

Abstract
Dexamethasone (1 mg/kg, i.p.) administered either 1 hr before or 1 hr after E. coli lipopolysaccharide (LPS, 4 mg/kg, i.p.) in conscious rats inhibited the subsequent (4 hrs) rise in plasma cytokine (interleukin [IL]‐1β, tumour necrosis factor [TNF]‐α), nitrate/nitrite (NO×), soluble intercellular adhesion molecule‐1 (sICAM‐1) concentration and lung/liver myeloperoxidase activity indicative of an anti‐inflammatory effect. Dexamethasone also reduced the LPS‐evoked rise in plasma hydrogen sulphide (H2S) concentration, liver H2S synthesizing activity and expression of cystathionine γ lyase (CSE) and inducible nitric oxide synthase (iNOS). Mifepristone (RU‐486) inhibited these effects. Dexamethasone (1–10 μM) reduced the LPS‐evoked release of IL‐1β, TNF‐α and L‐selectin, decreased expression of CSE and iNOS and diminished nuclear factor κB (NF‐κB)‐DNA binding in isolated rat neutrophils. In contrast, NaHS (100 μM) increased L‐selectin release from rat neutrophils. Dexamethasone also reduced LPS‐induced up‐regulation of CSE in foetal liver cells. 6‐amino‐4‐(4‐phenoxyphenylethylamino) quinazoline (QNZ, 10 nM), a selective inhibitor of transcription via the NF‐κB pathway, abolished LPS‐induced up‐regulation of CSE expression. We propose that inhibition of CSE expression and reduction in formation of the pro‐inflammatory component of H2S activity contributes to the anti‐inflammatory effect of dexamethasone in endotoxic shock. Whether H2S plays a part in the anti‐inflammatory effect of this steroid in other forms of inflammation such as arthritis or asthma warrants further study.
Keywords: hydrogen sulphide, steroids, inflammation
Introduction
Glucocorticoids have been extensively used as anti‐inflammatory drugs to treat a wide variety of inflammatory disorders including septic shock. Glucocorticoids interact with specific glucocorticoid receptors [1] to suppress the expression of numerous pro‐inflammatory mediators including cytokines, chemokines, adhesion molecules [2] and enzymes such as inducible nitric oxide synthase (iNOS) and cyclooxygenase‐2 [3, 4] which synthesise nitric oxide and prostanoids, respectively. These various effects are achieved by a variety of mechanisms most notably switching off transcription of pro‐inflammatory molecules due to inhibition of transduction via nuclear factor κB (NF‐κB) and activator protein (AP‐1) and also by switching on gene expression for anti‐inflammatory mediators such as annexin‐1, interleukin (IL)‐10 and IκBα (inhibitor of NF‐κB) [5, 6].
It is now becoming increasingly apparent that hydrogen sulphide (H2S) formed from cysteine by the enzyme, cystathionine γ lyase (CSE), exerts complex effects on inflammation. Evidence has been presented that H2S exhibits pro‐inflammatory activity as demonstrated, for example, by its ability to dilate blood vessels in vitro and in vivo[7]. Furthermore, administration of NaHS (an H2S donor) to mice [8] provokes an inflammatory reaction as shown by increased tissue myeloperoxidase (MPO) activity (a marker for tissue leucocyte infiltration) and histologically by the presence of accumulated leucocytes extravascularly in the lung. Recent evidence has also unveiled a hyperalgesic effect of NaHS injected into the rat hindpaw [9] again indicative of a pro‐inflammatory action. Finally, DL‐propargylglycine, an irreversible inhibitor of CSE, exhibits anti‐inflammatory activity in a range of animal models of inflammation [8, 10, 11]. In stark contrast, NaHS has been reported to inhibit leucocyte adhesion to gastric mucosal blood vessels [12] perhaps suggestive of an anti‐inflammatory effect.
H2S also promotes neutrophil apoptosis and scavenges nitric oxide, peroxynitrite (ONOO.) and hypochlorous acid (HOCl) [13, 14, 15] all of which might be expected to alleviate inflammation. In addition, S‐diclofenac (an H2S‐releasing derivative of the non‐steroidal anti‐inflammatory drug, diclofenac) exhibits more pronounced anti‐inflammatory activity in both endotoxic shock [16] and against carrageenan‐induced hindpaw oedema [17] in the rat than does diclofenac whose effect was proposed to be due to release of H2S from the parent molecule. Finally, ADT‐OH (a slow releasing H2S donor) also exhibits anti‐inflammatory activity in endotoxic shock [16]. Thus, it seems likely that H2S is able to exhibit both pro‐ and anti‐inflammatory components of action.
Bearing in mind the controversial nature of the part played by H2S in inflammation as well as continuing uncertainties about the precise mechanism of the anti‐inflammatory effect of glucocorticoids, we considered it of value to investigate the role of the L‐cysteine/H2S system in the effect of dexamethasone in a model of endotoxic shock in the rat and to investigate the mechanisms involved in greater detail in isolated cells. Since the ‘active’ constituents responsible for the biological effects of H2S are currently unknown, we herein use the term H2S to reflect the sum of species that will be present at physiological pH, 37°C (H2S, HS−, S2 −, etc).
Materials and methods
Lipopolysaccharide (LPS)‐induced endotoxic shock in the rat
All experiments were approved by the animal ethics committee of National University of Singapore and carried out in accordance with established Guiding Principles for Animal Research. Male Sprague‐Dawley rats (250–300 g) were injected with bacterial endotoxin LPS (E. coli, serotype O127:B8; 4 mg/kg, i.p.) as described previously [8]. Control animals received sterile saline (1 ml/kg, i.p.). Animals were killed by anaesthetic overdose 4 hrs after LPS/saline injection. Blood was removed by cardiac puncture into heparinized (50 units/ml) tubes and lung and liver snap frozen in liquid nitrogen. Dexamethasone (1 mg/kg, i.p.) or vehicle (ethanol/olive oil 5/95 v/v, 1 ml/kg) was administered either 1 hr before (i.e.‘prophylactic’) or 1 hr after (i.e.‘therapeutic’) LPS injection. In some experiments, animals received mifepristone (RU‐486, 6.6 mg/kg, i.p.) 30 min. prior to injection of dexamethasone (1 mg/kg, i.p.) which, in turn, was administered 1 hr before LPS (4 mg/kg, i.p.). LPS was chosen for the present experiments since it induces a very well characterized pro‐inflammatory effect both in experimental animals and also in man. In addition, we have previously shown that exposure of rats to LPS for 4 hrs is sufficient to elicit pronounced systemic pro‐inflammatory effects [16].
Isolation of rat peripheral neutrophils and culture of foetal liver cells
Rat neutrophils were isolated according to a previously published method [18]. Cells were resuspended in RPMI 1640 medium supplemented with 10% FCS, L‐glutamine, penicillin and streptomycin and consistently yielded >95% polymorphonuclear cells (by morphology in Giemsa stain) and >98% viability (by trypan blue dye exclusion). Cells (106 cells/ml) were exposed to LPS (1 μg/ml, 37°C, 4 hrs) in the presence or absence of dexamethasone (1–10 μM), vehicle (dimethylsulfoxide) or NaHS (100 μM) and thereafter harvested for measurement of cytokines, L‐selectin and CSE/iNOS expression. In some experiments, LPS‐treated neutrophils were incubated with 6‐amino‐4‐(4‐phenoxyphenylethylamino) quinazoline (QNZ, 10 nM). Human foetal liver cells were isolated from second‐trimester prostaglandin‐induced abortuses and cultured in Dulbecco’s modified Eagle medium with 10% v/v foetal bovine serum and 1% w/v penicillin/streptomycin as described elsewhere [19]. Cells (106 cells/ml) were exposed to LPS (1 μg/ml, 37°C, 4 hrs) in the presence or absence of dexamethasone (1–10 μM).
Assay of plasma and tissue H2S concentration and tissue H2S synthesis from exogenous cysteine
Biosynthesis of H2S (defined herein as a HS−, S− plus H2S) in liver homogenate was measured as described previously [8]. Briefly, excised liver was weighed, homogenized (1/20 v/v) in ice‐cold 100 mM potassium phosphate buffer (pH 7.4) and incubated with L‐cysteine (10 mM; 20 μl), pyridoxal‐5‐phosphate (2 mM; 20 μl) and physiological saline (30 μl). After incubation (37°C, 30 min.), zinc acetate (1% w/v, 250 μl) was injected to trap generated H2S, followed by trichloroacetic acid (10% w/v, 250 μl). N,N dimethyl‐p‐phenylenediamine sulphate (20 mM; 133 μl) in 7.2 M HCl was added followed by FeCl3 (30 mM; 133 μl) in 1.2 M HCl, and absorbance (670 nm) of aliquots of the resulting solution (300 μl) was determined. Essentially, in this assay, H2S is liberated from aqueous media by contact with trichloroacetic acid and then reacts with zinc acetate to form zinc sulphide. After treatment with an acid solution of N,N‐dimethyl‐p‐phenylenediamine and ferric chloride, methylene blue is formed over about 10 min. and is stable for several hours. The absorbance of methylene blue is then determined. The H2S concentration of each sample was calculated against a calibration curve of NaHS (3.125–250 μM). Aliquots (100 μl) of rat plasma were also assayed for H2S using the same procedure.
Assay of tissue myeloperoxidase activity, nitrate/nitrite (NO×), IL‐1β, TNF‐α, L‐selectin and sICAM‐1
NO× was determined spectrophotometrically in aliquots (80 μl) of plasma using the Griess reagent as described elsewhere [8]. Neutrophil sequestration in liver and lung from dexamethasone‐ and vehicle‐treated rats subjected to LPS‐induced endotoxic shock was quantified by measuring tissue MPO activity as described previously [8]. Plasma or neutrophil culture supernatant IL‐1β, TNF‐α, sICAM‐1 and L‐selectin were determined by ELISA using commercially available kits (R&D Systems, Inc., Minneapolis, MN, USA) according to the manufacturer’s instructions.
Measurement of CSE and iNOS by Western blotting
Liver (0.15 g) from control and LPS‐exposed animals was homogenized in 4 ml of ice cold homogenization buffer (250 mM sucrose, 20 mM Tris, 1 mM dithiothreitol). The homogenates were centrifuged at 4°C, 9000 ×g for 30 min. Supernatants were collected and centrifuged at 4°C, 14,000 ×g for 30 min. Neutrophils were lysed with a NP‐40 lysate buffer (Nonidet P‐40, 0.1% SDS in phosphate buffered saline (PBS)) supplemented with a protease inhibitor cocktail (10 μl/1ml, Hoffmann‐La Roche Ltd, Grenzacherstrasse, Basel, Switzerland) as described previously [20]. Aliquots of rat liver, LPS‐exposed neutrophils or human foetal liver cell lysates were subjected to Western blotting for CSE and iNOS as described previously [13] using an enhanced chemiluminescence detection kit (Amersham Biosciences, Amersham, Buckinghamshire, UK) followed by analysis using a Kodak Image Analyser (IS440CF, NEN Life Science, Boston, MA, USA). Antibodies to iNOS and to CSE were obtained from Caymen Chemicals Ltd., MI, USA and Brand Abnova Ltd., Taiwan, respectively. Protein concentration was determined using a commercial kit (Dc protein assay, Bio‐Rad Ltd, Hercules, CA, USA) and 30 μg total protein was analysed for each assay.
Assay of NF‐κB/DNA binding in neutrophils
Nuclear extract was prepared as described elsewhere [21]. Briefly, neutrophils were spun down (2000 ×g, 2 min., 4°C) and the resulting pellets were resuspended with a hypotonic buffer A (HEPES 10 mM, pH 7.5, KCl 10 mM, ethylene glycol tetraacetic acid (EGTA) 0.1 mM, ethylenediaminetetraacetic acid [EDTA] 0.1 mM, pH 8.0) containing a protease inhibitor cocktail (2 mM DFP [diisopropylfluorophosphate], 1 mM AEBSF [aminoethylbenzene sulfonyl fluoride], 1 mM PMSF [phenylmethylsulfonyl fluoride], 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 0.5 mM benzamidine, 1 mM dithiothreitol). Cells were kept on ice for 10 min., followed by vortex and centrifuge at 1000 ×g, 10 min., 4°C. The resulting nuclei pellet was washed again with buffer A, and the hypertonic, high salt buffer C was then added (HEPES 20 mM, pH 7.5, NaCl 0.4 M, EDTA 1 mM, EGTA 1 mM, glycerol 20%) together with the protease inhibitor cocktail. After centrifugation (14,000 ×g, 10 min., 4°C), the supernatant was collected and referred as the nuclear fraction. Protein measurements were done using the Bio‐Rad assay (Bio‐Rad Ltd). The nuclear extracts (10 μg) were assayed in duplicate for activity using a TransAM™ NF‐κB p65, assay kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer’s instructions. The OD450 was read on a 96‐well microplate reader (Tecan System, Inc., Mannedorf, Switzerland).
Statistics
Data show mean ± S.E. with the number of observations indicated in parentheses. Statistical analysis was by one‐way anova followed by post hoc Tukey test. A P‐value of <0.05 was taken to indicate a statistically significant difference.
Results
Effect of dexamethasone on LPS‐evoked inflammation in the conscious rat
Administration of LPS in conscious rats resulted 4 hrs thereafter in pronounced rises in plasma TNF‐α, IL‐1β, sICAM‐1 and NO× concentrations and elevated lung and liver MPO activity (Fig. 1), all of which are indicative of the presence of a systemic inflammatory response. Administration of dexamethasone (but not vehicle) significantly (P < 0.05) attenuated the LPS‐evoked rise in all of the plasma and tissue inflammatory parameters measured (Fig. 1). Mifepristone injection in these animals reduced the ability of dexamethasone to decrease the LPS‐evoked rise in plasma TNF‐α and IL‐1β (Fig. 1A and B). No difference was apparent in the effectiveness of dexamethasone administered either 1 hr before (i.e.‘prophylactic’) or 1 hr after (i.e.‘therapeutic’) LPS injection. An increase in plasma H2S concentration (Fig. 2A) alongside augmented liver H2S biosynthesis from exogenous cysteine (Fig. 2B) was also apparent in animals 4 hr after LPS injection. Dexamethasone administered either 1 hr before or 1 hr after LPS reduced the rise in plasma H2S concentration and the increase in LPS‐evoked liver H2S synthesizing activity. Pre‐treatment of animals with mifepristone reduced the inhibitory effect of dexamethasone on the LPS‐evoked rise in plasma H2S concentration and increase in liver H2S synthesizing activity.
Figure 1.
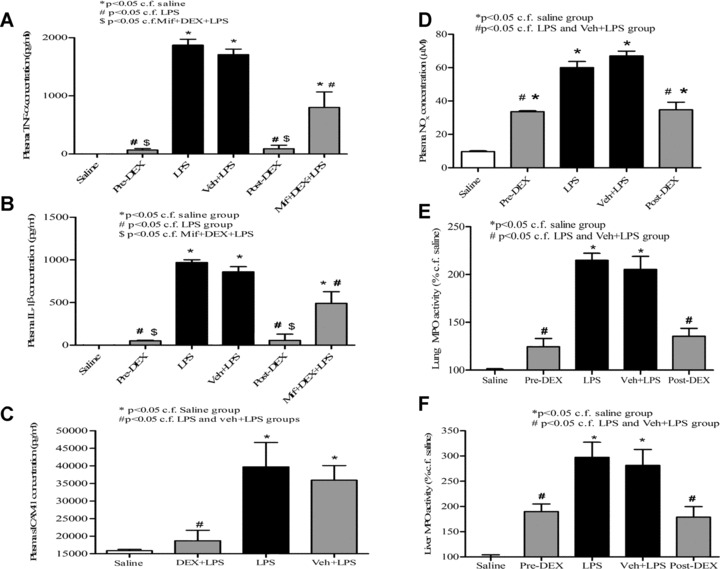
Effects of dexamethasone (1 mg/kg, i.p.) administered either 1 hr before (pre‐DEX) or 1 hr after (post‐DEX) injection of lipopolysaccharide (LPS) (4 mg/kg, i.p.) on (A) plasma TNF‐α, (B) plasma IL‐1β, (C) plasma sICAM‐1, (D) plasma NO×, (E) lung myeloperoxidase (MPO) activity and (F) liver MPO activity all determined 4 hrs after LPS injection. Absolute MPO activity in control (saline‐injected animals) was 14.7 ± 0.8 and 1.4 ± 0.05 OD405/μg DNA (n= 6) for lung and liver, respectively. ‘Saline’ indicates data from animals injected with saline (1 ml/kg, i.p.) in place of LPS. ‘Veh + LPS’ indicates data obtained from animals injected with vehicle for dexamethasone (ethanol/olive oil 5/95 v/v, 1 ml/kg) 1 hr before LPS (4 mg/kg, i.p.) administration. Results are mean ± S.E., n= 6.
Figure 2.
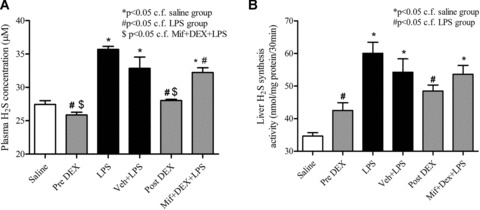
Effects of dexamethasone (1 mg/kg, i.p.) administered either 1 hr before (pre‐DEX) or 1 hr after (post‐DEX) lipopolysaccharide (LPS) (4 mg/kg, i.p.) injection on (A) plasma H2S concentration and (B) H2S synthesis from added L‐cysteine in liver homogenates in samples obtained 4 hrs after LPS injection. ‘Saline’ indicates data from animals injected with saline (1 ml/kg, i.p.) in place of LPS. ‘Veh + LPS’ indicates data obtained from animals injected with vehicle for dexamethasone (ethanol/olive oil 5/95 v/v, 1 ml/kg) 1 hr before LPS (4 mg/kg, i.p.) administration. ‘Mif+DEX+LPS’ indicates data from animals pre‐treated with mifepristone. Results are mean ± S.E., n= 6.
In order to investigate the mechanism(s) underlying the effect of dexamethasone on liver H2S synthesizing activity in LPS‐treated rats, experiments were undertaken to measure liver CSE protein expression. LPS injection resulted 4 hrs thereafter in significant increases in liver CSE (and iNOS) protein expression. Pre‐ treatment of animals with dexamethasone abolished the LPS‐evoked expression of both CSE and iNOS in the liver and these effects were partially antagonized by mifepristone (Fig. 3). In separate control experiments, dexamethasone (1–100 μM) did not affect liver H2S synthesizing activity in vitro (e.g. 100 μM; 43.3 ± 0.5 nmol H2S formed /mg protein c.f. 42.2 ± 0.3 nmol/mg protein in the absence of drug, n= 5, P > 0.05).
Figure 3.
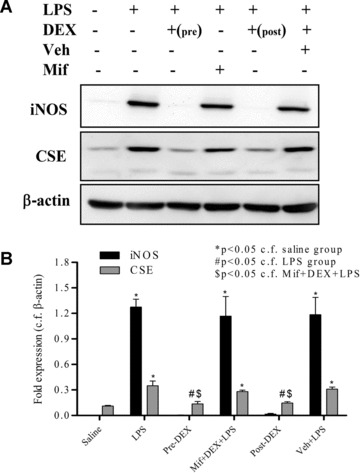
Effects of dexamethasone (1 mg/kg, i.p.) administered 1 hr before lipopolysaccharide (LPS) (Pre‐DEX; 4 mg/kg, i.p.) injection on liver expression of CSE and iNOS protein expression as determined by Western blotting. β‐actin was used as control protein. Livers were obtained from animals 4 hrs after LPS injection. Representative gel photos are shown in (A) and quantitative data are shown in (B). ‘Saline’ indicates data from animals injected with saline (1 ml/kg, i.p.) in place of LPS. ‘Veh + LPS’ indicates data obtained from animals injected with vehicle for dexamethasone (ethanol/olive oil 5/95 v/v, 1 ml/kg) 1 hr before LPS (4 mg/kg, i.p.) administration. ‘Mif+DEX+LPS’ indicates data from animals pre‐treated with mifepristone. Results are mean ± S.E., n= 6.
Effect of dexamethasone on LPS‐treated primary rat neutrophils and human foetal liver cells
Exposure of rat neutrophils to LPS (but not vehicle) resulted 4 hrs thereafter in significant increases in TNF‐α, IL‐1β and L‐selectin release into the medium. Pre‐treatment of neutrophils with dexamethasone inhibited the LPS‐evoked rise in release of both cytokines and L‐selectin (Fig. 4A–C). Dexamethasone, at the highest concentration tested (10 μM) inhibited TNF‐α, IL‐1β and L‐selectin generation by 75.2 ± 10.5%, 86.1 ± 7.6%, respectively, and 33.9 ± 8.7% (all n= 6, P < 0.05). In contrast, exposure to NaHS (100 μM) increased L‐selectin shedding from LPS‐exposed rat neutrophils (Fig. 4D).
Figure 4.
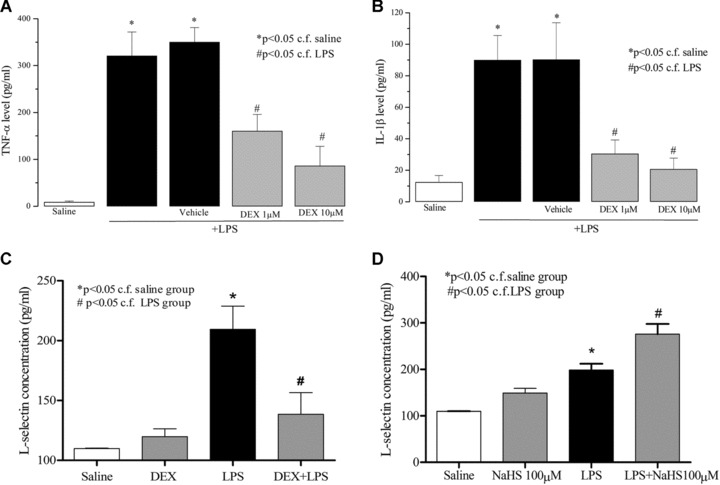
Effects of dexamethasone (DEX; 1 and 10 μM) on TNF‐α (A), IL‐1β (B) and L‐selectin (C) release from lipopolysaccharide (LPS) (1 μg/ml, 37°C, 4 hrs) challenged isolated rat neutrophils. Effect of NaHS (100 μM) on L‐selectin release from saline/LPS challenged isolated rat neutrophils (D). ‘Saline’ indicates data obtained from sampling of culture medium in the presence of saline (vehicle for LPS). ‘Vehicle’ indicates data obtained from LPS‐challenged neutrophils in the presence of vehicle for dexamethasone. Results are mean ± S.E., n= 6.
LPS (but not vehicle) also significantly increased neutrophil expression of both CSE and iNOS. LPS‐induced expression of iNOS was greater than that of CSE in these cells. Dexamethasone abolished the LPS‐induced expression of both CSE and iNOS in isolated neutrophils (Fig. 5). In separate experiments, LPS (but not vehicle) also increased CSE expression in human foetal liver cells. Dexamethasone (10 μM) again abolished LPS‐induced CSE expression in human foetal liver cells. In these experiments, human foetal liver cells did not express iNOS before or after treatment with LPS and thus the effect of dexamethasone on this enzyme could not be determined in these cells (Fig. 6).
Figure 5.
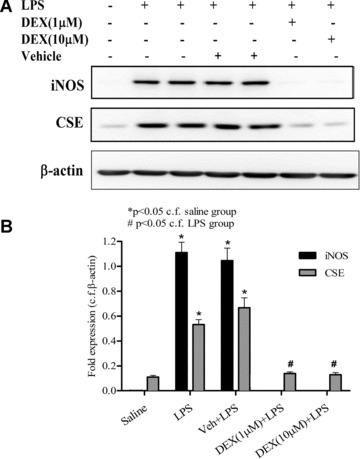
Effects of dexamethasone (1 and 10 μM) on expression of CSE and iNOS proteins as determined by Western blotting in lipopolysaccharide (LPS) (1 μg/ml, 37°C, 4 hrs) challenged isolated rat neutrophils. β‐actin was used as control protein. Representative gel photos are shown in (A) and quantitative data are shown in (B). ‘Saline’ indicates data obtained from sampling of culture medium in the presence of saline (vehicle for LPS). ‘Veh + LPS’ indicates data obtained from LPS‐challenged neutrophils in the presence of vehicle for dexamethasone. Results are mean ± S.E., n= 6.
Figure 6.
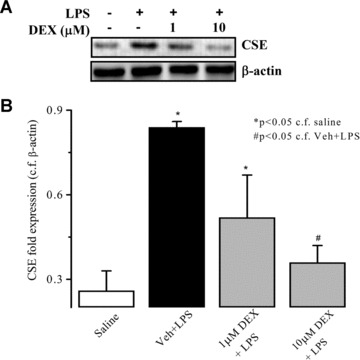
Effects of dexamethasone (1 and 10 μM) on expression of CSE and iNOS proteins as determined by Western blotting in lipopolysaccharide (LPS) (1 μg/ml, 37°C, 4 hrs) challenged human foetal liver cells. β‐actin was used as control protein. Representative gel photos are shown in (A) and quantitative data are shown in (B). ‘Saline’ indicates data obtained from sampling of culture medium in the presence of saline (vehicle for LPS). ‘Veh + LPS’ indicates data obtained from LPS‐challenged liver cells in the presence of vehicle for dexamethasone. Results are mean ± S.E., n= 4.
In an attempt to gain further information about the mechanism(s) underlying the ability of dexamethasone to inhibit LPS‐induced expression of CSE, further experiments were undertaken to monitor changes in NF‐κB‐p65 activation in LPS‐challenged neutrophils. LPS significantly induced NF‐κB‐p65 activation in incubated neutrophils whose effect was abolished by dexamethasone (1 and 10 μM) (Fig. 7). QNZ (NF‐κB transcription inhibitor) abolished LPS‐induced CSE overexpression in incubated neutrophils (Fig. 7B and C).
Figure 7.
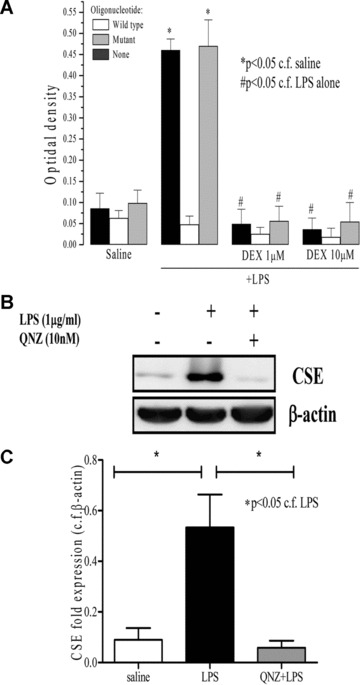
Effects of dexamethasone (DEX; 1 and 10 μM) on lipopolysaccharide (LPS) (1 μg/ml, 37°C, 4 hrs) challenged rat neutrophil NF‐κB‐p65 DNA‐binding activity (A). Effect of quinazoline (10 nM) on expression of CSE protein as determined by Western blotting in LPS (1 μg/ml, 37°C, 4 hrs) challenged isolated rat neutrophils. β‐actin was used as control protein. ‘Saline’ indicates data obtained from sampling of culture medium in the presence of saline (vehicle for LPS). Representative gel photos are shown in (B) and quantitative data are shown in (C). Results are mean ± S.E., n= 6.
Discussion
We show here that LPS injection increased plasma TNF‐α, IL‐1β, sICAM‐1 and NO× concentrations as well as lung and liver MPO activity and iNOS expression in rats 4 hrs thereafter. These various effects are indicative of the induction of a systemic inflammatory response i.e. endotoxic shock. Administration of dexamethasone either 1 hr before or 1 hr after LPS injection decreased all markers of inflammation thereby testifying to the potent anti‐inflammatory activity of this steroid as previously noted both in isolated cells and animals exposed to LPS [e.g. 22] as well as in a variety of animal models of inflammation [e.g. 23, 24]. The present data also confirm and extend our previous observation that LPS administered to either conscious mice [8] or rats [16] elevates both plasma H2S concentration and tissue H2S synthesizing activity most likely by increasing tissue expression of CSE enzyme.
Dexamethasone also abolished the LPS‐induced rise in plasma H2S concentration and significantly (>50%) inhibited tissue H2S biosynthesis in these animals. This effect is secondary to inhibition of LPS‐induced up‐regulation of tissue CSE protein expression. Interestingly, dexamethasone was equally effective in reducing the activity of the L‐cysteine/H2S system when administered either 1 hr before or 1 hr after LPS implying that this action of the steroid may contribute to its therapeutic use. The effect of dexamethasone on LPS‐evoked up‐regulation of CSE (and iNOS) was reduced by pre‐treating animals with mifepristone prior to steroid injection. Mifepristone has previously been reported to act as a potent antagonist of glucocorticoid receptors [25] and, as such, the present data indicate that the ability of dexamethasone to down‐regulate the LPS‐evoked increase in H2S formation was steroid receptor mediated.
In order to investigate the underlying mechanism of action of dexamethasone in more detail we studied its effect on LPS‐challenged rat neutrophils and human foetal liver cells. We report here that CSE is present in rat neutrophils, that LPS up‐regulates CSE expression in neutrophils and that co‐incubation with dexamethasone reduced the LPS‐evoked changes of TNF‐α, IL‐1β and L‐selectin in these cells. Dexamethasone also inhibited the expression of CSE (and iNOS) in LPS‐challenged neutrophils and of CSE in human foetal liver cells. Inhibition of the expression of both iNOS and CSE in rat neutrophils was accompanied by inhibition of NF‐κB activation as evidence by reduced NF‐κB‐DNA binding. In addition, we show that QNZ, a potent and selective inhibitor of NF‐κB transcriptional activation [6] abolished the increase in CSE expression in LPS‐challenged neutrophils again providing further evidence that CSE expression in neutrophils is largely under the control of NF‐κB and adding to our hypothesis that dexamethasone inhibits CSE expression in vivo thereby reducing tissue H2S formation in these animals by blocking NF‐κB activation via a steroid receptor dependent pathway.
Although a direct effect of dexamethasone on tissue/neutrophil CSE expression is the most likely explanation for the present data alternative mechanisms of action should be considered. Firstly, as reported previously and confirmed in the present study, dexamethasone, and indeed other steroids, reduce LPS‐up‐regulated tissue iNOS expression and thereby decrease nitric oxide biosynthesis. It is conceivable that reduced CSE expression in tissues/ cells exposed to dexamethasone may be secondary to inhibition of nitric oxide formation. If this is the case then exogenous nitric oxide would be expected to augment CSE expression in LPS‐injected rats. In this respect, we have previously reported that nitric oxide (released from the donor, nitroflurbiprofen) reduces (not increases) tissue H2S synthesizing activity/CSE expression in LPS‐injected rats [26]. As such, it seems unlikely that reduced nitric oxide formation contributes to the ability of dexamethasone to down‐regulate CSE expression in these tissues/cells. Secondly, we failed to detect any inhibitory effect of dexamethasone on liver H2S generation from added L‐cysteine in vitro thereby suggesting that this steroid does not directly inhibit CSE enzyme activity.
The demonstration that CSE is present in rat neutrophils is also of interest. Although there have, to the best of our knowledge, been no published reports of endogenous H2S release from neutrophils, it is known that the function of these cells are affected by exogenous H2S. For example, NaHS inhibited aspirin‐induced leukocyte adherence in rat mesenteric venules likely via activation of KATP channels [27]. In stark contrast, in a mouse model of septic shock (cecal ligation puncture), NaHS administration up‐regulated neutrophil rolling and attachment in mesenteric vessels [28]. In the present study, we show that H2S increases L‐selectin shedding from LPS‐exposed neutrophils suggesting a possible stimulatory effect of this gas on neutrophil function. Although the precise effect of H2S on neutrophil function is clearly complex and warrants further study, our finding that rat neutrophils contain CSE raises the intriguing possibility that, (i) H2S may act as an autocrine mediator in this cell and (ii) neutrophils may be a target for the effect of dexamethasone on H2S (as well as nitric oxide) formation.
Conclusions
We propose that the anti‐inflammatory effect of dexamethasone in this model of endotoxic shock may be due, at least in part, to inhibition of the formation of pro‐inflammatory H2S most probably by reduced NF‐κB‐mediated tissue/neutrophil CSE expression. Whether this effect of dexamethasone is shared by other anti‐inflammatory steroids and whether such an effect also contributes to other anti‐inflammatory effects of this class of drugs (e.g. in arthritis or asthma) warrants further investigation.
Acknowledgements
We thank King’s College, University of London, Peninsula Medical School, Universities of Exeter and Plymouth and the National University of Singapore for their financial support.
References
- 1. Rhen T, Cidlowski JA. Anti‐inflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med . 2005; 353: 1711–23. [DOI] [PubMed] [Google Scholar]
- 2. Barnes PJ, Adcock I. Anti‐inflammatory actions of steroids: molecular mechanisms. Trends Pharmacol Sci . 1993; 14: 436–41. [DOI] [PubMed] [Google Scholar]
- 3. Korhonen R, Lahti A, Hamalainen M, et al . Dexamethasone inhibits inducible nitric‐oxide synthase expression and nitric oxide production by destabilizing mRNA in lipopolysaccharide‐treated macrophages. Mol Pharmacol . 2002; 62: 698–704. [DOI] [PubMed] [Google Scholar]
- 4. Lasa M, Brook M, Saklatvala J, et al . Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen‐ activated protein kinase p38. Mol Cell Biol . 2001; 21: 771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barnes PJ, Karin M. Nuclear factor‐kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med . 1997; 336: 1066–71. [DOI] [PubMed] [Google Scholar]
- 6. Tobe M, Isobe Y, Tomizawa H, et al . A novel structural class of potent inhibitors of NF‐kappa B activation: structure‐activity relationships and biological effects of 6‐aminoquinazoline derivatives. Bioorg Med Chem . 2003; 11: 3869–78. [DOI] [PubMed] [Google Scholar]
- 7. Zhao W, Zhang J, Lu Y, et al . The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J . 2001; 20: 6008–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li L, Bhatia M, Zhu YZ, et al . Hydrogen sulfide is a novel mediator of lipopolysaccharide‐induced inflammation in the mouse. FASEB J . 2005; 19: 1196–8. [DOI] [PubMed] [Google Scholar]
- 9. Kawabata A, Ishiki T, Nagasawa K, et al . Hydrogen sulfide as a novel nociceptive messenger. Pain . 2007; 132: 74–81. [DOI] [PubMed] [Google Scholar]
- 10. Bhatia M, Wong FL, Fu D, et al . Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J . 2005; 19: 623–5. [DOI] [PubMed] [Google Scholar]
- 11. Mok YYP, Atan MS, Cheung YP, et al . Role of hydrogen sulphide in haemorragic shock in the rat: protective effect of inhibitors of hydrogen sulphide biosynthesis. Br J Pharmacol . 2004; 143: 881–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fiorucci S, Antonelli E, Distrutti E, et al . Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti‐inflammatory nonsteroidal drugs. Gastroenterology . 2005; 129: 1210–24. [DOI] [PubMed] [Google Scholar]
- 13. Whiteman M, Armstrong JS, Chu SH, et al . The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’ J Neurochem . 2004; 90: 765–8. [DOI] [PubMed] [Google Scholar]
- 14. Whiteman M, Cheung NS, Zhu YZ, et al . Hydrogen sulphide: a novel inhibitor of hypochlorous acid‐mediated oxidative damage in the brain Biochem Biophys Res Commun . 2005; 326: 794–8. [DOI] [PubMed] [Google Scholar]
- 15. Whiteman M, Li L, Kostetski I, et al . Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem Biophys Res Commun . 2006; 343: 303–10. [DOI] [PubMed] [Google Scholar]
- 16. Li L, Rossoni G, Sparatore A, et al . Anti‐inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic Biol Med . 2007; 42: 706–19. [DOI] [PubMed] [Google Scholar]
- 17. Sidhapuriwala J, Li L, Sparatore A, et al . Effect of S‐diclofenac, a novel hydrogen sulfide releasing derivative, on carrageenan‐induced hindpaw oedema formation in the rat. Eur J Pharmacol . 2007; 569: 149–54. [DOI] [PubMed] [Google Scholar]
- 18. Sun J, Ramnath RD, Bhatia M. The neuropeptide substance P upregulates chemokine and chemokine receptor expression in primary mouse neutrophils. Am J Physiol . 2007; 293: C696–704. [DOI] [PubMed] [Google Scholar]
- 19. Tan TCH, Sit KH, Wong KP. UDP‐ glucuronyltransferase activity toward harmol in human liver and human fetal liver cells in culture. Anal Biochem . 1990; 185: 44–50. [DOI] [PubMed] [Google Scholar]
- 20. Dragon S, Saffar AS, Shan L, et al . IL‐17 attenuates the anti‐apoptotic effects of GM‐CSF in human neutrophils. Mol Immunol . 2008; 45: 160–8. [DOI] [PubMed] [Google Scholar]
- 21. Choi M, Rolle S, Wellner M, et al . Inhibition of NF‐κB by a TAT‐NEMO‐ binding domain peptide accelerates constitutive apoptosis and abrogates LPS‐delayed neutrophil apoptosis. Blood . 2003; 102: 2259–67. [DOI] [PubMed] [Google Scholar]
- 22. Fierro IM, Nascimento‐DaSilva V, Arruda MA, et al . Induction of NOS in rat blood PMN in vivo and in vitro: modulation by tyrosine kinase and involvement in bactericidal activity. J Leukoc Biol . 1999; 65: 508–14. [DOI] [PubMed] [Google Scholar]
- 23. Desouza IA, Franco‐Penteado CF, Camargo EA, et al . Inflammatory mechanisms underlying the rat pulmonary neutrophil influx induced by airway exposure to staphylococcal enterotoxin type A. Br J Pharmacol . 2005; 146: 781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsao CM, Ho ST, Chen A, et al . Low‐dose dexamethasone ameliorates circulatory failure and renal dysfunction in conscious rats with endotoxemia. Shock . 2004; 21: 484–91. [DOI] [PubMed] [Google Scholar]
- 25. Fan J, Gong XQ, Wu J, et al . Effect of glucocorticoid receptor (GR) blockade on endotoxemia in rats. Circ Shock . 1994; 42: 76–82. [PubMed] [Google Scholar]
- 26. Anuar F, Whiteman M, Siau JL, et al . Nitric oxide‐releasing flurbiprofen reduces formation of proinflammatory hydrogen sulfide in lipopolysaccharide‐treated rat. Br J Pharmacol . 2006; 147: 966–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zanardo RC, Brancaleone V, Distrutti E, et al . Hydrogen sulfide is an endogenous modulator of leukocyte‐mediated inflammation. FASEB J . 2006; 20: 2118–20. [DOI] [PubMed] [Google Scholar]
- 28. Zhang H, Zhi L, Moochhala SM, et al . Endogenous hydrogen sulfide regulates leukocyte trafficking in cecal ligation and puncture‐induced sepsis. J Leukoc Biol . 2007; 82: 894–905. [DOI] [PubMed] [Google Scholar]