

Abstract
p53 apoptosis effector related to PMP‐22 (PERP) is a transcriptional target gene of p53 tumour suppressor that is specifically induced during apoptosis and not during cell cycle arrest. In primary uveal melanoma (UM), the most common intraocular malignancy in adults that has a reportedly unaffected signalling pathway upstream of and including p53, PERP expression is down‐regulated in the metastatic monosomy 3‐type tumours, compared with the less aggressive disomy 3‐type tumours. Here, we demonstrate experimentally, by the use of full‐length PERP‐green fluorescent protein (GFP) fusions and real‐time confocal microscopy, the intracellular targeting and plasma membrane localization of PERP in living UM cells and show that expression of PERP induces caspase‐mediated apoptosis in UM cells. Induction of PERP expression in GFP‐PERP‐transfected UM cells leads to increased levels of cleaved caspase‐8 forms, as well as to reduction of its full‐length substrate Bid, but not to detectable processing of caspase‐9. The levels of mature caspase‐8, ‐9 and ‐3 proteins significantly correlate with PERP expression levels in primary UMs. Transcriptional profiling of PERP and caspase‐8 in tumour specimens indicates that the positive association of PERP and caspase‐8 proteins is a consequence of post‐translational processing, most likely at the level of caspase‐8 cleavage, and not of increased transcription of pro‐caspase‐8. We conclude that PERP expression leads to activation of an extrinsic receptor‐mediated apoptotic pathway, with a possible subsequent engagement of the intrinsic apoptotic pathway. The findings underline the apoptotic pathway mediated by PERP as a critical mechanism employed by UM tumours to modulate susceptibility to apoptosis.
Keywords: apoptosis, PERP, p53, caspase, uveal melanoma, transfection, green fluorescent protein
Introduction
Cell death by apoptosis is critical for the health of multi‐cellular organisms being involved both in the development and in the maintenance of tissue homeostasis, as well as in the elimination of abnormal cells [1]. The transcription factor p53 is part of a complex signalling network involved in the regulation of cellular responses to oncogenic stress, including DNA damage, which performs its tumour suppression role by either inducing cell cycle arrest (in G1 and/or G2) or eliminating abnormal cells by apoptosis [2, 3, 4]. The function of p53 as a tumour suppressor is underlined by the presence of its mutations in approximately half of human cancers [5], whereas the p53 pathway is compromised by other means in the majority of other cancers [3, 6]. The mechanism by which p53 activates cell cycle arrest is far better characterized [7, 8, 9] than the mechanisms of p53‐dependent apoptosis, and hence, the molecular factors influencing and adjusting the choice between cell cycle arrest and apoptosis in different cell types and cellular states are of major interest.
p53 apoptosis effector related to PMP‐22 (PERP) was originally identified as a novel candidate effector in the p53‐dependent apoptotic pathway, being a transcriptional target gene of p53 that is specifically induced during apoptosis and not during cell cycle arrest [10]. The induction of apoptosis by expression of PERP was described in fibroblasts, thymocytes and neurons [10, 11, 12] and in early zebrafish embryos [13]. However, functional studies thus far have only addressed the requirement of PERP to mediate stress‐induced apoptosis [10, 11, 12, 13], leaving the mechanism of PERP action undefined.
More recently, we identified PERP as an important molecular determinant of an aggressive type of tumour in which p53 mutations are rare. Specifically, the expression of PERP was down‐regulated at both the transcriptional and the protein level in the aggressive, highly metastatic (monosomy 3)‐type primary uveal melanoma (UM) tumours, compared with the less aggressive (disomy 3)‐type tumours [14]. UM, the most common primary intraocular tumour in adults [15], has a high mortality rate primarily due to liver metastases [16, 17, 18]. The notorious resistance of UMs to radiotherapy [19] and various forms of chemotherapy [20, 21]‐treatments acting by inducing p53‐dependent apoptosis in cancer cells [22, 23, 24]‐can only be partly explained by a low proliferation rate, suggesting that resistance to apoptosis is an intrinsic feature of UM. It is thus accepted that the p53‐dependent apoptotic pathway is functionally impaired in UM, despite the reported infrequency of p53 mutations, inactivation or loss of heterozygosity and the apparently undamaged upstream signalling to p53 [25, 26, 27]. Hence, reports of functional defects downstream of p53 [20] point to a likely de‐regulation of a downstream effector of p53, such as PERP, as a strategy adopted by UM to acquire resistance to apoptosis.
The PERP gene (also initially designated THW [28]) was additionally reported to be down‐regulated in metastasizing cutaneous melanoma, pancreas and mammary carcinoma cell lines and tumours of the ovary, uterus and breast, compared with the respective nonmetastasizing cell lines and normal tissues [28]. A tumour suppressor role for PERP was consequently suggested based on this down‐regulation and on the chromosomal map location of the PERP gene (6q24), for which loss of heterozygosity was described in several tumours [29, 30, 31], including cutaneous melanoma [32]. However, any characterization of PERP as a tumour suppressor related to the apoptotic ability of the cells remains severely restricted as long as the mechanism of activation of cell death by PERP is not clear and the apoptotic pathway engaged by PERP expression remains unknown.
The present study aimed to assess the functional role of PERP in UM in relation to the ability of this type of cancer to undergo apoptosis and initiate the characterization of the cell death mechanism(s) mediated by PERP. We report for the first time the fate of newly synthesized PERP protein in living cells and demonstrate the increased susceptibility of UM cells in vitro to apoptosis induced by elevated levels of PERP protein. Furthermore, we present evidence that PERP‐induced apoptosis is mediated by the caspase‐dependent pathway and report a strong association between the levels of PERP expression and caspase activation, both in UM cells in vitro and in primary UM tumours.
Materials and methods
cDNA cloning and construction of green fluorescent protein (GFP)‐tagged PERP
The full‐length open‐reading frame (ORF) of PERP was amplified by PCR from a clone containing the human PERP cDNA (Genbank accession No: BC010163, I.M.A.G.E. ID: 3639866, Gene Collection [MGC] Clones; Geneservice, Ltd., Cambridge, UK) using oligonucleotide primers designed to incorporate flanking attB1 and attB2 sites into the forward and reverse primers, respectively, to allow subsequent recombination with the Gateway donor vector pDONR221 (Invitrogen, Paisley, UK). The primers (custom‐synthesized by MWG‐Biotech, Ebersberg, Germany) were: for PERP‐N‐ terminus fusion, forward primer PERP53attB‐F (5′‐GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGATCCGCTGCGGCCTGGCCTG‐3′) and reverse primer PERP53attB‐R (5′‐GGGGACCACTTTGTACAAGAAAGCTGGGTCTTAGGCAGATGTGTAGAAGTAC‐3′), where attB1 and the ATG initiation codon are shown in bold and underlined, respectively, in the forward primer, and the attB2 and stop codon are shown in bold and underlined, respectively, in the reverse primer; for PERP C‐terminus fusion, forward primer PERP47attB‐F (5′‐GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAGGAGATAGAACCATGATCCGCTGCGGCCTGGCCTG‐3′) and reverse primer PERP47attB‐R (5′‐GGGGACCACTTTGTACAAGAAAGCTGGGTCGGCAGATGTGTAGAAGTAC‐3′), where attB1 and the ATG initiation codon are shown in bold and underlined, respectively, in the forward primer, and the attB2 site is shown in bold in the reverse primer.
The final composition of the PERP PCR reaction was 10 ng template plasmid DNA, 0.6 μM forward primer, 0.6 μM reverse primer, 0.3 mM dNTPs, 1 mM MgSO4, 1X Pfx DNA polymerase buffer (Invitrogen) and 2.5 units of Platinum Pfx DNA polymerase (Invitrogen). The PCR conditions were 1 cycle of 94°C for 2 min., followed by 35 cycles of 94°C for 30 sec., 60°C for 1 min. and 68°C for 1 min. PCR products were analysed by agarose gel electrophoresis, revealing a single amplified product of approximately 640 bp (PERP ORF plus attB sites), which was purified using the Qiagen Gel Purification System (Qiagen, West Sussex, UK), following the manufacturer’s protocol. An attL‐containing entry clone (pENTRY) was generated from a BP recombination reaction (Invitrogen) composed of 50 fmol of attB‐flanked PERP PCR product, attP‐containing donor vector (pDONR221, 150 ng; Invitrogen) and BP Clonase™ II enzyme mix (Invitrogen), performed at 25°C for 18 hrs. One‐tenth of the BP recombination reaction was used to transform competent DH5‐αEscherichia coli (E. coli) cells, and pENTRY clones were selected on LB/kanamycin agar plates (50 μg/ml). pENTRY clones were verified by custom sequencing (The Sequencing Service, School of Life Sciences, University of Dundee, Scotland) to ensure no errors had been introduced by PCR. LR recombination reactions between the attL‐containing PERP pENTRY clone (150 ng) and the attR‐containing GFP‐expressing destination vectors, pcDNA‐DEST53 or pcDNA‐DEST47 (150 ng each; Invitrogen) were performed at 25°C for 3 hrs with LR Clonase™ II enzyme mix (Invitrogen), according to the manufacturer’s recommendations, to generate pGFP‐PERP and pPERP‐GFP fusion constructs, respectively. The resulting expression clones were selected on LB‐ampicillin agar plates (100 μg/ml) following transformation of DH5‐αE. coli cells with one‐tenth of the LR recombination reaction mixture. The recombinant expression clones pGFP‐PERP and pPERP‐GFP were purified using a Qiagen Endofree Plasmid Maxi kit and procedure. The sequences of pGFP‐PERP and pPERP‐GFP constructs were determined to verify that the PERP ORF was in frame with the C‐ and N‐terminus, respectively, of the GFP ORF following the LR recombination reaction.
Cell culture and transfection
Three human UM cell lines were used in this study: OCM‐1 [33], kindly provided by Dr. Dan Albert, University of Wisconsin‐Madison, Madison, WI, USA; 92‐1 [34] and MEL202 [35], kindly provided by Dr. Martine Jager, University Hospital Leiden, Leiden, The Netherlands. All tissue culture media and supplements were from Invitrogen, unless otherwise stated.
The UM cell lines were grown in RPMI‐1640 medium with 2 mM L‐glutamine and 25 mM HEPES supplemented with 10% v/v heat‐inactivated foetal calf serum (FCS; Biosera, East Sussex, UK), 1 mM sodium pyruvate and 1% v/v non‐essential amino acids. The cells were grown at 37°C in controlled humidity and 5% CO2 atmosphere, with changes of medium every 2 to 3 days. Normal development of cultures was assessed daily by phase‐contrast microscopy. The cultures were routinely passaged by gentle dissociation with Trypsin‐Versene (Cambrex, Berkshire, UK), followed by re‐seeding at a split ratio of 1:6. For confocal microscopy and fluorescence microscopy, the cells were seeded in 3 ml medium on Iwaki 35‐mm dishes (Barloworld Scientific, Staffordshire, UK) at a density of 2 × 105 cells per dish. After 24 hrs, the cells were transfected with the appropriate plasmid using ExGen 500 in vitro transfection reagent (Fermentas Life Sciences, York, UK), following the manufacturer’s recommendations, using 3 μg DNA and 9.87 μl ExGen 500 (6 equivalents) per 35‐mm dish, prior to analysis by confocal imaging and Western immunoblotting.
Confocal imaging of live transfected cells
Live cell imaging of transfected cells was performed in a humidity‐controlled stage incubator, where cells were maintained at 37°C with 5% CO2 for the duration of the experiment. The cells were imaged with a Zeiss LSM 510 confocal microscope with a Pan‐Apochromat 63×/1.4 oil objective (Carl Zeiss Ltd., Welwyn Garden City, UK). Excitation of GFP was achieved with a 488‐nm argon laser, and the emitted light was collected through a 505‐ to 530‐nm bandwidth filter. The Alexa Fluor 647 annexin‐V conjugate dye (Molecular Probes, Inc., Eugene, OR, US) used for cell viability assays, as described below, was excited at 633 nm, and its emission was detected at 650 nm, whereas propidium iodide (PI; Sigma‐Aldrich, Poole, UK) was excited at 543 nm, followed by detection at 580–620 nm. Images were collected at 512 × 512 or 1024 × 1024 pixel resolution. Serial Z‐plane sections (0.1–0.5 μm thickness) of transfected cells were concatenated for reconstruction of whole cells. Cell viability counts were observed under a 20× magnification. Data acquisition and analysis were carried out with Zeiss LSM 510 software, version 3.2.
Determination of cell viability and apoptotic cell morphology
The cells were incubated with Alexa Fluor 647 annexin‐V conjugate dye at a dilution of 1:500 to detect externalization of phosphatidylserine (PS) as an indicator of early‐stage apoptosis, and PI at a final concentration of 2 μg/ml to identify cells that had lost membrane integrity. In order to quantify cell death, including cells committed to apoptosis, cells that were positive for both annexin‐V and PI staining in addition to cells that were positive for annexin‐V staining only were considered nonviable.
Cell counts were determined over 20 viewing fields between 24 and 72 hrs after transfection. The percentage of nonviable cells was calculated by averaging the number of nonviable cells in the transfected (or nontransfected) cell population relative to the total number of transfected (or nontransfected) cells in all viewing fields. The cell counts from three independent transfection experiments were averaged and the standard deviation was calculated.
Caspase inhibitors
The irreversible pan‐caspase inhibitor, Z‐VAD‐FMK (MBL International Corporation, Woburn, MA, USA), and caspase inhibitor negative control, Z‐FA‐FMK (BD Biosciences Pharmingen, San Diego, CA, USA), were dissolved in DMSO and added directly to the cell culture medium 4 hrs after transfection at a final concentration of 40 μM. The final concentration of DMSO in culture medium was less than 2μl/ml. Cells not treated with caspase inhibitor (untreated) were incubated with an equivalent volume of DMSO to control for the effects of inhibitor carrier.
UM primary tumour specimens
UM specimens were from patients treated by primary enucleation or local resection at the Ocular Oncology Centre, Royal Liverpool University Hospital, Liverpool, UK, with no prior radiation or chemotherapy. Freshly dissected tumour specimens were snap‐frozen in liquid nitrogen and stored at −80°C, prior to protein and RNA isolation for analysis by Western immunoblotting and real‐time quantitative PCR, respectively. Of the total 224 UM specimens processed for this study, 165 specimens successfully passed the required quality control and suitability criteria for downstream application, resulting in 101 specimens processed for protein studies only, 33 specimens for combined protein and RNA experiments and 31 specimens subjected to RNA isolation only. The study was approved by the Liverpool Research Ethics Committee. Informed consent was obtained from each patient prior to sample collection.
Western immunoblot analysis
Total protein was purified from tumour tissue homogenates using the AllPrep® DNA/RNA/Protein mini kit (Qiagen) and diluted 1:4 with Laemmli sample buffer for Western blot analysis. For the preparation of lysates of cultured UM cells, cells grown on 35‐mm tissue culture dishes were washed with phosphate buffered saline (PBS) and lysed in 100 μl of lysis buffer (0.128 M β‐mercaptoethanol, 40 mM Tris, 10% v/v glycerol, 1% w/v SDS and 0.01% w/v bromophenol blue; all materials from Sigma‐Aldrich). Floating cells were recovered from the pooled media and PBS washes by centrifugation and combined with the cell lysate. The extracts were passed four times through a 25‐gauge needle to reduce sample viscosity.
Proteins in cell lysates or from tumour tissue homogenates were boiled for 10 min. and resolved by electrophoresis in 10% SDS‐polyacrylamide gels, alongside molecular weight markers (Precision Plus Protein Standards; Bio‐Rad Laboratories, Hemel Hempstead, UK) and subjected to Western blot analysis, as described previously [14]. The primary antibodies used were: anti‐PERP polyclonal (1:1000 dilution; Abcam, Cambridge, UK), anti‐GFP polyclonal (1:100 dilution; Living Colours™ Clontech, Palo Alto, CA, USA), anti‐caspase‐8 monoclonal (clone 1‐3, 1:1000 dilution; Calbiochem, Merck Biosciences, Nottingham, UK), anti‐caspase‐9 monoclonal (clone 96‐2‐22, 1:100 dilution; Upstate, Lake Placid, NY, USA), anti‐caspase‐3 polyclonal (clone H‐277, 1:100 dilution; Santa Cruz Biotechnology, Inc., CA, USA), anti‐Bid polyclonal (clone FL‐195, 1:100 dilution; Santa Cruz Biotechnology) and anti‐glyceraldehyde phosphate dehydrogenase (GAPDH, 1:5000 dilution; Abcam).
Immunocomplexes were detected with horseradish peroxidase‐conjugated secondary antibody (Sigma‐Aldrich) by a chemiluminescent method (ECL™, Amersham Pharmacia Biotech, Little Chalfont, UK), followed by exposure to autoradiography film. Blots were stripped by incubation in stripping solution (62.5 mM Tris‐HCl, pH 6.8, 100 mM β‐mercaptoethanol, 2% w/v SDS) at 65°C for 30 min., followed by 2 × 10 min. washes in TBS‐T buffer (20 mM Tris‐HCl, 150 mM NaCl, pH 7.6, plus 0.1% Tween‐20) at room temperature, prior to reprobing with antibody. Relative intensities of the bands were obtained by densitometry using ImageJ (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, MD, USA; http://rsb.info.nih.gov/ij/, 1997–2007.)
Real‐time quantitative PCR
Total RNA was isolated from primary tumour specimens using Qiagen AllPrep® DNA/RNA/Protein mini kit. First strand of cDNA was synthesized from 2 μg of DNase‐treated total RNA using SuperScript™ II RNaseH‐ negative reverse transcriptase (Invitrogen) and random hexamers, following the manufacturer’s protocol. The RNA template was removed from the cDNA by incubating the reaction mixture with 2 units of E.coli RNaseH (Promega) at 37°C for 20 min. Quantitative real‐time PCR was performed with qPCR Core Kit for SYBR® Green I (Eurogentec, Southampton, UK) and Rox Passive Reference (Eurogentec) with 1/20th of the reverse transcription reaction. Each 20 μl PCR reaction contained 1× reaction buffer, 3.5 mM MgCl2, 200 μM each dNTP, 0.5 μM forward primer, 0.5 μM reverse primer, 0.6 μl diluted SYBR® Green, 0.8 μl diluted Rox and 0.5 U HotGoldStar polymerase (Eurogentec). All amplifications were performed in triplicate with a Stratagene MX3005P® QPCR System (Stratagene, La Jolla, CA, USA). PCR conditions consisted of an initial denaturing step at 95°C for 10 min., followed by 35 cycles of denaturation at 94°C for 10 sec., annealing at 60°C for 20 sec. and extension at 72°C for 15 sec., followed by one dissociation/melt segment at 95°C for 1 min., 55°C for 30 sec. and 95°C for 30 sec., during which melting curve data were collected. Absolute quantification was based on gene‐specific standard curves, constructed by amplification of serially diluted gene‐specific oligo template. Lactate dehydrogenase A (LDHA)‐specific amplification was performed under similar conditions and was used as an endogenous control for normalization. Control reactions with no template were performed in all rounds of amplification as a negative control.
The gene‐specific primer sets and templates used were as follows: PERP forward primer, 5′‐GGCTTCATCATCCTGGTGAT; PERP reverse primer, 5′‐ACAGCAGCCAAGGCAAGGAG; PERP oligo template, 110 bp spanning the two primer sites above and two exons (accession number NM_022121); caspase‐8 forward primer, 5′‐AAGAGTCTGTGCCCAAATCAAC, caspase‐8 reverse primer, 5′‐ GAGAGTCCGAGATTGTCATTACC; caspase‐8 oligo template, 107 bp spanning the two primer sites and two exons (accession number NM_001228); LDHA forward primer, 5′‐GATGATGGATCTCCAACATG, LDHA reverse primer, 5′‐CCCAGCCGTGATAATGACCA and LDHA oligo template, 109 bp spanning the respective primers and two exons (accession number NM_005566). Primers and oligo probes were custom‐synthesized (MWG‐Biotech).
Results
Expression and subcellular localization of GFP‐tagged PERP fusion proteins
To characterize trafficking and subcellular localization of newly synthesized PERP protein in living cells, 92‐1, OCM‐1 and MEL202 cell lines were transiently transfected with pGFP‐PERP and pPERP‐GFP constructs and monitored in real time by confocal fluorescence microscopy. Fluorescence was first detected approximately 17 hrs after transfection, and the transfection efficiency achieved, evaluated at 24 hrs after transfection, ranged between 20 and 25% for 92‐1 and MEL202 cells, and between 30 and 40% for OCM‐1 cells. Control transfections with the vector plasmid pEGFP‐N1 encoding GFP on its own and mock transfections (no exogenous DNA) were performed for all cell lines.
All cell lines transfected with pGFP‐PERP construct showed marked plasma membrane‐associated fluorescence, with additional perinuclear localization in the cytoplasm, previously identified as the endoplasmic reticulum (ER)/Golgi apparatus [36, 37], and with no detectable fluorescence signal in the nucleus (Fig. 1A). In contrast, cells transfected with pEGFP‐N1 and expressing GFP only presented a diffuse fluorescence, evenly distributed throughout the cytoplasm and in the nucleus, except the nucleoli, indicating that the plasma membrane localization of the GFP‐PERP fusion protein is specified by PERP. Cells transfected with pPERP‐GFP construct showed fluorescence concentrated perinuclearly that lacked plasma membrane localization (Fig. 1A), suggesting that the presence of the fluorescent tag at the C‐terminus of PERP interferes with the processing of PERP by ER, thus impairing its trafficking to the plasma membrane. To account for the differences in the subcellular localization of GFP‐PERP and PERP‐GFP fusion proteins, the respective amino acid sequences were analysed using two topology prediction programs, hmmtop [38] and TMpred [39]. The analysis confirmed that the predicted topology of GFP‐PERP consists of four transmembrane helices and is similar to that of PERP alone, whereas the PERP‐GFP fusion protein lacked clear prediction of such transmembrane domains. Consequently, all further in vitro functional experiments were based on the use of the pGFP‐PERP fusion construct. The expression and stability of the GFP‐PERP fusion protein were assessed by immunoblot analysis of transfected cells (Fig. 1B). The fusion protein detected by both anti‐PERP and anti‐GFP antibodies in lysates of all three UM cell lines investigated was of the expected molecular weight (approximately 48 kD) of PERP (21 kD) fused to GFP (27 kD), thus confirming the nature and integrity of the de novo‐synthesized fusion protein.
Figure 1.
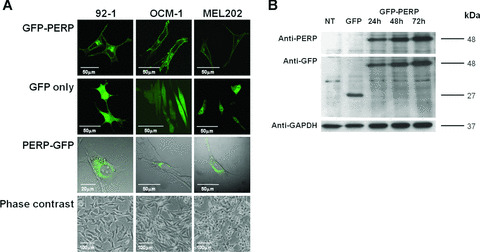
(A) Confocal images showing transiently transfected living 92‐1, OCM‐1 and MEL202 cells expressing GFP‐PERP, GFP only and PERP‐GFP at 24 hr after transfection. GFP‐PERP fusion protein is targeted to the plasma membrane, although the presence of the fluorescent tag at the C‐terminus of PERP impairs this targeting (bright field is shown for pPERP‐GFP transfected cells to illustrate the lack of plasma membrane targeting of the PERP‐GFP fusion protein). Phase‐contrast images illustrate the characteristic cell types of the cell lines used, specifically, 92‐1 cells being predominantly epithelioid, OCM‐1 being predominantly spindle cell type and MEL202 being mixed (epithelioid and spindle) cell type, which are features that assist in the prediction of metastatic disease. (B) Lysates of cells transfected with pGFP‐PERP were analysed at 24, 48 and 72 hrs after transfection by Western blotting using anti‐PERP and anti‐GFP antibody. Nontransfected (NT) and pEGFP‐N1 (GFP only)‐transfected cells served as controls. GFP‐PERP fusion protein of the expected 48 kD size was detected by both antibodies in pGFP‐PERP‐transfected cells only, whereas the 27 kD GFP protein was detected only by the anti‐GFP antibody in pEGFP‐N1‐transfected cells. The same blot was sequentially probed with anti‐GAPDH antibody to confirm equal loading. GFP‐PERP fusion protein was identically detected in all UM cells studied (blot shown is of MEL202 cell lysates).
Effect of PERP expression on UM cell viability
To initiate the functional study of PERP in UM, we determined the effect of PERP expression on cell viability. For this purpose, 92‐1, OCM‐1 and MEL202 UM cells were transfected with pGFP‐PERP and their viability assessed by annexin‐V binding and PI staining, monitored between 24 and 72 h after transfection. In all three cell lines, cell death was significantly higher in transfected cells expressing GFP‐PERP protein than in nontransfected cells or cells expressing GFP‐only (Student’s t‐test, P≤ 0.009; Fig. 2A). The highest number of nonviable cells was detected in the pGFP‐PERP‐transfected population of 92‐1 cells (57.45 ± 4.18%), followed by OCM‐1 (41.71 ± 2.05%) and MEL202 (38.75 ± 3.45%). PERP‐induced cell death was thus significantly higher in 92‐1 cells compared with OCM‐1 (t‐test, P= 0.02) and MEL202 (t‐test, P= 0.009). The pGFP‐PERP‐transfected cells presented hallmarks of apoptosis such as membrane blebbing, nuclear fragmentation and formation of apoptotic bodies (Fig. 2B and C). Nontransfected cells in the same cell population had normal appearance and lacked morphological characteristics of apoptosis (bright field in Fig. 2C).
Figure 2.
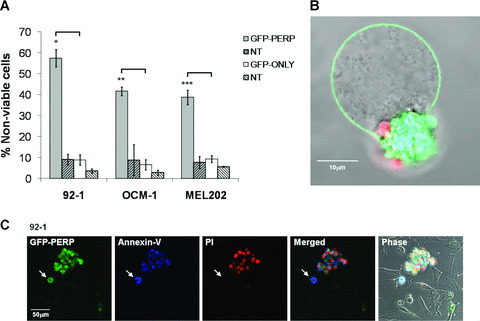
Effect of PERP expression on UM cell viability. 92‐1, OCM‐1 and MEL202 cells were transiently transfected with either pGFP‐PERP or pGFP‐only and monitored by fluorescence microscopy in the presence of annexin‐V and PI. (A) The number of viable and nonviable cells in 20 fields were counted between 24 and 72 hrs after transfection, taking into account the sequential binding of annexin‐V and PI. The mean percentage of transfected (or nontransfected) cells dead in the transfected (or nontransfected) cell population, with standard deviation from three independent transfections, is shown. t‐test, *P= 0.002, **P= 0.004 and ***P= 0.009 compared with pGFP‐only transfected cells. (B) MEL202 cell expressing GFP‐PERP (green) showing morphological features of apoptosis, including blebbing of the plasma membrane and DNA stained with PI (red) packaged into apoptotic bodies that have lost membrane integrity. (C) 92‐1 UM cells expressing GFP‐PERP (green) showing positive annexin‐V binding (blue) and PI staining (red). A cell committed to apoptosis that has not yet lost membrane integrity is indicated (arrow). The bright field image shows nontransfected cells in the same cell population, which do not present the apoptotic characteristics.
Effect of caspase inhibition on PERP‐induced cell death
To determine whether the observed PERP‐induced cell death was mediated through the activation of a caspase‐dependent apoptotic pathway, cells transfected with pGFP‐PERP were treated with the pan‐caspase inhibitor, Z‐VAD‐FMK. As a negative control treatment, pGFP‐PERP‐transfected cells were incubated with Z‐FA‐FMK, a compound structurally similar to the pan‐caspase inhibitor that, however, lacks the inhibitory effect on caspase‐mediated apoptosis [40]. Cell viability was assessed by confocal fluorescence microscopy following addition of annexin‐V and PI, as described in the Methods section.
Treatment with Z‐VAD‐FMK significantly attenuated overall cell death in all three cell lines expressing GFP‐PERP fusion protein, compared both with nontreated transfected cells and with cells treated with the negative control inhibitor Z‐FA‐FMK (Fig. 3A). Specifically, the number of nonviable cells expressing GFP‐PERP was reduced by the caspase inhibitor to 18.79 ± 0.54% for 92‐1 cells (t‐test, P= 0.004), 12.60 ± 1.69% for OCM‐1 cells (t‐test, P= 0.001) and 7.27 ± 4.45% for MEL202 cells (t‐test, P= 0.008). There was no significant difference in cell viability between nontreated pGFP‐PERP‐transfected cells and transfected cells treated with the control inhibitor Z‐FA‐FMK (t‐test, P= 0.35 for 92‐1 cells, P= 0.81 for MEL202, P= 0.29 for OCM‐1). The normal morphology and viability of nontransfected cells in the same culture further supported the finding that the observed apoptosis was PERP‐induced, whereas the inhibition of PERP‐induced apoptosis by the caspase inhibitor was clearly reflected by the reversal of the specific apoptotic cell morphology (Fig. 3B). Taken together, these data indicated that PERP‐induced cell death occurs via an apoptotic pathway mediated by caspases.
Figure 3.
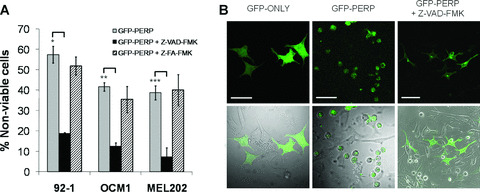
Effect of caspase inhibitor on GFP‐PERP‐induced cell death. (A) 92‐1, OCM‐1 and MEL202 cells were transiently transfected with pGFP‐PERP and incubated in the presence of Z‐VAD‐FMK (pan‐caspase inhibitor) or Z‐FA‐FMK (negative control), starting at 4 hrs after transfection. The cells were monitored by fluorescence microscopy in the presence of annexin‐V and PI between 24 and 72 hrs after transfection. The mean percentage of nonviable transfected cells in the cell population, with standard deviation from three independent transfections, is shown. pGFP‐PERP‐transfected cells treated with caspase inhibitor presented a significantly lower percentage of nonviable cells compared with nontreated GFP‐PERP‐expressing cells (t‐test, *P= 0.004, **P= 0.001 and ***P= 0.008). (B) Morphology of 92‐1 cells expressing GFP only, GFP‐PERP (note rounding up of most transfected cells and not of nontransfected cells) and GFP‐PERP in the presence of Z‐VAD‐FMK (significantly fewer rounded up cells). The images are also representative of morphology observed for OCM‐1 and MEL202 cells. Scale bar 50 μm.
Detection of specific caspase activation in response to PERP expression
To further investigate the nature of specific caspases activated in the apoptosis induced by PERP expression, lysates of cells transfected with pGFP‐PERP were analysed by Western blotting alongside lysates of appropriate control cells. Cleaved caspase‐8 products—including 44, 41 and 28 kD proteins, previously described as intermediate cleavage products [41, 42]—were detected in all UM cells expressing GFP‐PERP protein from 24 hrs after transfection (Fig. 4). In addition, the processing of Bid, the downstream component of the caspase‐8‐initiated death‐signalling pathway, was examined using an antibody to its full‐length. The levels of full‐length Bid (∼22 kD) were reduced in cells expressing GFP‐PERP compared with cells expressing GFP‐only and nontransfected cells, suggesting that cleavage of Bid had taken place. In contrast, there was no significant difference in the levels of the 46 kD precursor and processed 34 kD form of caspase‐9 detected in control and GFP‐PERP‐expressing cells, suggesting that PERP expression does not directly lead to caspase‐9 processing.
Figure 4.
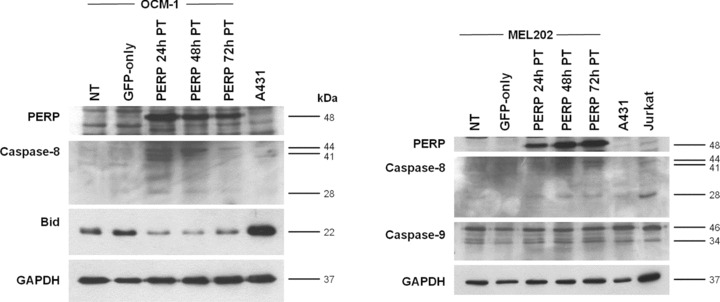
Caspase activation following PERP expression. Cell lysates were prepared from pGFP‐PERP‐transfected UM cells at 24, 48 and 72 hrs after transfection. Lysates from nontransfected (NT) and pEGFP‐N1‐transfected (GFP‐only) cells were harvested 48 hrs after transfection and served as controls. A431 and Jurkat cell lysates served as positive controls for the specified antibodies, as recommended by the manufacturers of the respective antibodies. Lysates were analysed by Western blotting for the processing of initiator caspase‐8 and ‐9 and the cleavage of Bid. Molecular weights of detected cleaved caspase proteins and full‐length Bid are indicated. Images referring to the same cell line are of the same blot probed sequentially with the specified antibodies. Detection of GAPDH was used to confirm equal loading of samples. Partially processed caspase‐8 proteins (44, 41 and 28 kD) were readily detected in UM cells expressing GFP‐PERP. In addition, full‐length Bid levels were depleted in cells expressing GFP‐PERP compared with control cells. With regard to caspase‐9, there was no difference in the levels of cleaved enzyme (34 kD) in cells expressing GFP‐PERP, GFP only, or nontransfected cells.
Evaluation of processing/activation of caspases in relation to PERP protein level in primary UM specimens
Having established the proapoptotic, caspase‐mediated function of PERP in experimental UM cell models, we investigated the status of specific caspases in primary UM tumours.
Relative levels of PERP protein, processed initiator caspase‐8 and ‐9 and effector caspase‐3 protein levels were evaluated by immunodetection in tissue lysates of 134 primary UM specimens. PERP was identified as a 21 kD band, as previously described [14]. Two forms of processed caspase‐8 were detected: the active cleaved form of caspase‐8 (18 kD, p18 subunit) and an intermediate caspase‐8 cleaved product (28 kD). The partially cleaved 34 kD form of caspase‐9 and the cleaved 17 kD form of caspase‐3 (p17 subunit) were also assessed (Fig. 5A). Data obtained from 31 individual specimens for which all caspase forms and PERP protein levels were determined indicated increased overall caspase processing accompanying increased PERP protein levels (Fig. 5B and C; Pearson’s correlation coefficient, calculated R 2 value = 0.627, critical R 2 value = 0.304 at P= 0.001, n= 31).
Figure 5.
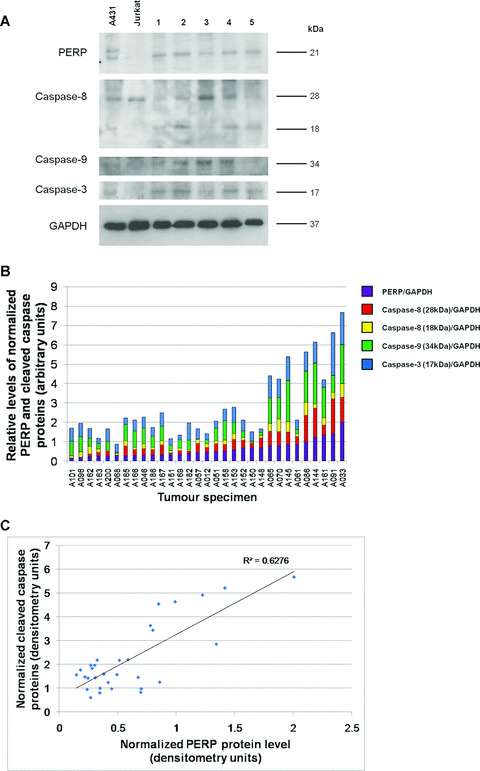
Relative levels of PERP and cleaved caspase proteins in primary uveal melanoma specimens. (A) Representative Western blot of tumour homogenates analysed alongside A431 and Jurkat cell lysates as positive controls for the specific antibodies used. Images are of the same blot probed sequentially with anti‐PERP antibody and antibodies to detect the cleaved/active forms of caspase‐8, ‐9 and ‐3. Anti‐GAPDH antibody was used to confirm equal loading of samples. Depleted levels of the partially processed 28 kD protein were detected in specimens in which there was evidence of further processing to the active protein (18 kD), whereas higher levels of the 28 kD form were detected in specimens presenting lower levels of 18 kD protein. Procaspase‐8 proteins (55/58 kD) were detected (not shown), but due to the strong intensity, were not sensitive enough for quantification of changes in protein level. (B) Graphical representation of the relative levels of PERP, partially cleaved caspase‐8 (28 kD), mature caspase‐8 (18 kD), cleaved caspase‐9 (34 kD) and mature caspase‐3 (17 kD) proteins, normalized to corresponding GAPDH protein level in 31 uveal melanoma homogenates, determined by Western blotting. Intensity of the corresponding protein bands was measured by densitometry on films of equivalent exposure, resulting from the sequential probing of the blots with the respective antibodies. The specimens are ranked in order of increasing PERP protein level. (C) Positive, significant rank correlation between overall processed caspases (‐8, ‐9 and ‐3) level and PERP protein (Pearson’s product‐moment correlation, critical R 2 value = 0.304 at P= 0.001, n= 31).
An analysis of the processed forms of individual caspases revealed the strongest correlation between normalized protein levels of PERP and the partially processed 28 kD caspase‐8 protein (Fig. 6A), and a significant correlation between PERP and the active (18 kD) caspase‐8 form. The correlation of PERP protein levels with cleaved caspase‐9 protein level (34 kD) appeared less stringent, although statistically significant (Fig. 6B), as was the correlation between PERP and active (17 kD) caspase‐3 protein levels (Fig. 6C).
Figure 6.
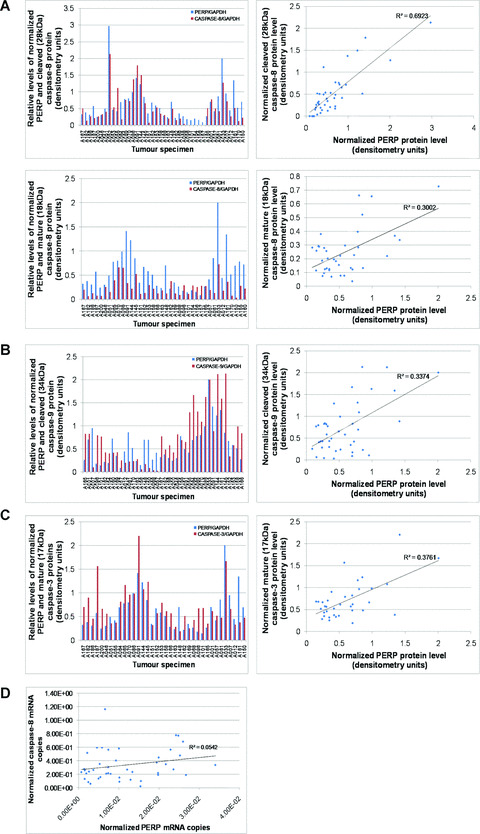
Evaluation of PERP protein expression and caspase activation in primary UM specimens. Comparison of PERP protein levels with (A) partially processed 28 kD caspase‐8 (critical R 2 value = 0.239 at P= 0.001, n= 42) and active 18 kD caspase‐8 (critical R 2 value = 0.261 at P= 0.001, n= 38), (B) cleaved 34 kD caspase‐9 (critical R 2 value = 0.249 at P= 0.001, n= 40) and (C) active 17 kD caspase‐3 (critical R 2 value = 0.273 at P= 0.001, n= 36). The levels of PERP protein and cleaved caspase proteins, determined by densitometry following Western blot analysis, were normalized to the corresponding GAPDH protein level in UM homogenates. The square of the Pearson product‐moment correlation was calculated in each case and significant and positive rank correlations were determined (calculated R 2 values indicated on graphs). (D) Evaluation of relative transcriptional levels of PERP and procaspase‐8 in primary UM specimens. Quantification of mRNA copy number of PERP and procaspase‐8 was achieved by real‐time PCR, as described in Methods; normalization was against specimen‐specific LDHA mRNA copy number. No significant correlation was found between PERP and caspase‐8 at the transcriptional level (Pearson’s product‐moment correlation, critical R 2 value = 0.098 at P= 0.05, n= 43).
PERP and pro‐caspase‐8 transcriptional levels were determined by quantitative real‐time RT‐PCR, measuring the respective mRNA copy numbers normalized to LDHA expression level as an endogenous control. Quality‐controlled data obtained from 43 primary UM specimens indicated that there was no significant correlation between PERP and caspase‐8 mRNA levels (Fig. 6D; Pearson’s correlation, calculated R 2 value = 0.054, critical R 2 value = 0.098 at P= 0.05, n= 43). The transcriptional profile of PERP and caspase‐8 thus suggested that the positive association of PERP and caspase‐8 proteins was a consequence of post‐translational processing, most likely at the level of caspase‐8 cleavage, and not of increased transcription of pro‐caspase‐8.
Discussion
The present study provides the experimental evidence for the intracellular targeting of the p53 effector PERP in living UM cells and demonstrates that PERP expression triggers the death of UM cells by apoptosis. Furthermore, the study establishes that PERP‐induced apoptosis is mediated via the caspase‐dependent pathway and provides evidence for the association between PERP expression and caspase‐8 cleavage/activation in UM cell lines and primary UM tumours. The findings advance the previous characterization of PERP as an important molecular determinant of aggressiveness in primary UM tumours [14] by identifying a functional pathway by which PERP can exert its pro‐apoptotic effect.
PERP is a direct p53 target gene, containing several p53 binding sites in its promoter region [10, 43], specifically induced upon DNA damage during apoptosis [10]. Targeting of PERP protein to the plasma membrane demonstrated by this study occurs via the ER/Golgi apparatus, most likely following the classical route for transmembrane proteins [44]. This distinct localization is in line with the in silico predicted subcellular targeting of PERP protein, as detailed in Results, as well as the putative tetraspan transmembrane topology of the PERP gene product suggested previously [10, 28], and sets PERP apart from other p53 target genes, such as the Bcl‐2‐related apoptotic effectors, that are functional mainly at the mitochondria.
The pro‐apoptotic effect of PERP expression in UM cell lines implies that the cell death machinery required for PERP‐induced apoptosis in these cells is intact and responsive to a threshold level of PERP protein. The demonstration that UM cells readily commit to apoptosis in response to enhanced PERP levels, taken together with our previous findings that PERP is down‐regulated in aggressive (monosomy 3) type of UM compared with disomy 3 tumours [14], supports the hypothesis that the lack of PERP is a major contributor to the inactivation of p53 apoptotic pathway in UM. Notably, PERP‐induced apoptosis is most prominent in 92‐1 cells, which are characterized by a high proliferation rate and metastatic potential [45, 46] and have an epithelioid morphology that is associated in UM in situ with a poorer prognosis compared with spindle or mixed phenotype [47], which are characteristic of OCM‐1 and MEL202 cell lines, respectively.
The strong positive association between PERP protein levels and processed forms of the initiator caspase‐8 in primary UM specimens, in addition to detection of caspase‐8 processing and cleavage of caspase‐8 proximal substrate Bid [48, 49] in response to exogenous PERP expression in UM cell lines, suggests that processing and subsequent activation of caspase‐8 have a role in PERP‐induced apoptosis. The fact that the most stringent correlation appears between PERP protein levels and the partial 28 kD caspase‐8 form strongly suggests that a threshold of PERP expression may be required for full processing of caspase‐8. These findings, together with plasma membrane localization of PERP, implicate a role for PERP in the extrinsic apoptotic pathway, which is mediated by cell surface death receptors and involves direct interaction between adaptor proteins, such as Fas‐ associated protein with death domain (FADD), and the ligand‐bound receptor complexes that trigger sequestration‐mediated autoactivation of initiator caspase‐8 [50, 51]. Indeed, intracellular recruitment of caspase‐8 to the death‐inducing signalling complex (DISC) at the cytoplasmic side of the plasma membrane accelerates its proteolytic activation following engagement of death receptors belonging to the tumour necrosis factor receptor (TNF‐R) family [52]. Our data indicating caspase‐8 activation support the previously suggested role for PERP as a novel cell death receptor. It remains to be discovered whether PERP plays an integral role in the relatively well documented death receptor pathways of Fas (CD95/APO‐1) and tumour necrosis factor (TNF)‐related apoptosis inducing ligand (TRAIL) (DR5/KILLER) receptors (reviewed in [52]), or is part of a novel signalling complex. Supporting these scenarios are reports that, similar to the regulation of PERP expression, p53 can transcriptionally regulate the expression of the aforementioned membrane death receptors [53, 54, 55, 56].
Consideration of how signalling by the TNF‐R superfamily is transmitted and regulated may offer some insight into the potential signalling mechanisms induced by PERP. Fas and TRAIL death receptors contain an intracellular death domain (DD) that plays a crucial role in the transduction of the apoptotic signal. This relatively well conserved region of approximately 80 residues is required for interaction between the receptor and FADD via DD motifs present in both proteins [57, 58], whereas the death effector domain (DED) of FADD recruits and interacts with the N‐terminal tandem DED of pro‐caspase‐8 to form the DISC, which initiates apoptosis [42, 50, 51, 59]. Crucially, DD or DED motifs are not present in the PERP protein sequence, thus there is no evidence to suggest any direct interaction between PERP and the components of the DISC via these previously described mechanisms. It is more likely, therefore, that PERP may participate in the extrinsic apoptotic pathway by a mechanism similar to that described for caspase‐8 activation in p53‐mediated, transcription‐independent apoptosis, which does not appear to require FADD [41].
An increased PERP expression in UM cell lines does not cause significant changes in caspase‐9 processing, suggesting that the intrinsic apoptotic pathway is not directly involved in PERP‐induced apoptosis. However, since cleaved or truncated Bid (tBid) is able to trigger the release of cytochrome c from the mitochondria, thereby initiating the intrinsic pathway of apoptosis [48, 49], a potential cross‐talk between PERP‐induced apoptosis via activated caspase‐8 and the intrinsic (mitochondrial) apoptotic pathway may exist. Detection of a partially processed form of caspase‐9 in primary tumour specimens, albeit less consistent compared with detection of caspase‐8 processed forms, supports the scenario of engagement of the mitochondrial apoptotic pathway, secondary to the activation of caspase‐8 following PERP expression. Z‐VAD‐FMK inhibits PERP‐induced apoptosis and, consistent with our proposed scenario, this pan‐caspase inhibitor is thought to inhibit a caspase at or near the apex of the apoptotic cascade [60] and has been shown to inhibit caspase‐8‐mediated apoptosis [59, 61] by preventing activation of related downstream events such as tBid‐induced apoptosis [48]. Further experiments addressing the effect on PERP‐mediated apoptosis by specific inhibition of caspase‐8, either with sufficiently specific inhibitors or through siRNA modulation, are likely to provide more details of the sequence of the apoptotic cascade engaged by PERP. With or without the involvement of mitochondrial apoptotic pathway, the positive association between PERP and mature caspase‐3 protein levels is of no surprise, given that both the extrinsic and the intrinsic pathways converge on the activation of downstream effector caspases [62].
De‐regulation of apoptosis has been implicated in UM pathogenesis [20, 63], and several studies indicated that p53‐dependent apoptosis is impaired in this tumour [25, 26]. However, p53 mutation or loss of heterozygosity is an infrequent occurrence in untreated UM [25, 27] and, therefore, cannot explain the functional inactivation of p53 in UM. The upstream signalling to p53 is also reportedly undamaged [20, 25]. Furthermore, the apoptotic caspase pathway required downstream of PERP is functional in all UM cells investigated by us. With the exception of neuroblastoma‐derived cell lines and primary neuroblastomas presenting deficiencies in caspase‐8 and ‐3 [64, 65], caspase deficiency is not widely reported in malignant cells. The demonstration of a pro‐apoptotic function for PERP in UM therefore highlights an important apoptotic junction that is de‐regulated in UM tumours and possibly in other cancer types as a mechanism to evade p53‐dependent apoptosis. The levels of PERP and downstream caspases may provide an important marker for assessing the ability of tumour cells to undergo apoptosis, as well as provide prospective therapeutic targets for treatments aimed at overcoming resistance to radiotherapy and/or chemotherapy.
Acknowledgements
The authors are grateful to Dan Albert and Martine Jager for their kind gifts of cell lines. The technical support for confocal microscopy and general computing given by Daniel Brotchie is acknowledged. The patients contributing to the study are gratefully acknowledged. The study was supported by The North West Cancer Research Fund, United Kingdom.
References
- 1. Hengartner MO. The biochemistry of apoptosis. Nature. 2000; 407: 770–6. [DOI] [PubMed] [Google Scholar]
- 2. Benchimol S. p53‐dependent pathways of apoptosis. Cell Death Differ. 2001; 8: 1049–51. [DOI] [PubMed] [Google Scholar]
- 3. Haupt S, Berger M, Goldberg Z, et al . Apoptosis—the p53 network. J Cell Sci. 2003; 116: 4077–85. [DOI] [PubMed] [Google Scholar]
- 4. Rozan LM, El‐Deiry WS. p53 downstream target genes and tumor suppression: a classical view in evolution. Cell Death Differ. 2007; 14: 3–9. [DOI] [PubMed] [Google Scholar]
- 5. Sherr CJ. Principles of tumor suppression. Cell. 2004; 116: 235–46. [DOI] [PubMed] [Google Scholar]
- 6. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000; 408: 307–10. [DOI] [PubMed] [Google Scholar]
- 7. Deng C, Zhang P, Harper JW, et al . Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995; 82: 675–84. [DOI] [PubMed] [Google Scholar]
- 8. El‐Deiry WS, Tokino T, Velculescu VE, et al . WAF1, a potential mediator of p53 tumor suppression. Cell. 1993; 75: 817–25. [DOI] [PubMed] [Google Scholar]
- 9. Hermeking H, Lengauer C, Polyak K, et al . 14‐3‐3 sigma is a p53‐regulated inhibitor of G2/M progression. Mol Cell. 1997; 1: 3–11. [DOI] [PubMed] [Google Scholar]
- 10. Attardi LD, Reczek EE, Cosmas C, et al . PERP, an apoptosis‐associated target of p53, is a novel member of the PMP‐22/gas3 family. Genes Dev. 2000; 14: 704–18. [PMC free article] [PubMed] [Google Scholar]
- 11. Chao C, Saito S, Kang J, et al . p53 transcriptional activity is essential for p53‐dependent apoptosis following DNA damage. EMBO J. 2000; 19: 4967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ihrie RA, Reczek E, Horner JS, et al . Perp is a mediator of p53‐dependent apoptosis in diverse cell types. Curr Biol. 2003; 13: 1985–90. [DOI] [PubMed] [Google Scholar]
- 13. Nowak M, Koster C, Hammerschmidt M. Perp is required for tissue‐specific cell survival during zebrafish develop‐ment. Cell Death Differ. 2005; 12: 52–64. [DOI] [PubMed] [Google Scholar]
- 14. Paraoan L, Gray D, Hiscott P, et al . Expression of p53‐induced apoptosis effector PERP in primary uveal melanomas: downregulation is associated with aggressive type. Exp Eye Res. 2006; 83: 911–9. [DOI] [PubMed] [Google Scholar]
- 15. Egan KM, Seddon JM, Glynn RJ, et al . Epidemiologic aspects of uveal melanoma. Surv Ophthalmol. 1988; 32: 239–51. [DOI] [PubMed] [Google Scholar]
- 16. Albert DM. The ocular melanoma story. LIII Edward Jackson Memorial Lecture: Part II. Am J Ophthalmol. 1997; 123: 729–41. [DOI] [PubMed] [Google Scholar]
- 17. Kivela T, Eskelin S, Kujala E. Metastatic uveal melanoma. Int Ophthalmol Clin. 2006; 46: 133–49. [DOI] [PubMed] [Google Scholar]
- 18. Damato B. Developments in the management of uveal melanoma. Clin Exp Ophthalmol. 2004; 32: 639–47. [DOI] [PubMed] [Google Scholar]
- 19. Soulieres D, Rousseau A, Tardif M, et al . The radiosensitivity of uveal melanoma cells and the cell survival curve. Graefes Arch Clin Exp Ophthalmol. 1995; 233: 85–9. [DOI] [PubMed] [Google Scholar]
- 20. Sun Y, Tran BN, Worley LA, et al . Functional analysis of the p53 pathway in response to ionizing radiation in uveal melanoma. Invest Ophthalmol Vis Sci. 2005; 46: 1561–4. [DOI] [PubMed] [Google Scholar]
- 21. Woll E, Bedikian A, Legha SS. Uveal melanoma: natural history and treatment options for metastatic disease. Melanoma Res. 1999; 9: 575–81. [PubMed] [Google Scholar]
- 22. Debatin KM, Poncet D, Kroemer G. Chemotherapy: targeting the mitochondrial cell death pathway. Oncogene. 2002; 21: 8786–803. [DOI] [PubMed] [Google Scholar]
- 23. Lewanski CR, Gullick WJ. Radiotherapy and cellular signalling. Lancet Oncol. 2001; 2: 366–70. [DOI] [PubMed] [Google Scholar]
- 24. Wesselborg S, Engels IH, Rossmann E, et al . Anticancer drugs induce caspase‐8/FLICE activation and apoptosis in the absence of CD95 receptor/ligand interaction. Blood. 1999; 93: 3053–63. [PubMed] [Google Scholar]
- 25. Brantley MA Jr, Harbour JW. Deregulation of the Rb and p53 pathways in uveal melanoma. Am J Pathol. 2000; 157: 1795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chana JS, Wilson GD, Cree IA, et al . c‐myc, p53, and Bcl‐2 expression and clinical outcome in uveal melanoma. Br J Ophthalmol. 1999; 83: 110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kishore K, Ghazvini S, Char DH, et al . p53 gene and cell cycling in uveal melanoma. Am J Ophthalmol. 1996; 121: 561–7. [DOI] [PubMed] [Google Scholar]
- 28. Hildebrandt T, Preiherr J, Tarbe N, et al . Identification of THW, a putative new tumor suppressor gene. Anticancer Res. 2000; 20: 2801–9. [PubMed] [Google Scholar]
- 29. Devilee P, van VM, van SP, et al . Allelotype of human breast carcinoma: a second major site for loss of heterozygosity is on chromosome 6q. Oncogene. 1991; 6: 1705–11. [PubMed] [Google Scholar]
- 30. Srikantan V, Sesterhenn IA, Davis L, et al . Allelic loss on chromosome 6Q in primary prostate cancer. Int J Cancer. 1999; 84: 331–5. [DOI] [PubMed] [Google Scholar]
- 31. Trent JM, Stanbridge EJ, McBride HL, et al . Tumorigenicity in human melanoma cell lines controlled by introduction of human chromosome 6. Science. 1990; 247: 568–71. [DOI] [PubMed] [Google Scholar]
- 32. Hildebrandt T, van Dijk MC, van Muijen GN, et al . Loss of heterozygosity of gene THW is frequently found in melanoma metastases. Anticancer Res. 2001; 21: 1071–80. [PubMed] [Google Scholar]
- 33. Kan‐Mitchell J, Mitchell MS, Rao N, et al . Characterization of uveal melanoma cell lines that grow as xenografts in rabbit eyes. Invest Ophthalmol Vis Sci. 1989; 30: 829–34. [PubMed] [Google Scholar]
- 34. de Waard‐Siebinga I, Blom DJ, Griffioen M, et al . Establishment and characterization of an uveal‐melanoma cell line. Int J Cancer. 1995; 62: 155–61. [DOI] [PubMed] [Google Scholar]
- 35. Verbik DJ, Murray TG, Tran JM, et al . Melanomas that develop within the eye inhibit lymphocyte proliferation. Int J Cancer. 1997; 73: 470–8. [DOI] [PubMed] [Google Scholar]
- 36. Paraoan L, White MR, Spiller DG, et al . Precursor cystatin C in cultured retinal pigment epithelium cells: evidence for processing through the secretory pathway. Mol Membr Biol. 2001; 18: 229–36. [DOI] [PubMed] [Google Scholar]
- 37. Paraoan L, Grierson I, Maden BE. Fate of cystatin C lacking the leader sequence in RPE cells. Exp Eye Res. 2003; 76: 753–6. [DOI] [PubMed] [Google Scholar]
- 38. Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001; 17: 849–50. [DOI] [PubMed] [Google Scholar]
- 39. Hofmann K, Stoffel W. TMbase‐a database of membrane spanning proteins segments. Biol Chem Hoppe Seyler. 1993; 374: 166. [Google Scholar]
- 40. McColl KS, He H, Zhong H, et al . Apoptosis induction by the glucocorticoid hormone dexamethasone and the calcium‐ATPase inhibitor thapsigargin involves Bc1‐2 regulated caspase activation. Mol Cell Endocrinol. 1998; 139: 229–38. [DOI] [PubMed] [Google Scholar]
- 41. Ding HF, Lin YL, McGill G, et al . Essential role for caspase‐8 in transcription‐independent apoptosis triggered by p53. J Biol Chem. 2000; 275: 38905–11. [DOI] [PubMed] [Google Scholar]
- 42. Medema JP, Scaffidi C, Kischkel FC, et al . FLICE is activated by association with the CD95 death‐inducing signaling complex (DISC). EMBO J. 1997; 16: 2794–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reczek EE, Flores ER, Tsay AS, et al . Multiple response elements and differential p53 binding control Perp expression during apoptosis. Mol Cancer Res. 2003; 1: 1048–57. [PubMed] [Google Scholar]
- 44. Cobbold C, Monaco AP, Sivaprasadarao A, et al . Aberrant trafficking of transmembrane proteins in human disease. Trends Cell Biol. 2003; 13: 639–47. [DOI] [PubMed] [Google Scholar]
- 45. Lopez‐Velasco R, Morilla‐Grasa A, Saornil‐Alvarez MA, et al . Efficacy of five human melanocytic cell lines in experimental rabbit choroidal melanoma. Melanoma Res. 2005; 15: 29–37. [DOI] [PubMed] [Google Scholar]
- 46. Marshall JC, Caissie AL, Callejo SA, et al . Cell proliferation profile of five human uveal melanoma cell lines of different metastatic potential. Pathobiology. 2004; 71: 241–5. [DOI] [PubMed] [Google Scholar]
- 47. McLean IW, Keefe KS, Burnier MN. Uveal melanoma. Comparison of the prognostic value of fibrovascular loops, mean of the ten largest nucleoli, cell type, and tumor size. Ophthalmology. 1997; 104: 777–80. [DOI] [PubMed] [Google Scholar]
- 48. Li H, Zhu H, Xu CJ, et al . Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998; 94: 491–501. [DOI] [PubMed] [Google Scholar]
- 49. Luo X, Budihardjo I, Zou H, et al . Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998; 94: 481–90. [DOI] [PubMed] [Google Scholar]
- 50. Martin DA, Siegel RM, Zheng L, et al . Membrane oligomerization and cleavage activates the caspase‐8 (FLICE/MACHalpha1) death signal. J Biol Chem. 1998; 273: 4345–9. [DOI] [PubMed] [Google Scholar]
- 51. Muzio M, Stockwell BR, Stennicke HR, et al . An induced proximity model for caspase‐8 activation. J Biol Chem. 1998; 273: 2926–30. [DOI] [PubMed] [Google Scholar]
- 52. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998; 281: 1305–8. [DOI] [PubMed] [Google Scholar]
- 53. Bennett M, Macdonald K, Chan SW, et al . Cell surface trafficking of Fas: a rapid mechanism of p53‐mediated apoptosis. Science. 1998; 282: 290–3. [DOI] [PubMed] [Google Scholar]
- 54. Muller M, Strand S, Hug H, et al . Drug‐induced apoptosis in hepatoma cells is mediated by the CD95 (APO‐1/Fas) receptor/ligand system and involves activation of wild‐type p53. J Clin Invest. 1997; 99: 403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Owen‐Schaub LB, Zhang W, Cusack JC, et al . Wild‐type human p53 and a temperature‐sensitive mutant induce Fas/APO‐1 expression. Mol Cell Biol. 1995; 15: 3032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wu GS, Burns TF, McDonald ER III, et al . KILLER/DR5 is a DNA damage‐inducible p53‐regulated death receptor gene. Nat Genet. 1997; 17: 141–3. [DOI] [PubMed] [Google Scholar]
- 57. Boldin MP, Varfolomeev EE, Pancer Z, et al . A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995; 270: 7795–8. [DOI] [PubMed] [Google Scholar]
- 58. Chinnaiyan AM, O’Rourke K, Tewari M, et al . FADD, a novel death domain‐containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995; 81: 505–12. [DOI] [PubMed] [Google Scholar]
- 59. Muzio M, Chinnaiyan AM, Kischkel FC, et al . FLICE, a novel FADD‐ homologous ICE/CED‐3‐like protease, is recruited to the CD95 (Fas/APO‐1) death‐inducing signaling complex. Cell. 1996; 85: 817–27. [DOI] [PubMed] [Google Scholar]
- 60. MacFarlane M, Cain K, Sun XM, et al . Processing/activation of at least four interleukin‐1beta converting enzyme‐like proteases occurs during the execution phase of apoptosis in human monocytic tumor cells. J Cell Biol. 1997; 137: 469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997; 326: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999; 68: 383–424. [DOI] [PubMed] [Google Scholar]
- 63. Ziangirova GG, Likhvantseva VG, Ukhov I, et al . Key role of apoptosis in progressing uveal melanoma. Izv Akad Nauk Ser Biol. 2003; 5: 527–33. Translated in Biology Bulletin. 2003; 30: 436–41. [PubMed] [Google Scholar]
- 64. Iolascon A, Borriello A, Giordani L, et al . Caspase 3 and 8 deficiency in human neuroblastoma. Cancer Genet Cytogenet. 2003; 146: 41–7. [DOI] [PubMed] [Google Scholar]
- 65. Teitz T, Lahti JM, Kidd VJ. Aggressive childhood neuroblastomas do not express caspase‐8: an important component of programmed cell death. J Mol Med. 2001; 79: 428–36. [DOI] [PubMed] [Google Scholar]