

Abstract
Caspy2, a zebrafish protease, is an active caspase for inducing apoptosis in mammalian cells. To investigate whether caspy2‐mediated apoptosis could be used in cancer therapy, its cDNA was amplified and cloned into eukaryotic expression vector pcDNA3.1+. The recombinant plasmid was mixed with cationic liposome and introduced into various tumour cell lines in vitro. Our data showed that caspy2 induced remarkable apoptosis of cancer cells in vitro. Treatment of mice‐bearing CT26 colon carcinoma or MethA fibrosarcoma with intratumoral injection of liposome‐caspy2 plasmid complex resulted in substantial killing of neoplastic cells and long‐term survivors. Apoptotic cells were widely distributed in caspy2‐treated tumour tissue. Infiltration of CD8+ T lymphocyte was also observed apparently in the tumour tissue after the treatment with caspy2. Tumour‐specific major histocompatibility complex (MHC) class I‐dependent CD8+ cytotoxic T lymphocyte activity was found by means of 51Cr release assay. In MethA model, the mice acquired a long‐time protective immunity against the parental tumour cell re‐challenge. These results indicated that caspy2 can act as both apoptosis inducer and immune response initiator, which may account for its extraordinary antitumour effect. Furthermore, in vivo depletion of CD8+ T lymphocytes could completely abrogate the antitumour immune activity, whereas the depletion of CD4+ cells showed partial abrogation. In this study, we developed a novel anticancer strategy that combines both induction of apoptosis and immune response in mice‐bearing tumours with a single caspy2 gene. This approach may provide an important way for treatment of cancer.
Keywords: apoptosis, immune response, plasmid DNA, cationic liposome, dual effect
Introduction
Apoptosis is a form of cell death mediated by an intracellular suicide program, which is often associated with characteristic morphological and biochemical changes, such as membrane blebbing, cell shrinkage, chromatin condensation and fragmentation of genomic DNA [1, 2]. Cancer cells have an elevated apoptotic threshold, and the induction of apoptosis in cancer cells has been referred to as an ‘Achilles’ heel approach [3]. Many types of cancer therapeutic agents currently in use induce apoptosis of cancer cells and thus regression of malignancies [4, 5]. Meanwhile it has long been suggested that apoptotic cell death is poorly immunogenic or even tolerogenic whereas necrotic cell death is truly immunogenic [6, 7]. The fate of apoptotic material is rapid clearance and degradation by phagocytes [8]. Nonetheless, it seems that the dichotomy between immunogenic necrosis versus tolerogenic apoptosis is an oversimplification. There is, however, growing evidence that apoptotic death was a critical trigger for the antigen‐processing pathway, and apoptotic cells were a preferred source of antigen for the induction of T‐cell immunity [9, 10, 11, 12]. Moreover, the capacity of apoptotic tumour cells to trigger the immune response was found to depend on the death‐inducing stimulus [13]. Therefore, apoptotic tumour cells can theoretically be a candidate for stimulation of antitumour immune response. In fact, several studies have applied for these approaches to improve the therapeutic efficacy of cancer [14, 15, 16, 17]. For example, both CD4+ and CD8+ T‐cell responses improved when the genes for mutated caspases 2 or 3 were co‐injected with the antigen‐carring plasmid [16].
In biochemical terms, apoptosis is frequently accompanied by the activation of a specific subset of cysteine kinases, the caspases [18]. Therefore, we decided to explore the possibility of inducing immunogenic tumour cell apoptosis by a zebrafish caspase, namely caspy2. Caspy2 is an active caspase by biochemical analysis and expression of caspy2 gene can induce apoptosis in mammalian cells [19]. In the present study, we prove that expression of caspy2 gene can induce apoptosis in tumour cells. Meanwhile, a specific antitumour cellular immune response could also be elicited. It is well accepted that the immune system can distinguish self from ‘altered self’ rather than the traditional non‐self [20]. In this study, caspy2 (as a foreign antigen) could modify the apoptotic tumour cells and help for the amplification of weaker responses against the endogenous tumour antigen of the tumour cells, which could favour tumour eradication.
Among the various antitumour therapeutic strategies that are currently regarded as promising, apoptosis induction and active immunotherapy with cancer vaccine are two prominent approaches [21, 22]. In our studies, a novel strategy that can both induce apoptosis and specific antitumour immunity in tumour cells through a single gene was developed.
Materials and methods
Cells lines
Murine colon adenocarcinoma cell CT26, murine fibrosarcoma cell MethA, murine Lewis lung carcinoma cell (LLC) and human lung adenocarcinoma cell A549 were purchased from the American Type Culture Collection and were maintained in Dulbecco’s modified Eagle media (DMEM) or Roswell Park Memorial Institute (RPMI)‐1640 supplemented with 10% foetal bovine serum.
Plasmid construction
The study protocol was approved by the institutional review board of Sichuan University (Chengdu, Sichuan, People’s Republic of China). Total RNA was isolated from zebrafish using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the instructions of the manufacturer. The open reading frame of caspy2 gene was cloned by RT‐PCR. The sequences of the primers for caspy2 gene were:
Forward Primer: 5′‐CGCGGATCCAAGATGGAGGATATTACCCAGCTGCT‐3′
Reverse Primer: 5′‐CGCGATATCTCACAGTCCAGGAAACAGGTAGAAC‐3′.
The amplified product was subcloned into pcDNA3.1+ expression vector (Invitrogen) to generate caspy2 gene recombined plasmid (p‐c). Verification of the recombinant construct was performed by DNA sequencing. An empty pcDNA3.1 (e‐p) was used as a control. The plasmids were purified using Endo‐free columns (Qiagen, Germantown, MD, USA) following the instructions of the manufacturer.
Preparation of cationic liposome and liposome‐DNA complex
Cationic liposome containing DOTAP (1,2‐dioleoyl‐3‐trimethylammoniumpropane) (Sigma D6182, Sigma‐Aldrich, Louis, MO, USA) in a 1:1 molar ratio with DOPE (dioleylphosphatidyl‐ethanolamine) (Sigma P1223) were prepared by solubilizing the lipids in chloroform and ethanol with 3:1volume ratio. The lipid mixture was then dried as a thin layer in a round‐bottomed flask under a stream of N2. Residual chloroform and ethanol was removed under high vacuum. The resulting lipid film was hydrated in 5% dextrose, and sonicated until completely solubilized. The liposome was extruded through 100 nm polycarbonate membrane to generate small unilaminar vesicles and the recovered liposome reagent was stored at 4°C. Before use, plasmid DNA was mixed with liposome in 1:5 ratio (W/W) to form a complex. This complex was used in all of the experiments below.
MTT colorimetric assay
Cell viability was monitored using 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (MTT) colorimetric assay, which is based on the ability of living cells to reduce MTT, a yellow, water‐soluble monotetrazolium salt, to an insoluble purple formazan [23]. To validate the influence of caspy2 on cell growth, CT26, LLC and A549 cells were seeded in 96‐well flat‐bottomed plates at a concentration of 2000–5000 cells per well in 100 μl medium. The next day, cells were transfected with p‐c or e‐p and cultivated in a CO2 incubator at 37°C for 48–72 hrs. Empty liposome and non‐transfected cells were used as controls. At the time of analysis, each well was supplemented with 50 μl of MTT solution (2 mg/ml) in complete media and incubated at 37°C for 4 hrs, then the medium and MTT solution were removed and 150 μl of dimethyl sulfoxide was added into each well. Absorbance was read at 540 nm using a microplate reader and the experiment was repeated three times. Media‐only treated cells served as the indicator of 100% cell viability.
Morphological analysis
CT26, LLC and A549 cells were cultured in six‐well plates and incubated at 37°C with 5% CO2 until 60–70% confluence was reached. Then the cells were transfected with plasmids (p‐c or e‐p), with liposome alone and untransfected cells as controls. The morphological analysis was performed 24–48 hrs after transfection. Cells were collected and rinsed with PBS, and then cells were resuspended in hypotonic propidium iodide (PI) solution containing 50 μg PI/ml in 0.1% sodium citrate plus 0.1% Triton X‐100 and then examined by fluorescence microscopy.
Sub‐G1 FACS analysis for apoptosis
CT26 cells were cultured in six‐well plates and incubated at 37°C with 5% CO2 until 60–70% confluence was reached, then the cells were transfected with plasmids as described above. Cells were harvested 48 hrs after transfection, and flow cytometric analysis was done to identify sub‐G1 cells/apoptosis cells as previous described [24]. Briefly, cells were suspended in 1 ml hypotonic fluorochrome solution containing 50 μg/ml PI in 0.1% sodium citrate plus 0.1% Triton X‐100, and cells were analysed by a flow cytometer (ESP Elite, Beckman Coulter, Fullerton, CA, USA). Apoptosis cells appeared in the cell cycle distribution as cells with DNA content less than that of G1 cells.
Detection of caspase‐3/7 activity
The activity of caspase‐3/7 was assayed by using a kit (Apo‐ONE Homogeneous Caspase‐3/7 assay system; Promega, Madison WI, USA) according to the manufacturer’s protocol. Briefly, cells were plated in 96‐well white plates at a concentration of 5 × 103 to 1 × 104 cells per well in 100 μl medium. The next day, cells were transfected with p‐c or e‐p and cultivated in a CO2 incubator at 37°C for 24–48 hrs. Empty liposome and non‐transfected cells were used as controls. At the time of analysis, 100 μl of apo‐ONE caspase‐3/7 reagent was added to each well of a white 96‐well plate comprising 100 μl of blank, control cells or treated cells in culture. The plate was incubated for 4 hrs at room temperature using a plate shaker at 400 rpm. After this time, fluorescence was measured using a Spectra Max M5 microplate reader (Molecular Devices Corporation, Sunnyvale, CA, USA) at excitation 485 nm and emission 520 nm.
Murine tumour models and treatment of established tumours
Animal studies were performed in accordance with institutional guidelines concerning animal use and care. All studies involving animals in connection with the present investigation were approved by the Animal Care and Use Committee of Sichuan University (Chengdu, Sichuan, People’s Republic of China).
Syngeneic CT26 Colon carcinoma and MethA fibrosarcoma models were established in 6–8‐week female BALB/c mice. Each type of cell (2 × 105 to 1 × 107) suspended in 100 μl of PBS was injected into the right flank of mice. Mice‐bearing tumours were treated at days 7–10 after the implantation of tumour cells when tumour size ranged between 15 and 20 mm3 in cross‐section. Meantime, mice were randomly allocated to one of the following groups (10 mice each group): lipid/p‐c complex, lipid/e‐p complex, empty lipid and PBS. Injections were distributed equally into each of four tumour quadrants twice weekly for 3 weeks. The tumour volume was determined by the following formula: tumour volume (mm3) =π/6 × length (mm) × width (mm)2.
Histopathological examination
Tumour tissues were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned in a thickness of 5 μm, and stained with haematoxylin and eosin. Liver, spleen, lung, kidney and brain from the mice were fixed in 4% neutral buffered formalin solution and embedded in paraffin for observation of potential side effects in treated mice.
In situ TUNEL assay
Terminal deoxynucleotidyltransferase‐mediated nick end labelling (TUNEL) assay was performed for detecting fluorescence of apoptotic cells in situ using the TUNEL kit (Boehringer Mannheim, Indianapolis, IN, USA) following the manufacturer’s protocol. Apoptotic cells were counted under a microscope (×400) in randomly selected fields. Five to six random fields totalling at least 1000 tumour cells were counted per slide. The apoptotic index (AI) was defined as follows: AI (%) = 100 × apoptotic cells/total tumour cells.
Immunofluorescence assay
Immunofluorescence staining was used for detection and analysis of lymphocytes infiltrated into tumour in situ. Anti‐CD3+ (fluorescein isothiocyanate [FITC] conjugate; eBioscience, San Diego, CA, USA) and CD8+ (Cy5PE conjugate; eBioscience) monoclonal antibodies were used to determine cytotoxic T lymphocytes (CTLs). Tumours were snap‐frozen, and 8‐μm sections were prepared in Tissue Tek (Sakura Finetek, Torrance, CA, USA) for immunofluorescence analysis. Sections were blocked [10% fatal bovine serum (FBS), 3% bovine serum albumin] for 30 min. before staining with anti‐CD3+ or anti‐CD8+ monoclonal antibody. Sections were washed three times between each reagent. Fluorescence was visualized, and images were captured with Olympus BX60 (Olympus, Tokyo, Japan).
In vitro cytotoxicity assay
The 51Cr release assay was performed as described elsewhere [25]. Briefly, mouse spleens were removed and homogenized. Erythrocytes were depleted by ammonium chloride–potassium lysis buffer. The single‐cell suspension was collected by passing splenocytes through 70‐μm cell strainers. Then, the cell suspension was incubated in nylon‐wool columns for 1 hr at 37°C. The enriched T cells were collected by washing cells through the columns with RPMI 1640. Syngeneic CT26 cells (5 × 106) were used as target cells by incubating at 37°C with 51Cr for 2 hrs. After washing several times, 51Cr‐labeled target cells (1 × 104) and serially diluted effector cells, at varying E:T ratios (40:1 to 5:1), were incubated in 200 μl of RPMI 1640 with 10% FBS in each well of 96‐well V‐bottomed plates. The plates were centrifuged at 500 ×g for 3 min. and incubated at 37°C for 4 hrs. The supernatant (50 μl) was harvested, and the activity was calculated by the formula:% cytotoxicity = ([experimental release‐spontaneous release]/[maximum release‐spontaneous release]) × 100. In the cytotoxicity inhibition assays, effectors cells or 51Cr‐labeled tumour cells were pre‐treated with monoclonal Antibody (mAb) at room temperature for 30 min., washed, and tested. mAbs used included anti‐CD4+ (10 μg/ml), anti‐CD8+ (10 μg /ml), anti‐H‐2Kb/H‐2Db (50 μg /ml) (BD PharMingen, San Diego, CA, USA). The above concentrations of mAb were effective in mediating their activity in preliminary experiments. Control cytotoxicity assay was performed in the presence of control mAb (anti‐H‐2Dd) or isotype IgG.
Tumour re‐challenge
To determine whether surviving mice in MethA model had developed a long‐term immunological memory, the animals were divided into two groups (five mice each group) and re‐challenged subcutaneously at the other flank with 1 × 106 parental MethA cells or 3 × 105 irrelevant syngeneic CT26 cells, respectively.
Antitumour vaccination and in vivo depletion of immune cell subsets
The colon carcinoma CT26 cell was transfected with lipid/p‐c complex in vitro. For the tumorigenicity assay, 1 × 106 treated or untreated CT26 cells were injected subcutaneously into 6–8‐week‐old female BALB/c mice. To investigate the protective antitumour immunity, 1 × 106 treated CT26 cells were inoculated subcutaneously in 100 μl PBS into 6‐week‐old female BALB/c mice into the left flank. The mice were re‐challenged with same live tumour cells injected into the opposite flank 7 days later. In some experiments, immune cell subsets were depleted as previous described [14]. Depletion of CD4+, CD8+ or NK cells was achieved by intraperitoneal injection of 500 μg anti‐CD4+ (clone GK 1.5, rat IgG), anti‐CD8+ (clone2.43, rat IgG) or anti‐NK (clone PK136) mAb, respectively, 4 days before challenge with dying (p‐c treated) tumour cells and 3 days before injection of live tumour cells (10 mice each group). Isotype IgG2a and IgG2b were used as controls [14].
Statistical analyses
For comparison of individual time‐points, anova and an unpaired Student’s t‐test were used. Survival analysis was computed by the Kaplan–Meier method and compared by the log‐rank test. P < 0.05 was considered statistically significant.
Results
Caspy2 inhibits growth of tumour cells via apoptosis induction in vitro
The expression of caspy2 in COS cells was confirmed by RT‐PCR and Western Blotting (data not shown). The biological activity of caspy2 was tested in cells including murine colon adenocarcinoma CT26 cell, murine Lewis lung carcinoma LLC cell and human lung adenocarcinoma A549 cell, which have been transiently transfected with lipid/p‐c complex. The caspy2 significantly inhibited the growth of tumour cells compared with controls (Fig. 1A).
Figure 1.
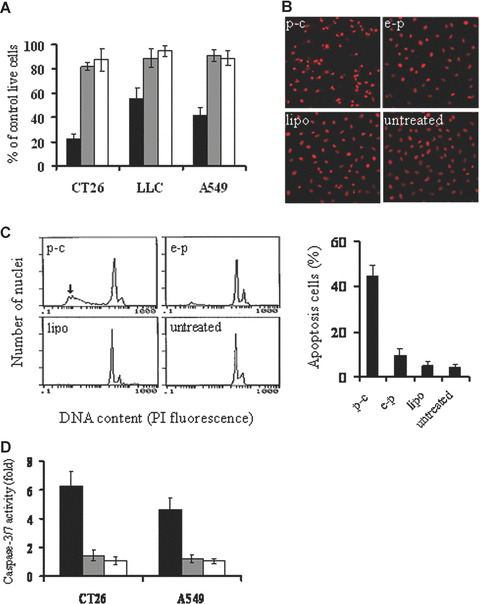
Induction of cancer cell apoptosis in vitro. (A) Viability of cancer cells by MTT assay. CT26, LLC and A549 cancer cell line were transfected with p‐c (black bars), e‐p (gray bars), or liposome alone (white bars). The transfected cells were cultured in 96‐well plates for 48–72 hrs and MTT assay was performed for observing the viability of the cells. Results are means ± S.D. of 6 wells and triplicate experiments. In each experiment, the media only treatment (untreated) represents 100% of cell viability. (B) Fluorescence‐microscopic appearance of PI‐stained nuclei in CT26 cells. Condensation of nuclear and apoptotic bodies were observed in p‐c treated group (Original magnification ×200). (C) Quantitative assessment of apoptotic cells by flow cytometry. CT26 cells were treated with p‐c or controls for 48 hrs, then cells were harvested from each group. Percentage of sub‐G1 cells (apoptotic cells) was identified by flow cytometry. Left: representative flow cytograph from three separate experiments. Arrowhead points to sub‐G1 peak in p‐c treated group. Right: Percentage of sub‐G1/apoptotic cells. Data are represented as mean ± S.D. of three independent experiments (P < 0.05). (D) Quantification of caspase‐3/7 activity in response to caspy2 treatment. CT26 and A549 cells were transfected with p‐c (black bars), e‐p (gray bars), or liposome alone (white bars). The transfected cells were cultured in 96‐well white plates for 24–48 hrs, and then the activity of caspase‐3/7 was analysed. Results are means ± S.E. of five wells and triplicate experiments, and plotted values indicate the fold increase in activity relative to untreated samples.
The type of cell death induced by caspy2 was also determined. Morphological changes characterized as apoptosis were observed in tumour cells transfected with p‐c after PI staining. Brightly red‐fluorescent condensed nuclei (intact or fragmented) and apoptotic bodies were observed by fluorescence microscopy focused on PI‐stained nuclei (Fig. 1B). Furthermore, a ladder‐like pattern of DNA fragments consisting of multiples of 180–200 base pairs was observed by agarose gel electrophoresis of chromosomal DNA extracted from caspy2‐treated cells (data not shown). Quantitative assessment of sub‐G1 cells by flow cytometry was further done to estimate the number of apoptotic cells. As shown in Fig. 1C, there was an apoptotic peak before the normal G1 peak of cell cycles in liposome/p‐c complex treated group and the percentage of apoptotic cells was significantly higher in the p‐c transfected cells than that in control groups. Next, in order to decide whether caspy2 induced apoptosis via caspase activation, the activity of caspase‐3/7 was analysed using a kit that detected the DEVDase activity of caspase‐3/7. Figure 1D shows that treatment of CT26 cells and A549 cells with liposome/p‐c complex significantly increased caspase activity. Taken together, our data indicated that caspy2 could directly induce apoptosis in tumour cells in vitro.
In vivo growth inhibition of the established tumours in mice
To determine the optimal dose of lipid/p‐c complex in therapeutic treatment, a pilot study was performed. A ratio of 1:5 (100 μg plasmid plus 500 μg liposome) was found to be most effective and was used in subsequent experiments.
Mice were treated with lipid/p‐c complex twice weekly for 3 weeks, beginning 7 days after tumour cell implantation. Treatment with lipid/p‐c complex resulted in retarded progression or complete regression of the established tumours in CT26 colon carcinoma or MethA fibrosarcoma models, respectively (Fig. 2A and B; P < 0.05). The survival of the tumour‐bearing mice treated with lipid/p‐c complex was also significantly greater than that of controls (Fig. 2C and D; P < 0.05).
Figure 2.
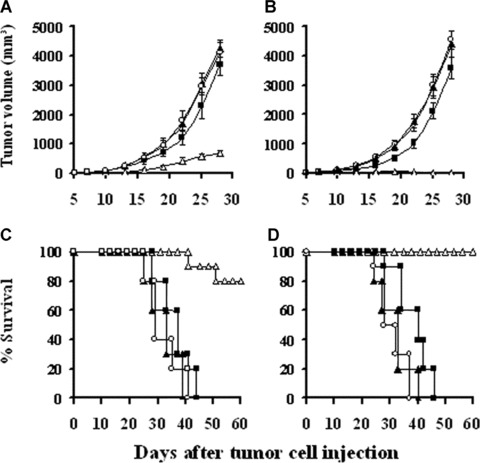
Induction of the therapeutic antitumour activity. Tumour‐bearing mice were injected peritumorally or intratumorally with 100 μl total volume of 100 μg p‐c/500 μg liposome complex (□), 100 μg e‐p/500 μg liposome complex (▪), 500 μg liposome alone (▴) or PBS (◯), respectively, twice a week for 3 weeks. It was evident that, compared with control groups, p‐c treated mice showed significant tumour volume decrease and survival time prolongation both in CT26 model (A, C) and MethA fibrosarcoma model (B, D) (P < 0.05). The survival rate of the mice was 80% and 100% at day 60 for CT26 Colon carcinoma and MethA fibrosarcoma, respectively.
The potential long‐term toxicity of administrating caspy2 was also investigated. No adverse consequences were observed in gross measures, such as weight loss, ruffling of fur, life span, behaviour or feeding. Furthermore, no pathologic changes of liver, kidney, lung, spleens, brain, etc. were found by the microscopic examination.
Histological analysis
Histological examination of tumour sections by haematoxylin and eosin staining showed that there were little or no tumour tissue necrosis and had normal capillaries surrounding nests of confluent tumour cells in control tumours. In contrast, those lipid/p‐c complex treated tumours showed remarkable response with necrosis (Fig. 3A–D). Meanwhile, lymphocyte infiltration exhibiting clustering‐like aggregation was found apparently in the margin of lipid/p‐c complex treated group (Fig. 3E). The infiltration of lymphocyte was also increased in the central regions from the experimental group (Fig. 3F).
Figure 3.
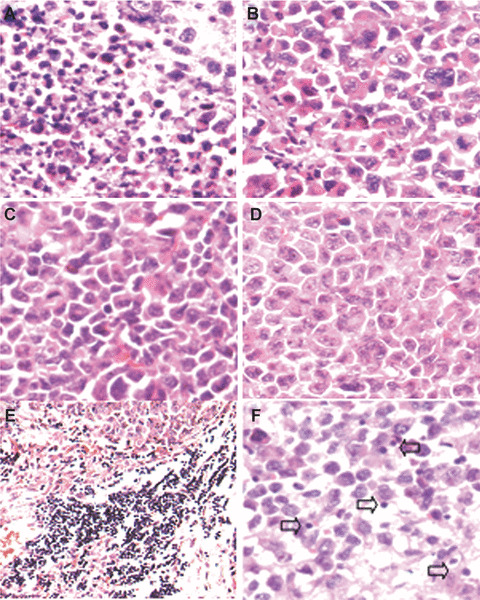
Histological analysis of tumour tissue. Histological examination of tumours by haematoxylin and eosin staining was performed. Images show paraffin‐embedded tumour sections from tumour‐bearing mice treated with p‐c (A), e‐p (B), liposome alone (C) or PBS (D). There were large areas of confluent tumour cells with little or no tumour tissue necrosis (B–D) but extensive necrosis (A), respectively. Meanwhile, lymphocyte infiltration was also enhanced both in the margin (E) and central regions in p‐c treated group (F).
Fluorescence analysis of intratumoral apoptosis and T‐cell infiltration
In view of apparent antitumour activity in BALB/c mice tumour models, it was decided to investigate the mechanism underlying caspy2 mediated cancer cell death. Towards this goal, we examined apoptosis‐related molecular markers on tumour sections. TUNEL assay was carried out to detect early DNA fragmentation associated with apoptosis. Many strongly positive nuclei identified as apoptosis were found in p‐c treated tumour tissue, whereas such nuclei were rare in e‐p treated or other control groups (Fig. 4).
Figure 4.
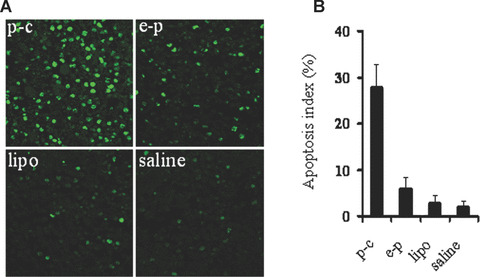
TUNEL staining of tumour tissue. (A) Representative images of TUNEL staining. TUNEL staining was performed using an in situ apoptotic cell detection kit at day 14 after the beginning of treatment. Apoptotic nuclei (green) were observed under a fluorescence microscope. Tumours treated with p‐c showed apparently increase in the number of apoptotic cells within the residual tumours compared with control groups. (B) Apoptotic index. Graph shows mean values in each group, and data are presented as mean ± S.E. (P < 0.05).
To further detect the infiltrated lymphocytes in tumours and determine their type, anti‐CD3+ and anti‐CD8+ monoclonal antibodies were used in immunofluorescence staining. The results showed that lymphocytes infiltration was apparently increased in the caspy2‐treated tumour than in control groups. Meanwhile, these infiltrated cells are mainly CD8+ CTLs (Fig. 5).
Figure 5.

Fluorescence staining of infiltrated lymphocytes. Tumours were snap‐frozen, and 8‐μm sections were prepared in Tissue Tek (Sakura Finetek). Anti‐CD3+ (FITC conjugate, green) monoclonal antibody was used to detect lymphocytes infiltrated into tumours in situ. Tumours of mice treated with p‐c (D) showed more infiltrated lymphocytes than control groups treated with e‐p (C), liposome alone (B) or PBS (A). To further determine the type of infiltrated lymphocytes, anti‐CD3+ and anti‐CD8+ (Cy5PE conjugate, red) monoclonal antibodies were used simultaneously to stain the p‐c treated tumour tissue (D and E, respectively), and the merged image (F) confirmed that the infiltrated lymphocytes were mainly CD8+ CTLs.
Induction of specific CTL in response to caspy2 treatment
The tumour killing activity of CTLs was observed. T cells isolated from spleens of lipid/p‐c complex treated mice showed higher cytotoxicity against parental tumour cells than those from control groups (Fig. 6A). Next, we observed that this cytotoxicity could be blocked by anti‐CD8+ or anti‐MHC I mAb, but not by anti‐CD4+ in vitro (Fig. 6B).
Figure 6.
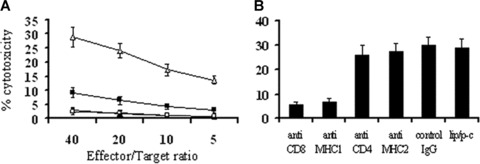
CTL‐mediated antitumour activity. (A) CTL‐mediated cytotoxicity in vitro. The cytotoxic activity of spleen lymphocytes was measured in 4‐hr 51Cr‐release assay. The enriched T lymphocytes were added to 51Cr‐labeled CT26 cells immediately after isolation from spleens. T cells derived from the spleens of p‐c (□) treated mice showed higher cytotoxicity against CT26 cells than those from e‐p treated (▪), liposome alone (▴) and PBS (◯) groups (P < 0.05). (B) Abrogation of CTL‐mediated cytotoxicity in vitro. CTL‐mediated cytotoxicity is abrogated by certain kinds of mAbs as described in Materials and Methods. The ratio of effector:target cells was at 40:1. The p‐c induced tumour cytotoxic activity can be blocked by anti‐CD8+ or anti‐MHC I (anti‐H‐2Kb/H‐2Db) mAb. Similar results can be found at other ratios.
Tumour cell re‐challenge
To examine whether a long‐term tumour‐specific protective immunity was established by the treatment with lipid/p‐c complex, the long‐term surviving mice were re‐challenged subcutaneously, 90 days after first injection of MethA, with parental MethA or irrelevant syngeneic tumour CT26. The results showed that detectable flank tumours were not observed when inoculation of MethA tumour cells into previously p‐c treated mice and all the mice survived healthily. However, when these mice were inoculated with CT26 the tumour grew vigorously (Fig. 7). In the MethA model, animals treated with caspy2 were ‘cured’ and exhibited long‐term protection against the parental tumour re‐challenge. These observations suggest that the intratumoral treatment of established MethA tumour model with caspy2 gene resulted in the development of a specific long‐term protective immune response.
Figure 7.
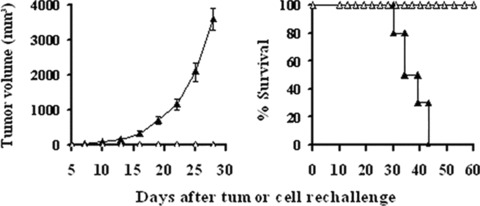
Development of specific and long‐term antitumour immune response. Recovered mice (n= 5) from p‐c treated group in MethA model were re‐challenged with 1 × 106 parental MethA cells (□) or 3 × 105 irrelevant syngeneic CT26 (▴)tumour cells in the left flank, the mice re‐challenged with CT26 developed large flank tumours and died soon, whereas none of mice re‐challenged with MethA had detectable flank tumours and all the mice survived healthily.
Apoptotic cell vaccination and function of T‐cell subsets in vivo
In the tumourigenicity assay, no tumour growth was found when lipid/p‐c complex treated dying cells were inoculated into immunocompetent BALB/c mice (data not shown). To explore the putative immunogenicity of dying cells and characterize the immune cell types, BALB/c mice were inoculated into one flank with p‐c treated CT26 cells and received an injection of live CT26 cells into the opposite flank 1 week later. Simultaneously, we selectively eliminated specific population of immune cell subsets in vivo in some mice. As shown in Fig. 8A, there was apparent protection from tumour evolution in mice vaccinated with p‐c treated cells and the survival time was significantly prolonged. Meanwhile, depletion of both CD8+ and CD4+ T lymphocytes could decrease the anticancer activity. The depletion of CD4+ lymphocytes showed partially abrogation of the antitumour activity, whereas the depletion of CD8+ lymphocytes displayed complete abrogation, indicating that CD8+ T cells are mainly responsible for immune response (Fig. 8B). In addition, the treatment with anti‐NK mAb or normal rat IgG showed no effect.
Figure 8.
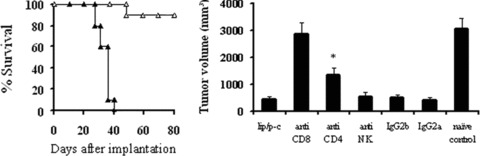
Function of T‐cell subsets in antitumour activity. (A) Immunogenic effect of p‐c treated tumour cells. BALB/c mice were injected into one flank with 1 × 106 p‐c treated CT26 tumour cells and into the opposite flank with 2 × 105 live CT26 cells 1 week later and the survival of mice were monitored. All mice vaccinated with p‐c treated cells (□) appeared to be protected and survival was significantly prolonged, whereas the mice implanted with naive CT26 cells (▴) died soon. (B) Depletion of the immune cell subsets in vivo. CD4+, CD8+ or NK cells were depleted by intraperitoneal injection of 500 μg anti‐CD4+ (clone GK 1.5, rat IgG), anti‐CD8+ (clone2.43, rat IgG) or anti‐NK (clone PK136) mAb, respectively, 4 days before challenge with p‐c treated dying CT26 tumour cells and 3 days before injection of live CT26 cells. Depletion of CD8+ T lymphocytes showed complete abrogation of the antitumour activity, whereas the depletion of CD4+ T lymphocytes showed partial abrogation. Data represent day 25 after naive tumour cell injection. Similar results can be found at other time‐points. Asterisks (*) indicate a significant difference in tumour volume (P < 0.05) between CD4+‐depleted and other groups.
Discussion
Either triggering apoptosis or specific immune response in cancer cells is one of the major approaches in anticancer therapies. Thus, the combination of apoptosis induction and active immunotherapy may be a new and potent strategy for cancer therapy. Indeed, researchers have applied for this strategy to improve the therapeutic efficacy of cancer [26, 27, 28]. For example, both apoptosis and tumour‐specific T‐cell immunity could be acquired by the treatment with anti‐DR5 antibody [26]. In the present study, we cloned cDNA of caspy2 gene and investigated its antitumour activity. We observed that the plasmid DNA encoding caspy2 gene could inhibit cellular growth and induce apoptosis in several murine and human tumour cells in vitro. Through the treatment with the recombinant plasmid of caspy2 gene, a significant delay in tumour growth and prolongation of life span was observed in the mice bearing the tumours. For MethA fibrosarcoma, the tumour was completely regressed and the mice acquired long‐term survival. TUNEL staining of the caspy2‐treated tumour sections showed significantly higher numbers of apoptotic cells than controls. These results consist with previous observation that the liposome‐DNA complex encoding caspy2 could induce apoptosis in human 293T cells [19]. Meanwhile, it has been reported that cytochrome c is released into the culture medium after activation of the intrinsic apoptotic caspase pathway in vitro and serum cytochrome c is a sensitive apoptotic marker in vivo reflecting therapy‐induced cell death burden [29, 30]. Therefore, we detected the serum cytochrome c levels of the mice during the time of animal experiments. But we failed to detect the release of cytochrome c in the sera of p‐c treated mice. This result may indicate that the mechanism of caspy2 induced apoptosis is not through activation of the intrinsic apoptotic pathway. Furthermore, caspy2 appeared to initiate a potent cellular immune response against tumour cells, suggesting that the induction of immune killing of tumour cells should play an important role in its therapeutic antitumour efficacy. To our knowledge, most cancer therapy agents that are used in clinical practice or in research usually show only one major antitumour activity. For example, known chemotherapeutic agents kill target cells by apoptosis, but most chemotherapies have been regarded as immunosuppressive. On the other hand, most therapeutic tumour vaccines do initiate immune responses against the tumour but cannot induce cell apoptosis directly. An agent that is able to concomitantly initiate both of these two effects, such as the caspy2 described here, is rarely found.
Tumours can induce tolerance to their tumour‐associated antigen through pathways that reflect natural mechanisms of tolerance induction to self‐tissue antigens express in the periphery [31]. The breaking of immune tolerance against self‐tumour antigen and induction of autoimmunity against tumours should be a useful approach for the treatment of tumour. Given that apoptosis is a physiological phenomenon affecting several million cells per second [6], it is tempting to assume that in immunological terms apoptosis must be either silent or tolerogenic. However, intramuscular DNA vaccines elicit improved CD4+ and CD8+ responses, when pro‐apoptotic genes (caspase‐2 or ‐3, CD95) are coinjected, indicating that apoptosis by itself is not by definition tolerogenic [16, 32]. In this study, expression of caspy2 (a foreign antigen) in tumour cells is intended to modify the apoptotic cells and therefore enhance the presentation ability of the modified apoptotic tumour cells to antigen‐presenting cells (APCs) and then cross‐priming through APCs, finally, induce a T‐cell response against the unmodified tumours [33]. Infiltration of lymphocyte was observed apparently in the tumour tissues after the treatment with lipid/p‐c complex. This finding indicates an onset of immune response in the local mass. In order to confirm the modification could lead to an immune recognition and killing of the unmodified tumour cells, a mouse model of vaccination with apoptotic cancer cells was established. In this study, apparent protection from tumour growth in mice could be obtained when mice were vaccinated with p‐c treated cells. Meanwhile, a tumour‐specific long‐term protective immunity from the mice treated with p‐c was also observed in our research. We observed that the ‘cured’ mice have no palpable tumours when they were re‐challenged with parental syngeneic tumour cells. However, when irrelevant tumour cells were inoculated into these ‘cured’ mice, the tumours grew vigorously.
In addition, we investigated the immune type of the tumour cells. Both MHC class I‐dependent CD8+ CTL activity and CD8+ T‐cell infiltration in tumour sections were found by 51Cr release assay and immunofluorescence staining, respectively. The depletion of CD8+ lymphocytes showed complete abrogation of the antitumour immune activity in vivo. Meanwhile, the depletion of CD4+ T lymphocytes showed partial abrogation. These findings suggest that MHC class I‐dependent CD8+ CTL‐mediated immune response may be mainly responsible for its antitumour immune activity and CD4+ T cells play an assistant role in the immune response. The systemic antitumour immune responses mainly depend on CD8+ T cells, indicating enhanced presentation of tumour antigens by cross‐presentation on CD8+ T cells. It has been found that APCs phagocytose apoptotic tumour cells results in tumour antigen access to the cytoplasm and cross‐presentation on MHC class I molecules [34]. We supposed that the altered apoptotic tumour cells could be enhanced in this transfer ability. Meanwhile, the antitumour responses partially depend on CD4+ T cell suggests CD4+ T cells also play a role in the mechanism of the immune response. Although CD8+ T cells can be activated in the absence of CD4+ T helper lymphocytes, optimal CD8+ T‐cell activation and long‐term immunological memory probably require coactivation of CD4+ T lymphocytes [35, 36, 37]. The added benefit of CD4+ T‐cell help is particularly apparent in tumour immunity [38, 39, 40]. Differentiated CD4+ T lymphocytes have been categorized into either T helper type 1 or type 2 (Th1 or Th2). Th1 cells are generated considered to provide cytokines help to CD8+ T cells [41]. Induction of optimal antitumour immunity, therefore, should involve activation of both CD4+ Th and CD8+ tumour‐specific T cells.
Taken together, the findings in our present study may provide a new strategy for the treatment of tumours both through the induction of apoptosis and tumour‐specific immune response by the treatment with the plasmid DNA encoding caspy2. In the early period when the immune response was not effectively established, the inhibition of tumour grow could be acquired by the induction of apoptosis, then a more powerful tumour‐specific immune response was elicited. This dual effect resulted in a better therapeutic efficacy. We believe that this strategy has the potential to be developed into a useful method for treatment of tumour.
Acknowledgements
This work is supported by The National Key Basic Research Program (973 Program) of China (2004CB518807 and 2004CB518805), Hi‐tech Research and Development Program (863 Program) of China (2007AA021008).
References
- 1. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide‐ranging implications in tissue kinetics. Br J Cancer . 1972; 26: 239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature . 2000; 407: 784–8. [DOI] [PubMed] [Google Scholar]
- 3. Bhutia S, Maiti TK. Targeting tumors with peptides from natural sources. Trends Biotechnol . 2008; 26: 210–7. [DOI] [PubMed] [Google Scholar]
- 4. Call JA, Eckhardt SG, Camidge DR. Targeted manipulation of apoptosis in cancer treatment. Lancet Oncol . 2008; 9: 1002–11. [DOI] [PubMed] [Google Scholar]
- 5. Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer . 2005; 5: 231–7. [DOI] [PubMed] [Google Scholar]
- 6. Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science . 1995; 267: 1456–62. [DOI] [PubMed] [Google Scholar]
- 7. Bellamy CO, Malcomson RD, Harrison DJ, et al . Cell death in health and disease: the biology and regulation of apoptosis. Semin Cancer Biol . 1995; 6: 3–16. [DOI] [PubMed] [Google Scholar]
- 8. Lauber K, Blumenthal SG, Waibel M, et al . Clearance of apoptotic cells: getting rid of the corpses. Mol Cell . 2004; 14: 277–87. [DOI] [PubMed] [Google Scholar]
- 9. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature . 1998; 392: 245–52. [DOI] [PubMed] [Google Scholar]
- 10. Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I‐restricted CTLs. Nature . 1998; 392: 86–9. [DOI] [PubMed] [Google Scholar]
- 11. Bellone M, Iezzi G, Rovere P, et al . Processing of engulfed apoptotic bodies yields T cell epitopes. J Immunol . 1997; 159: 5391–9. [PubMed] [Google Scholar]
- 12. Leitner WW, Restifo NP. DNA vaccines and apoptosis: to kill or not to kill J Clin Invest . 2003; 112: 22–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zitvogel L, Casares N, Péquignot MO, et al . The immune response against dying tumor cells. Adv Immunol . 2004; 84: 131–79. [DOI] [PubMed] [Google Scholar]
- 14. Casares N, Pequignot MO, Tesniere A, et al . Caspase‐dependent immunogenicity of doxorubicin‐induced tumor cell death. J Exp Med . 2005; 202: 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nowak AK, Lake RA, Marzo AL, et al . Induction of tumor cell apoptosis in vivo increases tumor antigen cross‐presentation, cross‐priming rather than cross‐tolerizing host tumor‐specific CD8 T cells. J Immunol . 2003; 170: 4905–13. [DOI] [PubMed] [Google Scholar]
- 16. Sasaki S, Amara RR, Oran AE, et al . Apoptosis‐mediated enhancement of DNA‐raised immune responses by mutant caspases. Nat Biotechnol . 2001; 19: 543–7. [DOI] [PubMed] [Google Scholar]
- 17. Hoffmann TK, Meidenbauer N, Dworacki G, et al . Generation of tumor‐specific T‐lymphocytes by cross‐priming with human dendritic cells ingesting apoptotic tumor cells. Cancer Res . 2000; 60: 3542–9. [PubMed] [Google Scholar]
- 18. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science . 1998; 281: 1312–6. [DOI] [PubMed] [Google Scholar]
- 19. Masumoto J, Zhou W, Chen FF, et al . Caspy, a zebrafish caspase, activated by ASC oligomerization is required for pharyngeal arch development. J Biol Chem . 2003; 278: 4268–76. [DOI] [PubMed] [Google Scholar]
- 20. Matzinger P. The danger model: a renewed sense of self. Science . 2002; 296: 301–5. [DOI] [PubMed] [Google Scholar]
- 21. Ding HF, Fisher DE. Induction of apoptosis in cancer: new therapeutic opportunities. Ann Med . 2002; 34: 451–69. [DOI] [PubMed] [Google Scholar]
- 22. Michaeli D. Vaccines and monoclonal antibodies. Semin Oncol . 2005; 32: 82–6. [DOI] [PubMed] [Google Scholar]
- 23. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods . 1983; 65: 55–63. [DOI] [PubMed] [Google Scholar]
- 24. Wei YQ, Zhao X, Kariya Y, et al . Induction of autologous tumor killing by heat treatment of fresh human tumor cells: involvement of gamma delta T cells and heat shock protein 70. Cancer Res . 1996; 56: 1104–10. [PubMed] [Google Scholar]
- 25. Mashino K, Sadanaga N, Tanaka F, et al . Effective strategy of dendritic cell‐based immunotherapy for advanced tumor‐bearing hosts: the critical role of Th1‐dominant immunity. Mol Cancer Ther . 2002; 1: 785–94. [PubMed] [Google Scholar]
- 26. Takeda K, Yamaguchi N, Akiba H, et al . Induction of tumor‐specific T cell immunity by anti‐DR5 antibody therapy. J Exp Med . 2004; 199: 437–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uno T, Takeda K, Kojima Y, et al . Eradication of established tumors in mice by a combination antibody‐based therapy. Nat Med . 2006; 12: 693–8. [DOI] [PubMed] [Google Scholar]
- 28. Selenko N, Maidic O, Draxier S, et al . CD20 antibody (C2B8)‐induced apoptosis of lymphoma cells promotes phagocytosis by dendritic cells and cross‐priming of CD8+ cytotoxic T cells. Leukemia . 2001; 15: 1619–26. [DOI] [PubMed] [Google Scholar]
- 29. Barczyk K, Kreuter M, Pryjma J, et al . Serum cytochrome c indicates in vivo apoptosis and can serve as a prognostic marker during cancer therapy. Int J Cancer . 2005; 116: 167–73. [DOI] [PubMed] [Google Scholar]
- 30. Renz A, Berdel WE, Kreuter M, et al . Rapid extracellular release of cytochrome c is specific for apoptosis and marks cell death in vivo. Blood . 2001; 98: 1542–8. [DOI] [PubMed] [Google Scholar]
- 31. Miller JF, Morahan G, Allison J. Extrathimic acquisition of tolerance by T lymphocytes. Cold Spring Harb Symp Quant Bio . 1989; 2: 807–13. [DOI] [PubMed] [Google Scholar]
- 32. Chattergoon MA, Kim JJ, Yang JS, et al . Targeted antigen delivery to antigen‐presenting cells including dendritic cells by engineered Fas‐mediated apoptosis. Nat Biotechnol . 2000; 18: 974–9. [DOI] [PubMed] [Google Scholar]
- 33. Plautz GE, Yang ZY, Wu BY, et al . Immunotherapy of malignancy by in vivo gene transfer into tumors. Proc Natl Acad Sci USA . 1993; 90: 4645–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Albert ML, Pearce SF, Francisco LM, et al . Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross‐present antigens to cytotoxic T lymphocytes. J Exp Med . 1998; 188: 1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bennett SR, Carbone FR, Karamalis F, et al . Induction of a CD8+ cytotoxic T lymphocyte response by cross‐priming requires cognate CD4+ T cell help. J Exp Med . 1997; 186: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu HM, Winter H, Urba WJ, et al . Divergent roles for CD4+ T cells in the priming and effector/memory phases of adoptive immunotherapy. J Immunol . 2000; 165: 4246–53. [DOI] [PubMed] [Google Scholar]
- 37. Machy P, Serre K, Baillet M, et al. Induction of MHC class I presentation of exogenous antigen by dendritic cells is controlled by CD4+ T cells engaging class II molecules in cholesterol‐rich domains. J Immunol . 2002; 168: 1172–80. [DOI] [PubMed] [Google Scholar]
- 38. Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumor immunity. Curr Opin Immunol . 1998; 10: 588–94. [DOI] [PubMed] [Google Scholar]
- 39. Ossendorp F, Mengede E, Camps M, et al . Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J Exp Med . 1998; 187: 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dolan BP, Gibbs KD, Ostrand‐Rosenberg S. Tumor‐specific CD4+ T cells are activated by “cross‐dressed” dendritic cells presenting peptide‐MHC class II complexes acquired from cell‐based cancer vaccines. J Immunol . 2006; 176: 1447–55. [DOI] [PubMed] [Google Scholar]
- 41. Romagnani S. The Th1/Th2 paradigm. Immunol Today . 1997; 18: 263–6. [DOI] [PubMed] [Google Scholar]