

Abstract
Pancreatic ductal adenocarcinoma remains one of the most challenging human cancers. Desmoplasia is predominant in this disease exhibiting a strong stromal reaction with an abundance of the cancer-associated fibroblasts (CAFs). We aimed in this study to investigate the reciprocal interaction between the tumor cells and the CAFs and its effect on tumor cells survival. We hypothesized that the survival of pancreatic cancer cell with aggressive phenotype is modulated by the Interactions between malignant pancreatic tumor cells and surrounding CAFs. To examine this, we utilized co-culture methods where tumor cells with different malignant potentials, HPAF (low) HPAF-CD11 (moderate/high) co-cultured with CAFs. CAFs-conditioned media increased the growth of HPAF-CD11 but not HPAF cells and increased CXCL8 levels highly in HPAF-CD11 and slightly in HPAF. The growth stimulatory effect and elevated CXCL8 level caused by CAFs-conditioned media were diminished by neutralizing the fibroblast growth factor-2 (FGF-2). In addition, conditioned media of HPAF-CD11 increased CAFs cell number whereas that of HPAF did not, and these effects were suppressed by neutralizing CXCL8. Furthermore, data from gene expression microarray study exhibited different expression profiles between HPAF and HPAF-CD11 when co-culture with CAFs. A significant increase in CXCL8 and FGF-2 expression was observed with HPAF-CD11/CAFs co-culture and to a lower extent with HPAF/CAFs co-culture. Together, these data demonstrate a paracrine bi-directional interaction between pancreatic tumor cells and the CAFs through CXCL8 and FGF-2 that helps the tumor growth. Future in-depth study of these pathways will assist in obtaining diagnostic and therapeutic tools for pancreatic ductal adenocarcinoma.
Keywords: Pancreatic tumor, Cell survival, FGF-2, CXCL8, Tumor progression
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancers, the fourth leading cause of cancer deaths in the United States [1]. The increased incidence of the disease, as well as the cellular and molecular complexity of the tumor, makes it very challenging to manage. A chance of cure exists for only a minority of the patients, those with locally limited and surgically resectable tumors [2]. At the time of diagnosis, the majority of PDAC patients present at advanced stages beyond surgical resection. Studying the complex cellular and molecular interaction between malignant cells and other cells in the tumor microenvironment can shed more light on how the diseases initiate, progresses and spreads.
Desmoplasia is of particular predominance in PDAC exhibiting a strong stromal reaction [3–5]. A consistently low ratio of the infiltrating adenocarcinoma component relative to this abundant desmoplastic response is unique to PDAC, in contrast to infiltrating carcinomas in other organ or tissue types [6, 7]. Typically, these invasive pancreatic tumors are composed of infiltrating adenocarcinoma surrounded by a predominance of dense fibrous (or desmoplastic) stroma [8], which itself contains proliferating cancer-associated fibroblasts (CAFs), small endothelial-lined vessels, inflammatory cells, and trapped residual atrophic parenchymal components of the organ invaded [9]. CAFs, represent the fibrotic component of the tumor microenvironment, are derived from cells of multiple origins including tissue-resident fibroblasts, bone marrow-derived mesenchymal cells, fibrocytes, and pancreatic stellate cells (PSCs) [10]. PSCs, in particular, have gained much attention more than other subsets of CAFs. PSCs, similar to other stellate cells found in other organs such as in liver, kidneys, and lungs, are known to modulate physiological functions by storing vitamin-A at their quiescent state and tissue maintenance and repair at the activated state [11–14]. In PDAC, activated PSCs have been described to be involved in tumorigenesis, therapy resistance, and metastasis [11, 15–18]. Interactions between the malignant cells and surrounding stromal CAFs have been suggested to play a critical role in tumor invasion and progression [19, 20]. Once tumor cells have spread to different microenvironments, their subsequent growth will depend on the compatibility of the “seed” with the “soil” that they encounter in the microenvironment [21, 22], which depend on the molecular interactions between cancer cells and the stromal cells in the different microenvironment [23, 24]. Invasive cancers do not exist in isolation. Rather, they arise from and intimately interact with non-neoplastic host cells [25, 26].
For long, CAFs have been regarded for their role in the formation of desmoplasia, by producing excessive amounts of extracellular matrix proteins [27]. Desmoplasia aids in acquiring resistance to current chemotherapy treatments [28–30]. Nonetheless, recent literature describes a vast network of CAFs interactions beyond the desmoplasia formation. Through their network of secreted factors, such as cytokine, chemokines and growth factors, CAFs can interact with the multiple components in the tumor microenvironment to modulate tumor progression in different malignancies [31–34].
Fibroblast growth factor (FGF)-2 is a member of the FGF family that control multiple cellular processes including proliferation, differentiation, survival, and motility [35]. In the context of cancer, FGF-2 has been shown to promote tumor progression [36]. Enhanced FGF-2 protein levels have been shown to correlate with shorter postoperative survival of patients with PDAC [37]. Furthermore, FGF-2 was linked to PDAC invasion via its activity in PSCs [38].
A member of the CXC chemokine family, CXCL8 signals through CXCR1 and CXCR2 chemokine receptors. These chemokines are known for their role in inflammation by recruiting inflammatory cells and inducing angiogenesis. In malignant tumors, sustained CXCL8 signaling is associated with immunosuppression, angiogenesis, and tumor growth; thus, essential to the progression of PDAC [39–41]. There is evidence that CXCL8 and FGF-2 are involved in tumor-stromal interaction [38, 42].
We hypothesized that the aggressive phenotype of PDAC depends on their interaction with CAFs, which involves FGF-2 and CXCL8. To test this hypothesis, we examined the effect of CAFs on pancreatic tumor cells with different malignant potential, HPAF (low) and HPAF-CD11 (moderate/high). HPAF-CD11 is derived from the parent cell line HPAF, where both cells show well-differentiation features and mutations in both Kras and TP53 [43–47]. We demonstrated that the aggressive phenotype of PDAC has a stronger bi-directional interaction with CAFs through paracrine factors such as FGF-2 and CXCL8.
Materials and Methods
Patient-Derived Cancer-Associated Fibroblasts (CAF) Cell Line
CAF cell line was derived from patient pancreatic tumor tissues. All procedures performed in studies involving human participants were in accordance with the ethical standards of the University of Nebraska Medical Center, Omaha, NE and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Cell Lines and Culture Conditions
The human pancreatic cancer cell lines (HPAF and HPAF-CD11) were kind gifts from Dr. Michel Hollingsworth (Eppley Cancer Center, University of Nebraska Medical Center, Omaha, NE) and Dr. S.K. Batra (Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center). Normal human fibroblast, BJ cell line, was obtained from ATCC. CAF was derived from the pancreatic tumor tissues. The pancreatic tumor was minced, and the fibroblasts were isolated by differential trypsinization, and subsequently immortalized using hTERT. HPAF were maintained in RPMI 1640 (Cellgro, Herndon, VA) supplemented with 5% fetal bovine serum (FBS), HPAF-CD11 were in DMEM (Cellgro, Herndon, VA) with 10% FBS, and both BJ and CAF cells were maintained in EMEM (Cellgro, Herndon, VA) with 10% FBS supplemented with streptomycin, and penicillin (complete media) at 37 °C in a humidified atmosphere containing 5% CO2. All the cell lines were free of mycoplasma as determined by MycoAlert Plus Mycoplasma Detection kit (Lonza) and pathogenic murine viruses. For cell line authentication, Human DNA Identification Laboratory, University of Nebraska Medical Center, Omaha, NE, USA performed the short tandem repeat (STR) tests [48]. Cultures were maintained for no longer than six weeks after recovery from frozen stocks.
PDAC Cells Cultured with Experimental Conditions
1 × 106 cells of normal fibroblasts (BJ) were seeded in the six-well plate and incubated with EMEM media for 24 h to generate fibroblasts monolayer. PDAC cells (1 × 105) were then seeded onto the fibroblasts monolayer and co-cultured in complete media. After a 24 h incubation, complete media was changed to serum-free media (day 0) and incubated for an additional 72 h (day 4).
Generation of CAFs-Conditioned Media and ECM
To analyze the effects of secreted factors from CAFs on tumor cells, CAFs were cultured with the respective media for 48 h. Cell-free conditioned media was collected and stored at −70 °C until used. To generate CAFs ECM, we used the methods established by Mizuguchi et al. [49]. Briefly, we seeded CAFs cell (1 × 105 cells) onto six-well plate and culture them with EMEM supplemented with 10% FBS. After the CAFs cell reach confluence (24hlater), we removed the media and washed the cells once with PBS, and then added 1 ml of the aqueous solution of 0.02 N ammonia to the cells, and incubated them at room temperature for 10 min to lyse the cells. We removed any remaining cellular debris from the culture plate by gentle pipetting and washed the resulting lysate over ten times Hanks’s balanced salt solution (HBSS).
PDAC cells (1 × 105) were seeded alone or on top of CAFs-derived ECM for 24 h with complete media. Next, at day 0, cells were washed and serum-free, serum containing or CAF-conditioned media were added accordingly for an additional 72 h (day 4). We counted the number of tumor cell and calculated the differences between Day 0 and Day 4. To examine the effect of neutralizing antibodies, we added human FGF (basic) antibody (American Diagnostica inc., Greenwich, CT) and human CXCL8 antibody (BD Pharmingen) at 10 μg/ml concentration on Day 0.
Enzyme-Linked Immunosorbent Assay (ELISA)
Cells (1 × 105) / well were grown in a six-well culture plate. After 24 h, media was replaced, and tumor cells either were incubated with standard media or conditioned media from CAFs. Oppositely, CAFs were incubated with standard media or conditioned media from tumor cells. After 48 h, supernatants were taken for ELISA analysis.
CXCL8 levels in culture supernatants were determined using an ELISA kit paired antibody purchased from Pierce Inc. (Woburn, MA), according to manufacturer instructions. Briefly, 100 μl of the primary monoclonal antibody against CXCL8 (2 μg/ml) was coated in Immulon plates in each well. After 1 h of incubation at 37 °C, the plates were washed and blocked for 1 h with blocking buffer (4% BSA in PBS). After washing the plates four times, 50 μl culture supernatants or standards at different concentrations (recombinant CXCL8 protein, Endogen Inc. Woburn, MA) and 50 μl of biotinylated CXCL8 Ab was added to each well. After 2 h of incubation, the plates were washed, and the immunoreactivity determined using the avidin-HRP-TMB detection system (Dako Labs. Denmark). The reactions were stopped by adding50 μl of 0.18 N H2SO4. Absorbance was determined using an ELISA microtiter plate reader (Bio-Tek Instruments Inc. Winooski, VT) at 450 nm. CXCL8 concentrations of the unknown samples were calculated using a plotted curve of the absorbance versus the concentration of CXCL8 in the standard wells.
For analyzing levels of FGF-2, we used direct ELISA. Samples and different concentrations of recombinant FGF-2 protein (for standard curve) was coated onto ELISA plate overnight. Following washing and blocking non-specific activity, 100 μl of anti-FGF-2 antibody (R & D System, Minneapolis MN) was added into each well. Following two hours of incubation, samples were incubated with biotinylated secondary antibody and immunoreactivity was determined using avidin-HRP-TMB detection system. A curve of the absorbance versus the concentration of FGF-2 in the standard wells was plotted to calculate FGF-2 concentrations in the unknown samples by comparing the absorbance of the samples to the standard curve.
Gene Expression Microarray
PDAC cell lines HPAF and HPAF-CD11 were cultured alone or on CAF monolayer. Next, nucleic acid was collected for cDNA microarray analysis using a set of two 10 K chips (Compugn/Sigma Genosys) that interrogate the full 18+ Compugen Human oligonucleotide at DNA Microarray core facility (UNMC). The library contains 18,861 oligos representing 17,260 unique genes. Raw fluorescent intensity values were collected to determine gene expression levels. Flagged artifacts and negative controls were removed from the series. The data was then normalized, and the channels (Cy3 and Cy5) were background subtracted. The normalized and background subtracted values were log2 transformed. The fold-change was calculated between the Cy3 and Cy5 channels. Emphasis was placed on genes demonstrating greater than 2 fold-change in expression between the two channels. A list of 169 chemokines and cytokines identified by the KEGG_CHEMOKINE_SIGNALING_PATHWAY and BIOCARTA_CYTOKINE_PATHWAY were identified in the dataset and used for differential expression where indicated. Cluster 3.0 was used to median-center the genes prior to heat map generation in Java TreeView.
Statistical Analysis
Statistical method and sample size (n; the number of replicates) are indicated in the figure legends. Statistical analysis was performed using Prism 7 (GraphPad) software. A two-tailed student’s t test was performed on the means of all parametric data. Statistical significance was defined as p < 0.05. Error bars on figures show standard error of the mean (SEM).
Results
Aggressive Pancreatic Tumor Cells Survived Following Co-Culture with Fibroblasts in the Absence of Serum
We examined the growth of HPAF and HPAF-CD11 cells in the presence or absence of serum-containing media when cultured with or without fibroblasts. Both HPAF and HPAF-CD11 cells showed serum dependency irrespective of their aggressive differential phenotype when cultured in the absence of fibroblasts (Fig. 1a and b). Further, the growth of HPAF cells was inhibited following co-culture with fibroblasts (BJ cells monolayer) in the presence of or absence of serum (Fig. 1c) as compared to HPAF cells cultured alone. The level of inhibition of HPAF cells growth following co-culture with fibroblasts was similar to that observed when HPAF cells alone were cultured in the absence of serum (Fig. 1a). In contrast, we observed an increased survival and growth of HPAF-CD11 cells following co-culture with fibroblasts (BJ cells monolayer). Interestingly the growth of HPAF-CD11 cells was enhanced in following co-culture with fibroblasts in the absence of serum (Fig. 1d).
Fig. 1.
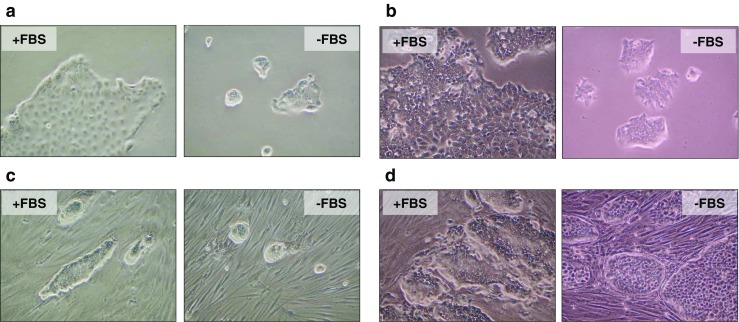
Fibroblasts promote the survival of aggressive pancreatic tumor cell. a Light microscope images of HPAF cells cultured in complete media or serum-free media. b Light microscope images of HPAF-CD11 cells cultured in complete media or serum-free media. c Light microscope images of HPAF cells cultured on top of fibroblasts (BJ) monolayer with complete media, or serum-free media. d Light microscope images of HPAF-CD11 cells cultured on top of fibroblasts (BJ) monolayer with complete media, or serum-free media
Survival of Tumor Cells Is Mediated by CAFs Conditioned Media
To evaluate whether the survival of HPAF-CD11 cells on the fibroblasts monolayer was mediated by the direct contact or paracrine factors, HPAF and HPAF-CD11 cells were incubated with CAF-conditioned media or extracellular matrix (ECM) generated from CAFs, and the increase in cell number was quantitated. No increase in the cell number was observed in HPAF cells in response to CAFs conditioned media, and further inhibition was detected with ECM culture (Fig. 2a). We observed a significant increase in the number of HPAF-CD11 cells following co-culture with both CAFs conditioned media and CAFs ECM (Fig. 2b). The increase in HPAF-CD11 cells cultured with CAFs conditioned media was greater than that observed in response to ECM but less than that observed with serum containing media (Fig. 2b). Next, we examined the dose-dependence of CAFs-conditioned media on the growth of HPAF-CD11 cell (Fig. 2c) and demonstrated that increasing concentrations of CAFs conditioned media increased the growth of HPAF-CD11 cells. Together, we perceive that CAFs-derived paracrine factors contribute to the survival of the aggressive PDAC cells.
Fig. 2.
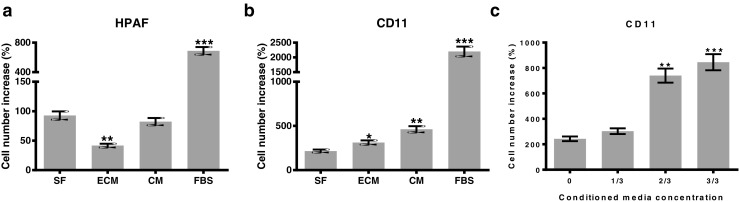
CAFs promotes the survival of tumor cells through paracrine contact. a and b Survival of HPAF cells (a) and HPAF-CD11 (b) following their culture with CAFs-ECM, CAFs-conditioned media, or complete media as compared to serum-free media. b Survival of HPAF-CD11 cells in increasing amounts of CAFs- conditioned media to serum-free media. (n = 3), student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001
CAFs Promote Tumor Cell Survival Via FGF-2
To determine the putative growth factors present in CAFs conditioned media, we examined the effect of the neutralizing antibodies of FGF-2 and CXCL8, which have been shown to be involved in the tumor-stromal interaction [38, 42], on the survival of HPAF-CD11 cells. Anti-FGF-2 antibody treatment significantly abrogated the increase of cell number of HPAF-CD11 following culture with CAFs conditioned media (Fig. 3a), while anti-CXCL8 antibody treatment did not (Fig. 3b). To confirm that FGF-2 is produced by CAFs, we performed ELISA on the CAF supernatant collected in serum-free media or complete media (Fig. 3c). These results indicate that FGF-2 but not CXCL8 in the CAFs conditioned media was involved in the survival of HPAF-CD11.
Fig. 3.
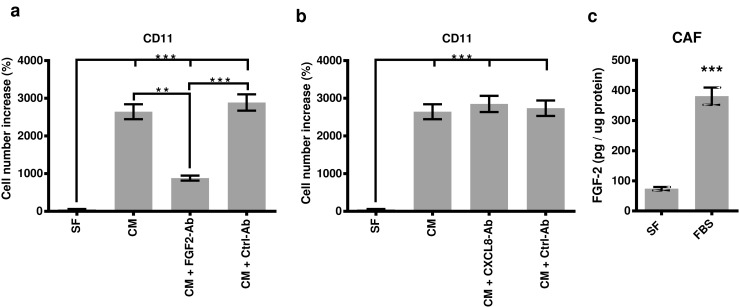
Effects of CAFs conditioned media on the survival of HPAF-CD11 is mediated by FGF-2. a Number increase of HPAF-CD11 in response to CAFs-conditioned media ± FGF-2 neutralizing antibody as compared to serum-free media. b Number increase of HPAF-CD11 in response to CAFs-conditioned media ± CXCL8 neutralizing antibody as compared to serum-free media. c FGF-2 concentrations, evaluated by ELISA, in the supernatant of CAF incubated in serum-free media or complete media. (n = 3), student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001
FGF-2 Secreted by CAFs Induces CXCL8 Production in Tumor Cells
CXCL8 and other CXC chemokines that signal through CXCR1/2 axis are known to be expressed in PDAC cells [50–52]. Autocrine and paracrine signaling through CXCR1/2 axis plays a vital role in the progression of PDAC by promoting tumor cells growth, angiogenesis, immunosuppression and chemotherapy resistance [39, 53–55]. To examine the involvement of FGF-2 in the CXCL8 production by PDAC cells, we determined CXCL8 production in HPAF and HPAF-CD11 cell after treatment with CAFs-conditioned media using ELISA. Our data shows that HPAF-CD11 produce more CXCL8 than their HPAF counterparts by comparing CXCL8 levels produced in serum-free media treatment as well as in response to CAFs-conditioned media treatment (Fig. 4a-b). By comparing the CXCL8 production in each cell in response to CAFs-conditioned media, we show that CAFs-conditioned media increased CXCL8 level in both HPAF and HPAF-CD11 cells, and neutralizing FGF-2 has lowered the CXCL8 inducing effect of the CAFs-conditioned media (Fig. 4c and d).
Fig. 4.
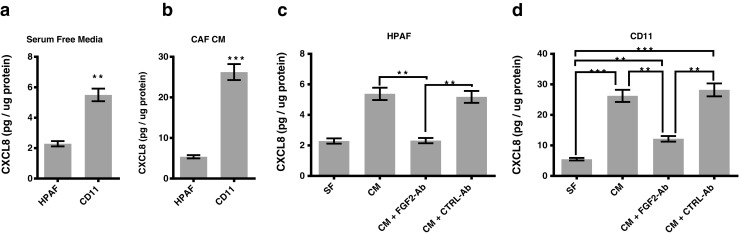
FGF-2 in the CAFs conditioned media promotes the CXCL8 production of pancreatic tumor cells. a CXCL8 concentration, evaluated by ELISA, in the supernatant of HPAF cells and b HPAF-CD11 after treatment with serum-free media or CAFs-conditioned media. b CXCL8 concentration, evaluated by ELISA, in the supernatant of HPAF cells after treatment with CAFs-conditioned media ± FGF-2 neutralizing. d CXCL8 concentration, evaluated by ELISA, in the supernatant of HPAF-CD11 cells after treatment with CAFs-conditioned media ± FGF-2 neutralizing. (n = 3), student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001
Effect of Tumor Cell Conditioned Media on the Survival of CAFs
Next, we examined the effect of the conditioned media from PDAC cells with different aggressiveness on the survival of CAFs. CAFs were incubated with the conditioned media of HPAF or and HPAF-CD11 for 1, 2, and 3 days, and the increase in cell number was quantitated. We observed a significant increase in the number of CAFs following co-culture with conditioned media of HPAF-CD11 cell at each time point but not with that of HPAF (Fig. 5a). To determine the putative growth factors present in the conditioned media of HPAF-CD11, we examined the effect of the neutralizing antibodies of CXCL8 (Fig. 5b). Anti-CXCL8 antibody treatment significantly abrogated the increase of cell number of CAFs following culture with HPAF-CD11 conditioned media. These results indicate that CXCL8 was involved in the survival of CAFs by the conditioned media of PDAC cells.
Fig. 5.

Effect of tumor cell conditioned media on the survival of CAFs. a Survival, increase in cell number, of CAFs incubated with the conditioned media of HPAF or and HPAF-CD11 for 1, 2, and 3 days, and the increase in cell number was quantitated. b Net growth, determined by the increase in cell number, of CAFs in HPAF-CD11 conditioned media ± CXCL8 neutralizing antibody. (n = 3), student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001
Aggressiveness-Dependent Gene Expression of Tumor Cells when Co-Cultured with CAF
Finally, we used gene expression microarray to explore the expressional differences between HPAF and HPAF-CD11 cells upon their co-cultured with CAF. Distinct gene expression profiles were observed for HPAF and HPAF-CD11 when compared alone or in co-culture. More focused look into the expression profile of paracrine factors revealed that CAFs are the major contributor of many cytokines and chemokines (Fig. 6a). Comparing HPAF/CAF co-culture to HPAF-CD11/CAF co-culture revealed upregulation of motility supporting gene ELMO1 in HPAF-CD11 co-culture, whereas, mainly cytokines and chemokines were upregulated in the HPAF co-culture (Fig. 6a). Targeted look into CXCL8 and FGF-2 expression exhibited that CAFs are the leading producer of CXCL8 and that CXCL8 was upregulated in the co-culture condition compared to tumor cells cultured alone for both HPAF and HPAF-CD11 (Fig. 6b). For FGF-2, only HPAF-CD11 co-culture exhibited significance upregulation of the gene compared to the tumor cells cultured alone (Fig. 6b). Together, these data demonstrate the versatility of CAFs and their ability to support tumor cells in an aggressive-dependent manner.
Fig. 6.
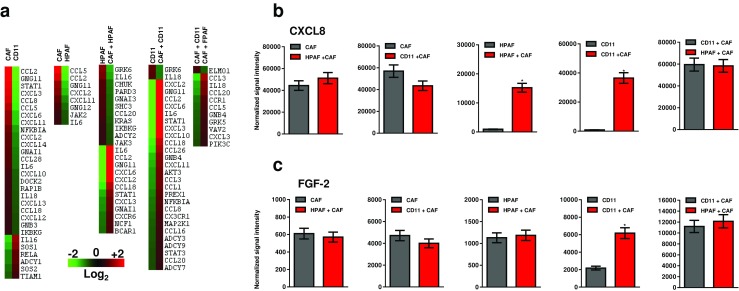
Differential gene expression of tumor cells cultured with CAFs. a Heat map of gene expression of HPAF and HPAF-CD11 cultured alone or with CAFs determined using gene expression microarray. Heat maps show chemokines and cytokine with >2-fold increase. b Expression of CXCL8, represented as normalized signal intensity, in HPAF and HPAF-CD11 cultured alone or with CAFs determined using gene expression microarray. c Expression of FGF-2, represented as normalized signal intensity, in HPAF and HPAF-CD11 cultured alone or with CAFs determined using gene expression microarray
Discussion
PDAC, one of the most malignant tumors, is often characterized by an abundant desmoplastic stroma. CAFs, which constitute a major stromal compartment in PDAC, have been shown to promote the invasive growth of several cancer types such as breast, prostate, and lung [31, 56]. CAFs are often only associated with excess extracellular matrix production; thus, their contribution to desmoplasia [27]. Recent studies have addressed the role of CAFs in pancreatic tumor aggressiveness. Non-irradiated CAFs significantly increased the invasive ability of pancreatic cancer cells and the invasiveness was further accelerated when they were co-cultured with irradiated CAFs [57]. Nitric Oxide released by CAFs has been shown to lead to the upregulation of IL-1β in pancreatic carcinoma cells, leading to the induction of chemotherapy resistance in these tumor cells [58]. CAFs can produce many paracrine factors including chemokines, cytokines and growth factor, which allow interaction and subsequent modulation of other cells in the tumor microenvironment [10]. This secretory role of CAFs remained under-investigated. In the present study, we demonstrated that pancreatic tumor with more aggressive phenotype could interact with CAFs more than non-aggressive cells. These data underscored the importance of the interaction with CAFs in the exertion of the malignant potential of the pancreatic tumor.
FGF-2 is expressed in pancreas cancer [59], as well as in many other malignant neoplasms [60, 61]. FGFs bind to a family of transmembrane tyrosine kinase receptors (FGFRs 1–4), and FGFR-1 and FGFR-4 are potent receptors for FGF-2 [62]. A member of the CXC chemokine family, CXCL8, its production has been correlated with tumor growth, immunosuppression, resistance to chemotherapy, angiogenesis, and increased metastatic potential of PDAC [40, 41].
In the current study, we demonstrate a bi-directional interaction between tumor cells and CAFs that creates a feedforward loop to promote the survival of the tumor cells in PDAC. The said interaction was more obvious with HPAF-CD11 cells that acquire more PDAC aggressive features. Culturing tumor cells on top of CAFs monolayer proved that the interaction between malignant cells and CAFs could promote survival and growth of PDAC cells. Nonetheless, culturing tumor cells in CAFs-derived conditioned media demonstrated that the survival stimulation effect of CAFs on malignant cells is mediated through paracrine factors rather than direct interaction. The use of neutralizing antibodies demonstrated that FGF-2 is the putative factor that stimulates malignant cells survival. Whereas it is clear that FGF-2 was present in CAFs-conditioned media, the difference in the expression of the appropriate FGF-2 receptors is possibly responsible for the difference between HPAF and HPAF-CD11. Moreover, CXCL8 has been shown to enhance endothelial cell proliferation and to regulate angiogenesis [63, 64]. In this study, we show that CAFs-conditioned media increased CXCL8 production by HPAF-CD11. Subsequently, CXCL8 may induce angiogenesis necessary for further tumor progression. On the other hand, HPAF cells that carry less aggressive potential produce less CXCL8. Therefore, the CXCL8 level induced by CAFs-conditioned media may be one of the determinants for malignant potential. Recently, CXCL8 has been shown to be produced by prostatic epithelial cells of benign prostatic hyperplasia which consists of slow but progressive growth of both epithelial and stromal cell and can act as a paracrine inducer of FGF-2 production by prostatic stromal cells in vitro [42]. In our study, conditioned media of the more aggressive PDAC cells, HPAF-CD11, stimulated and maintained the survival of CAFs through the secretion of CXCL8. Therefore, pancreatic tumor cell-derived CXCL8, released as a consequence of FGF-2 stimulation, may act on CAFs to stimulate further FGF-2 production. On the other hand, we have shown that CAFs derived FGF-2 can act as a paracrine inducer of CXCL8 production by pancreatic tumor cells.
Looking into the differential gene expression profiles of HPAF and HPAF-CD11 upon their co-culture with CAFs can reveal the extent of CAFs contribution to tumor progression. CAFs appear to have a high baseline of several cytokines and chemokines including CXCL8. An interesting observation is that HPAF-CD11 cell/CAFs co-culture upregulates ELMO1 gene that has been associated with motility [65, 66]. If we put this together with the ability of CXCL8 to induce angiogenesis, we can assume that CAFs can contribute to tumor cells spread to other organs.
In conclusion, interactions between pancreatic tumor cells and CAFs promote the survival of tumor cells with aggressive potentials and promote CXCL8 production. CXCL8, released as a consequence of FGF-2 stimulation, act on CAFs to stimulate further FGF-2 production. Thus, such bi-directional interactions between pancreatic tumor cells and CAFs help the tumor growth in different microenvironments, which leads to the pancreatic tumor progression and spread.
Acknowledgments
We thank Dr. M. Hollingsworth and Dr. S.K. Batra from University of Nebraska Medical Center, Omaha, NE for providing the pancreatic cancer cell lines, and the pancreatic CAFs cell line. This work was supported in part by grants U54CA163120, R01CA228524, and Cancer Center Support Grant (P30CA036727) from the National Cancer Institute, National Institutes of Health. Mohammad Awaji as a graduate student was supported by a scholarship from King Fahad Specialist Hospital-Dammam and the Saudi Arabian Cultural mission in the USA, and a pre-doctoral fellowship from the University of Nebraska Medical Center.
Compliance with Ethical Standards
Conflict of Interest
The authors declare no potential conflicts of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Warshaw AL, Fernandez-del Castillo C. Pancreatic carcinoma. N Engl J Med. 1992;326(7):455–465. doi: 10.1056/NEJM199202133260706. [DOI] [PubMed] [Google Scholar]
- 3.Kuniyasu H, Abbruzzese JL, Cleary KR, Fidler IJ. Induction of ductal and stromal hyperplasia by basic fibroblast growth factor produced by human pancreatic carcinoma. Int J Oncol. 2001;19(4):681–685. doi: 10.3892/ijo.19.4.681. [DOI] [PubMed] [Google Scholar]
- 4.Iacobuzio-Donahue CA, Ryu B, Hruban RH, Kern SE. Exploring the host desmoplastic response to pancreatic carcinoma: gene expression of stromal and neoplastic cells at the site of primary invasion. Am J Pathol. 2002;160(1):91–99. doi: 10.1016/S0002-9440(10)64353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watanabe I, Hasebe T, Sasaki S, Konishi M, Inoue K, Nakagohri T, et al. Advanced pancreatic ductal cancer: fibrotic focus and beta-catenin expression correlate with outcome. Pancreas. 2003;26(4):326–333. doi: 10.1097/00006676-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Seymour AB, Hruban RH, Redston M, Caldas C, Powell SM, Kinzler KW, et al. Allelotype of pancreatic adenocarcinoma. Cancer Res. 1994;54(10):2761–2764. [PubMed] [Google Scholar]
- 7.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 8.Kloppel G, Lingenthal G, von Bulow M, Kern HF. Histological and fine structural features of pancreatic ductal adenocarcinomas in relation to growth and prognosis: studies in xenografted tumours and clinico-histopathological correlation in a series of 75 cases. Histopathology. 1985;9(8):841–856. doi: 10.1111/j.1365-2559.1985.tb02870.x. [DOI] [PubMed] [Google Scholar]
- 9.Ryu B, Jones J, Hollingsworth MA, Hruban RH, Kern SE. Invasion-specific genes in malignancy: serial analysis of gene expression comparisons of primary and passaged cancers. Cancer Res. 2001;61(5):1833–1838. [PubMed] [Google Scholar]
- 10.Öhlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211(8):1503–1523. doi: 10.1084/jem.20140692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moir JA, Mann J, White SA. The role of pancreatic stellate cells in pancreatic cancer. Surg Oncol. 2015;24(3):232–238. doi: 10.1016/j.suronc.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117(1):50–59. doi: 10.1172/JCI30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Apte MV, Wilson JS, Lugea A, Pandol SJ. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology. 2013;144(6):1210–1219. doi: 10.1053/j.gastro.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Apte M, Pirola RC, Wilson JS. Pancreatic stellate cell: physiologic role, role in fibrosis and cancer. Curr Opin Gastroenterol. 2015;31(5):416–423. doi: 10.1097/MOG.0000000000000196. [DOI] [PubMed] [Google Scholar]
- 15.Xu Z, Vonlaufen A, Phillips PA, Fiala-Beer E, Zhang X, Yang L, et al. Role of pancreatic stellate cells in pancreatic Cancer metastasis. Am J Pathol. 2010;177(5):2585–2596. doi: 10.2353/ajpath.2010.090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. 2012;11(7):1282–1290. doi: 10.4161/cc.19679. [DOI] [PubMed] [Google Scholar]
- 17.McCarroll JA, Naim S, Sharbeen G, Russia N, Lee J, Kavallaris M, et al. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front Physiol. 2014;5:141. doi: 10.3389/fphys.2014.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zambirinis CP, Levie E, Nguy S, Avanzi A, Barilla R, Xu Y, et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J Exp Med. 2015;212(12):2077–2094. doi: 10.1084/jem.20142162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grey AM, Schor AM, Rushton G, Ellis I, Schor SL. Purification of the migration stimulating factor produced by fetal and breast cancer patient fibroblasts. Proc Natl Acad Sci U S A. 1989;86(7):2438–2442. doi: 10.1073/pnas.86.7.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camps JL, Chang SM, Hsu TC, Freeman MR, Hong SJ, Zhau HE, et al. Fibroblast-mediated acceleration of human epithelial tumor growth in vivo. Proc Natl Acad Sci U S A. 1990;87(1):75–79. doi: 10.1073/pnas.87.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hart IR. 'Seed and soil' revisited: mechanisms of site-specific metastasis. Cancer Metastasis Rev. 1982;1(1):5–16. doi: 10.1007/BF00049477. [DOI] [PubMed] [Google Scholar]
- 22.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8(2):98–101. [PubMed] [Google Scholar]
- 23.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 24.Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncol. 2002;3(1):53–57. doi: 10.1016/s1470-2045(01)00622-2. [DOI] [PubMed] [Google Scholar]
- 25.Maehara N, Matsumoto K, Kuba K, Mizumoto K, Tanaka M, Nakamura T. NK4, a four-kringle antagonist of HGF, inhibits spreading and invasion of human pancreatic cancer cells. Br J Cancer. 2001;84(6):864–873. doi: 10.1054/bjoc.2000.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian LW, Mizumoto K, Maehara N, Ohuchida K, Inadome N, Saimura M, et al. Co-cultivation of pancreatic cancer cells with orthotopic tumor-derived fibroblasts: fibroblasts stimulate tumor cell invasion via HGF secretion whereas cancer cells exert a minor regulative effect on fibroblasts HGF production. Cancer Lett. 2003;190(1):105–112. doi: 10.1016/s0304-3835(02)00517-7. [DOI] [PubMed] [Google Scholar]
- 27.Apte MV, Wilson JS. Mechanisms of pancreatic fibrosis. Dig Dis. 2004;22(3):273–279. doi: 10.1159/000082799. [DOI] [PubMed] [Google Scholar]
- 28.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic Cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Provenzano PP, Cuevas C, Chang AE, Goel VK, Hoff V, Daniel D, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2012;62(1):112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Micke P, tman A. Tumour-stroma interaction: cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer. 2004;45:S163–S175. doi: 10.1016/j.lungcan.2004.07.977. [DOI] [PubMed] [Google Scholar]
- 32.Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol. 2014;25:61–68. doi: 10.1016/j.semcancer.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 33.De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol. 2003;200(4):429–447. doi: 10.1002/path.1398. [DOI] [PubMed] [Google Scholar]
- 34.Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R, et al. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, MSP- and HGF-mediated signaling networks. Oncogene. 2005;24(32):5053–5068. doi: 10.1038/sj.onc.1208685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basilico C, Moscatelli D. The FGF family of growth factors and oncogenes. Adv Cancer Res. 1992;59:115–165. doi: 10.1016/s0065-230x(08)60305-x. [DOI] [PubMed] [Google Scholar]
- 36.Polnaszek N, Kwabi-Addo B, Peterson LE, Ozen M, Greenberg NM, Ortega S, et al. Fibroblast growth factor 2 promotes tumor progression in an autochthonous mouse model of prostate cancer. Cancer Res. 2003;63(18):5754–5760. [PubMed] [Google Scholar]
- 37.Kleeff J, Kothari NH, Friess H, Fan H, Korc M. Adenovirus-mediated transfer of a truncated fibroblast growth factor (FGF) type I receptor blocks FGF-2 signaling in multiple pancreatic cancer cell lines. Pancreas. 2004;28(1):25–30. doi: 10.1097/00006676-200401000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Coleman SJ, Chioni A-M, Ghallab M, Anderson RK, Lemoine NR, Kocher HM, et al. Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol Med. 2014;6(4):467–481. doi: 10.1002/emmm.201302698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purohit A, Varney M, Rachagani S, Ouellette MM, Batra SK, Singh RK. CXCR2 signaling regulates KRAS(G(1)(2)D)-induced autocrine growth of pancreatic cancer. Oncotarget. 2016;7(6):7280–7296. doi: 10.18632/oncotarget.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Q, Li A, Tian Y, Wu JD, Liu Y, Li T, et al. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016;31:61–71. doi: 10.1016/j.cytogfr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saintigny P, Massarelli E, Lin S, Ahn YH, Chen Y, Goswami S, et al. CXCR2 expression in tumor cells is a poor prognostic factor and promotes invasion and metastasis in lung adenocarcinoma. Cancer Res. 2012;73(2):571–582. doi: 10.1158/0008-5472.CAN-12-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giri D, Ittmann M. Interleukin-8 is a paracrine inducer of fibroblast growth factor 2, a stromal and epithelial growth factor in benign prostatic hyperplasia. Am J Pathol. 2001;159(1):139–147. doi: 10.1016/S0002-9440(10)61681-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim YW, Kern HF, Mullins TD, Koriwchak MJ, Metzgar RS. Characterization of clones of a human pancreatic adenocarcinoma cell line representing different stages of differentiation. Pancreas. 1989;4(3):353–362. doi: 10.1097/00006676-198906000-00013. [DOI] [PubMed] [Google Scholar]
- 44.Egami H, Takiyama Y, Chaney WG, Cano M, Fujii H, Tomioka T, et al. Comparative studies on expression of tumor-associated antigens in human and induced pancreatic cancer in Syrian hamsters. Int J Pancreatol. 1990;7(1–3):91–100. doi: 10.1007/BF02924224. [DOI] [PubMed] [Google Scholar]
- 45.Batra SK, Metzgar RS, Hollingsworth MA. Isolation and characterization of a complementary DNA (PD-1) differentially expressed by human pancreatic ductal cell tumors. Cell Growth Differ. 1991;2(8):385–390. [PubMed] [Google Scholar]
- 46.Wang QJ, Knezetic JA, Schally AV, Pour PM, Adrian TE (1996) Bombesin may stimulate proliferation of human pancreatic cancer cells through an autocrine pathway. Int J Cancer 68(4):528–534. https://doi.org/10.1002/(SICI)1097-0215(19961115)68:4<528::AID-IJC20>3.0.CO;2-# [DOI] [PubMed]
- 47.Ding XZ, Fehsenfeld DM, Murphy LO, Permert J, Adrian TE. Physiological concentrations of insulin augment pancreatic cancer cell proliferation and glucose utilization by activating MAP kinase, PI3 kinase and enhancing GLUT-1 expression. Pancreas. 2000;21(3):310–320. doi: 10.1097/00006676-200010000-00014. [DOI] [PubMed] [Google Scholar]
- 48.Saxena S, Purohit A, Varney ML, Hayashi Y, Singh RK. Semaphorin-5A maintains epithelial phenotype of malignant pancreatic cancer cells. BMC Cancer. 2018;18(1):1283. doi: 10.1186/s12885-018-5204-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mizuguchi H, Utoguchi N, Mayumi T. Preparation of glial extracellular matrix: a novel method to analyze glial-endothelial cell interaction. Brain Res Protocol. 1997;1(4):339–343. doi: 10.1016/s1385-299x(97)00008-1. [DOI] [PubMed] [Google Scholar]
- 50.Le X, Shi Q, Wang B, Xiong Q, Qian C, Peng Z, et al. Molecular regulation of constitutive expression of interleukin-8 in human pancreatic adenocarcinoma. J Interf Cytokine Res. 2000;20(11):935–946. doi: 10.1089/10799900050198372. [DOI] [PubMed] [Google Scholar]
- 51.Takamori H, Oades ZG, Hoch RC, Burger M, Schraufstatter IU. Autocrine growth effect of IL-8 and GRO?? On a human pancreatic Cancer cell line, Capan-1. Pancreas. 2000;21(1):52–56. doi: 10.1097/00006676-200007000-00051. [DOI] [PubMed] [Google Scholar]
- 52.Frick VO, Rubie C, Wagner M, Graeber S, Grimm H, Kopp B, et al. Enhanced ENA-78 and IL-8 expression in patients with malignant pancreatic diseases. Pancreatology. 2008;8(4–5):488–497. doi: 10.1159/000151776. [DOI] [PubMed] [Google Scholar]
- 53.Strieter RM, Burdick MD, Mestas J, Gomperts B, Keane MP, Belperio JA. Cancer CXC chemokine networks and tumour angiogenesis. Eur J Cancer. 2006;42(6):768–778. doi: 10.1016/j.ejca.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 54.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6(237):237ra267–237ra267. doi: 10.1126/scitranslmed.3007974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan TS, Hsu CC, Pai VC, Liao WY, Huang SS, Tan KT, et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J Exp Med. 2016;213(13):2967–2988. doi: 10.1084/jem.20151665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD, Rowley DR. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin Cancer Res. 2002;8(9):2912–2923. [PubMed] [Google Scholar]
- 57.Ohuchida K, Mizumoto K, Murakami M, Qian LW, Sato N, Nagai E, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004;64(9):3215–3222. doi: 10.1158/0008-5472.can-03-2464. [DOI] [PubMed] [Google Scholar]
- 58.Muerkoster S, Wegehenkel K, Arlt A, Witt M, Sipos B, Kruse ML, et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 2004;64(4):1331–1337. doi: 10.1158/0008-5472.can-03-1860. [DOI] [PubMed] [Google Scholar]
- 59.Yamanaka Y, Friess H, Buchler M, Beger HG, Uchida E, Onda M, et al. Overexpression of acidic and basic fibroblast growth factors in human pancreatic cancer correlates with advanced tumor stage. Cancer Res. 1993;53(21):5289–5296. [PubMed] [Google Scholar]
- 60.Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R, et al. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res. 1997;57(5):963–969. [PubMed] [Google Scholar]
- 61.Feng S, Wang F, Matsubara A, Kan M, McKeehan WL. Fibroblast growth factor receptor 2 limits and receptor 1 accelerates tumorigenicity of prostate epithelial cells. Cancer Res. 1997;57(23):5369–5378. [PubMed] [Google Scholar]
- 62.Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271(25):15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 63.Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170(6):3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 64.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14(21):6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 65.Sanui T. DOCK2 regulates Rac activation and cytoskeletal reorganization through interaction with ELMO1. Blood. 2003;102(8):2948–2950. doi: 10.1182/blood-2003-01-0173. [DOI] [PubMed] [Google Scholar]
- 66.Grimsley CM, Kinchen JM, Tosello-Trampont A-C, Brugnera E, Haney LB, Lu M, et al. Dock180 and ELMO1 proteins cooperate to promote evolutionarily conserved Rac-dependent cell migration. J Biol Chem. 2003;279(7):6087–6097. doi: 10.1074/jbc.M307087200. [DOI] [PubMed] [Google Scholar]