

Abstract
Pain is a multidimensional experience and negative affect, or how much the pain is “bothersome”, significantly impacts the sufferers' quality of life. It is well established that the κ opioid system contributes to depressive and dysphoric states, but whether this system contributes to the negative affect precipitated by the occurrence of chronic pain remains tenuous. Using a model of persistent pain, we show by quantitative real-time-PCR, florescence in situ hybridization, Western blotting and GTPgS autoradiography an upregulation of expression and the function of κ opioid receptors (KORs) and its endogenous ligand dynorphin in the mesolimbic circuitry in animals with chronic pain compared with surgical controls. Using in vivo microdialysis and microinjection of drugs into the mesolimbic dopamine system, we demonstrate that inhibiting KORs reinstates evoked dopamine release and reward-related behaviors in chronic pain animals. Chronic pain enhanced KOR agonist-induced place aversion in a sex-dependent manner. Using various place preference paradigms, we show that activation of KORs drives pain aversive states in male but not female mice. However, KOR antagonist treatment was effective in alleviating anxiogenic and depressive affective-like behaviors in both sexes. Finally, ablation of KORs from dopamine neurons using AAV-TH-cre in KORloxP mice prevented pain-induced aversive states as measured by place aversion assays. Our results strongly support the use of KOR antagonists as therapeutic adjuvants to alleviate the emotional, tonic-aversive component of chronic pain, which is argued to be the most significant component of the pain experience that impacts patients' quality of life.
SIGNIFICANCE STATEMENT We show that KORs are sufficient to drive the tonic-aversive component of chronic pain; the emotional component of pain that is argued to significantly impact a patient's quality of life. The impact of our study is broadly relevant to affective disorders associated with disruption of reward circuitry and thus likely contributes to many of the devastating sequelae of chronic pain, including the poor response to treatment of many patients, debilitating affective disorders (other disorders including anxiety and depression that demonstrate high comorbidity with chronic pain) and substance abuse. Indeed, coexisting psychopathology increases pain intensity, pain-related disability and effectiveness of treatments (Jamison and Edwards, 2013).
Keywords: aversion, chronic pain, dopamine, emotion, negative affect, opiate
Introduction
Systems involved in pain processing interact extensively with other affective and motivational systems (Elman et al., 2013). This is most evident in the epidemiological evidence that shows that chronic pain is second only to bipolar disorder as a major cause of suicide among all medical illnesses (Asmundson and Katz, 2009; Elman et al., 2013). Risk factors for suicide in chronic pain patients include pain severity, pain type, catastrophizing, poor sleep, perception of disability, presence of comorbid depression, and access to analgesics (Hassett et al., 2014). Mood disorders are highly comorbid in chronic pain patients, where the prevalence of depression ranges between 30 and 80%, depending on the pain etiology (Bair et al., 2003; Howe and Sullivan, 2014). It is consistently reported that chronic pain patients with comorbid psychopathology exhibit increased pain intensity and increased pain-related disability (Jamison and Edwards, 2013; Martel et al., 2014). For example, intravenous morphine is 40% more effective in chronic pain patients without coexisting psychopathology (Jamison and Edwards, 2013).
The Analgesic, Anesthetic, and Addiction Clinical Trial Translations Innovations Opportunities and Networks public–private partnership with the U.S. Food and Drug Administration, and the American Pain Society have joined together to develop an evidence-based chronic pain classification system termed the ACTTION-APS Pain Taxonomy (Fillingim et al., 2014). Part of this taxonomy recognizes the multidimensional and biopsychosocial nature of chronic pain, which is subject to comorbidities that impact the experience of chronic pain. It was argued that the reliance on reflex behavioral outcomes in animal pain models that measure changes in sensory nociceptive thresholds (mechanical, thermal, chemical) has led to poor predictive translational outcomes. As such, the myriad of promising candidate analgesics identified by preclinical pain studies relying on these sensory outcomes have failed in clinical trials (Mogil and Crager, 2004; Cobos and Portillo-Salido, 2013; Percie du Sert and Rice, 2014; Yaksh et al., 2015). Thus, capturing ongoing pain and negative affective in chronic pain models are necessary for future analgesic drug development. This is emphasized by research findings that show that negative affect not only exacerbates the pain experience, it also is a predictor of opioid prescription misuse leading to an opioid use disorders (Evans and Cahill, 2016).
Activation of κ opioid receptors (KORs) produces negative affect. For example, KOR agonists produce dysphoric effects and elicit psychotomimetic properties in humans, as well as elicit place aversion and depressive-like affective behaviors in rodents (Shippenberg et al., 1993; Knoll and Carlezon, 2010; Chavkin and Koob, 2016). Indeed, KOR agonists produce signs of anxiety, fear, and depression in animals and humans (Chartoff and Mavrikaki, 2015). One mechanism implicated in κ-mediated aversion is the modulation of mesolimbic dopamine circuitry, where KORs are expressed on dopamine terminals. Activation of KORs following systemic agonist treatment reduces dopamine release (Chefer et al., 2013). Ablation of KORs from dopamine neurons (Van't Veer and Carlezon, 2013) or KORs on BLA glutamatergic neurons that project to the medial PFC (Tejeda et al., 2015) results in an anxiolytic phenotype, suggesting that these circuits are critical to the expression of negative affective-like behavior. Others, and we identified that mesolimbic circuitry dysfunction, including that occurring in dopamine neurotransmission, precipitates mood disorders, impairs motivated behavior, and likely contributes to chronic pain (Taylor, 2013; Cahill et al., 2014a,b; Yalcin and Barrot, 2014; Taylor et al., 2016; Cahill and Taylor, 2017).
Considering that the circuitry involved in pain processing and affective/motivational systems overlaps extensively, we investigated specifically whether KOR contributes to the aversive nature of chronic pain. In a rodent model of chronic neuropathic pain, we show that the endogenous tone of the KOR system within mesolimbic dopaminergic circuitry is robustly increased. Importantly, we show that KOR blockade or elimination of KOR in midbrain dopamine neurons alleviates a tonic-aversive component of chronic neuropathic and inflammatory pain, which is sex-dependent. However, attenuation of depressive and anxiogenic affective-like behaviors by KOR antagonism is not sex-dependent, suggesting a dichotomy of mechanisms between affective dimensions of chronic pain and the ongoing tonic-aversive states.
Materials and Methods
Animals.
Male and female C57BL/6J wild-type (WT) mice were obtained (Jackson Laboratories) at 8 weeks of age. Male Long–Evans rats (250–300 g) were obtained from Charles River Laboratories. Mice acing one functional copy of the pre-proenekephalin (PENK) gene (König et al., 1996; C57BL/6 background) were bred to generate PENK knock-out (KO) mice and WT littermates. The PENK KO and control mice were used beginning at 9–10 weeks of age. KOR conditional KO mice (Ehrich et al., 2015) were obtained from JAX laboratories at 9–10 weeks of age. This latter transgenic mouse strain was on a mixed 129/C57 background.
Mice were housed in groups of two to four per cage on a 12 h reverse light/dark cycle with food and water available ad libitum. Long–Evans rats (250–300 g) were housed in pairs on a 12 h reverse light/dark cycle with food and water available ad libitum. Rodents were allowed to habituate to their housing environments for 1 week before handling. Experiments were conducted in the dark phase between 9:00 and 16:00 h. To the extent possible, experimenters handling mice were blind to surgery, genotype, sex, and drug treatment. Importantly, all tests were performed by experimenters' blind to experimental condition. Mice were assigned to experimental conditions in a randomized block design so that factors such as time of day were counterbalanced over the experimental conditions. For experiments that had large numbers of groups, we completed experiments using successive cohorts ensuring that experiments were performed for all conditions within each cohort. All replications were balanced with respect to experimental groups. All procedures were pre-approved by the University of California, Irvine Institutional Animal Care and Use Committee, the University of California, Los Angeles Chancellor's Animal Research Council, or the Canadian Council on Animal Care and the Queen's University or University of Calgary Animal Care Committees.
Drugs.
The U50,488, U69,593, and naloxone were obtained from Sigma-Aldrich and dissolved in 0.9% sterile saline. The JDTic, a highly specific, long-acting KOR antagonist (Deehan et al., 2012; Munro et al., 2012; Wu et al., 2012; Chavkin and Martinez, 2015) was obtained from Dr. F. Ivy Carroll (Research Triangle Institute International) and dissolved in 0.9% sterile saline. The AAV-TH-cre and AAV-GFP were obtained from Dr. Caroline Bass at the University of Buffalo (Gompf et al., 2015).
Peripheral nerve injury neuropathic pain model.
Mice and rats were randomly assigned into either [pain]-naive (no surgery but exposed to general anesthetic and hindlimb shaving), sham, or peripheral nerve injury (PNI) surgery groups. Before surgery, all animals received acetaminophen (∼1 mg) and were subsequently anesthetized with gaseous isoflurane (∼2.5% in O2). An ∼1 cm incision was made in the upper left hind leg. For PNI, the sciatic nerve was constricted with polyethylene tubing (PE20 for mice and PE90 for rats, 2 mm length for both species). Animals in the sham group received a similar surgery but without nerve ligation. The pain-naive control group received equivalent time under isoflurane anesthesia, hindlimb shaving and all pharmaceutical treatments but did not undergo skin incision.
Complete Freund's adjuvant inflammatory pain model.
Mice were briefly anesthetized with 1–2% isoflurane (v/v) and 30 μl of undiluted complete Freund's adjuvant (CFA; Sigma-Aldrich) was injected into the intraplantar surface of the left hindpaw. Control animals received an equivalent volume of intraplantar sterile saline.
AAV injection.
Isoflurane-anesthetized mice were mounted on a stereotaxic alignment system. A small incision was made to expose the skull surface and to visualize bregma. Two holes were drilled in the skull overlying the ventral tegmental area (VTA). A 35-gauge bevel-tipped Hamilton syringe was lowered bilaterally from the surface of the skull into the VTA and virus was injected (0.3 μl/side at 0.1 μl/min). Viruses were injected bilaterally using coordinates from bregma; AP: −3.2, ML: +0.7, DV: −4.6. The needle was left in place for 5–7 min before being slowly removed. The incision was closed with 5-0 silk sutures and animals recover for at least 3 weeks to allow adequate time for viral expression before conducting sham or peripheral nerve injuries. The TH-iCre-AAV2/10 was synthesized by and obtained from Dr. Caroline Bass (University of Buffalo) and was recently shown to be effective in knocking down KORs (Illiano et al., 2017). The AAV2/10 virus is highly neurotrophic, is easily titrated, and has previously been used by various groups to drive high levels of expression in both extremely small and large brain regions of both rats and mice.
KOR agonist-stimulated GTPγS autoradiography.
Brains were collected from naive, sham, and PNI mice 2 weeks postsurgery. Brains were snap-frozen with isopentane at −30°C and stored at −80°C until further processing. On day of processing, brains were sectioned coronally using a cryostat (20-μm-thick sections) at −20°C. Sections were thaw-mounted on Superfrost charged slides. Sections were pre-incubated in assay buffer (50 mm Tris-HCl, 3 mm MgCl2, 0.2 mm EGTA, 100 mm NaCl, 2 mm GDP, 1 μm DPCPX, pH 7.4) for 15 min. Agonist-stimulated KOR activity was determined by incubating brain sections in [35S]GTPγS (40 pm) with U69,593 (10 μm) for 1 h at RT. After incubation, slides were washed two times in ice-cold wash buffer (50 mm Tris-HCl, pH 7.4) followed by a brief wash in ice-cold deionized water (30 s). Slides were air-dried and exposed to Kodak Biomax film together with [14C] standards for 2 d. Films were developed using a Kodak GBX Developer and RapidFix solution. Films were digitally analyzed and binding quantified using a MicroComputer Image Device (MCID) normalized to a [14C] standard curve in dpm/mg (MCID, Imaging Research). Data from the resulting agonist-stimulated samples were compared with non-agonist-treated brain samples to determine the percentage activation of KOR above basal.
Western immunoblotting for KOR-P antibody specificity.
Brains were collected from naive WT or KOR KO mice 1 h after treatment with vehicle or KOR agonist (U50, 488 1–30 mg/kg, i.p.) and snap-frozen with isopentane at −50°C and stored at −80°C until sectioning. Brains were sectioned coronally using a cryostat (150-μm-thick section) at −20°C. Sections were mounted on Superfrost charged slides, and tissue punches (1 mm diameter) were taken using a disposable biopsy plunger to isolate nucleus accumbens (NAc) tissue. Tissue punches were homogenized in lysis buffer (50 mm Tris-base, 4 mm EDTA, pH 7.4) with protease inhibitor cocktail (Pierce Biotech; ThermoFisher Scientific). Samples were then centrifuged at 10,000 × g to remove DNA/debris, and supernatant protein was extracted and stored at −20°C. Protein samples were mixed with NuPAGE LDS Loading Buffer and reducing reagent (Novex), heated to 70°C for 10 min, and stored at −20°C for gel electrophoresis. Gel electrophoresis of protein samples was conducted using an Invitrogen SDS-PAGE gel box, NuPAGE MES Running Buffer, and Bis-Tris Mini Gels (Novex). Samples were loaded alongside PAGE-Ruler Plus Prestained Protein Ladders (Fisher Scientific) and bands were separated at 120 V for 2 h.
Proteins from gels were transferred to nitrocellulose (0.45 μm pore size; Novex) using a Bio-Rad protein transfer box at 4°C and 250 mA for 50 min. Membranes were blocked with 5% nonfat dried reconstituted milk and TBS-T (1%) at RT for 1 h. Membranes were subsequently incubated with phosphor-KOR antibodies produced in-house (Bruchas laboratory, University of Washington, Seattle) at 1:1000 dilution in antibody buffer (2% BSA, 2% gelatin from cold-water fish, TBS-T) overnight at 4°C with gentle shaking. After primary antibody incubation, membranes were washed 3 times with TBS-T (10 min per wash). Membranes were then incubated with GOXCH HRP-conjugated anti-chicken secondary antibodies (Novex) at 1:4000 dilution in 5% milk and TBS-T (1%) for 90 min at RT with gentle shaking. The membranes were washed again three times with TBS-T (10 min per wash), followed by 1 min revelation of membranes using GE Healthcare ECL-Plus substrate (GE Healthcare). The membranes were visualized using a Li-Cor Odyssey Fc Imager. Membranes were then washed and antibody-stripped using glycine stripping buffer (200 mm glycine, pH 2.6) with shaking at RT for 1 h, then washed three times with TBS-T (10 min per wash). Membranes were re-probed with β-actin antibody (Abcam) at 1:4000 dilution overnight and 4°C and subsequent anti-rabbit HRP-conjugated secondary antibody (Life Technologies) at 1:4000 dilution for 1 h. Band intensities were quantified using Li-Cor Image software and normalized to β-actin control bands.
Western immunoblotting of phosphorylated KOR.
Brains were collected from sham and PNI mice 2 weeks postsurgery and snap-frozen with isopentane at −50°C and stored in −80°C until ready to be sectioned. Brains were coronal-sectioned via cryostat (150 μm thick) at −20°C, mounted on Superfrost charged slides, and tissue punches (1 mm diameter) were taken using a disposable biopsy plunger for medial prefrontal cortex (mPFC), NAc, bed nucleus stria terminalis (BNST), amygdala (AMYG), hippocampus (HIPP), thalamus (THAL), VTA, and dorsal raphe nucleus (DRN). Tissue punches were homogenized in lysis buffer (50 mm Tris-Base, 4 mm EDTA, pH 7.4) with protease inhibitor cocktail (Pierce Biotech; ThermoFisher Scientific), centrifuged at 10,000 × g to remove DNA/debris, and supernatant protein was extracted and stored at −20°C. Protein samples were mixed with NuPAGE LDS Loading Buffer and reducing reagent (Novex), heated to 70°C for 10 min, and stored at −20°C to be used for gel electrophoresis. Gel electrophoresis of protein samples was conducted using Invitrogen SDS-PAGE gel box, NuPAGE MES Running Buffer and Bis-Tris Mini Gels (Novex), and loaded alongside PAGE-Ruler Plus Prestained Protein Ladder (Fisher Scientific), run at 120 V for 2 h. Proteins from gels were transferred onto nitrocellulose (0.45 μm pore size; Novex) using Bio-Rad protein transfer box at 4°C and 250 mA for 50 min. Membranes were blocked with 5% milk and TBS-T (1%) at RT for 1 h. Membranes were subsequently incubated with pKOR antibodies made in-house (Bruchas laboratory, Washington University, St. Louis) at 1:1000 dilution in antibody buffer (2% BSA, 2% gelatin from cold-water fish, TBS-T) overnight at 4°C with gentle shaking. After primary antibody incubation, membranes were washed 3× with TBS-T (10 min per wash) and GOXCH HRP-conjugated anti-chicken secondary antibodies (Novex) were then incubated at 1:4000 dilution in 5% milk and TBS-T (1%) for 90 min at RT with gentle shaking. The membranes were washed again 3× with TBS-T (10 min per wash), followed by 1 min revelation of membranes using GE Healthcare ECL-Plus substrate (GE Healthcare). The membranes were visualized with Li-Cor Odyssey Fc Imager. Membranes were then washed and antibody-stripped using glycine stripping buffer (200 mm glycine, pH 2.6) with shaking at RT for 1 h, then washed 3× with TBS-T (10 min per wash). Membranes were re-probed with β-actin antibody (Abcam) with 1:4000 dilution overnight at 4°C, and subsequent anti-rabbit HRP-conjugated secondary antibody (Life Technologies) with 1:4000 dilution for 1 h. Band intensities were quantified using Li-Cor Image software and normalized to beta-actin control bands.
Real-time quantitative PCR.
Brains were collected from sham and PNI mice 8 weeks postsurgery and were coronal-sectioned via cryostat (150 μm thick) at −20°C and mounted on Superfrost charged slides (Fisher Scientific). Tissue punches (1 mm diameter) were taken using a disposable biopsy plunger (Miltex) for mPFC, NAc, BNST, AMYG, HIPP, THAL, VTA, and DRN. Total RNA was collected from the brain tissue punches via Trizol extraction method (Ambion Life Technologies). RNA was converted to cDNA using 100 U of M-MulV Reverse Transcriptase, 1 μm Oligo d(T)23VN, and 2 mm dNTP mix (New England Biolabs), annealed at 70°C and inactivated at 95°C. Real-time qPCR was conducted using primer sets for dynorphin (DYN), KOR, and β-actin control genes. Using, 96-well optical plates (Applied Biosystems), cDNA and PerfeCTa SYBR Green FastMix containing the primer sets (Quanta Biosciences) were loaded and run on ABI ViiA7 fast block qPCR machine using cycling conditions in the PerfeCTa SYBR Green FastMix manual. Cycle threshold outputs were calculated and normalized to the actin housekeeping gene to compute ΔCT. Relative expression levels were determined by normalizing sham and neuropathic groups to three age-matched naive (non-surgery) mice brain samples via ΔΔCT method.
RNAscope multiplex fluorescent in situ hybridization.
Brains were collected from pain-naive, sham, and PNI mice 2 weeks postsurgery. Brains were perfused with 4% paraformaldehyde in saline and snap-frozen with isopentane at −30°C and stored in −80°C until further processing. Brains were coronal-sectioned via cryostat (18 μm thick) at −20°C, and thaw-mounted on Superfrost charged slides. Custom fluorescent probe labels were designed to fit complementary sequences on mRNA strands for κ opioid receptor gene (Oprk1) carrying fluorescent AlexaFluor 488, tyrosine hydroxylase (TH) or pro-enkephalin (Penk) carrying Atto555, and glutamate decarboxylase 1 (Gad1) or pro-dynorphin (Pdyn) carrying Atto647. Brain slices were incubated with 4% paraformaldehyde and then dehydrated with 50, 70, and 100% ethanol washes for 5 min each. Slides were then incubated overnight at −20°C in 100% ethanol. Slides were then taken out and dried for 5 min and a hydrophobic barrier was drawn around the brain slices. Probes were activated in oven at 40°C for 10 min. Slides were then incubated with probes following the RNAScope Multiplex processing kit (ACDBiosciences). Slides were then coverslipped and sealed with nail polish and stored in dark at −20°C until visualization. Slides were visualized using a Nikon Ti-E wide-field inverted fluorescence microscope and NIS Elements software setup. Image fluorescence intensities were quantified using FIJI (ImageJ) software.
Microdialysis probe insertion.
Fast microdialysis to detect extracellular dopamine levels was performed essentially as previously described (Yang et al., 2013, 2015). At zeitgeber time (ZT)10–ZT12, each subject was briefly (1–3 min) anesthetized using isoflurane for insertion of a CMA/7 microdialysis probe (1 mm) into a guide cannula (AP: +1.5, ML: +0.8, DV: −3.5). Immediately after insertion, regular artificial CSF (aCSF; 147 mm NaCl (Fluka, 73575), 3.5 mm KCl (Fluka, 05257), 1.0 mm CaCl2 (Aldrich, 499609), 1.0 mm NaH2PO4, 2.5 mm NaHCO3 (Fluka, 88208), 1.2 mm MgCl2 (Sigma-Aldrich, 449172), pH 7.3 ± 0.03 at room temperature) was continuously perfused through the probe at 2 μl/min for 30–60 min followed by a 0.3 μl/min flow rate for an additional 12–14 h to allow recovery from probe insertion.
Morphine-induced dopamine overflow.
For all mice, at ZT1–ZT2 the day after probe insertion, the aCSF flow rate through the probes was increased to 2 μl/min for 60–120 min before collecting dialysate samples for analysis. The dialysate sampling rate was 5 min/sample. Basal samples were collected for 60 min and each animal's average baseline level used to calculate percentage evoked release as the mean of the sample values between 30 and 45 min (3 values) following the beginning of sampling. At the end of the 60 min basal sampling period, mice received an intraperitoneal injection of 10 mg/kg morphine. Morphine-induced dopamine overflow was sampled for an additional 250 min. The KOR antagonist JDTic (10 mg/kg, i.p.) was administered 24 h before morphine injection.
HPLC.
Analysis was performed using an Eicom integrated high-performance liquid chromatography (HPLC) system (HTEC-500, Eicom) with an Insight autosampler and two Eicom EAS-20s online autoinjectors. Chromatographic separation was achieved using an Eicompak PP-ODS II stationary phase (4.6 mm, i.d. × 30 mm, 2 μm, particle diameter) and a phosphate buffered mobile phase with 96 mm NaH2PO4 (Fluka, 17844), 3.8 mm Na2HPO4 (Fluka, 71633), pH 5.4, 1.8–2.5% MeOH (EMD, MX0475–1), 50 mg/L EDTA.Na2 (Fluka, 03682), and 500 mg/L sodium decanesulfonate (TCI, I0348) in water purified via a Milli-Q Synthesis A10 system (EMD Millipore). The column temperature was maintained at 20–21°C. The volumetric flow rate was 350–500 μl/min. Electrochemical detection was performed using an Eicom WE-3G graphite working electrode with an applied potential of +450 mV versus a Ag/AgCl reference electrode. Dopamine (Sigma-Aldrich, H8502) standards were prepared in aCSF. Standard curves, which were verified daily, encompassed physiological dopamine concentration ranges (0, 7.8, 15.6, 31.2, 62.5, 125, 250, 500, 1000 nm using 20 μl sample volumes). The limit of detection was ≤60 amol and the practical limit of quantification was ≤120 amol using a 20 μl sample volume. All dialysate samples for in vivo experiments were collected at 5 min intervals at a dialysate flow rate of 2 μl/min using EAS-20s autoinjectors. Samples were injected immediately onto the HPLC system in an online configuration.
Drug-induced conditioned place preference/aversion.
This test was conducted using an unbiased, counter-balanced three chamber apparatus as previously described (Cahill et al., 2013; Taylor et al., 2015a). Each box (28 × 28 × 19 cm) was divided into two equal-sized conditioning chambers separated by a neutral compartment. The two chambers were distinguished with visual and tactile cues. Mice were placed in the apparatus and allowed free access to both chambers. The time spent in each chamber was recorded using an infrared CCD camera attached to a computer running behavioral tracking software (Noldus, EthoVision). Mice were conditioned to drug over 6 or 8 d (half vehicle and half drug) counterbalanced for chamber and order of drug injection. Post-conditioning was performed without treatment (day following conditioning), in which animals were again allowed to explore the whole box freely for 15 or 30 min. Time spent and activity in each compartment were recorded during the sessions. Preference scores were calculated using the following formula: [Time in Test(paired) − Time in Test(unpaired)] − [Time at baseline(paired) − time at baseline(unpaired)].
Single chamber place conditioning.
Single chamber conditioning was used to assess ongoing pain in two models of chronic pain. This test was conducted using an unbiased, counter-balanced three chamber apparatus. Each box (28 × 28 × 19 cm) was divided into two equal-sized conditioning chambers separated by a neutral compartment. The two chambers were distinguished with visual and tactile cues. Mice were placed in the apparatus and allowed free access to both chambers. The time spent in each chamber was recorded using an infrared CCD camera attached to a computer running behavioral tracking software (Noldus, EthoVision). Mice received an injection of saline daily and were immediately returned to their home cage or placed in one of the conditioning chambers for 30 min. Mice were counterbalanced to chamber and order of home cage versus conditioning chamber. Post-conditioning was performed the day following conditioning, in which animals were allowed to freely explore the entire apparatus for 30 min. Time spent and activity in each compartment were recorded during the sessions and the time (15–30 min depending on the experiment) in each compartment was used for calculating preference scores. Preference scores were calculated using the following formula: [Time in Test(paired) − Time in Test(unpaired)] − [Time at baseline(paired) − time at baseline(unpaired)]. For PNI mice, conditioning was commenced either 7 or 14 d following nerve injury, whereas for inflammatory pain, testing commenced 3 d following induction of inflammation.
Locomotor activity.
Locomotor activity was measured in an open-field apparatus. The apparatus consisted of a Plexiglas box arena (28 cm2 and 19 cm high) with an open top. The distance traveled and velocity were recorded using an infrared CCD camera attached to a computer running behavioral tracking software (Noldus, EthoVision). Mice were habituated to test boxes for 3 d before surgery to induce neuropathic pain or control surgery. One hour before surgery, mice received an injection of vehicle or the KOR antagonist JDTic (10 mg/kg, i.p.). On Day 6 and/or Day 11 postsurgery, animals received an injection of vehicle or the KOR agonist U50,488 (1, 5, or 10 mg/kg, i.p.) and were placed immediately in the apparatus for 20 min to test the ability of JDTic to block KOR agonist-induced hypolocomotion.
Intra-VTA DAMGO and conditioned place preference.
Male Long–Evans rats were anesthetized with isoflurane and mounted on a stereotaxic frame to allow implantation of a bilateral intra-VTA cannulae (CMA; coordinates from bregma: AP: −5.4, ML: +0.75, DV: −8.0). Rats were used instead of mice to ensure accurate targeting of drug to the VTA. We have demonstrated comparable systemic opioid place preference responses between rats and mice in control and chronic pain models (Cahill et al., 2013). Animals were individually housed and allowed to recover for 7 d after intra-VTA cannula insertion, at which point the animals received a sham or peripheral nerve injury. Seven days after nerve injury, place conditioning was performed as described above. The apparatus consisted of two large compartments of equal size (45 × 45 × 30 cm) joined by a gray tunnel (18 × 18 × 30 cm). Chambers were distinguished with textual and visual cues. Groups were assigned so that any innate bias to side was balanced between treatment groups, as described above. During the conditioning sessions, animals received four trials with [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO; 1 ng/0.5 μl/side infused at a rate of 0.25 μl/min) and four trials with sterile saline, injected directly into the VTA, and were confined to the chambers for 30 min. On the post-conditioning day, animals were allowed free access to both chambers in a drug-free state. The time spent in the drug-paired chamber was measured over 30 min.
Light/dark test.
The apparatus consisted of light and dark sides (25 cm3) separated by a guillotine door. The lit compartment was brightly illuminated (1000 lux). Mice were placed in the dark compartment at the beginning of the test, and the time spent in the lit compartment was recorded over 5 min. Activity and location was tracked by Noldus EthoVision software.
Forced swim test.
The apparatus for this test was a tank (20 cm diameter × 35 cm high) filled with water to 15 cm for males, and 10 cm for females. Water temperature was maintained at 28°C. This test evaluates depressive-like behavior in terms of the total time mice spend immobile when forced to swim in an inescapable water tank (Porsolt et al., 1977). A mouse was considered to be immobile once swimming and escape behaviors have ceased, and it floated belly-down with its head above the water. The test session lasted 6 min but only the last 4 min of the test were scored. Water in the tank was replaced with clean, 28°C water following each trial. Videos were recorded for all mice and subsequently scored by two independent observers blind to experimental condition.
Experimental design and statistical analyses.
Data are either expressed either as scatter plots for raw data and bar graphs for mean ± SEM or as medians with 25 and 75% quartiles with minimum and maximum data points. Data are analyzed by either GraphPad Prism v7.0 or PAWS Statistics 18 (SPSS). All behavioral data were shown to fit assumptions of a general linear model and data were subjected to factorial ANOVAs. To determine sex differences, we conducted three-way ANOVAs, with surgery and sex as between animal factors and time as the within animal factor. For conditioned place preference data, the amount of time (s) spent in the drug-paired and vehicle-paired compartments on the post-conditioning test day is presented, as well as the preference score. To determine the presence of place aversion induced by ongoing pain, we also performed a one-sample t test to determine whether means were significantly different versus a hypothetical value of 0 (where 0 is no preference). A two-way ANOVA with drug dose and group as the between-subject factors was used to examine differences in the magnitude of preference or aversion across neuropathic pain, sham, and naive groups. Two-way ANOVAs were also used to determine the effects of the KOR antagonist on locomotor activity, time spent in the light compartment in the light dark test, or time spent immobile in the forced swim test, and differences in extracellular dopamine concentrations. Either Tukey's or Sidak post hoc analysis was used to correct for multiple comparisons.
Results
Chronic pain induces changes in KOR-mediated place aversion and hypolocomotion
To investigate the behavioral/affective relationship between KOR systems and chronic pain states, we used the conditioned place preference(CPP)/conditioned place aversion (CPA) paradigm to test for preference or aversion to the KOR agonist U50,488 (1–10 mg/kg, i.p.; Fig. 1a) in mice with peripheral nerve injury (PNI). Post-conditioning testing in a drug-free state revealed that U50,488 produced a dose-dependent place aversion in male but not female pain-naive mice (Fig. 1b–e). At a dose that did not produce place aversion in pain-naive or sham surgical control mice, U50,488 (1 mg/kg, i.p.) produced place aversion in male PNI mice (Fig. 1c,e). None of the doses of U50,488 produced a significant place aversion in female mice regardless of the presence of chronic pain. Examples of heat maps of KOR-mediated place aversion are shown in Figure 1f.
Figure 1.

Enhanced KOR mediated place aversion in chronic pain animals. a, Timeline for CPA to KOR agonist (U50,488) in control (naive and sham surgery) and PNI mice. b, In female mice, U50,488 (U50, 1–10 mg/kg, i.p.) did not produce a place aversion in pain-naive, sham, and PNI. A one-way ANOVA of U50,488 dose revealed no significant effect at any dose (1 mg dose: F(2,23) = 0.216, p = 0.808, 5 mg dose: F(2,37) = 1.296, p = 0.286, 10 mg dose: F(2,22) = 0.003 p = 0.997). One-sample t test for difference from a theoretical value of zero (no preference) revealed that none of the dose tested produced a place aversion independent of surgery (1 mg dose in naive: t = 0.468, p = 0.643, sham: t = 0.542, p = 0.602, PNI: t = 0.465, p = 0.655; 5 mg dose in naive t = 0.767, p = 0.468, sham: t = 1.733, p = 0.103, PNI: t = 1.78, p = 0.094; 10 mg dose in naive: t = 1.578, p = 0.153, sham: t = 1.268, p = 0.252, PNI: t = 0.974, p = 0.358). c, In male mice, U50,488 (1–10 mg/kg, i.p.) dose-dependently produced place aversion in pain-naive and sham male mice, but unlike female mice, all doses produced place aversion in male chronic pain mice. A one-way ANOVA of U50,488 dose revealed a significant effect at 1 and 5 mg doses in PNI mice (1 mg dose: F(2,24) = 9.102, p = 0.0011, 5 mg dose: F(2,37) = 26.19, p < 0.0001, 10 mg dose: F(2,22) = 0.465 p = 0.634). One-sample t test for difference from a theoretical value of zero (no preference) revealed that all doses produced a place aversion in PNI mice (1 mg dose in naive: t = 0.021, p = 0.984, sham: t = 2.301, p = 0.0504, PNI: t = 4.426, p = 0.0022; 5 mg dose in naive t = 2.823, p = 0.0166, sham: t = 10.83, p < 0.0001, PNI: t = 19.66, p < 0.0001; 10 mg dose in naive: t = 3.14, p = 0.0164, sham: t = 2.528, p = 0.0353, PNI: t = 2.531, p = 0.0392). d, Dose–response curves for female mice. A two-way ANOVA revealed no significant effect of surgery (F(2,82) = 0.558, p = 0.572), dose (F(2,82) = 1.743, p = 0.181), or interaction (F(4,82) = 0.227, p = 0.852). e, Dose–response curves for male mice. A two-way ANOVA revealed a significant effect of surgery (F(2,85) = 6.707, p = 0.002) and an interaction (F(4,85) = 4.746, p = 0.0017), but not dose (F(2,85) = 2.392, p = 0.0976). For all datasets, #compares to sham and *compares to naive. Data are expressed as mean ± SEM. N = 7–16 per group. f, Heat map of representative data male U50,488 (U50) 5mg/kg dose.
We also investigated KOR-mediated activity effects and potential enhancement in chronic pain states. The KOR agonist U50,488 produced a dose-dependent decrease in distance traveled in male mice compared with saline for all groups tested (Fig. 2d). In female mice, the lowest dose of U50,488 (1 mg/kg) did not induce hypolocomotion in either of the surgical groups (Fig. 2b), whereas there was a significant effect of this dose in pain-naive but not sham or PNI male mice (Fig. 2c). At the intermediate dose of 5 mg/kg, U50,488 produced hypolocomotion in both male and female PNI groups, but not the pain-naive groups. At the highest dose of U50,488 (10 mg/kg), hypolocomotion was evident in all surgical groups for both sexes. Interestingly, the intermediate dose (5 mg/kg) produced a significant effect in male sham but not female sham mice. Examples of hypolocomotion activity traces to the 5 mg dose are shown in Figure 2e.
Figure 2.

Dose-dependent KOR-mediated hypolocomotion. a, Timeline for KOR-agonist-induced hypolocomotion. b, c, The presence of chronic pain enhanced U50-induced hypolocomotion in both male and female mice at the 5 mg/kg dose (middle). A two-way ANOVA in female mice (for the 5 mg dose) demonstrated a significant effect of surgery F(2,35) = 16.62, p < 0.0001, treatment F(1,35) = 26.99, p < 0.0001 and an interaction F(2,35) = 4.231, p = 0.0226. A two-way ANOVA in male mice (for the 5 mg dose) demonstrated a significant effect of treatment F(1,43) = 20.36, p < 0.0001, but no effect of surgery F(2,43) = 2.956, p = 0.063 and no interaction F(2,43) = 1.783, p = 0.1804. Data are expressed as mean =/− SEM. N = 4–8 per group. d, U50,488 (1–10 mg/kg i.p.) produced a dose-dependent effect on locomotor activity in female and male mice. For female mice, a two-way ANOVA revealed a significant effect of surgery F(2,52) = 12.72, p < 0.0001, dose F(2,52) = 39.54, p < 0.0001and an interaction F(4,52) = 2.62, p = 0.045. For male mice, a two-way ANOVA revealed a significant effect of dose F(2,50) = 45.36, p < 0.0001 and interaction F(4,50) = 2.82, p = 0.0345, but not dose F(2,50) = 0.048, p = 0.9528. For all datasets, *compares saline to U50,488 treatment. e, Representative activity tracks of U50,488 (5 mg/kg, i.p.) -induced locomotion in male mice.
To further examine the effects of chronic pain on KOR-mediated effects, we took advantage of the long-lasting KOR antagonist effects of JDTic, which can last up to 2 weeks following a single injection (Deehan et al., 2012). Resensitization of KOR-induced behaviors requires synthesis of new KOR protein to produce subsequent KOR-induced effects. We reasoned that if there was an increase in KOR activation in chronic pain states that KORs may turn over faster and predicted that the duration of effects produced by JDTic would be shorter in chronic pain states. Here, JDTic (or vehicle) was injected 24 h before sham or PNI surgery (Fig. 3). At 6 or 11 d postsurgery, mice were injected with the KOR agonist U50,488 (10 mg/kg, i.p.) or saline and locomotor activity was determined.
Figure 3.
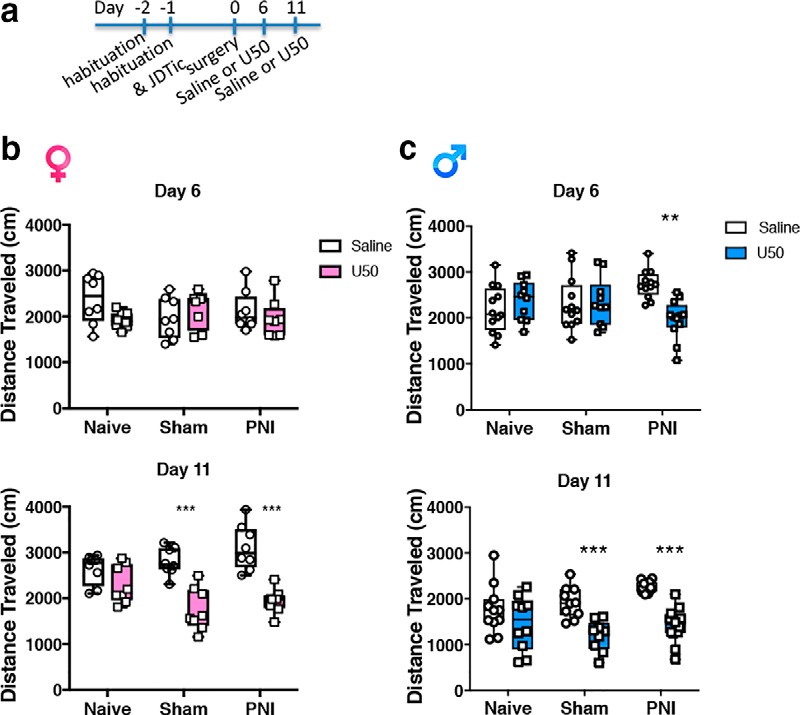
Chronic pain enhances recovery of KOR-mediated effects after JDTic. a, Timeline for KOR agonist-induced hypolocomotion at either 6 or 11 d post-KOR antagonist administration (JDTic 10 mg/kg, i.p.). All mice were injected with the KOR antagonist JDTic (10 mg/kg, i.p.) 24 h before surgery. b, KOR blockade prevented KOR agonist U50,488 (U50 10 mg/kg, i.p.) -induced hypolocomotion in naive, sham, and PNI female mice at 6 d following KOR antagonist administration. A two-way ANOVA in female mice on Day 6 demonstrated no significant effect of surgery F(2,42) = 0.3691, p = 0.6936, or treatment F(1,42) = 1.57, p = 0.2172 and no interaction F(2,42) = 2.471, p = 0.0967. Thus, at 6 d post-JDTic administration, KOR agonist-induced hypolocomotion was blocked in female mice independent of the presence of chronic pain. c, In contrast, the KOR antagonist prevented U50-induced hypolocomotion in naive and sham but not PNI male mice. A two-way ANOVA in male mice on Day 6 demonstrated no significant effect of surgery F(2,62) = 0.128, p = 0.8801, or treatment F(1,61) = 2.417, p = 0.1243 but there was an interaction F(2,62) = 6.856, p = 0.0020. Thus, JDTic blocked KOR agonist-induced hypolocomotion in naive and sham male mice but not chronic pain male mice. At Day 11, JDTic continued to block U50,488-induced effects in both male and female naive mice, but did not prevent hypolocomotion of the KOR agonist in either sham or PNI female or male mice. A two-way ANOVA in female mice on Day 11 demonstrated a significant effect of surgery F(2,42) = 058.11, p < 0.0001 and an interaction F(2,42) = 5.42, p = 0.0081 but no effect of treatment F(1,42) = 1.754, p = 0.1856. A two-way ANOVA in male mice on Day 11 demonstrated no significant effect of surgery F(2,57) = 2.816, p = 0.0682 or interaction F(2,57) = 2.629, p < 0.0809, but an effect of treatment F(1,57) = 36.67, p < 0.0001. Data are expressed as median with 25 and 75% quartiles, minimum and maximum values with all data points included for N = 7–12 per group. *Compares saline to U50 within a surgery group.
As hypothesized, KOR agonist-induced hypolocomotion (U50,488 10 mg/kg, i.p.) was blocked at 6 d following JDTic administration in both male and female pain-naive and sham mice (Fig. 3), confirming the long-lasting blocking effects of JDTic. Similarly, KOR agonist-induced hypolocomotion was blocked at 6 d post-JDTic injection in female pain mice (Fig. 3b). However, JDTic failed to block U50,488 hypolocomotion in male PNI mice (Fig. 3c). At 11 d post-JDTic injection, U50,488-induced hypolocomotion was evident in both males and female sham and PNI mice, but not in pain-naive mice. Thus, the KOR agonist-induced hypolocomotion recovers faster after KOR antagonist treatment in male, but not female PNI animals compared with surgical controls. Together, these data demonstrate that KOR behavioral sensitivity and turnover is increased in chronic neuropathic pain states, particularly in male mice. This increased function of the KOR system is consistent with evidence that KOR-induced hypolocomotion (Heidbreder et al., 1993; Karper et al., 2000), aversion (Van't Veer et al., 2013) and reduced dopamine release in the ventral striatum (Leitl et al., 2014a) are critically mediated by regulation of dopaminergic circuitry.
Chronic pain-induced changes in κ opioid system expression and function in the nucleus accumbens
To determine the extent to which chronic pain alters expression and function of KORs and expression of the KOR endogenous ligand DYN in the mesolimbic circuitry, we performed ex vivo analysis of mRNA expression using RNAscope multiplex in situ hybridization and KOR G-protein coupling via GTPγS autoradiography. We observed a profound increase in KOR (Oprk1) mRNA expression (Fig. 4a,b), where KOR mRNA expression in the contralateral NAc was increased in male, but not female PNI mice compared with pain-naive and sham control mice. This increase in KOR mRNA remained elevated for at least 8 weeks post-PNI as determined by quantitative real-time PCR (Table 1). Pdyn mRNA was significantly increased in the contralateral (right) NAc of both male and female mice at both 2 weeks (Fig. 4a,b) and 8 weeks (Table 2) post-PNI. Similar sex differences were evident in KOR activation as measured by GTPγS autoradiography. Activation of KORs, quantified by GTPγS autoradiography in the NAc core and shell 2 weeks post-PNI was increased in male (Fig. 4c) but not female PNI mice (Fig. 4d) compared with pain-naive or sham control groups. Examples of heatmap autoradiograms of [35S]GTPγS binding are shown in Figure 4, c and d, where warm (red) colors reflect more agonist-stimulated KOR activity and cold (blue) colors indicate less KOR activity.
Figure 4.

KOR upregulation and function within the nucleus accumbens of chronic pain animals. a, In multicolor images, Oprk1 (white), Pdyn (green), and pro-enkephalin (pink) gene expression was detected with RNAscope multiplex florescent in situ hybridization in the NAc of pain-naive, sham and PNI mice. Scale bar, 25 μm. b, In the PNI model of neuropathic pain, the expression of Oprk1 transcript was increased in male, but not female mice compared with pain naive and sham controls. Tissue was collected 2 weeks post-induction of pain by PNI. One-way ANOVAs revealed a significant effect in male (F(2,12) = 19.26, p < 0.001), but not female (F(2,18) = 3.483, p = 0.054) PNI mice. *Compares naive to PNI, # compares sham to PNI. In contrast, Pdyn transcript was increased in both male and female PNI mice. One-way ANOVAs revealed a significant effect in male (F(2,12) = 10.83, p < 0.01) and female (F(2,17) = 3.511) PNI mice compared with pain-naive but not sham control mice. *Compares naive to PNI. Data are expressed as mean ± SEM. N = 12 per group. c, Representative autoradiograms and quantification of KOR agonist-induced [35S]GTPγs binding in coronal brain sections from naive, sham and PNI male mice. Increases in KOR agonist (U69,593) stimulated [35S]GTPγs binding are evident in the NAc of PNI male mice compared with both pain-naive and sham controls. Increases in binding were evident in both the core (*compares to naive, and # compares to sham, one-way ANOVA, F(2,21) = 5.455, p = 0.0124) as well as the shell (*compares to naive, # compares to sham, one-way ANOVA, F(2,21) = 5.988, p = 0.0088) of the NAc. d, Representative autoradiograms and quantification of KOR agonist-induced [35S]GTPγs binding in coronal brain sections from naive, sham and PNI female mice. No differences in KOR agonist (U69,593) stimulated [35S]GTPγs binding are evident between pain-naive, sham and PNI female mice in either the NAc core (one-way ANOVA, F(2,21) = 0.1296, p = 0.8972) or NAc shell (one-way ANOVA, F(2,21) = 0.1669, p = 0.8474). Data are expressed as mean ± SEM for N = 8 per surgical group. For all data presented, tissue was collected 2 weeks post-induction of pain by PNI.
Table 1.
Quantification of Oprk1 gene expression in brain regions of 8 week postsurgery sham and chronic pain mice
| Sham 2ddCT | Sham SEM | PNI 2ddCT | PNI SEM | |
|---|---|---|---|---|
| Male | ||||
| NAC-ipsi | 52.09 | 2.97 | 170.54* | 3.00 |
| NAC-con | 260.3 | 4.16 | 359.54* | 3.88 |
| VTA-ipsi | 50.63 | 5.85 | 156.49* | 4.73 |
| VTA-con | 76.17 | 4.90 | 173.29* | 4.94 |
| Female | ||||
| NAC-ipsi | 2.258 | 0.38 | 1.353 | 0.45 |
| NAC-con | 3.739 | 0.41 | 3.610 | 0.40 |
| VTA-ipsi | 0.49 | 0.433 | 0.08 | 0.401 |
| VTA-con | 0.09 | 0.363 | 0.65 | 0.397 |
Brain regions are also subcategorized by hemispheres in terms of ipsilateral (ipsi) and contralateral (contra) to PNI. Statistical significance is denoted with asterisk where p < 0.05. A total of 10 sham and 10 PNI mice were used in these experiments.
Table 2.
Quantification of Pdyn gene expression in brain regions of 8 week postsurgery sham and PNI mice
| Sham 2ddCT | Sham SEM | PNI 2ddCT | PNI SEM | |
|---|---|---|---|---|
| Male | ||||
| NAC-ipsi | 0.379 | 1.244 | 4.690 | 1.371 |
| NAC-con | 1.042 | 1.521 | 10.89* | 1.418 |
| VTA-ipsi | 0.286 | 1.134 | 1.755 | 1.410 |
| VTA-con | 2.215 | 1.093 | 5.535 | 1.454 |
| Female | ||||
| NAC-ipsi | 6.751 | 1.898 | 33.5 | 15.23 |
| NAC-con | 0.069 | 0.006 | 6.19* | 0.432 |
| VTA-ipsi | 0.18 | 0.029 | 5.633** | 1.455 |
| VTA-con | 0.34 | 0.025 | 7.488*** | 0.0959 |
Brain regions are also subcategorized by hemispheres in terms of ipsilateral (ipsi) and contralateral (contra) to PNI. Statistical significance is denoted with asterisk where p < 0.05. A total of 10 sham and 10 PNI mice were used in these experiments.
To provide further evidence that chronic pain upregulates endogenous KOR system activity, we determined changes in expression of the phosphorylated state of KORs (in the absence of exogenous agonist), as a measure of tonic KOR activation in the PNI pain state. Using a phospho-specific KOR (KOR-P) antibody, we determined KOR-P levels in various brain regions using Western blotting techniques. Antibody specificity was confirmed by showing a dose-dependent increase in KOR agonist-stimulated KOR-P expression in the NAc of pain-naive animals (Fig. 5a) and by the absence of signal in membranes prepared from KOR KO male mice (Fig. 5b). The phosphorylated state of KORs was increased in the contralateral NAc of PNI male mice (Fig. 5c). There was also an increase in phosphorylated KOR in the contralateral NAc of sham mice, which we believe likely reflect effects because of postoperative pain. Together, these data show that chronic pain associated with PNI increases the expression and function of the KORs in the NAc of male, but not female mice.
Figure 5.

Chronic pain increases the phosphorylated state of the KOR. a, Characterization of KOR-P antibody. Systemic administration of the KOR agonist U50,488 (U50) dose-dependently increased the phosphorylated state of the receptor (KOR-P) in the NAc of naive mice. b, The molecular species identified by the KOR-P antibody was absent in homogenates of brains from KOR KO−/− mice. N = 4–6 per group. c, In the absence of exogenously administered KOR agonist, the “activated” state of the KOR (phosphorylated) was significantly increased in the contralateral (contra) NAc of male PNI and sham animals when normalized to pain-naive tissue. Tissue was collected 2 weeks post-induction of pain by PNI. *Compares to ipsilateral (ipsi) sham (t = 2.249) or PNI (t = 2.139). Data are expressed as mean ± SEM for N = 4–10 per group.
KOR regulation of mesolimbic dopamine and reward in chronic pain states
Kappa opioid receptors are known to modulate mesolimbic dopaminergic circuitry by inhibiting dopamine release at dopaminergic nerve terminals within the NAc (Spanagel et al., 1992; Ebner et al., 2010; Cahill et al., 2014b; Ehrich et al., 2015; Chartoff et al., 2016). Preclinical and human subject research have identified strong correlations between dysfunction of this mesolimbic circuitry and chronic pain (Taylor et al., 2016). Our prior studies have shown that opioid-evoked dopamine release is disrupted in a model of neuropathic pain (Taylor et al., 2014, 2015a). Using in vivo microdialysis in awake, freely-moving mice, we asked whether blocking KORs would recover the loss of opioid-induced dopamine release in the NAc. No significant differences in basal dopamine levels between pain and control animals were observed (Table 3). Consistent with our previous findings, systemic administration of morphine did not evoke dopamine release above basal levels in male PNI mice (Fig. 6c). However, when these mice were pretreated with JDTic, morphine-induced dopamine release recovered to levels similar to the sham group. This finding is concordant with prior reports of blocking KOR recovered μ opioid-evoked dopamine release in a model of inflammatory (formalin) pain (Narita et al., 2005).
Table 3.
Basal dopamine levels in the NAc as detected by in vivo microdialysis
| Sham | PNI | PNI + JDTic |
|---|---|---|
| 0.20938 | 0.02728 | 0.10100 |
| 0.22982 | 0.12125 | 0.21516 |
| 0.12838 | 0.05175 | 0.48927 |
| 0.12442 | 1.32625 | 0.79104 |
| 0.65862 | 0.31025 | 0.47336 |
| 0.08182 | — | 0.23101 |
| — | — | 0.63812 |
Mice underwent sham or PNI to induce neuropathic pain 14 d prior to assessing basal dopamine levels by in vivo microdialysis. JDTic was administered 24 h prior to conducting microdialysis measurements. Each data point is a mean of eight measurements taken consecutively in 5 min samples. Data are raw data values. N = 5–7 per group. One-way ANOVA showed no difference between groups: F(2,15) = 0.4558, p = 0.642.
Figure 6.

KOR regulates opioid-induced reward and dopamine release. a, Time course for drug treatment and conditioning with intra-VTA DAMGO. b, CPP of intra-VTA μ opioid agonist DAMGO (1 ng/bilateral injection) in sham and PNI mice. Intra-VTA DAMGO produced a CPP in sham but not PNI mice. Pretreatment of mice with KOR antagonist JDTic (10 mg/kg, i.p.) did not alter DAMGO CPP in sham animals but recovered DAMGO CPP in pain animals. Data are expressed mean ± SEM. N = 8–9 per group. Two-way ANOVA revealed a significant effect of surgical group (F(1,29) = 5.184, p < 0.05), treatment (F(1,29) = 7.483, p < 0.05) and a significant interaction (F(1,29) = 4.233, p < 0.05). *p < 0.05 compares sham to PNI in vehicle-treated mice. Images depict cannula placement for DAMGO delivery. c, Morphine (10 mg/kg, i.p.) failed to stimulate extracellular dopamine in PNI male mice, but treatment with KOR antagonists JDTic (10 mg/kg, i.p.) restored morphine-evoked extracellular dopamine levels. Microdialysis experiments were conducted 2 weeks post-induction of pain by PNI. A two-way ANOVA revealed a significant effect of time (F(51,627) = 2.364, p < 0.0001) and surgery (F(2,627) = 76.56, p < 0.0001) but no interaction (F(102,627) = 0.994, p = 0.513. Images depict location of microdialysis fiber location.
Seeking pain relief is a motivational state that engages circuitry involved in affect and salience (Navratilova and Porreca, 2014; Borsook et al., 2016; Taylor et al., 2016). The mesolimbic circuitry plays a central role in motivational processes and incorporates dopaminergic neurons in the VTA that project to the NAc (Berridge and Kringelbach, 2013, 2015; Zarrindast and Khakpai, 2015). Next, we asked whether KOR blockade restores opioid reward in a CPP paradigm, as hypodopaminergic states contribute to chronic pain (Borsook et al., 2016; Taylor et al., 2016) and mood disorders (Elman et al., 2013). The rewarding effects of intra-VTA injection of the μ-opioid agonist DAMGO were determined to isolate reward expression to the mesolimbic dopaminergic circuitry.
We and others previously showed that intra-VTA μ opioid injection is sufficient to produce a CPP (McBride et al., 1999; Narita et al., 2010), but this preference is blunted in chronic neuropathic pain states (Narita et al., 2005). Male rats were conditioned to intra-VTA DAMGO beginning 7 d post-nerve injury (Fig. 6a), a time point when mechanical allodynia is robust. JDTic administration had no effect on opioid reward in sham animals (Fig. 6b). Consistent with our previous data (Taylor et al., 2015a), intra-VTA DAMGO did not elicit a CPP in chronic pain animals. Remarkably, JDTic treatment in neuropathic pain animals restored DAMGO-induced CPP (Fig. 6b). Together, these results demonstrate that KORs contribute to the loss of μ opioid-evoked dopamine release and reward-related behaviors in chronic neuropathic pain.
Chronic pain induced changes in κ opioid system expression and function in the ventral tegmental area
KORs are located presynaptically on dopamine neurons in the NAc as well as postsynaptically within the NAc. Because KOR inhibition by JDTic recovered morphine-evoked dopamine release in male PNI mice (Fig. 6c) we determined whether pain-induced changes in the KOR system were evident in dopamine cell bodies projecting from the VTA to the NAc. Fluorescent in situ hybridization enabled identification of KOR mRNA (Oprk1) expression in various cell types via colabeling of heterogeneous populations of GABAergic and dopaminergic neurons within the VTA (Fig. 7a,b) of sham and PNI male and female mice at 2 weeks following induction of the pain state. We found that Oprk1 mRNA levels were increased in PNI male and female mice compared with sham: Oprk1 levels were primarily localized to TH+ (dopamine)-containing neurons, but not glutamic acid decarboxylase (GAD1+, GABA) neurons. Using, quantitative real-time PCR, we determined that increases in KOR mRNA were sustained for at least 8 weeks post-PNI in male, but not female PNI mice (Table 1). Sex differences were also evident in the GTPγS autoradiography functional assay, where KOR agonist-stimulated [35S]GTPγS binding was increased in the VTA of male, but not female PNI mice compared with sham controls (Fig. 7c,d). The mechanisms responsible for the upregulation of the KOR system within mesolimbic circuitry of chronic pain remain unclear; however, these data demonstrate that chronic neuropathic pain causes a sex-dependent increase in KOR expression and function.
Figure 7.

KOR expression and function is increased in the midbrain of chronic pain animals. Oprk1 gene expression (white) was detected with RNAscope multiplex florescent in situ hybridization in both GABA (green; identified with GAD) and TH (red, male; pink, female) positive neurons in the VTA of female (a) and male (b) mice. The PNI model of neuropathic pain increased the expression of Oprk1 mRNA in the VTA of pain male and female compared with sham surgical controls. Data are expressed as mean ± SEM for N = 6–8 per surgical condition. Male: two-way ANOVA reveled a significant effect of gene (F(2,30) = 9.539, p < 0.001), but not surgery (F(1,30) = 1.33, p = 0.258). There was also a significant interaction (F(2,30) = 9.33, p < 0.001). Female: two-way ANOVA reveled a significant effect of gene (F(2,90) = 59.37, p < 0.001), and surgery (F(1,90) = 5.52, p < 0.05) but not an interaction (F(2,90) = 1.933, p = 0.1507). *Compares sham to PNI. c, Representative autoradiograms and quantification of KOR agonist-induced [35S]GTPγs binding in coronal brain sections from naive, sham, and PNI male and female mice. d, Increases in KOR agonist U69593-stimulated [35S]GTPγs binding are evident in the VTA of male, but not female PNI mice. *Compares to naive and sham. Data are expressed as mean ± SEM for N = 8 per surgical group.
Negative reinforcement is driven by KOR blockade
Although KOR blockade recovered mesolimbic dopamine levels and opioid reward in chronic pain states, it was important to directly assess whether KOR contributes to the tonic-aversive component of chronic pain. The long-acting properties of the KOR antagonist JDTic prevent it from being used in a CPP paradigm because of the requirement of learning drug effects with contextual cues. We thus, decided to use the nonselective opioid antagonist naloxone (Codd et al., 1995) in pre-pro-enkephalin KO mice. Naloxone produces a CPA in WT mice (Dymshitz and Lieblich, 1987) but this aversion is driven by endogenous enkephalin, where the CPA is absent in PENK KO mice (Shoblock and Maidment, 2007). Using this transgenic mouse with genetic loss of enkephalin, we therefore asked whether naloxone would produce a CPP via naloxone's ability to block KORs. Thus, naloxone via KOR blockade may produce expression of negative reinforcement by removing the tonic aversive state of pain. This approach was chosen as available short acting KOR antagonists did not prevent KOR-induced hypolocomotion in our pilot studies.
Consistent with previous findings, naloxone produced a CPA in pain-naive WT mice that was absent in the PENK KO mice (Fig. 8b). Naloxone also produced a CPA in chronic pain PNI WT mice. However, naloxone elicited a place preference in chronic pain PENK KO mice (Fig. 8b). To determine whether the CPP produced by naloxone was because of antagonism of KORs, we pretreated mice with the KOR antagonist JDTic before conditioning with naloxone. Because expression of a CPP is dependent upon the learned association of drug effects with contextual cues, the acute KOR blocking effects of naloxone would be prevented by pretreatment with the long-acting KOR antagonist JDTic. In this experiment, JDTic prevented naloxone-induced CPP in PNI PENK KO mice but had no effect on naloxone CPA in WT animals (Fig. 8c). These data suggest that the naloxone-induced CPP in chronic pain PENK KO mice was associated with KOR blockade by naloxone. These findings further contribute to the idea that KORs contribute to the ongoing tonic aversive nature of chronic pain.
Figure 8.

The tonic-aversive component of chronic pain is driven by κ opioid receptor activation. a, Time Course of drug treatment and conditioning. b, In naive mice: naloxone (NLX) produced a CPA in WT mice, but not in PENK KO mice. In a model of chronic neuropathic pain induced by PNI, naloxone produced a CPA in WT mice, but produced a CPP in PENK KO mice. Heatmaps of naloxone CPA or CPP are from representative data. Two-way ANOVA revealed a significant effect of genotype (F(1,35) = 113.2, p < 0.001), treatment (F(1,35) = 23.39, p < 0.001), but not interaction (F(1,35) = 0.0295, p = 0.864). *Compares genotype within PNI groups and compares genotype in surgery control mice or between control and PNI in the PENK KO groups. c, In PENK KO mice, administration of the KOR antagonist JDTic (10 mg/kg, i.p.) prevented naloxone CPP in chronic pain animals. Two-way ANOVA revealed a significant effect of surgery (F(1,37) = 4.826, p < 0.05), treatment (F(1,37) = 3.795, p < 0.05), and a significant interaction (F(1,37) = 5.325, p < 0.05). *Compares saline-treated control and PNI groups or saline and JDTic treatments in PNI groups. In PENK WT littermate mice, administration of the KOR antagonist JDTic (10 mg/kg, i.p.) had no effect on naloxone CPA in surgical control or chronic pain animals. Two-way ANOVA revealed no significant effect of surgery (F(1,32) = 0.1763, p = 0.677) or treatment (F(1,32) = 0.6015, p = 0.443). However, all groups showed a significant effect compared with a theoretical value of 0 demonstrating all groups demonstrated a CPA to naloxone (t = 5.52–10.88, **p < 0.01 to ***p < 0.001). Data are expressed as medians with 25 and 75% quartiles, minimum and maximum values with all data points included; N = 5–18 per group. d, Time course of treatment and conditioning paradigm. e, Systemic administration of morphine produced a CPP in sham mice that was not altered by pretreatment with the KOR antagonist JDTic (10 mg/kg, i.p.); however, place preference to morphine was blocked by JDTic in chronic pain mice. Two-way ANOVA revealed a significant effect of surgery (F(1,30) = 4.955, p < 0.05) and treatment (F(1,30) = 8.579, p < 0.01), but not an interaction (F(1,30) = 2.985, p = 0.0943). *Compares saline to JDTic in PNI and JDTic sham and PNI groups. Data are expressed as median with 25 and 75% quartiles, minimum and maximum values with all data points included; N = 8–10 per group.
To provide further evidence that KORs contributed to the tonic-aversive component of chronic pain, we used two additional approaches. Previous research reported that analgesic treatment via a peripheral nerve block or systemic administration of analgesics drugs prevents the expression of morphine-induced CPP (negative reinforcement) in chronic pain animals because of the absence of motivation to seek pain relief (Navratilova et al., 2015). Here, we showed that JDTic effectively blocked expression of CPP produced by systemic administration of morphine in chronic pain, but not sham animals (Fig. 8e). Thus, alleviation of the tonic-aversive component of chronic pain by KOR blockade was sufficient to prevent expression of morphine CPP in pain but not sham animals. We suggest that this outcome results from the primary motivation for morphine CPP being associated with relief from the tonic-aversive component of pain.
KOR mediates a sex-dependent tonic aversive component of chronic pain
Next, we took advantage of a conditioning paradigm where animals were conditioned to only one chamber of the CPP apparatus. Male and female PNI, but not sham, mice showed a CPA to the conditioning chamber, demonstrating the ongoing tonic-aversive component of pain can be captured using this protocol (Fig. 9a,b). In separate cohorts of mice, administration of JDTic 24 h before the beginning of conditioning prevented place aversion in male, but not female PNI mice (Fig. 9a). To test the generalizability of KOR contributions to the ongoing tonic-aversive component of pain in other models, we performed similar experiments in male mice injected with CFA to induce chronic inflammatory pain. Using this model, CFA also produced a place aversion that was blocked by JDTic (Fig. 9b). It is noteworthy that KOR inhibition had no effect on mechanical withdrawal thresholds in sham or chronic pain male or female mice (Fig. 9c). These findings further implicate KOR involvement in pain circuitry is restricted to the affective but not sensory dimension of chronic pain.
Figure 9.

Sex-dependent KOR involvement in the ongoing tonic-aversive component of chronic pain. a, Time course and treatment regimen of single chamber conditioning to capture place aversion of sham and chronic pain (PNI) mice. Mice received saline (i.p.) injections daily in their home cage or in one conditioning chamber. Sham animals conditioned to the CPP apparatus did not show place aversion or preference to the chamber where they received saline but PNI male and female mice developed an aversion to the conditioning chamber. This aversion demonstrates ongoing tonic aversive component of chronic pain. Treating male, but not female mice with the KOR antagonist JDTic (10 mg/kg, i.p.) prevented aversion to the conditioning chamber. Male: two-way ANOVA revealed a significant effect of treatment (F(1,28) = 9.923, p < 0.01), an interaction (F(1,28) = 4.781, p < 0.05), and an effect of surgery (F(1,28) = 3.581, p < 0.05). *p < 0.05 compares saline-treated control and PNI groups as well as control surgery and PNI in vehicle-treated groups. Female: two-way ANOVA revealed a significant effect of surgery (F(1,28) = 8.346, p < 0.01), but no interaction (F(1,28) = 0.150, p = 0.701) or treatment effect (F(1,28) = 0.241, p = 0.627). *Compares saline-treated control and PNI groups as well as control surgery and PNI in vehicle-treated groups. Data are expressed as median with 25 and 75% quartiles, minimum and maximum values with all data points included; N = 8 per group. b, Time course and treatment regimen of conditioning to capture place aversion after CFA. Sham animals conditioned to CPP apparatus did not show place aversion or preference to chamber that they received saline but CFA male mice developed an aversion to the conditioning chamber. Treating mice with KOR antagonist JDTic (10 mg/kg, i.p.) prevented this aversive state. Two-way ANOVA revealed a significant effect of surgery (F(1,28) = 5.281, p < 0.05) and treatment (F(1,28) = 12.76, p < 0.01), but no interaction (F(1,28) = 2.192, p = 1.499). *Compares saline-treated control and PNI groups. Data are expressed as median with 25 and 75% quartiles, minimum and maximum values with all data points included; N = 8 per group. c, Mechanical withdrawal thresholds determined at 2 weeks postsurgery were significantly lower in PNI compared with sham mice. Administration of JDTic (10 mg/kg, i.p.) 24 h before testing did not alter withdrawal thresholds in PNI or sham mice. Two-way ANOVA revealed a significant effect of surgery (F(1,28) = 111.5, p < 0.001), but not drug treatment (F(1,28) = 0.356, p = 0.556), and there was no significant interaction (F(1,28) = 0.167, p = 0.686). Data are expressed as median with 25 and 75% quartiles, minimum and maximum values with all data points included; N = 8 per group.
We examined the circuitry involved in the tonic-aversive component of pain using KOR conditional KO (cKO) mice. Here, AAV-TH-cre or control virus (AAV-eGFP) was injected into the VTA of KOR cKO mice. As an additional control, AAV-TH-cre was injected into WT C57BL/6 mice 3 weeks before inducing chronic neuropathic pain followed by single chamber conditioning. The RNAscope technique confirmed elimination of KORs from TH+ dopaminergic neurons in KOR cKO, but not WT mice (Fig. 10). The PNI-induced place aversion was present in WT mice injected with TH-cre-virus as well as in KOR cKO mice injected with control virus. However, place aversion was absent in KOR cKO mice where KOR expression was significantly reduced within VTA TH+ neurons (Fig. 10). Together, these results provide strong evidence that KORs in dopaminergic mesolimbic circuits contribute to the tonic-aversive component of chronic pain.
Figure 10.

Deleting KOR from mesolimbic dopamine prevents an ongoing pain aversive state. Time course of conditioning and AAV-TH-cre treatment in KOR cKO or C57BL/6 mice. Elimination of KOR from VTA TH+ neurons prevented place aversion in male PNI mice. t(12) = 3.46, *p < 0.05. Data are expressed as median with 25 and 75% quartiles, minimum and maximum values with all data points included for N = 4–8 per group. Bottom, Represented images demonstrate the absence of KOR mRNA in the VTA of TH-cre/KORloxP but not control mouse groups.
KOR antagonists attenuate negative affect in chronic pain states but is not sex dependent
The findings described above suggest sex differences in the involvement of KORs in the affective dimension of pain. We therefore determined whether similar sex differences were evident in other measures of affect. Others have reported anti-depressant-like and anti-anxiety-like effects of KOR antagonists in rodent models of affective-like behavior (Crowley and Kash, 2015). The PNI model of chronic pain induces both anxiogenic and depressive phenotypes in mice (Yalcin et al., 2011). The administration of JDTic increased percentage time in the light compartment of the light/dark test (Fig. 11a) and decreased immobility time in the forced-swim test (Fig. 11b) in PNI male and female mice, demonstrating that inhibition of KORs improves anxiety-like and affective-like deficits in behavior of chronic pain animals. Nonetheless, we did not observe sex-dependent effects in these tests. Thus, there is a divergence in mechanisms underlying the ongoing tonic-aversive component of pain compared with other measures of affect often precipitated by pain.
Figure 11.

No sex-dependent effect of KOR antagonists to block pain-induced affective like behavior. a, JDTic (10 mg/kg, i.p.) significantly increased the percentage time in the light compartment of the light dark anxiety-like affective-like test of both male and female PNI mice. Male: two-way ANOVA reveals a significant effect of surgery (F(1,76) = 9.997, p = 0.0023) and an interaction (F(1,76) = 5.747, p = 0.0190), but not of treatment (F(1,76) = 0.0513, p = 0.8214). Female: two-way ANOVA reveals a significant effect of surgery (F(1,44) = 6.787, p = 0.0125) and an interaction (F(1,44) = 10.08, p = 0.0027), but not of treatment (F(1,44) = 1.663, p = 0.2040). Data are expressed as raw data and mean ± SEM for N = 10–24 per group. Male: #compares sham to PNI JDTic treatment, female: *compares sham to PNI saline-treated mice, $compares PNI saline to PNI JDTic treatment. b, JDTic (10 mg/kg, i.p.) significantly decreased the immobility time in the forced swim depressive-like affective-like test of male and female PNI mice. Male: two-way ANOVA reveals a significant effect of surgery (F(1,72) = 9.077, p = 0.0036) and treatment (F(1,72) = 23.57, p < 0001), and an interaction (F(1,72) = 4.659, p = 0.0125). Female: two-way ANOVA reveals a significant effect of surgery (F(1,44) = 8.147, p = 0.0066) and treatment (F(1,44) = 14.4, p = 0.0004), but not an interaction (F(1,44) = 0.867, p = 0.3569). Data are expressed as raw data and mean ± SEM for N = 10–24 per group. Male: *compares PNI to naive, #compares sham to PNI, $compares to PNI saline to PNI JDTic-treated mice. Female: #compares sham to PNI JDTic-treated mice.
Discussion
The present study provides evidence that chronic pain increases KOR expression and function within the mesolimbic circuitry that contributes to a tonic-aversive component of pain in a sex-dependent manner. We have previously shown that KOR agonist-induced anti-nociception in male, but not female mice, was mediated by engagement of stress pathways, as analgesia was blocked by anxiolytics (Taylor et al., 2015b). Sex differences in KOR function have been reported in human and animal studies. Indeed, PET studies in humans show that KOR binding is greater in men than women in multiple brain regions, including areas associated with pain affect such as the anterior cingulate cortex (Vijay et al., 2016). Differences between sexes can occur as a result of hormonal differences, either during development or in adulthood, or as a result of genetic differences due to the complement of genes expressed on the X or Y chromosomes. Because the type of gonadal hormones is closely tied to the complement of sex chromosomes (XX vs XY), it has been difficult to separate the effects of each of these variables. However, both hormones and sex chromosomes have been implicated in KOR-mediated sex differences. For example, the sex-dependent differences in sensitivity to the depressive effects of KOR agonists in a model of intracranial self-stimulation was independent of estrous cycle state in females or gonadectomy in males (Russell et al., 2014). In contrast, KOR-mediated anti-nociception was enhanced by hormone treatment (Dawson-Basoa and Gintzler, 1996) and the sex-specific effects of KOR-mediated anti-nociception in male but not female mice was shown to be mediated by estrous phase (Abraham et al., 2018).
Considerable evidence suggests that the KOR system within the NAc underlies negative affective states and heightens stress reactivity in various psychiatric disorders. For example, DYN expression is increased in the ventral striatum of suicidal individuals and in animal models of depression (Hurd et al., 1997; Peckys and Hurd, 2001). Similarly, the aversive effects produced by KOR agonists are partially mediated by their role in the NAc, where they modulate dopamine transmission from the VTA (Van't Veer et al., 2013). However, no prior study has shown the tonic-aversive component of chronic pain is mediated by KORs. Our study demonstrates that KOR modulation of dopamine release underlies the tonic aversive component of pain, but it unclear whether this modulation is because of regulation of dopamine release within the NAc or other projection sites. We previously demonstrated that intra-VTA opioid reward and evoked dopamine release within the NAc are blunted in chronic pain states (Taylor et al., 2015a). Here we show that the blunted release and reward are recovered by KOR antagonists, however the source of DYN responsible for KOR activation within the NAc remains unclear. We show that Pdyn mRNA is increased in the NAc of chronic pain mice suggesting a likely source is DYN-expressing medium spiny neurons (Al-Hasani et al., 2015). However, DYN regulation of dopamine may occur via another system; it was recently hypothesized that another DYN source may be orexin neurons projecting from the lateral hypothalamus (Tejeda and Bonci, 2019). It is important to note that KOR activation does not contribute to aversion to an acute pain stimulus. Neither administration of a KOR agonist or antagonist prevented visceral pain (acetic acid)-induced place aversion (Bagdas et al., 2016). Additionally, acute inflammatory or visceral pain was reported to reduce dopamine release in the NAc and to depress intracranial self-stimulation (Leitl et al., 2014a,b). However, neither effect was reversed by pretreatment with a KOR antagonist. We propose that the KOR system may only be engaged following tissue or nerve injury that induces a prolonged or chronic, but not acute pain states.
The possibility exists that the ability of KOR inhibition to recover dopamine release and opioid reward may increase risk of opioid misuse and addiction. Although a complex issue, KOR antagonism or absence of KORs increases drug-induced dopamine release (Chefer et al., 2005; Doyon et al., 2006), as well as increases nicotine, methamphetamine or ethanol reward as determined by conditioned place preference and self-administration studies (Matsuzawa et al., 1999; Mitchell et al., 2005; Whitfield et al., 2015). Nevertheless, KOR antagonists are being developed for the treatment of addiction because of their ability to block stress and cue-induced relapse, where the rationale for such treatment is driven in part by the ability of KOR antagonists to block stress-related pathways (Crowley and Kash, 2015; Chavkin and Koob, 2016).
It is not clear whether restoring dopamine release and dopamine-dependent reward would increase addiction potential in chronic pain states, as reward is not solely dependent on dopamine. More importantly, opioid use for pain treatment is associated with negative reinforcement rather than increasing hedonic tone. For example, rodents in chronic pain that undergo pain treatment with gabapentin or other analgesics do not show place preference for morphine (Navratilova et al., 2015). In clinical studies, opioid misuse and addiction was shown to be lower in chronic pain patients than the general population receiving opioids, if susceptible individuals (comorbid mood disorder or psychopathology, genetic susceptibility or previous substance misuse) were separated from the cohort (Fishbain et al., 2008). Because hypo-dopaminergic tone in chronic pain impairs motivated behavior and likely contributes to the occurrence of psychopathology commonly comorbid with chronic pain (Carroll and Carlezon, 2013; Taylor et al., 2015b, 2016), the ability of KOR antagonists to restore dopamine signaling may represent a novel approach to manage affective dimensions of chronic pain. This mechanism may underlie the effectiveness of buprenorphine (a partial μ opioid agonist with KOR antagonist properties) in alleviating chronic pain and improving quality of life (Uberall and Müller-Schwefe, 2013), and may also explain why buprenorphine when combined with naloxone for treatment of opioid addiction in chronic pain patients also decreased subjective pain (Worley et al., 2015).
Conclusions
We have discovered in two animal models of chronic pain that the expression and function of KOR systems are increased in mesolimbic brain structures. Whereas the affective and sensory components of pain are mediated by overlapping circuitry, limbic structures including mesolimbic circuitry are engaged to process the emotional component of pain. Considerable evidence suggests that the κ opioid systems within the NAc underlies negative affective states and heightens stress reactivity in various psychiatric disorders. Here we show that κ opioid receptors are sufficient to drive the tonic-aversive component of chronic pain; this emotional component of pain is argued to significantly impact patients' quality of life (compared with the sensory component).
These findings represent a significant contribution to understanding mechanisms of chronic pain because there is considerable research interest in validating peripherally restricted κ agonists as a novel analgesics, whereas our data suggest that κ blockade might prove effective. A case in point is that opioids remain one of our most effective pharmacotherapeutic agents in treating pain, which is likely because of their ability to blunt both the sensory and emotional components of pain. In fact, many patients indicate they still feel pain when taking opioids but the pain no longer bothers them. Thus, novel analgesic treatments must focus on alleviating the emotional component of pain if we are ever to replace opioids for treating long-term pain. The impact of the present study is also broadly relevant to affective disorders associated with disruption of reward circuitry and thus, likely contributes to many of the devastating sequelae of chronic pain, including the poor response to treatment of come patients, debilitating affective disorders and substance abuse. Because KORs are upregulated in areas of the CNS involved in mood, our findings likely bear on other disorders, including anxiety disorders and depression that demonstrate high comorbidity with chronic pain. Moreover, KORs are known to be involved in stress-induced relapse of drugs of abuse. Therefore, KOR antagonists may reduce prescription opioid misuse in pain patient populations.
Footnotes
This work was supported by a generous gift from Shirley Hatos to C.M.C., start-up funds from the University of California Irvine (C.M.C.), the Shirley and Stefan Hatos Foundation (C.J.E., A.M.W.T.), NIH DA005010 (C.J.E., A.M.W.T.), and Canadian Institutes of Health Research (C.M.C., M.C.O., MOP 123298; T.T., MOP133523); by the Cousins Center for Psychoneuroimmunology and NIH K99DA040016 to A.M.W.T.; by the UCI Graduate Division Public Impact Fellowship and the School of Medicine Outstanding Student Fellowship to S.L., and by a Predoctoral Fellowship from the Basque Government to I.I.-L. We thank Patrick Hullihen, Priscilla Orellana, Eric Thai, Vicki Tea, Jimmy Nguyen, Lauren Wong, Jacob Alderete, Reem Karmouta, and Angeli Mae C. Nazareth for technical assistance, and Dr. Michael Bruchas at University of Washington for the generous gift from KOR-P antibody.
The authors declare no competing financial interests.
References
- Abraham AD, Schattauer SS, Reichard KL, Cohen JH, Fontaine HM, Song AJ, Johnson SD, Land BB, Chavkin C (2018) Estrogen regulation of GRK2 inactivates kappa opioid receptor signaling mediating analgesia, but not aversion. J Neurosci 38:8031–8043. 10.1523/JNEUROSCI.0653-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, Pyo CO, Park SI, Marcinkiewcz CM, Crowley NA, Krashes MJ, Lowell BB, Kash TL, Rogers JA, Bruchas MR (2015) Distinct subpopulations of nucleus accumbens dynorphin neurons drive aversion and reward. Neuron 87:1063–1077. 10.1016/j.neuron.2015.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmundson GJ, Katz J (2009) Understanding the co-occurrence of anxiety disorders and chronic pain: state-of-the-art. Depress Anxiety 26:888–901. 10.1002/da.20600 [DOI] [PubMed] [Google Scholar]
- Bagdas D, Muldoon PP, AlSharari S, Carroll FI, Negus SS, Damaj MI (2016) Expression and pharmacological modulation of visceral pain-induced conditioned place aversion in mice. Neuropharmacology 102:236–243. 10.1016/j.neuropharm.2015.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bair MJ, Robinson RL, Katon W, Kroenke K (2003) Depression and pain comorbidity: a literature review. Arch Intern Med 163:2433–2445. 10.1001/archinte.163.20.2433 [DOI] [PubMed] [Google Scholar]
- Berridge KC, Kringelbach ML (2013) Neuroscience of affect: brain mechanisms of pleasure and displeasure. Curr Opin Neurobiol 23:294–303. 10.1016/j.conb.2013.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge KC, Kringelbach ML (2015) Pleasure systems in the brain. Neuron 86:646–664. 10.1016/j.neuron.2015.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsook D, Linnman C, Faria V, Strassman AM, Becerra L, Elman I (2016) Reward deficiency and anti-reward in pain chronification. Neurosci Biobehav Rev 68:282–297. 10.1016/j.neubiorev.2016.05.033 [DOI] [PubMed] [Google Scholar]
- Cahill CM, Taylor AM (2017) Neuroinflammation: a co-occurring phenomenon linking chronic pain and opioid dependence. Curr Opin Behav Sci 13:171–177. 10.1016/j.cobeha.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, Xue L, Grenier P, Magnussen C, Lecour S, Olmstead MC (2013) Changes in morphine reward in a model of neuropathic pain. Behav Pharmacol 24:207–213. 10.1097/FBP.0b013e3283618ac8 [DOI] [PubMed] [Google Scholar]
- Cahill CM, Cook C, Pickens S (2014a) Migraine and reward system: or is it aversive? Curr Pain Headache Rep 18:410. 10.1007/s11916-014-0410-y [DOI] [PubMed] [Google Scholar]
- Cahill CM, Taylor AM, Cook C, Ong E, Morón JA, Evans CJ (2014b) Does the kappa opioid receptor system contribute to pain aversion? Front Pharmacol 5:253. 10.3389/fphar.2014.00253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll FI, Carlezon WA Jr (2013) Development of kappa opioid receptor antagonists. J Med Chem 56:2178–2195. 10.1021/jm301783x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoff EH, Mavrikaki M (2015) Sex differences in kappa opioid receptor function and their potential impact on addiction. Front Neurosci 9:466. 10.3389/fnins.2015.00466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoff EH, Ebner SR, Sparrow A, Potter D, Baker PM, Ragozzino ME, Roitman MF (2016) Relative timing between kappa opioid receptor activation and cocaine determines the impact on reward and dopamine release. Neuropsychopharmacology 41:989–1002. 10.1038/npp.2015.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, Koob GF (2016) Dynorphin, dysphoria, and dependence: the stress of addiction. Neuropsychopharmacology 41:373–374. 10.1038/npp.2015.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, Martinez D (2015) Kappa antagonist JDTic in phase 1 clinical trial. Neuropsychopharmacology 40:2057–2058. 10.1038/npp.2015.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefer VI, Czyzyk T, Bolan EA, Moron J, Pintar JE, Shippenberg TS (2005) Endogenous kappa-opioid receptor systems regulate mesoaccumbal dopamine dynamics and vulnerability to cocaine. J Neurosci 25:5029–5037. 10.1523/JNEUROSCI.0854-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefer VI, Bäckman CM, Gigante ED, Shippenberg TS (2013) Kappa opioid receptors on dopaminergic neurons are necessary for kappa-mediated place aversion. Neuropsychopharmacology 38:2623–2631. 10.1038/npp.2013.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos EJ, Portillo-Salido E (2013) “Bedside-to-bench” behavioral outcomes in animal models of pain: beyond the evaluation of reflexes. Curr Neuropharmacol 11:560–591. 10.2174/1570159X113119990041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codd EE, Shank RP, Schupsky JJ, Raffa RB (1995) Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J Pharmacol Exp Ther 274:1263–1270. [PubMed] [Google Scholar]
- Crowley NA, Kash TL (2015) Kappa opioid receptor signaling in the brain: circuitry and implications for treatment. Prog Neuropsychopharmacol Biol Psychiatry 62:51–60. 10.1016/j.pnpbp.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson-Basoa ME, Gintzler AR (1996) Estrogen and progesterone activate spinal kappa-opiate receptor analgesic mechanisms. Pain 64:608–615. 10.1016/0304-3959(96)87175-2 [DOI] [PubMed] [Google Scholar]
- Deehan GA Jr, McKinzie DL, Carroll FI, McBride WJ, Rodd ZA (2012) The long-lasting effects of JDTic, a kappa opioid receptor antagonist, on the expression of ethanol-seeking behavior and the relapse drinking of female alcohol-preferring (P) rats. Pharmacol Biochem Behav 101:581–587. 10.1016/j.pbb.2012.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon WM, Howard EC, Shippenberg TS, Gonzales RA (2006) Kappa-opioid receptor modulation of accumbal dopamine concentration during operant ethanol self-administration. Neuropharmacology 51:487–496. 10.1016/j.neuropharm.2006.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymshitz J, Lieblich I (1987) Opiate reinforcement and naloxone aversion, as revealed by place preference paradigm, in two strains of rats. Psychopharmacology 92:473–477. [DOI] [PubMed] [Google Scholar]
- Ebner SR, Roitman MF, Potter DN, Rachlin AB, Chartoff EH (2010) Depressive-like effects of the kappa opioid receptor agonist salvinorin A are associated with decreased phasic dopamine release in the nucleus accumbens. Psychopharmacology 210:241–252. 10.1007/s00213-010-1836-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, Zweifel LS, Kieffer BL, Phillips PE, Chavkin C (2015) Kappa opioid receptor-induced aversion requires p38 MAPK activation in VTA dopamine neurons. J Neurosci 35:12917–12931. 10.1523/JNEUROSCI.2444-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elman I, Borsook D, Volkow ND (2013) Pain and suicidality: insights from reward and addiction neuroscience. Prog Neurobiol 109:1–27. 10.1016/j.pneurobio.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CJ, Cahill CM (2016) Neurobiology of opioid dependence in creating addiction vulnerability. F1000Research 5:1748. 10.12688/f1000research.8369.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillingim RB, Bruehl S, Dworkin RH, Dworkin SF, Loeser JD, Turk DC, Widerstrom-Noga E, Arnold L, Bennett R, Edwards RR, Freeman R, Gewandter J, Hertz S, Hochberg M, Krane E, Mantyh PW, Markman J, Neogi T, Ohrbach R, Paice JA, et al. (2014) The ACTTION-American pain society pain taxonomy (AAPT): an evidence-based and multidimensional approach to classifying chronic pain conditions. J Pain 15:241–249. 10.1016/j.jpain.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbain DA, Cole B, Lewis J, Rosomoff HL, Rosomoff RS (2008) What percentage of chronic nonmalignant pain patients exposed to chronic opioid analgesic therapy develop abuse/addiction and/or aberrant drug-related behaviors? A structured evidence-based review. Pain Med 9:444–459. 10.1111/j.1526-4637.2007.00370.x [DOI] [PubMed] [Google Scholar]
- Gompf HS, Budygin EA, Fuller PM, Bass CE (2015) Targeted genetic manipulations of neuronal subtypes using promoter-specific combinatorial AAVs in wild-type animals. Front Behav Neurosci 9:152. 10.3389/fnbeh.2015.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassett AL, Aquino JK, Ilgen MA (2014) The risk of suicide mortality in chronic pain patients. Curr Pain Headache Rep 18:436. 10.1007/s11916-014-0436-1 [DOI] [PubMed] [Google Scholar]
- Heidbreder CA, Goldberg SR, Shippenberg TS (1993) The kappa-opioid receptor agonist U-69593 attenuates cocaine-induced behavioral sensitization in the rat. Brain Res 616:335–338. 10.1016/0006-8993(93)90228-F [DOI] [PubMed] [Google Scholar]
- Howe CQ, Sullivan MD (2014) The missing “P” in pain management: how the current opioid epidemic highlights the need for psychiatric services in chronic pain care. Gen Hosp Psychiatry 36:99–104. 10.1016/j.genhosppsych.2013.10.003 [DOI] [PubMed] [Google Scholar]
- Hurd YL, Herman MM, Hyde TM, Bigelow LB, Weinberger DR, Kleinman JE (1997) Prodynorphin mRNA expression is increased in the patch vs matrix compartment of the caudate nucleus in suicide subjects. Mol Psychiatry 2:495–500. 10.1038/sj.mp.4000319 [DOI] [PubMed] [Google Scholar]
- Illiano P, Bass CE, Fichera L, Mus L, Budygin EA, Sotnikova TD, Leo D, Espinoza S, Gainetdinov RR (2017) Recombinant adeno-associated virus-mediated rescue of function in a mouse model of dopamine transporter deficiency syndrome. Sci Rep 7:46280. 10.1038/srep46280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamison RN, Edwards RR (2013) Risk factor assessment for problematic use of opioids for chronic pain. Clin Neuropsychol 27:60–80. 10.1080/13854046.2012.715204 [DOI] [PubMed] [Google Scholar]
- Karper PE, Nazarian A, Crawford CA, Drago J, McDougall SA (2000) Role of dopamine D1 receptors for kappa-opioid-mediated locomotor activity and antinociception during the preweanling period: a study using D1 receptor knockout mice. Physiol Behav 68:585–590. 10.1016/S0031-9384(99)00223-1 [DOI] [PubMed] [Google Scholar]
- Knoll AT, Carlezon WA Jr (2010) Dynorphin, stress, and depression. Brain Res 1314:56–73. 10.1016/j.brainres.2009.09.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- König M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ, Zimmer A (1996) Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature 383:535–538. 10.1038/383535a0 [DOI] [PubMed] [Google Scholar]
- Leitl MD, Onvani S, Bowers MS, Cheng K, Rice KC, Carlezon WA Jr, Banks ML, Negus SS (2014a) Pain-related depression of the mesolimbic dopamine system in rats: expression, blockade by analgesics, and role of endogenous κ-opioids. Neuropsychopharmacology 39:614–624. 10.1038/npp.2013.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitl MD, Potter DN, Cheng K, Rice KC, Carlezon WA Jr, Negus SS (2014b) Sustained pain-related depression of behavior: effects of intraplantar formalin and complete Freund's adjuvant on intracranial self-stimulation (ICSS) and endogenous kappa opioid biomarkers in rats. Mol Pain 10:62. 10.1186/1744-8069-10-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel MO, Dolman AJ, Edwards RR, Jamison RN, Wasan AD (2014) The association between negative affect and prescription opioid misuse in patients with chronic pain: the mediating role of opioid craving. J Pain 15:90–100. 10.1016/j.jpain.2013.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa S, Suzuki T, Misawa M, Nagase H (1999) Different roles of mu-, delta- and kappa-opioid receptors in ethanol-associated place preference in rats exposed to conditioned fear stress. Eur J Pharmacol 368:9–16. 10.1016/S0014-2999(99)00008-4 [DOI] [PubMed] [Google Scholar]
- McBride WJ, Murphy JM, Ikemoto S (1999) Localization of brain reinforcement mechanisms: intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res 101:129–152. 10.1016/S0166-4328(99)00022-4 [DOI] [PubMed] [Google Scholar]
- Mitchell JM, Liang MT, Fields HL (2005) A single injection of the kappa opioid antagonist norbinaltorphimine increases ethanol consumption in rats. Psychopharmacology 182:384–392. 10.1007/s00213-005-0067-7 [DOI] [PubMed] [Google Scholar]
- Mogil JS, Crager SE (2004) What should we be measuring in behavioral studies of chronic pain in animals? Pain 112:12–15. 10.1016/j.pain.2004.09.028 [DOI] [PubMed] [Google Scholar]
- Munro TA, Berry LM, Van't Veer A, Béguin C, Carroll FI, Zhao Z, Carlezon WA Jr, Cohen BM (2012) Long-acting kappa opioid antagonists nor-BNI, GNTI and JDTic: pharmacokinetics in mice and lipophilicity. BMC Pharmacol 12:5. 10.1186/1471-2210-12-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Kishimoto Y, Ise Y, Yajima Y, Misawa K, Suzuki T (2005) Direct evidence for the involvement of the mesolimbic kappa-opioid system in the morphine-induced rewarding effect under an inflammatory pain-like state. Neuropsychopharmacology 30:111–118. 10.1038/sj.npp.1300527 [DOI] [PubMed] [Google Scholar]
- Narita M, Matsushima Y, Niikura K, Narita M, Takagi S, Nakahara K, Kurahashi K, Abe M, Saeki M, Asato M, Imai S, Ikeda K, Kuzumaki N, Suzuki T (2010) Implication of dopaminergic projection from the ventral tegmental area to the anterior cingulate cortex in mu-opioid-induced place preference. Addict Biol 15:434–447. 10.1111/j.1369-1600.2010.00249.x [DOI] [PubMed] [Google Scholar]
- Navratilova E, Porreca F (2014) Reward and motivation in pain and pain relief. Nat Neurosci 17:1304–1312. 10.1038/nn.3811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova E, Atcherley CW, Porreca F (2015) Brain circuits encoding reward from pain relief. Trends Neurosci 38:741–750. 10.1016/j.tins.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckys D, Hurd YL (2001) Prodynorphin and kappa opioid receptor mRNA expression in the cingulate and prefrontal cortices of subjects diagnosed with schizophrenia or affective disorders. Brain Res Bull 55:619–624. 10.1016/S0361-9230(01)00525-1 [DOI] [PubMed] [Google Scholar]
- Percie du Sert N, Rice AS (2014) Improving the translation of analgesic drugs to the clinic: animal models of neuropathic pain. Br J Pharmacol 171:2951–2963. 10.1111/bph.12645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M (1977) Depression: a new animal model sensitive to antidepressant treatments. Nature 266:730–732. 10.1038/266730a0 [DOI] [PubMed] [Google Scholar]
- Russell SE, Rachlin AB, Smith KL, Muschamp J, Berry L, Zhao Z, Chartoff EH (2014) Sex differences in sensitivity to the depressive-like effects of the kappa opioid receptor agonist U-50488 in rats. Biol Psychiatry 76:213–222. 10.1016/j.biopsych.2013.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippenberg TS, Bals-Kubik R, Herz A (1993) Examination of the neurochemical substrates mediating the motivational effects of opioids: role of the mesolimbic dopamine system and D-1 vs. D-2 dopamine receptors. J Pharmacol Exp Ther 265:53–59. [PubMed] [Google Scholar]
- Shoblock JR, Maidment NT (2007) Enkephalin release promotes homeostatic increases in constitutively active mu opioid receptors during morphine withdrawal. Neuroscience 149:642–649. 10.1016/j.neuroscience.2007.05.011 [DOI] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS (1992) Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci U S A 89:2046–2050. 10.1073/pnas.89.6.2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM. (2013) Pain and reward: how the affective–motivational system is perturbed in chronic pain. Postdoc J 1:75–88. 10.14304/SURYA.JPR.V1N2.8 [DOI] [Google Scholar]
- Taylor AM, Murphy NP, Evans CJ, Cahill CM (2014) Correlation between ventral striatal catecholamine content and nociceptive thresholds in neuropathic mice. J Pain 15:878–885. 10.1016/j.jpain.2014.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Castonguay A, Taylor AJ, Murphy NP, Ghogha A, Cook C, Xue L, Olmstead MC, De Koninck Y, Evans CJ, Cahill CM (2015a) Microglia disrupt mesolimbic reward circuitry in chronic pain. J Neurosci 35:8442–8450. 10.1523/JNEUROSCI.4036-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Roberts KW, Pradhan AA, Akbari HA, Walwyn W, Lutfy K, Carroll FI, Cahill CM, Evans CJ (2015b) Anti-nociception mediated by a kappa opioid receptor agonist is blocked by a delta receptor agonist. Br J Pharmacol 172:691–703. 10.1111/bph.12810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Becker S, Schweinhardt P, Cahill C (2016) Mesolimbic dopamine signaling in acute and chronic pain: implications for motivation, analgesia, and addiction. Pain 157:1194–1198. 10.1097/j.pain.0000000000000494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Bonci A (2019) Dynorphin/kappa-opioid receptor control of dopamine dynamics: implications for negative affective states and psychiatric disorders. Brain Res 1713:91–101. 10.1016/j.brainres.2018.09.023 [DOI] [PubMed] [Google Scholar]
- Tejeda HA, Hanks AN, Scott L, Mejias-Aponte C, Hughes ZA, O'Donnell P (2015) Prefrontal cortical kappa opioid receptors attenuate responses to amygdala inputs. Neuropsychopharmacology 40:2856–2864. 10.1038/npp.2015.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uberall MA, Müller-Schwefe GH (2013) Long-term treatment of chronic pain with low-dose 7-day buprenorphine transdermal patch: observational data from elderly patients of pain relief and quality of life. MMW Fortschr Med 155:87–96. 10.1007/s15006-013-2188-y [DOI] [PubMed] [Google Scholar]
- Van't Veer A, Bechtholt AJ, Onvani S, Potter D, Wang Y, Liu-Chen LY, Schütz G, Chartoff EH, Rudolph U, Cohen BM, Carlezon WA Jr (2013) Ablation of kappa-opioid receptors from brain dopamine neurons has anxiolytic-like effects and enhances cocaine-induced plasticity. Neuropsychopharmacology 38:1585–1597. 10.1038/npp.2013.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van't Veer A, Carlezon WA Jr (2013) Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology 229:435–452. 10.1007/s00213-013-3195-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay A, Wang S, Worhunsky P, Zheng MQ, Nabulsi N, Ropchan J, Krishnan-Sarin S, Huang Y, Morris ED (2016) PET imaging reveals sex differences in kappa opioid receptor availability in humans, in vivo. Am J Nucl Med Mol Imaging 6:205–214. [PMC free article] [PubMed] [Google Scholar]
- Whitfield TW Jr, Schlosburg JE, Wee S, Gould A, George O, Grant Y, Zamora-Martinez ER, Edwards S, Crawford E, Vendruscolo LF, Koob GF (2015) κ Opioid receptors in the nucleus accumbens shell mediate escalation of methamphetamine intake. J Neurosci 35:4296–4305. 10.1523/JNEUROSCI.1978-13.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley MJ, Heinzerling KG, Shoptaw S, Ling W (2015) Pain volatility and prescription opioid addiction treatment outcomes in patients with chronic pain. Exp Clin Psychopharmacol 23:428–435. 10.1037/pha0000039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC (2012) Structure of the human kappa-opioid receptor in complex with JDTic. Nature 485:327–332. 10.1038/nature10939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Woller SA, Ramachandran R, Sorkin LS (2015) The search for novel analgesics: targets and mechanisms. F1000Prime Rep 7:56. 10.12703/P7-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin I, Barrot M (2014) The anxiodepressive comorbidity in chronic pain. Curr Opin Anaesthesiol 27:520–527. 10.1097/ACO.0000000000000116 [DOI] [PubMed] [Google Scholar]
- Yalcin I, Bohren Y, Waltisperger E, Sage-Ciocca D, Yin JC, Freund-Mercier MJ, Barrot M (2011) A time-dependent history of mood disorders in a murine model of neuropathic pain. Biol Psychiatry 70:946–953. 10.1016/j.biopsych.2011.07.017 [DOI] [PubMed] [Google Scholar]
- Yang H, Thompson AB, McIntosh BJ, Altieri SC, Andrews AM (2013) Physiologically relevant changes in serotonin resolved by fast microdialysis. ACS Chem Neurosci 4:790–798. 10.1021/cn400072f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Sampson MM, Senturk D, Andrews AM (2015) Sex- and SERT-mediated differences in stimulated serotonin revealed by fast microdialysis. ACS Chem Neurosci 6:1487–1501. 10.1021/acschemneuro.5b00132 [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Khakpai F (2015) The modulatory role of dopamine in anxiety-like behavior. Arch Iran Med 18:591–603. [PubMed] [Google Scholar]