J. N. (2008) The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321, 702–705 [DOI] [PubMed] [Google Scholar]
- 52.Laedermann C. J., Abriel H., Decosterd I. (2015) Post-translational modifications of voltage-gated sodium channels in chronic pain syndromes. Front. Pharmacol. 6, 263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gold M. S., Weinreich D., Kim C. S., Wang R., Treanor J., Porreca F., Lai J. (2003) Redistribution of Na(V)1.8 in uninjured axons enables neuropathic pain. J. Neurosci. 23, 158–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skouloudaki K., Walz G. (2012) YAP1 recruits c-Abl to protect angiomotin-like 1 from Nedd4-mediated degradation. PLoS One 7, e35735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benn S. C., Costigan M., Tate S., Fitzgerald M., Woolf C. J. (2001) Developmental expression of the TTX-resistant voltage-gated sodium channels Nav1.8 (SNS) and Nav1.9 (SNS2) in primary sensory neurons. J. Neurosci. 21, 6077–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abriel H., Staub O. (2005) Ubiquitylation of ion channels. Physiology (Bethesda) 20, 398–407 [DOI] [PubMed] [Google Scholar]
- 57.Pickart C. M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533 [DOI] [PubMed] [Google Scholar]
- 58.Bráz J. M., Basbaum A. I. (2010) Differential ATF3 expression in dorsal root ganglion neurons reveals the profile of primary afferents engaged by diverse noxious chemical stimuli. Pain 150, 290–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Inan S., Dun N. J., Cowan A. (2009) Inhibitory effect of lidocaine on pain and itch using formalin-induced nociception and 5′-guanidinonaltrindole-induced scratching models in mice: behavioral and neuroanatomical evidence. Eur. J. Pharmacol. 616, 141–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eisele F., Kuhlmann J., Waldmann H. (2002) Synthesis and membrane binding properties of a lipopeptide fragment from influenza virus a hemagglutinin. Chemistry 8, 3362–3376 [DOI] [PubMed] [Google Scholar]
- 61.Nelson A. R., Borland L., Allbritton N. L., Sims C. E. (2007) Myristoyl-based transport of peptides into living cells. Biochemistry 46, 14771–14781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tressel S. L., Koukos G., Tchernychev B., Jacques S. L., Covic L., Kuliopulos A. (2011) Pharmacology, biodistribution, and efficacy of GPCR-based pepducins in disease models. Methods Mol. Biol. 683, 259–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.François-Moutal L., Wang Y., Moutal A., Cottier K. E., Melemedjian O. K., Yang X., Wang Y., Ju W., Largent-Milnes T. M., Khanna M., Vanderah T. W., Khanna R. (2015) A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain 156, 1247–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ward N. E., O’Brian C. A. (1993) Inhibition of protein kinase C by N-myristoylated peptide substrate analogs. Biochemistry 32, 11903–11909 [DOI] [PubMed] [Google Scholar]
- 65.Carroll L. S., Woolf R., Ibrahim Y., Williams H. J., Dwyer S., Walters J., Kirov G., O’Donovan M. C., Owen M. J. (2016) Mutation screening of SCN2A in schizophrenia and identification of a novel loss-of-function mutation. Psychiatr. Genet. 26, 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferentinos P., Rivera M., Ising M., Spain S. L., Cohen-Woods S., Butler A. W., Craddock N., Owen M. J., Korszun A., Jones L., Jones I., Gill M., Rice J. P., Maier W., Mors O., Rietschel M., Lucae S., Binder E. B., Preisig M., Tozzi F., Muglia P., Breen G., Craig I. W., Farmer A. E., Müller-Myhsok B., McGuffin P., Lewis C. M. (2014) Investigating the genetic variation underlying episodicity in major depressive disorder: suggestive evidence for a bipolar contribution. J. Affect. Disord. 155, 81–89 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.