

Group A Streptococcus (GAS) (Streptococcus pyogenes) is an important human pathogen associated with significant global morbidity and mortality for which there is no safe and efficacious vaccine. The T antigen, a protein that polymerizes to form the backbone of the GAS pilus structure, is a potential vaccine candidate.
KEYWORDS: T antigen, group A Streptococcus, tee type, vaccine
ABSTRACT
Group A Streptococcus (GAS) (Streptococcus pyogenes) is an important human pathogen associated with significant global morbidity and mortality for which there is no safe and efficacious vaccine. The T antigen, a protein that polymerizes to form the backbone of the GAS pilus structure, is a potential vaccine candidate. Previous surveys of the tee gene, which encodes the T antigen, have identified 21 different tee types and subtypes such that any T antigen-based vaccine must be multivalent and carefully designed to provide broad strain coverage. In this study, the crystal structures of three two-domain T antigens (T3.2, T13, and T18.1) were determined and found to have remarkable structural similarity to the previously reported T1 antigen, despite moderate overall sequence similarity. This has enabled reliable modeling of all major two-domain T antigens to reveal that T antigen sequence variation is distributed along the full length of the protein and shields a highly conserved core. Immunoassays performed with sera from immunized animals and commercial T-typing sera identified a significant cross-reactive antibody response between T18.1, T18.2, T3.2, and T13. The existence of shared epitopes between T antigens, combined with the remarkably conserved structure and high level of surface sequence divergence, has important implications for the design of multivalent T antigen-based vaccines.
INTRODUCTION
The Gram-positive bacterium group A Streptococcus (GAS) (Streptococcus pyogenes) is a major human pathogen and a significant cause of global morbidity and mortality (1). Disease ranges from superficial impetigo and pharyngitis to severe invasive disease and the poststreptococcal sequelae of acute rheumatic fever (ARF), rheumatic heart disease (RHD), and poststreptococcal glomerulonephritis (PSGN). The highest burden of GAS disease is due to RHD, with an estimated worldwide prevalence of 33.4 million and over 300,000 deaths each year (2). ARF is a major cause of acquired heart disease in low-income countries and in indigenous populations in certain high-income countries such as New Zealand and Australia (3, 4).
Historically, GAS was classified using two serological markers, known as the “M” and “T” Lancefield antigens. These antigens, originally described by Rebecca Lancefield in the 1950s, have subsequently been shown to correspond to the fibrillar M protein and the pilin T antigen, respectively (5, 6). The GAS M protein is a major virulence factor with amino acid heterogeneity at the N terminus. Its antigenic diversity formed the basis for M serotyping, later replaced by emm typing that involves sequencing the hypervariable region of the emm gene (7). Using this method, over 200 GAS emm types have been identified (8). The T antigen is the backbone protein (BP) of the GAS pilus, and tee typing, which involves sequencing the entire tee gene, can be used as a supplementary typing tool (9, 10). However, unlike emm typing and the M protein, the regions of the T antigen that correspond to antigen heterogeneity (and, thus, form the basis of tee typing) are poorly understood at a structural level.
The GAS pilus backbone consists of multiple copies of the T antigen, which polymerize to form the pilus fiber, and one or two accessory proteins (APs); AP1 is the adhesin at the pilus tip, and AP2 is found at the base of most pilus types and is a cell wall anchor. GAS pilin genes, together with genes encoding pilus assembly sortases, are found in a single operon within the FCT (fibronectin-binding, collagen-binding, T antigen) region (9, 11, 12). Overall, the tee gene shows less genetic variability than the emm gene: previous surveys of the tee gene in GAS strains have identified 18 major tee types, 3 of which can be further split into subtypes (13, 14). These studies, although of a relatively small scale (39 and 100 GAS strains, respectively), appear to have captured the vast majority of tee type diversity. As evidence, a recent whole-genome sequencing (WGS) study of over 1,400 invasive GAS isolates in the United States clustered GAS into 21 different tee types (15). Unfortunately, the annotation of tee types in this WGS study differed from those reported previously (13, 14), which highlights a need to harmonize tee typing nomenclature to avoid future confusion.
To date, the protein structures of two T antigens have been solved by X-ray crystallography, and both show a modular architecture of immunoglobulin (Ig)-like domains (16, 17). The T1 antigen (Spy0128) possesses two Ig-like domains, while the T6 antigen comprises three Ig-like domains. Each of the IgG-like domains of these two structures contains a core of conserved residues that facilitate the autocatalytic formation of an intramolecular isopeptide bond (16, 17). These bonds provide the pili with their characteristic protease resistance and tensile strength (18–21). Outside this core, there is a high degree of sequence variability. The structure of the T6 antigen revealed that the central Ig-like domains were largely concealed by highly divergent decorations in the form of a series of variant loops or extensions. The positioning of these hypervariant loops and extensions is conserved in other three-domain Gram-positive bacterial pili (17), suggesting that the generation of strain-specific antibody responses to three-domain T antigens may be driven by these variant loops. Indeed, the largest of these extensions in T6 was shown to be immunogenic in individuals with GAS-associated ARF (17).
Previous surveys of the tee gene and encoded T antigens suggest that the vast majority of T antigens have two domains as per the T1 structure. Indeed, our previous phylogenetic study predicted that 16 of the 21 tee types and subtypes encode T antigens comprised of two domains (14). Furthermore, GAS strains expressing the two-domain T antigens are the most diverse in terms of the number of associated emm types and are associated with significant disease (9, 14). Unlike T6, the T1 antigen structure lacks large hypervariant extensions. With very little variation in the predicted size of two-domain T antigens, it is less clear which structural features are capable of generating type-specific immune responses. Moreover, T1 is evolutionarily distinct from all the other two-domain T antigens, being encoded by the FCT-2 operon (14). In contrast, the remaining 15 two-domain T antigens are encoded by FCT-3/4/7/8 operons, suggesting that T1 might not necessarily be structurally representative of all two-domain T antigens.
The aim of this study is to understand the variability of two-domain T antigens, which represent the most prevalent GAS tee types globally (13, 14), and determine the structural features that contribute to type-specific and cross-reactive antibody responses to these T antigens. Understanding the structural features that are the basis of a unique “tee type” will inform molecular epidemiological and vaccine studies for GAS. The pilus and T antigen are attractive GAS vaccine candidates, with previous small-animal studies showing that anti-AP1 antibodies inhibit bacterial adhesion while anti-T antigen antibodies can reduce adhesion and promote killing (5, 22). The intramolecular isopeptide bonds also provide exceptional protein stability, which is advantageous for vaccine development. Furthermore, compared with the >220 emm types identified, and the challenges associated with developing a type-specific M-protein-based vaccine that will provide global coverage, the limited diversity found within T antigens is an advantage for vaccine design (23). It has been predicted that a vaccine comprised of 18 T antigens would provide near-global coverage (14). Our study has used a combination of X-ray crystallography and homology modeling to show that despite a large range of sequence heterogeneity, two-domain T antigens exhibit remarkable structural conservation. This, in conjunction with specific cross-reactivity patterns observed between two-domain T antigens, will inform the rational design of T antigen-based vaccines.
RESULTS
Reactivity profile of T18 antisera.
We and others have previously shown that humans are capable of generating both type-specific and cross-reactive immune responses to T antigens (17, 24). However, information regarding the tee type of the infecting GAS strain was lacking in these previous studies, limiting the ability to understand how antigen diversity was driving these responses. In this study, we use the T18.1 antigen found in emm18, emm71, and emm217 strains as a model two-domain antigen to comprehensively investigate the nature of type-specific and cross-reactive T antigen antibody responses. This builds on a previous study that included just three recombinant two-domain T antigens (T1, T18.1, and T28.1), in which type-specific and weak cross-reactive immune responses were observed (22). FVB/n mice and New Zealand White (NZ White) rabbits were immunized with Lactococcus lactis carrying the complete T18.1 pilus and recombinant monomeric T18.1, respectively. Sera from these animals were screened by an enzyme-linked immunosorbent assay (ELISA) against a panel of 14 recombinant T antigens, covering the major tee types known to be circulating, in order to elucidate reactivity patterns (see Fig. S1 in the supplemental material) (14, 15).
As shown in Fig. 1A, sera from each of the immunized animals reacted strongly to T18.1 and the closely related T18.2 subtype. The sera from each of these immunized animals also exhibited strong reactivity to T3.2 and moderate reactivity with T13. Some sera also showed reactivity to T5, T11, and T28.1, although reactivity to these T antigens was not conserved in serum from all animals. Cross-reactive antibodies were also observed in commercially generated T3-typing sera, which reacted with T3.2, T18.1, and T18.2. Although T-typing sera are generally type specific, “T-typing cross-reactivity patterns” have previously been observed between FCT-3/4 T antigens, indicating that some T-typing sera bind to conserved cross-reactive epitopes on the surface of T antigens (13, 25). In contrast, the T1-typing serum reacted only with the antigenically distinct T1 antigen, with no observed cross-reactivity to other T antigens in the panel. This suggests that the T antigens that are more closely related to each other by sequence, including T3.2, T13, T18.1, and T18.2, share cross-reactive epitopes that are not present on the evolutionarily distinct T1 antigen.
FIG 1.

Reactivity of T18.1-immunized animal sera and T-typing sera to the T antigen panel measured by an ELISA. (A) The sera were titrated against the T antigen panel in a 5-fold dilution series beginning at 1:100. The endpoint titer of each serum against each T antigen was defined as the highest serum dilution above the value for the negative control (absorbance of preimmune rabbit serum plus 3 standard deviations). In the heat map, values for each serum sample were normalized to have the highest endpoint titer as 1.0 (shown in red), and positivity was defined as a normalized endpoint titer of >0.03, which equates to the signal of the negative control. (B) Binding of the affinity-purified cross-reactive antibodies to T3.2, T13, and T18.1. (i) T18.1-immunized rabbit serum passed over T18.1-coupled resin; (ii) T18.1-immunized rabbit serum passed over T3.2-coupled resin; (iii) T18.1-immunized rabbit serum passed over T13-coupled resin.
To further investigate cross-reactivity, antibodies from the T18.1-immunized rabbit were affinity purified using resin coupled with T18.1, T3.2, or T13. The purified antibodies were titrated, and their reactivity to T3.2, T13, and T18.1 was determined by an ELISA (Fig. 1B). The titration of the T18.1 purified antiserum (passed over T18.1-resin) confirmed that immunization with T18.1 evokes a strong type-specific response and a lesser, but significant, cross-reactive antibody response against T3.2 and T13. This indicates that the surface of T18.1 features distinct type-specific epitopes that are unique to T18.1 as well as cross-reactive epitopes that are found on T3.2 and T13. The titration curves for the T3.2 purified antibodies show overlapping reactivity with T18.1 but slightly reduced reactivity with T13. Similarly, the titration curves for the T13 purified antibodies show overlapping reactivity with T18.1 but slightly reduced activity with T3.2. This suggests differences in the specificity and/or avidity of the T3.2 and T13 cross-reactive antibodies induced by vaccination with T18.1, such that there is cross-reactivity between the three T antigens and additional cross-reactive epitopes that are shared between only two of the three T antigens. To determine whether the T antigen-specific antibodies bind to epitopes present on the surface of polymerized pili, flow cytometry was performed with L. lactis expressing full T1 and T18.1 pili. The antisera from animals vaccinated with T18.1, together with T3-typing sera and affinity-purified T18.1, T3.2, and T13 antisera, all resulted in a notable peak shift when incubated with L. lactis expressing T18.1. This confirms that these T antigen-specific serum antibodies bind to the pilus surface (Fig. S2).
Following the identification of this striking T3.2/T13/T18.1 cross-reactivity pattern, the structures of these three T antigens were solved and compared to the structure of T1 in order to explore the structural diversity of T antigens and investigate the features that contribute to the observed cross-reactivity pattern.
Structure determination.
The T antigen backbone pilins from S. pyogenes are expressed and exported to the extracellular environment as preproteins, which are subsequently processed into mature pilins with the removal of an N-terminal signal peptide and the cleavage of the variant C-terminal sortase recognition motif (QVPTG) by a specialized pilin sortase. All T antigen constructs used for structural analysis encompass the mature pilin, ending at a conserved aspartic acid residue directly before the sortase recognition motif (RD-TQVPTG).
T13 was crystallized in space group P212121, and the structure was solved by molecular replacement using the T1 (Spy0128) structure as a model (16, 26). The structure was refined using data to a 1.90-Å resolution (R = 20.5%; Rfree = 23.6) (see Table 1 for full details). As in the T1 structure, each T13 monomer consists of two Ig-like β-clasp domains, an N-terminal domain, and a C-terminal domain (Fig. 2). There are two molecules per asymmetric unit positioned antiparallel to each other. There is poor density for the first N-terminal residue of the mature pilin construct and the preceding two vector-derived residues (GAE) in each monomer. In addition, there are three external loops with either poor or uninterpretable electron density in each monomer. These loops (residues 45 to 48, 124 to 127, and 168 to 172) are positioned at the top of the N-terminal domain and at the base of the C-terminal domain and would sit at the interface between two successive pilin domains in the polymerized stalk of the pilus and very likely become ordered upon pilus assembly.
TABLE 1.
Data collection and refinement statisticsd
| Parameter | Value for antigen |
||
|---|---|---|---|
| T3.2 | T13 | T18.1 | |
| Data collection statistics | |||
| Wavelength (Å) | 0.95468 | 0.95468 | 0.95468 |
| No. of images | 360 | 360 | 360 |
| Oscillation angle (°) | 1.0 | 1.0 | 1.0 |
| Resolution range (Å) | 45.26–1.80 (1.84–1.80) | 48.03–1.90 (1.94–1.90) | 44.83–1.75 (1.78–1.75) |
| Total no. of observations | 186,142 (10,813) | 909,951 (57,678) | 189,630 (9,186) |
| No. of unique reflections | 47,406 (2,751) | 61,489 (3,877) | 51,303 (2,572) |
| Redundancy | 3.9 (3.9) | 14.8 (14.8) | 3.7 (3.6) |
| Space group | P1 | P212121 | P1 |
| Unit cell dimensions | |||
| Axial lengths (Å) | a = 32.05, b = 60.57, c = 72.79 | a = 41.77, b = 114.97, c = 128.71 | a = 40.59, b = 50.68, c = 67.92 |
| Angles (°) | α = 90.77, β = 91.77, γ = 104.1 | α = 90, β = 90, γ = 90 | α = 103.54, β = 93.53, γ = 90.03 |
| Completeness (%) | 96.4 (95.4) | 100 (100) | 96.7 (89.1) |
| Mean I/σ(I) | 19.7 (3.8) | 10.4 (1.2) | 9.4 (1.5) |
| Rmerge (%)b | 0.039 (0.296) | 0.256 (2.8) | 0.091 (0.654) |
| CC1/2a | 0.999 (0.937) | 0.997 (0.461) | 0.995 (0.636) |
| Refinement statistics | |||
| Resolution range (Å) | 24.48–1.80 (1.85–1.80) | 48.03–1.90 (1.94–1.90) | 40.54–1.75 (1.78–1.75) |
| Molecules per AU | 2 | 2 | 2 |
| Solvent content (%) | 45 | 61 | 40.16 |
| Rwork (%)c | 20.0 | 20.5 | 23.4 |
| Rfree (%)c | 25.0 | 23.6 | 27.7 |
| No. of protein atoms | 4,356 | 4,046 | 4,567 |
| No. of water molecules | 588 | 270 | 354 |
| RMSD from ideal geometry | |||
| Bonds (Å) | 0.008 | 0.009 | 0.010 |
| Angles (°) | 1.376 | 1.426 | 1.361 |
| Mean B-factor (Å2) | |||
| Protein | 25.1 | 29.1 | 17.3 |
| Water | 31.6 | 30.8 | 19.8 |
| Residues in the Ramachandran plot (MolProbity) | |||
| Ramachandran most favored (%) | 97.5 | 98.1 | 98.53 |
| Ramachandran outliers (%) | 0 | 0 | 0 |
CC1/2, correlation coefficient (47).
.
Rwork and , where Rfree was calculated over 5% of amplitudes that were chosen at random and not used in the refinement.
Numbers in parentheses indicate values for the outermost shell. AU, asymmetric unit.
FIG 2.

Conserved structure of S. pyogenes T antigens. The cartoon depicting the structures of four sequence-diverse T antigens shows a conserved, virtually identical tandem CnaB-type Ig-like domain configuration. The key residues involved in intramolecular isopeptide bond formation and the pilin lysine (K) are shown in stick mode. An adjacent topology diagram shows the N-terminal (blue) and C-terminal (red) domains. The isopeptide bond position in each domain is depicted as a horizontal black bar. N, N terminus; C, C terminus.
T3.2 was crystallized in space group P1 and solved by molecular replacement using the T13 structure as a model. The data were collected to a resolution of 1.6 Å but were extremely anisotropic and thus restricted to 1.8 Å during refinement (R = 20.0%; Rfree = 24.9) (Table 1). There were two antiparallel molecules in the asymmetric unit. Each two-domain monomer is fully modeled with only the first two N-terminal residues of the mature T antigen construct and two vector-derived residues (GAET) lacking interpretable electron density. T18.1 was crystallized in space group P1 and solved by molecular replacement using the T13 structure as a model. The structure was refined to a 1.80-Å resolution (R = 23.4%; Rfree = 27.7) (Table 1) with two antiparallel molecules per asymmetric unit. All residues except for an N-terminal loop (residues 124 to 130) have clearly interpretable electron density in each monomer (Fig. 2).
T antigen structure.
The T3.2, T13, and T18.1 proteins have a conserved modular structure typical of Gram-positive backbone pilins (BPs). They are folded into two tandem Ig-like domains arranged linearly to form an elongated structure ∼90 Å long and 20 to 30 Å wide. The domains are CnaB-type Ig-like folds similar to the reported two-domain BP structure from S. pyogenes: Spy0128 or T1 (Protein Data Bank [PDB] accession number 3B2M). The most striking aspect is the similarity of the four T antigen structures (Fig. 3A). An overlay of the four structures (T1, T3.2, T13, and T18.1) shows an almost identical Cα trace, with an overall root mean standard deviation (RMSD) of 1.34 Å over 261 aligned residues among the four structures (PDBeFold [http://www.ebi.ac.uk/msd-srv/ssm]). This is remarkable given the sequence diversity, with a pairwise sequence identity of only 34 to 58% between the structures. Cross-structure statistics for sequence identity and structure RSMD are shown in Table 2.
FIG 3.

Structural overlay of T antigens. (A) Ribbon diagram showing the almost identical Cα trace, with an overall RMSD of 1.34 Å over 261 aligned residues, among the four structures of T18.1 (green), T13 (cyan), T3.2 (blue), and T1 (magenta). The key residues involved in intramolecular isopeptide bond formation and the pilin lysine (K) are shown in stick mode. Views are at 0° and 90° rotations around the y axis. (B) Stereodiagram of the conserved intramolecular isopeptide bonds. The isopeptide bonds of both the N-terminal domain (top) and the C-terminal domain (bottom) are buried in the hydrophobic core of the β-clasp. For clarity, only T18.1 residues are numbered. The isopeptide bond forms between a buried lysine (K15) and asparagine (N153) in the N-terminal domain and between lysine (K164) and asparagine (N279) in the C-terminal domain. The conserved hydrophobic core surrounding the isopeptide bonds is largely composed of bulky aromatics. A strictly conserved buried glutamate (E102 and E237) is essential for isopeptide bond formation.
TABLE 2.
Cross-structure statisticsa
| T antigen | RMSD (% sequence identity) compared to: |
|||
|---|---|---|---|---|
| T18.1 | T3.2 | T13 | T1 | |
| T18.1 | 0.91 (57.9) | 1.39 (54.4) | 1.45 (34.1) | |
| T3.2 | 0.91 (57.9) | 1.15 (57.9) | 1.39 (37.5) | |
| T13 | 1.391 (54.4) | 1.15 (57.9) | 1.66 (36.0) | |
| T1 | 1.45 (34.1) | 1.39 (37.5) | 1.66 (36.0) | |
Shown are pairwise comparisons of root mean standard deviations (average distance in angstroms between the atoms of superimposed T antigens) and amino acid sequence identities of the four experimentally determined T antigen structures.
A distinctive feature of Gram-positive BPs is the presence of intramolecular isopeptide bonds (16, 27). In the CnaB fold, these covalent cross-links form between the first and last strands of each domain. First described in S. pyogenes BP T1 (Spy0128), these intramolecular cross-links have subsequently been found in many Gram-positive pilin domains and in some repeated “stalk” domains of other Gram-positive multidomain cell surface adhesins. In each T antigen structure, there is clearly defined continuous electron density linking the side chains of a buried lysine and a buried asparagine residue in both CnaB domains. As in other pilins, the isopeptide bonds are buried in the hydrophobic core of the β-clasp, stacked against aromatic residues (Fig. 3B). All the T antigen isopeptide bonds have a cis configuration, allowing the NH and O moieties of the isopeptide bond to form a bidentate hydrogen bond interaction with a buried glutamate carboxyl group. The environment surrounding the isopeptide bond is stringently conserved. The only variations are conservative substitutions at T18.1 residues Y32 and F17 in the N-terminal domain and a V277F change in the T1 C-terminal domain (Fig. 3B).
Another highly conserved structural feature is an Ω-loop that interrupts β-strand H of the N-terminal domain (Fig. 2). This structure positions a strictly conserved pilin lysine at the base of a cleft that accommodates the sortase motif of the preceding domain. During pilus maturation, a specific sortase cleaves the variant QVPTG sortase motif between threonine and glycine, reforming an amide bond between C-terminal threonine carboxylate generated by sortase cleavage and the ε-amino group of the pilin lysine from the next pilin subunit (28–31). This transpeptidase reaction covalently links successive T antigens to form the homopolymeric shaft of the pilus.
Fimbria-like packing.
The previously reported T1 crystal structure mimics the putative natural fimbria assembly, with successive molecules stacking head to tail and the C-terminal strand of the preceding pilin 16 Å from the ε-amino group of the pilin lysine (16). This corresponds to the predicted distance for the FEVPT peptide that occupies the cleft in the mature pilus. Of the three T antigen structures determined in this study, only the T18.1 structure formed pilus-like fimbria structures in its crystal packing, with the N terminus of each monomer packed against the C terminus of each adjacent pilin. However, examination of the interface between successive T18.1 domains revealed an unnatural positioning of the pilin domains that would prevent the alignment of the C-terminal sortase motif with the N-terminal cleft in the proceeding domain. Thus, the crystal packing for T18.1 was deemed a crystallization artifact. To examine how the T18.1, T3.2, and T13 antigens would assemble, the T1 crystal arrangement was utilized. An overlay of each of the three pilin structures on the T1 packing suggests that all pilins assemble in the same way, with the base of the C-terminal domain forming a wedge that slots into a shallow V-shaped groove at the top of the N-terminal domain of the next pilin (Fig. 4).
FIG 4.
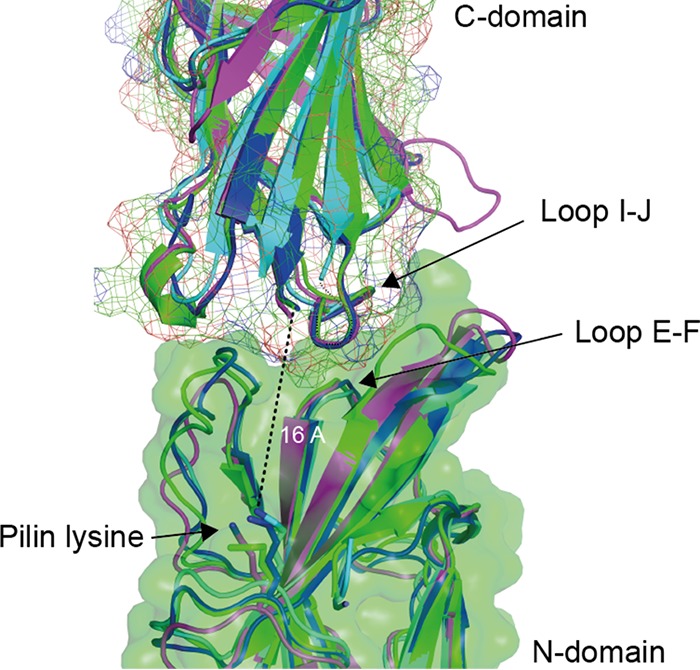
Conserved pilus packing arrangement. The similarity between the pilin structures suggests that all pilins assemble in the same manner. Pilin structures were overlaid on the T1 (Spy0128) crystal packing, which positions the C-terminal strand of each pilin at the top of the sortase-binding cleft in the N-terminal domain of the next pilin, 16 Å from the pilin lysine. A structurally conserved loop in the C-terminal domain (loop I-J) forms a wedge that packs on top of a conserved loop (loop E-F) at the base of a cleft at the top of the N-terminal domain.
Antigenic variation.
While the core of the pilin is remarkably conserved, the surface that is presented to the immune system is highly divergent between pilins. A comparison of the four T antigen structures reveals that the surface variation is not restricted to any single region, but rather, the sequence drift is uniform along the whole pilin (Fig. 5). An amino acid sequence alignment of these four T antigens also reveals regions of surface-accessible, conserved sequence along the length of the T antigen (Fig. S3). As expected, T3.2, T13, and T18.1 feature patches of similar or identical residues that are not shared with T1 and are likely locations of cross-reactive epitopes on the otherwise sequence-diverse surface. The overall sequence identity between the pilins ranges from 58% between T18.1 and T3.2 down to 34% between T18.1 and T1. However, this identity is reduced in an analysis of surface-exposed residues to 41% between T18.1 and T3.2 and 23% between T18.1 and T1. If only residues that are fully solvent exposed are compared, then the identity is reduced even further to 34% between T18.1 and T3.2 and 20% between T18.1 and T1. Surface- and solvent-exposed residues are believed to be predisposed to substitution as a means of escaping immune recognition (17, 32, 33).
FIG 5.

Phylogenetic tree showing surface divergence between pilins from each major clade. An alignment of both experimentally solved and modeled structures reveals that pilins from each clade present extremely diverse surfaces to the immune system. The surface of each pilin was compared with that of T18.1, with residues identical to those of T18.1 shown in green and residues that differ from those of T18.1 shown in blue. The pilin lysine is in red. Each pilin is viewed at 0° and 90° rotations around the y axis.
To determine if the high surface divergence seen in the four crystal structures is indicative of all T antigens, we modeled a representative T antigen from each major clade identified in our previous phylogenetic study (14). The highly conserved structural core of T antigens revealed in the present study means that all two-domain T antigens can be reliably predicted using homology modeling. We used PHYRE2 (Protein Homology/Analogy Recognition Engine V 2.0) homology modeling with the T18.1 structure as a template. All models had a probability score of 100%, and manual inspection revealed no major inconsistencies. Importantly, key residues involved in isopeptide bond formation were positioned correctly, requiring only minor editing to obtain the correct geometry. The conserved residues surrounding the isopeptide bonds were also correctly positioned, giving confidence that the homology-modeled structures are accurate representations of the T antigens. Consistent with the experimentally solved structures, the modeled structures revealed high sequence identity (74 to 86%) for buried residues but high sequence variability at the surface. All modeled structures display variation of surface-exposed residues over the entire pilin surface. Interestingly, even T antigens from closely related clades have high surface variation. Of the T antigens analyzed, only T18.1 and T18.2 share a high surface identity (82%), with only 20 to 34% identity between T18.1 and all remaining T types (Table 3).
TABLE 3.
Surface identity shared between T18.1 and representative T antigens from all major clades
| T18.1 residues | % identity to residues of: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| T18.2 | T28.2 | T3.2 | T5 | T12 | T13 | T10 | T11 | T1 | |
| Buried | 94 | 86 | 87 | 80 | 79 | 76 | 86 | 74 | 57 |
| Surface | 84 | 42 | 41 | 36 | 43 | 43 | 29 | 35 | 23 |
| Fully solvent exposed | 82 | 33 | 34 | 27 | 28 | 31 | 23 | 28 | 20 |
DISCUSSION
This study describes the crystal structures for three two-domain T antigens (T18.1, T3.2, and T13). Analysis of these T antigen structures, and comparison with the previously reported T1 (Spy0128) structure (16), revealed a highly conserved core, coated in variant surface residues distributed over the length of the T antigens. This contrasts with the M protein, where variability is primarily located at the N terminus of the protein (34). It follows that emm typing involves sequencing the region of the emm gene that encodes the hypervariable N terminus, while tee typing requires sequencing the entire tee gene (13–15). Furthermore, unlike the Lancefield M-typing sera that react with the hypervariable N-terminal region in an M-type-specific manner, the Lancefield T-typing sera must contain antibodies that bind to type-specific regions located along the length of T antigens.
The distribution of surface variation along the length of T antigens of different T types has implications for vaccine design. While multivalent M-protein vaccines have been designed based on the 50 amino acids that comprise the N-terminal hypervariable region of the M protein (35), the same approach cannot be taken for T antigens. Both the structures and modeling presented in this study clearly show that there is no dominant region of “type-specific sequence” located on the T antigen structures. This makes it difficult to identify which segments of a T antigen should be spliced together in a multivalent manner to ensure representation of each major T type in a candidate vaccine. However, given that a T antigen vaccine is predicted to need to contain only 18 T types for near-global coverage, the conserved modular structure of T antigens revealed by this, and the previously reported T1 and T6 structures (16, 17), it may be possible to design and synthesize multivalent vaccines based on entire T antigens or T antigen domains. Not only would such an approach ensure that all potential protective epitopes are represented, but the resulting vaccine candidate would harbor intramolecular isopeptide bonds, providing increased stability.
Sera from immunized animals and T-typing sera revealed that T18.1, T3.2, and T13 harbor cross-reactive epitopes in this study. A comparison of the surface-exposed residues of these three T antigens revealed multiple possible locations for the cross-reactive epitopes that were unable to be further defined due to the use of polyclonal sera. Nevertheless, a previous study also observed cross-reactivity between Lancefield T-typing sera and distinct T antigens (13), and it is possible that a systematic investigation of T antigen-induced cross-reactivity patterns may lead to a reduced number of antigens required to achieve global coverage.
In summary, the determination of three new two-domain T antigen structures has enabled reliable modeling of all major two-domain T antigens. This has revealed that T antigen sequence variation is distributed along the length of the protein and shields a highly conserved core. Thus, in order to capture and detect this variation, tee typing involves sequencing the entire tee gene. The distribution of sequence variation and observed cross-reactive antibody patterns between distinct T antigens have important implications for vaccine design.
MATERIALS AND METHODS
Cloning, expression, and purification of T antigens.
The tee genes comprising the extracellular region of the T antigens were PCR amplified (PrimeStar; Clontech) from genomic DNA of S. pyogenes strains using gene-specific primers (see Table S1 in the supplemental material). The amplified fragments were digested with the appropriate restriction enzymes and cloned into an Escherichia coli expression vector, and the sequence confirmed by Sanger sequencing (Table S1). For crystallization experiments, T antigens were cloned into pProExHTa (Invitrogen) or MBP-pProExHTa (36).
For recombinant protein expression, E. coli BL21(λDE3) cells were transformed with tee plasmids and grown in either 2× yeast-tryptone (YT) or Luria-Bertani (LB) broth supplemented with 100 μg/ml ampicillin at 37°C until the optical density at 600 nm (OD600) reached 0.6. The cultures were induced with 0.3 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) at 18°C for 16 h, and the cells were harvested by centrifugation. Cell pellets were resuspended in lysis buffer (50 mM Tris-Cl [pH 8.0], 300 mM NaCl, 10 mM imidazole) supplemented with complete protease inhibitor cocktail minitablets (EDTA free; Roche) and stored at −20°C. Cells were lysed using a cell disruptor (Constant Cell Disruption Systems) at 124 kPa. Insoluble matter was sedimented by centrifugation (30,000 × g at 4°C for 30 min), and the soluble phase was loaded onto a HiTrap chelating 5-ml column (GE Healthcare). Bound protein was washed with wash buffer (50 mM Tris-Cl [pH 8.0], 150 mM NaCl, 20 mM imidazole) and eluted in a gradient with elution buffer (50 mM Tris-Cl [pH 8.0], 150 mM NaCl, 500 mM imidazole). The His6 affinity tag was cleaved from the recombinant protein with a 1:100 ratio of recombinant tobacco etch virus (rTEV) protease to His6 and concurrently dialyzed against a solution containing 10 mM Tris-Cl (pH 8.0), 100 mM NaCl, and 1 mM β-mercaptoethanol at 4°C for 16 h. Recombinant T antigen was separated from the rTEV-His6 protease and uncleaved protein by immobilized-metal affinity chromatography (IMAC). The unbound protein containing recombinant T antigen was concentrated using a 10-kDa-molecular-weight-cutoff (MWCO) protein concentrator (VivaScience) and purified by size exclusion chromatography on a Superdex S200 10/300 column (GE Healthcare) in crystallization buffer (10 mM Tris-Cl [pH 8.0], 100 mM NaCl). All purified recombinant T antigens eluted in a single peak that corresponds to a monomer of approximately 30 kDa and was approximately 99% pure, as indicated by SDS-PAGE. T1 antigen was purified as previously described (16).
Preparation and characterization of T18 antisera.
Antisera were generated and/or obtained from three different sources for this study. To generate polyclonal antiserum to T18.1, five FVB/n mice were intranasally immunized on days 0, 14, and 28 with 1 × 108 CFU/mouse of Lactococcus lactis constitutively expressing the T18.1 pilus operon, as previously described (22). Serum was collected on day 52. A New Zealand White rabbit was immunized subcutaneously with 100 μg of recombinant T18.1 (rT18.1) with incomplete Freund’s adjuvant (1:1). The rabbit received three identical boosts on days 14, 28, and 42. Preimmune serum was collected at day 0, and immune serum was collected on day 56. All animal vaccinations were conducted using procedures approved by the University of Auckland animal ethics committee. Finally, commercial T-typing antisera (rabbit) were obtained from Denka Seiken (Tokyo, Japan).
T antigen-specific sera were affinity purified from the serum obtained from the rabbit immunized with rT18.1. Affinity columns for three T antigens (T18.1, T3.2, and T13) were generated by covalently coupling 150 μg of the recombinant T antigen to resin using a MicroLink protein-coupling kit (Thermo Fisher Scientific). A 300-μl sample of serum from the rT18.1-immunized rabbit was incubated with the resin for 2 h at room temperature to allow antibody binding. The resin was then washed three times with coupling buffer (0.1 M sodium phosphate, 0.15 M NaCl [pH 7.2]; Thermo Fisher Scientific) to remove unbound antibodies. Bound antibody was eluted using elution buffer (pH 2.8; Thermo Fisher Scientific), and the pH of the eluted solution was immediately neutralized with 1 M Tris buffer (pH 9). The concentration of antibody in each eluent was determined using a NanoDrop 2000 instrument (Thermo Fisher Scientific).
For enzyme-linked immunosorbent assays (ELISAs), Nunc-immuno Maxisorb plates (Thermo Fisher Scientific) were coated with recombinant T antigen (5 μg/ml) in phosphate-buffered saline (PBS) (pH 7.2) and blocked with 5% skim milk in PBS-T (PBS supplemented with 0.1% Tween 20). Animal sera or affinity-purified antibodies were serially diluted in 5% skim milk in PBS-T and incubated at room temperature for 1 h. The plates were washed with PBS-T and incubated with horseradish peroxidase-conjugated anti-mouse (GE Healthcare) or anti-rabbit (Abcam) antibodies for detection. The endpoint titer was defined as the highest serum dilution above the control (absorbance of preimmune rabbit serum plus 3 times the standard deviation). Graphs and heat maps were prepared using GraphPad Prism (version 7c).
Flow cytometry.
L. lactis expressing T1- or T18.1-containing pili were grown in GM17 medium for 16 h at 30°C. Cells were harvested by centrifugation at 5,000 × g for 5 min and resuspended in fluorescence-activated cell sorter (FACS) blocking buffer (PBS–3% fetal bovine serum [FBS]–5 mM EDTA) at an OD600 of 0.4. The cell suspension was sonicated in a water bath for 2 min and then incubated on ice for 30 min. The blocked cells were harvested by centrifugation and resuspended in FACS buffer (PBS–1% FBS–5 mM EDTA) at an OD600 of 0.4. Aliquots of cells were harvested by centrifugation, and the supernatant was removed. The cells were resuspended in FACS buffer containing animal serum (diluted 1:100) or affinity-purified antibodies (250 nM) and incubated at 37°C for 30 min. The cells were washed with FACS buffer and harvested by centrifugation. The cells were stained with anti-rabbit IgG(H+L)-Alexa Fluor 647 or anti-mouse IgG(H+L)-Alexa Fluor 647 diluted 1:1,000 in FACS buffer for 30 min on ice. The cells were washed with FACS buffer, resuspended in 500 μl FACS buffer, and transferred to FACS tubes. Binding was analyzed by flow cytometry using an LSR II flow cytometer (BD).
Crystallization.
Vapor diffusion crystallization trials were carried out at 18°C using an Oryx4 crystallization robot (Douglas Instruments) and a locally compiled crystallization screen (37). Initial crystals (rT3.2, rT13, and rT18.1) were grown in a 0.1-μl sitting-drop format and subsequently optimized in a hanging-drop vapor diffusion format. The crystals of T13 used for X-ray data collection grew by mixing a 1-μl protein solution (17 mg/ml in 10 mM Tris-Cl [pH 8.0]–100 mM NaCl) with 1 μl precipitant (20% [wt/vol] polyethylene glycol 6000 [PEG 6000], 0.2 M morpholinepropanesulfonic acid [MOPS] [pH 7.3]). Crystals of rT3.2 were grown by mixing a 1-μl protein solution (40 mg/ml in 10 mM Tris-Cl [pH 8.0]–100 mM NaCl) with 1 μl precipitant (0.2 M NH4H2PO4, 20% [wt/vol] PEG 3350), and crystals of rT18.1 were grown by mixing a 1-μl protein solution (140 mg/ml in 10 mM Tris-Cl [pH 8.0]–100 mM NaCl) with 1 μl precipitant [10% (wt/vol) PEG 20000, 20% (vol/vol) poly(ethylene glycol) methyl ether 550, 0.02 M 1,6-hexanediol, 0.02 M 1-butanol, 0.02 M 1,2-propanediol, 0.02 M 2-propanol, 0.02 M 1,4-butanediol, 0.02 M 1,3-propanediol, 0.1 M Bicine-Tris base (pH 8.5)]. All crystals were grown at 18°C and reached the optimum size within 4 days.
Data collection and structure determination.
Crystals of T antigens were flash-cooled in liquid nitrogen with a cryoprotectant consisting of the same precipitant in which the crystals were grown, with the addition of 20% glycerol. X-ray diffraction data were recorded at the Australian Synchrotron on the MX1 beamline (ADSC Quantum 210r detector). All data sets were integrated using XDS (38), reindexed using POINTLESS (39), and scaled using SCALA (39). The structure of T13 was determined by molecular replacement using the T1 structure (PDB accession number 3B2M) as a model. The T13 model was subsequently used in the molecular placement of T3.2 and T18.1. The T antigen structures were initially subjected to a round of autobuilding with ArpWarp (40) and subsequently refined using iterative cycles of manual building in COOT (41) and refinement with Buster (42) and/or REFMAC (43). The quality of each T antigen model was inspected using the program MOLPROBITY (44). Data collection and refinement statistics are shown in Table 1. All figures were generated using PyMOL.
Calculation of surface-exposed residues.
Any T antigens that were not experimentally solved were first modeled using PHYRE2, using the T18.1 structure as the template (45). The solvent-accessible surface areas and atomic solvation energies of the T antigens were calculated with GETAREA (46). With GETAREA, each residue in a structure is assigned as being buried, surface exposed, or fully solvent exposed. All structures were structurally aligned with T18.1, and residues in each of the three groups (buried, surface exposed, or fully solvent exposed) are presented as percent identities with T18.1 residues.
Accession number(s).
Atom coordinates and structure factors have been deposited in the worldwide Protein Data Bank (wwPDB) with PDB accession numbers 6BBW for T3.2, 6BBT for T13, and 6N0A for T18.1.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the Maurice Wilkins Centre and the Health Research Council of New Zealand. J.M.R. received doctoral scholarship funding from the University of Auckland and the Maurice Wilkins Centre, and J.M.L. is a New Zealand Heart Foundation Research Fellow.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00205-19.
REFERENCES
- 1.Carapetis JR, McDonald M, Wilson NJ. 2005. Acute rheumatic fever. Lancet 366:155–168. doi: 10.1016/S0140-6736(05)66874-2. [DOI] [PubMed] [Google Scholar]
- 2.Watkins DA, Johnson CO, Colquhoun SM, Karthikeyan G, Beaton A, Bukhman G, Forouzanfar MH, Longenecker CT, Mayosi BM, Mensah GA, Nascimento BR, Ribeiro ALP, Sable CA, Steer AC, Naghavi M, Mokdad AH, Murray CJL, Vos T, Carapetis JR, Roth GA. 2017. Global, regional, and national burden of rheumatic heart disease, 1990-2015. N Engl J Med 377:713–722. doi: 10.1056/NEJMoa1603693. [DOI] [PubMed] [Google Scholar]
- 3.Noonan S, Zurynski YA, Currie BJ, McDonald M, Wheaton G, Nissen M, Curtis N, Isaacs D, Richmond P, Ramsay JM, Elliott EJ, Carapetis JR. 2013. A national prospective surveillance study of acute rheumatic fever in Australian children. Pediatr Infect Dis J 32:e26–e32. doi: 10.1097/INF.0b013e31826faeb3. [DOI] [PubMed] [Google Scholar]
- 4.Gurney JK, Stanley J, Baker MG, Wilson NJ, Sarfati D. 2016. Estimating the risk of acute rheumatic fever in New Zealand by age, ethnicity and deprivation. Epidemiol Infect 144:3058–3067. doi: 10.1017/S0950268816001291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mora M, Bensi G, Capo S, Falugi F, Zingaretti C, Manetti AG, Maggi T, Taddei AR, Grandi G, Telford JL. 2005. Group A Streptococcus produce pilus-like structures containing protective antigens and Lancefield T antigens. Proc Natl Acad Sci U S A 102:15641–15646. doi: 10.1073/pnas.0507808102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischetti VA. 1989. Streptococcal M protein: molecular design and biological behavior. Clin Microbiol Rev 2:285–314. doi: 10.1128/CMR.2.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beall B, Facklam R, Thompson T. 1996. Sequencing emm-specific PCR products for routine and accurate typing of group A streptococci. J Clin Microbiol 34:953–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. 2009. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect Dis 9:611–616. doi: 10.1016/S1473-3099(09)70178-1. [DOI] [PubMed] [Google Scholar]
- 9.Koller T, Manetti AGO, Kreikemeyer B, Lembke C, Margarit I, Grandi G, Podbielski A. 2010. Typing of the pilus-protein-encoding FCT region and biofilm formation as novel parameters in epidemiological investigations of Streptococcus pyogenes isolates from various infection sites. J Med Microbiol 59:442–452. doi: 10.1099/jmm.0.013581-0. [DOI] [PubMed] [Google Scholar]
- 10.Luca-Harari B, Darenberg J, Neal S, Siljander T, Strakova L, Tanna A, Creti R, Ekelund K, Koliou M, Tassios PT, van der Linden M, Straut M, Vuopio-Varkila J, Bouvet A, Efstratiou A, Schalen C, Henriques-Normark B, Strep-EURO Study Group, Jasir A. 2009. Clinical and microbiological characteristics of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol 47:1155–1165. doi: 10.1128/JCM.02155-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kratovac Z, Manoharan A, Luo F, Lizano S, Bessen DE. 2007. Population genetics and linkage analysis of loci within the FCT region of Streptococcus pyogenes. J Bacteriol 189:1299–1310. doi: 10.1128/JB.01301-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kreikemeyer B, Gamez G, Margarit I, Giard JC, Hammerschmidt S, Hartke A, Podbielski A. 2011. Genomic organization, structure, regulation and pathogenic role of pilus constituents in major pathogenic streptococci and enterococci. Int J Med Microbiol 301:240–251. doi: 10.1016/j.ijmm.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Falugi F, Zingaretti C, Pinto V, Mariani M, Amodeo L, Manetti AGO, Capo S, Musser JM, Orefici G, Margarit I, Telford JL, Grandi G, Mora M. 2008. Sequence variation in group A Streptococcus pili and association of pilus backbone types with Lancefield T serotypes. J Infect Dis 198:1834–1841. doi: 10.1086/593176. [DOI] [PubMed] [Google Scholar]
- 14.Steemson JD, Moreland NJ, Williamson D, Morgan J, Carter PE, Proft T. 2014. Survey of the bp/tee genes from clinical group A streptococcus isolates in New Zealand—implications for vaccine development. J Med Microbiol 63:1670–1678. doi: 10.1099/jmm.0.080804-0. [DOI] [PubMed] [Google Scholar]
- 15.Chochua S, Metcalf BJ, Li Z, Rivers J, Mathis S, Jackson D, Gertz RE Jr, Srinivasan V, Lynfield R, Van Beneden C, McGee L, Beall B. 2017. Population and whole genome sequence based characterization of invasive group A streptococci recovered in the United States during 2015. mBio 8:e01422-17. doi: 10.1128/mBio.01422-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang HJ, Coulibaly F, Clow F, Proft T, Baker EN. 2007. Stabilizing isopeptide bonds revealed in Gram-positive bacterial pilus structure. Science 318:1625–1628. doi: 10.1126/science.1145806. [DOI] [PubMed] [Google Scholar]
- 17.Young PG, Moreland NJ, Loh JM, Bell A, Atatoa Carr P, Proft T, Baker EN. 2014. Structural conservation, variability, and immunogenicity of the T6 backbone pilin of serotype M6 Streptococcus pyogenes. Infect Immun 82:2949–2957. doi: 10.1128/IAI.01706-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang HJ, Baker EN. 2009. Intramolecular isopeptide bonds give thermodynamic and proteolytic stability to the major pilin protein of Streptococcus pyogenes. J Biol Chem 284:20729–20737. doi: 10.1074/jbc.M109.014514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alegre-Cebollada J, Badilla CL, Fernández JM. 2010. Isopeptide bonds block the mechanical extension of pili in pathogenic Streptococcus pyogenes. J Biol Chem 285:11235–11242. doi: 10.1074/jbc.M110.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang B, Xiao S, Edwards SA, Grater F. 2013. Isopeptide bonds mechanically stabilize Spy0128 in bacterial pili. Biophys J 104:2051–2057. doi: 10.1016/j.bpj.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker EN, Squire CJ, Young PG. 2015. Self-generated covalent cross-links in the cell-surface adhesins of Gram-positive bacteria. Biochem Soc Trans 43:787–794. doi: 10.1042/BST20150066. [DOI] [PubMed] [Google Scholar]
- 22.Loh JMS, Lorenz N, Tsai CJ, Khemlani AHJ, Proft T. 2017. Mucosal vaccination with pili from group A Streptococcus expressed on Lactococcus lactis generates protective immune responses. Sci Rep 7:7174. doi: 10.1038/s41598-017-07602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raynes JM, Young PG, Proft T, Williamson DA, Baker EN, Moreland NJ. 2018. Protein adhesins as vaccine antigens for group A Streptococcus. Pathog Dis 76:fty016. doi: 10.1093/femspd/fty016. [DOI] [PubMed] [Google Scholar]
- 24.Manetti AG, Zingaretti C, Falugi F, Capo S, Bombaci M, Bagnoli F, Gambellini G, Bensi G, Mora M, Edwards AM, Musser JM, Graviss EA, Telford JL, Grandi G, Margarit I. 2007. Streptococcus pyogenes pili promote pharyngeal cell adhesion and biofilm formation. Mol Microbiol 64:968–983. doi: 10.1111/j.1365-2958.2007.05704.x. [DOI] [PubMed] [Google Scholar]
- 25.Johnson DR, Kaplan EL, VanGheem A, Facklam RR, Beall B. 2006. Characterization of group A streptococci (Streptococcus pyogenes): correlation of M-protein and emm-gene type with T-protein agglutination pattern and serum opacity factor. J Med Microbiol 55:157–164. doi: 10.1099/jmm.0.46224-0. [DOI] [PubMed] [Google Scholar]
- 26.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang HJ, Baker EN. 2012. Structure and assembly of Gram-positive bacterial pili: unique covalent polymers. Curr Opin Struct Biol 22:200–207. doi: 10.1016/j.sbi.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Ton-That H, Schneewind O. 2003. Assembly of pili on the surface of Corynebacterium diphtheriae. Mol Microbiol 50:1429–1438. doi: 10.1046/j.1365-2958.2003.03782.x. [DOI] [PubMed] [Google Scholar]
- 29.Ton-That H, Schneewind O. 2004. Assembly of pili in Gram-positive bacteria. Trends Microbiol 12:228–234. doi: 10.1016/j.tim.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Kang HJ, Middleditch M, Proft T, Baker EN. 2009. Isopeptide bonds in bacterial pili and their characterization by X-ray crystallography and mass spectrometry. Biopolymers 91:1126–1134. doi: 10.1002/bip.21170. [DOI] [PubMed] [Google Scholar]
- 31.Budzik JM, Marraffini LA, Souda P, Whitelegge JP, Faull KF, Schneewind O. 2008. Amide bonds assemble pili on the surface of bacilli. Proc Natl Acad Sci U S A 105:10215–10220. doi: 10.1073/pnas.0803565105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deitsch KW, Lukehart SA, Stringer JR. 2009. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat Rev Microbiol 7:493–503. doi: 10.1038/nrmicro2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ernst JD. 2017. Antigenic variation and immune escape in the MTBC. Adv Exp Med Biol 1019:171–190. doi: 10.1007/978-3-319-64371-7_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanderson-Smith M, De Oliveira DM, Guglielmini J, McMillan DJ, Vu T, Holien JK, Henningham A, Steer AC, Bessen DE, Dale JB, Curtis N, Beall BW, Walker MJ, Parker MW, Carapetis JR, Van Melderen L, Sriprakash KS, Smeesters PR, M Protein Study Group. 2014. A systematic and functional classification of Streptococcus pyogenes that serves as a new tool for molecular typing and vaccine development. J Infect Dis 210:1325–1338. doi: 10.1093/infdis/jiu260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dale JB, Fischetti VA, Carapetis JR, Steer AC, Sow S, Kumar R, Mayosi BM, Rubin FA, Mulholland K, Hombach JM, Schodel F, Henao-Restrepo AM. 2013. Group A streptococcal vaccines: paving a path for accelerated development. Vaccine 31:B216–B222. doi: 10.1016/j.vaccine.2012.09.045. [DOI] [PubMed] [Google Scholar]
- 36.Ting YT, Harris PW, Batot G, Brimble MA, Baker EN, Young PG. 2016. Peptide binding to a bacterial signal peptidase visualized by peptide tethering and carrier-driven crystallization. IUCrJ 3:10–19. doi: 10.1107/S2052252515019971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moreland N, Ashton R, Baker HM, Ivanovic I, Patterson S, Arcus VL, Baker EN, Lott JS. 2005. A flexible and economical medium-throughput strategy for protein production and crystallization. Acta Crystallogr D Biol Crystallogr 61:1378–1385. doi: 10.1107/S0907444905023590. [DOI] [PubMed] [Google Scholar]
- 38.Kabsch W. 2010. Xds. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 40.Langer G, Cohen SX, Lamzin VS, Perrakis A. 2008. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc 3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G. 2012. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. 2015. The Phyre2 Web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fraczkiewicz R, Braun W. 1998. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem 19:319–333. doi:. [DOI] [Google Scholar]
- 47.Karplus PA, Diederichs K. 2012. Linking crystallographic model and data quality. Science 336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.