

Abstract
p53 regulates the expression of genes involved in cell cycle control, apoptosis and DNA damage repair. Here we demonstrate that DUSP11 (dual specificity phosphatase 11), a member of the protein tyrosine phosphatase family that binds to RNA‐RNP complexes and RNA splicing factors, is a p53 target gene. Consistent with this, the expression of DUSP11 is induced in a p53‐dependent manner after treatment with DNA damaging agents. Chromatin immunoprecipitation analysis showed that p53 binds to 2 putative p53 DNA binding sites in the promoter region of DUSP11. Colony formation and proliferation assays demonstrated that the ectopic expression of wild‐type, but not catalytical inactive, DUSP11 leads to growth arrest. Furthermore inhibition of DUSP11 expression by shRNA increases the proliferation of normal and DNA damaged cells in tissue culture. Finally we show that the splicing factor SAM68 (Src‐associated protein in mitotic cells) binds to DUSP11 in vitro and in vivo. Taken together these results suggest that DUSP11 contributes to p53‐dependent inhibition of cell proliferation and that it might be involved in regulating RNA splicing through SAM68.
Keywords: p53, DNA damage, cellular proliferation, RNA splicing
Introduction
The product of the tumour suppressor gene, TP53, is activated by a variety of cellular stimuli including DNA damage, oncogene activation and nucleotide depletion [1]. p53 functions as a transcription factor and it regulates the transcription of genes involved in cell cycle arrest, senescence and apoptosis [1]. While the ectopic expression of some p53 target genes, such as PUMA[2] or CDKN1A (p21) [3] is sufficient to initiate cell death or cell cycle arrest, the expression of other p53 regulated genes like CASP6 (caspase 6) or BID does not lead to apoptosis or growth arrest. Thus, it is believed that p53 controls the cellular response to aberrant signalling and genotoxic stress by stimulating the expression of a large number of genes and cellular pathways. In this way the extent of cellular damage and the physiological state of the cell will determine if a cell proliferates, arrests or dies [4].
p53 has been suggested to participate in several other cellular pathways and checkpoints in addition to those described above. For example, there is evidence for a role of p53 in the mitotic spindle checkpoint. For instance, the spindle checkpoint component Rit42, a gene coding for a microtubule‐associated protein, is a p53‐target gene [5]. Moreover, based on results showing the presence of aberrant transcripts of the putative tumour suppressor gene TSG101 in cells with mutations of TP53, Turpin and colleagues have suggested that p53 inhibits alternative splicing [6]. In agreement with this, the induction of p53 in cancer cells leads to a significant decrease of abnormal transcripts levels of the TSG101[7].
Here, we demonstrate that DUSP11 (dual specificity phosphatase 11) is a novel p53‐regulated gene. DUSP11, also known as PIR1 (phosphatase interacting with RNA and ribonucleoprotein 1), encodes a 40‐kD dual specificity phosphatase, being part of the protein tyrosine phosphatase family (PTP) [8]. PTPs are involved in the regulation of important cellular processes such as signal transduction, cell cycle progression and tumour suppression [9]. In particular, DUSP11 is able to dephosphorylate tyrosyl‐phosphorylated poly (GluTyr), serine/threonine residues and RNA trinucleotides [8, 9]. It can bind directly to RNA and ribonucleoproteins in vitro. Ectopically expressed DUSP11 localizes to the nucleus and it co‐localizes with SC35, a mammalian splicing factor [9]. Interestingly, it has been shown that DUSP11 mRNA levels are low in several human cell lines lacking p53 [9]. Furthermore, a mutant in the catalytic active site Cys 152 has been identified that is completely unable to exert any phosphatase activity in vitro[8, 9]. Since wild‐type DUSP11 shows a stronger de‐phosphorylation activity on RNA substrates in in vitro de‐phosphorylation experiments, Deshpande and colleagues proposed that DUSP11 could be involved in the control of RNA metabolism [8]. Here we show that DUSP11 is a physiological target gene of p53 that contributes to p53‐dependent growth arrest.
Materials and methods
Constructs
DUSP11 was cloned by PCR amplification of cDNA isolated from WI38 human fibroblasts, based on the sequence present in the database (NCBI accession numbers: NM_003584). The full‐length cDNA of DUSP11 was cloned into pCR2.1 TOPO cloning vector (Invitrogen, Carlsbad, CA, USA) and then subcloned into pCMVneoBam, pCMVHAneoBam, pBabepuro, pBabeHApuro, pcDNAFlag and pCMVMYEGFP.
pCMVp53 was a gift from Karen Vousden, pCMVHAp21 was obtained by cloning the full‐length human cDNA of p21 into pCMVHA vector. pBabeHAE2F1 was constructed cloning the full‐length human cDNA of E2F1 in pBabeHA. Mouse (in pCMVSPORT6) and human (in pOTB7) forms of SAM68, were kindly provided by Dr. Harald Koënig. They were sub‐cloned into pcDNA3.1+ expression vector (Invitrogen). Dr. Harald Koënig also furnished mouse Sam68 cloned in pGEX‐3X vector (Amersham‐Pharmacia Biotech, GE Healthcare, Chalfont St Giles, UK), the human form was cloned into the same vector.
The shRNA constructs for DUSP11 were prepared in the MSCV‐based pLMP retroviral vector [10]. The p21 shRNA construct was prepared in the pRetroSuper‐based retroviral vector [11].
Cloning of probes for Northern blotting
Primers were designed using Oligo 4.1 (Primer Analysis Software). Total RNA was prepared from cycling U2OS cells using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. One microgram of RNA was used for cDNA synthesis using the Superscript II Reverse Transcriptase (Invitrogen). PCR fragments were cloned into pCR2.1 using a TA cloning kit (Invitrogen). The identity of the PCR fragments was verified by sequencing.
Real‐time quantitative PCR (qPCR)
Quantitative PCR was performed on an ABI PRISM7700 Sequence detection system. All the reactions were performed in triplicates. Quantification was performed using the comparative CT method as described in the manufacturer procedures manual. ACTIN was used as normalization control. All primers were designed with Primer Express 1.0 software (Applied Biosystems Foster City, CA, USA) following the manufacturer’s suggested conditions.
Northern blotting
U2OS p53ER infected cells were grown, for 4 or 8 hrs, with or without 600 nM 4‐hydroxytamoxifen (OHT) and/or 10 μg/ml cycloheximide (CHX) to inhibit protein synthesis. Cells were harvested in guanidium thiocyanate 4 M, sodium acetate 20 mM pH 5.2, sarkosyl 0.5%, 1,4‐Dithiothreitol (DTT) 0.1 mM and lysed by 10 passages through a 20‐gauge needle. RNA was isolated by cesium chloride (CsCl) ultracentrifugation method as described [12]. Poly A+ RNA was isolated with the Oligotex reagents from Qiagen (Hilden, Germany) using a batch protocol as described by the manufacturer. A total of 1–4 μg of polyA+ RNA was resolved by electrophoresis on a 1% gel containing 1.9% formaldehyde and 1× MOPS/ethylenediaminetetraacetic acid (EDTA) buffer and then transferred to a nylon membrane. We sequentially hybridized the blot with 200 base pairs 32P‐labelled probes (Strip‐EZTM DNA kit, Ambion, Austin, TX, USA) specific for the genes of interest: APAF1, DUSP11, STRAIT 11499, EST1, p21 (CDKN1A) and GAPDH as a loading control. The same protocol was used to perform Northern blots from RKO and RKO + E6 cells treated or not with the specific DNA damaging agent.
Chromatin immunoprecipitation (ChIP)
U2OS cells were cross‐linked by the addition of paraformaldehyde to 1% final concentration. The reaction was stopped by addition of glycine, cells were washed in tris‐buffered saline (TBS) and harvested in SDS buffer. Following centrifugation, cells were resuspended in Immunoprecipitation Buffer and chromatin was sonicated to an average size of 500–1000 bp. Lysates were subsequently pre‐cleared with protein G Sepharose beads (GE Healthcare). Pre‐cleared chromatin was incubated at 4°C overnight with an antibody specific for p53 (DO‐1) or with an unrelated anti‐Flag antibody. All the reactions were performed in triplicates. Immunocomplexes were recovered with protein G Sepharose beads. After extensive washes immunocomplexes were eluted from the beads, cross‐link reversed and material recovered by phenol/chloroform extraction and ethanol precipitation. DNA was resuspended in 200 μl of water. A total of 7.5 μl of DNA was used per real‐time qPCR reaction with 0.5 μl of a 10‐μl primer mix in a total volume of 25 μl SYBR Green Reaction Mix (Perkin Elmer, Waltham, MA, USA). Acetylcholine muscarinic receptor (AchR) was used as a negative control gene. Promoter primers for AchR were a gift of Bruno Amati. Promoter primers for p21 (CDKN1A) were as described [13]. Promoter primers for DUSP11 were designed with Primer Express 1.0 software.
Cell culture
All cell lines were cultured at 37°C in a humid atmosphere containing 5% CO2. U2OS, U2OSp53ER, U2OSpRetroSuper (U2OSpRS) and U2OSp RetroSuperp53 (U2OSpRSp53) were grown in Dulbecco’s modified Eagles medium (DMEM) supplemented with 10% South American FBS (foetal bovine serum), 2 mM L‐glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. Puromycin (2.5 μg/ml) was added to the medium used for U2OSp53ER, U2OSpRetroSuper and U2OSpRetroSuperp53 cells. RKO, RKO+E6, WI38, Phoenix ecotropic, 293 cells and mouse embryonic fibrolasts (MEFs) wild‐type, were grown in DMEM, supplemented with 10% North American FBS, 2 mM L‐glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. Neomycin (540 μg/ml) was added to the medium used for RKO and RKO + E6 cells. RKO cells and RKO cells expressing the human papillomavirus type 16 E6 protein were gifts from Karen Vousden.
Transfections
A total of 10 μg DNA was diluted in 439 μl of ddH2O and mixed to 61 μl of 2 M CaCl2. The mixture was added, drop‐wise, to 500 μl of 2× HEPES buffered saline (HBS). After 15 min. incubation, the precipitate was added to cells plated on 10 cm dishes and removed after 8–16 hrs, depending on cell lines.
Retroviral infections
Retroviruses were produced by transfecting the ecotropic Phoenix helper cell line with 10 μg of DNA. Supernatants were collected 48 hrs after transfection, filtered and used to infect cells. Supernatants were removed approximately 3 hrs after infection and a second cycle of infection was repeated. Forty‐eight hours after infections, selection was performed. Selection was maintained for four days and surviving cells were used for further experiments.
Colony formation assay
U2OS cells were transfected or infected with the indicated plasmids. Transfected cells were plated at high dilution, in triplicate, in six‐well plates and selected with the required antibiotic; the media was changed every 3–4 days. After 2 weeks the ability to form colonies was assayed: cells were fixed and stained with crystal violet. After extensive washing, crystal violet was resolubilized in 10% acetic acid and quantified at 595 nm as a relative measure of cell number [14].
MTT assay
MEFs infected with the different constructs, were plated, in triplicate, into 12‐well plates. MTT 0.5 mg/ml was added to each well and left, in the dark, at 37°C for 2 hrs. After aspiration of the medium, dimethylsulfoxide was added to each well and crystals were dissolved pipetting up and down. A total of 200 μl of this solution was transferred to plate reader and absorbance was measured at 550 nm.
SDS‐PAGE and Western blotting
Cells were lysed in concentrated JS buffer [50 mM HEPES pH 7.5, 150 mM NaCl, 1% glycerol, 0.01% Triton X‐100, 1.5 mM MgCl2, 25 mM ethylene glycol tetraacetic acid (EGTA)] plus inhibitors (Aprotinin 10 μg, Leupetin 10 μg, 1 mM NaF, 1 mM PMSF). Equal amounts of proteins were loaded onto poly‐acrylamide gels for electrophoresis. SDS‐PAGE was performed as described [15].
The proteins were then transferred to nitrocellulose (GE Healthcare). After blocking, filters were incubated with the specific antibody. The bound secondary antibody was revealed using the enhanced chemiluminescence method (GE Healthcare).
Antibodies
DO‐1: mouse monoclonal anti‐human p53 (Santa Cruz Biotechnology, Paso Robles, CA, USA); F3165: mouse monoclonal anti Flag (Sigma‐Aldrich, St. Louis, MO, USA); p21 (187): mouse monoclonal anti‐human p21 (Santa Cruz Biotechnology); Anti‐actin α: mouse monoclonal anti‐human actin (Sigma); anti‐HA: mouse monoclonal antibody [Berkeley Antibody Company (BAbCO), Richmond, CA, USA; SAM68 (C20): rabbit polyclonal anti‐human and antimouse SAM68 (Santa Cruz Biotechnology); anti‐RAS: mouse monoclonal antibody anti‐human H‐RAS (BD Biosciences, San Jose, CA, USA); FL‐393: rabbit polyclonal antibody anti‐human, antimouse and anti‐rabbit p53 (Santa Cruz Biotechnology); DUSP11 affinity purified rabbit polyclonal antibody (ProteinTech Group, Chicago, IL, USA); anti‐vinculin: mouse monoclonal anti‐human vinculin clone hVIN‐1. Mouse Ascite Fluid (Sigma).
Co‐immunoprecipitations (Co‐IP)
A total of 293 cells were transfected with pCMVHA, DUSP11 and pCMV empty vectors, respectively, as described [12]. Transfected cells were lysed in concentrated JS lysis buffer plus inhibitors, the amount of lysates, corresponding to 1 mg of protein, was pre‐cleared for 30 min. at 4°C with 40 μl of protein A Sepharose beads (GE Healthcare). After centrifugation, supernatants were collected and incubated on ice for 1 hr with the specific antibody. This step was followed by 1‐hr rotation with 40 μl of protein A Sepharose beads at 4°C. After extensive washes in NET buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 0.1% Triton X‐100, 1 mM EDTA) the bound proteins were resolved by SDS‐PAGE. Western blots were performed as described above using the indicated antibodies.
Expression of GST‐fusion proteins
Proteins were expressed from Escherichia coli as described [16]. After binding to Glutathione‐SepharoseTM 4B beads (Amersham Biosciences) the proteins were washed and then denaturated with 1× protein sample buffer. The size of the proteins was evaluated by Coomassie staining of SDS‐poly‐acrylamide gels.
In vitro binding experiments
In vitro translations were carried out with the TNT coupled reticulocyte lysate system (Promega, Madison, WI, USA) in accordance with the instructions provided by the manufacturer. The synthesized polypeptide pcDNAFlag DUSP11, labelled with [35S] methionine, was mixed with the following GST (glutathione S transferase) fusion proteins: GST, GSTmouseSam68 or GSThumanSAM68. The same procedure was applied for the empty vector control: pcDNAFlag. The GST fusion proteins and any associated proteins were recovered using glutathione‐sepharose beads (Amersham Biosciences) washed three times with p19 buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 0.1% Tween‐20), resolved on 13% SDS‐poly‐acrylamide gels and detected by autoradiography.
Results
To identify genes regulated by p53 we have previously performed microarray analysis using human U2OS osteosarcoma cells expressing p53 fused to a modified form of the estrogen receptor ligand‐binding domain (p53ER) [17]. The p53ER fusion protein is activated upon addition of OHT [17, 18, 19]. Using this technology we have reported on the identification of APAF1 (apoptosis protease activating factor 1) as a p53 target gene [17].
Here we report on the preliminary characterization of another p53 target gene DUSP11. To confirm that activation of p53 leads to increased levels of DUSP11, Northern blots were performed on polyA+ RNA harvested from U2OSp53ER cells treated with OHT, the protein synthesis inhibitor CHX or with both CHX and OHT (CHX/OHT) (Fig. 1A). The mRNA levels of DUSP11, two other p53‐induced genes (STRAIT11499 and EST1), identified in our microarray screenings, as well as the known p53 target genes p21 (CDKN1A) and APAF1 were determined. The mRNA levels of all the tested genes were significantly increased following p53 activation. This increase was independent of de novo protein synthesis indicating that p53 directly regulates the transcription of the tested genes (Fig. 1A). The mRNA levels of p21 (CDKN1A) and DUSP11 were also determined following p53 activation by real‐time qPCR (Fig. 1B). Consistent with the Northern blot analysis, DUSP11 and p21 (CDKN1A) transcript levels were increased 2.5‐ to 4‐fold, respectively, following OHT treatment.
Figure 1.
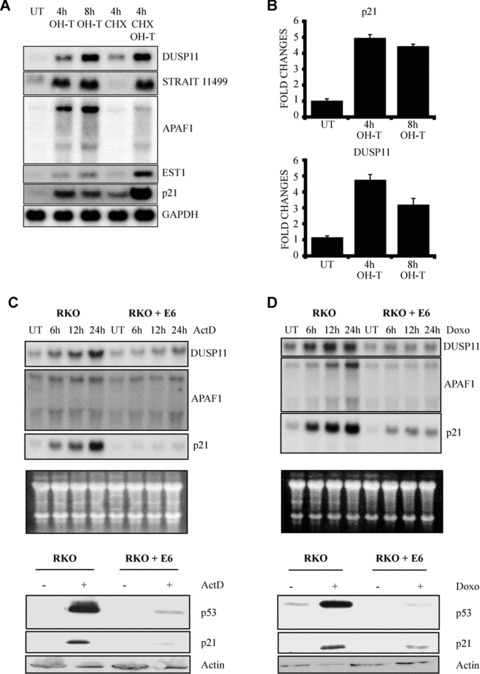
p53‐dependent activation of DUSP11. (A) Northern blot analysis for the expression of DUSP11, STRAIT11499, APAF1, EST1 and p21 (CDKN1A) mRNA isolated from U2OSp53ER cells. Cells were incubated with 4‐hydroxytamoxifen (OHT), cycloheximide (CHX) or both for the indicated times. UT: untreated cells. (B) qPCR analysis for the expression of p21 (CDKN1A) and DUSP11. U2OSp53ER cells were incubated with OHT, for the indicated times. UT: untreated cells. (C) Actinomycin D (ActD) treatment induces the expression of DUSP11, APAF1 and p21 (CDKN1A) mRNA, in a p53‐dependent manner. Upper part: Northern blot analysis of mRNA isolated from RKO and RKO + E6 cells. Cells were treated with 5 nM ActD for the indicated times. UT: untreated cells. The blots were probed for expression of DUSP11, APAF1 and p21 (CDKN1A). Total RNA is shown as a loading control. Lower part: Western blot analysis of cell lysates prepared from RKO cells or from RKO cells expressing E6. Cells were grown in the presence (+) or absence (−) of 5 nM ActD for 24 hrs before lysis. The blots were probed with anti‐p53, anti‐p21 and anti‐actin antibodies. (D) Doxorubicin treatment induces the expression of DUSP11, APAF1 and p21 (CDKN1A) mRNA, in a p53‐dependent manner. Upper part: Northern blot analysis of mRNA isolated from RKO and RKO + E6 cells. Cells were treated with 0.3 μg/ml Doxorubicin (Doxo) for the indicated times. UT: untreated cells. The blots were probed for expression of DUSP11, APAF1 and p21 (CDKN1A). Total RNA is shown as a loading control. Lower part: Western blot analysis of cell lysates prepared from RKO cells or from RKO cells expressing E6. Cells were grown in the presence (+) or absence (−) of 0.3 μg/ml Doxorubicin (Doxo) for 24 hrs before lysis. The blots were probed with anti‐p53, anti‐p21 and anti‐actin antibodies.
Because of the potential intriguing role as a regulator of RNA metabolism we decided to focus on DUSP11[8]. To understand whether DUSP11 is a physiological target of p53, we determined DUSP11 mRNA levels following DNA damage. We used actinomycin D (ActD) and doxorubicin as DNA damaging agents. ActD acts by binding to DNA CpG sequences, thereby interfering with DNA replication and transcription [20], while doxorubicin inhibits the function of topoisomerase II [21]. Human colorectal carcinoma (RKO) cells were treated with ActD and doxorubicin for 24 hrs. As shown in Fig. 1C and 1D, treatment of RKO cells with the two DNA damaging agents led to a significant increase in DUSP11 mRNA levels, similar to the two known p53 target genes, APAF1 and p21 (CDKN1A).
To test if p53 is required for the increase in DUSP11 expression following DNA damage, we used RKO cells expressing the human papilloma virus type 16 E6 protein [22]. This cell line expresses non‐detectable levels of p53, which are not significantly increased upon DNA damage (Fig. 1C, D). Consistent with this, p21 does not accumulate in the RKO‐E6 cell line following DNA damage and apoptosis is strongly impaired (Fig. S1). In contrast to the RKO cells, DUSP11 did not accumulate in RKO‐E6 cell line suggesting that DUSP11 is a physiological target of p53.
To confirm the activation of DUSP11 by physiological levels of p53, in a different cell line, we measured DUSP11 mRNA levels following DNA damage treatment of human U2OS cells with wild‐type (U2OS pRS) or down‐regulated levels of p53 (U2OSpRSp53). These cells are stably infected with pRetroSuper shRNA vector that specifically targets p53 [11]. Western blots of U2OSpRS and U2OSpRSp53 cells, treated with 5 nM ActD for 24 hrs, showed a p53‐dependent increase in p21 levels (data not shown). Supporting the notion that DUSP11 is a physiological target of p53, the DNA damaging agents (ActD, IR and UV) led to a robust p53‐dependent increase in DUSP11 mRNA (Fig. 2) as well as protein levels (Fig. 3).
Figure 2.
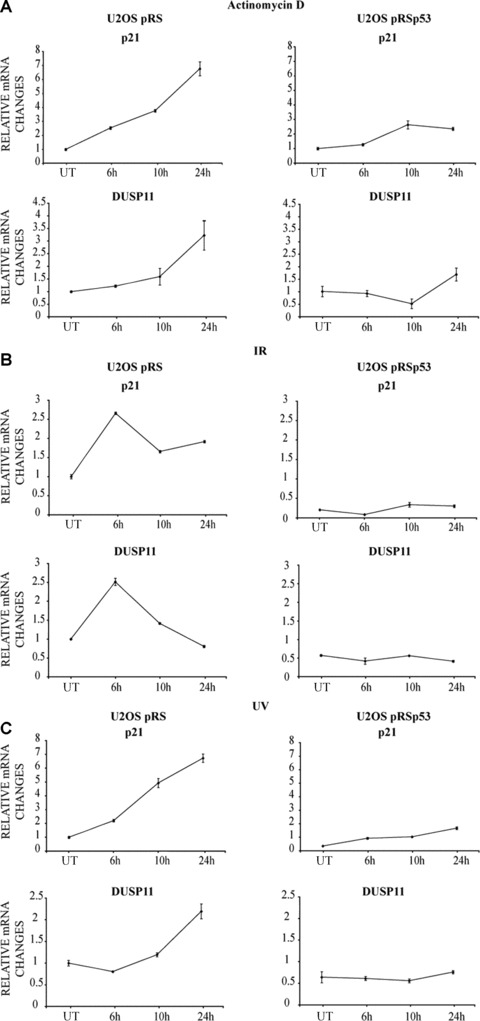
DNA damaging agents results in p53‐dependent increase in DUSP11 expression. (A) qPCR analysis of DUSP11 expression after actinomycin D (ActD) of U2OS pRetroSuper and U2OS pRetroSuperp53 infected cells. Cells were incubated with 5 nM ActD for the indicated times. UT: untreated cells. Specific primers were used to analyse the expression of DUSP11 and p21 (CDKN1A). β‐ACTIN levels were used for normalization. (B) qPCR analysis of DUSP11 expression after ionizing radiation (IR) of U2OS pRetroSuper and U2OS pRetroSuperp53 infected cells. Cells were incubated after 6 Gy ionizing radiation (IR) treatment for the indicated times. UT: untreated cells. Specific primers were used to analyse the expression of DUSP11 and p21 (CDKN1A). β‐ACTIN levels were used for normalization. (C) qPCR analysis of DUSP11 expression after UV of U2OS pRetroSuper and U2OS pRetroSuperp53 infected cells. Cells were incubated after 15 J/m2 UV treatment for the indicated times. UT: untreated cells. Specific primers were used to analyse the expression of DUSP11 and p21 (CDKN1A). β‐ACTIN levels were used for normalization.
Figure 3.
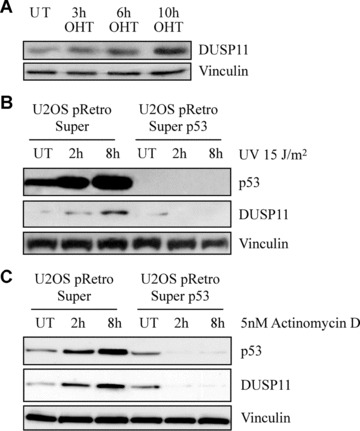
DNA damage leads to p53‐dependent increase of DUSP11 expression. (A) Western blot analysis of cell lysates prepared from U2OSp53ER cells treated with OHT as indicated. UT: untreated cells. The blots were probed with anti‐DUSP11, anti‐p53 and anti‐vinculin antibodies. (B) Western blot analysis of cell lysates prepared from U2OSpRetroSuper and U2OSpRetroSuperp53 cells, either before (UT) or after 2 and 8‐hr treatment with 15 J/m2 UV. The blots were probed as described above. (C) Western blot analysis of cell lysates prepared from U2OSpRetroSuper and U2OSpRetroSuperp53 cells, either before (UT) or after 2 and 8‐hr treatment with 5 nM actinomycin D. The blots were probed as described above.
To understand the mechanism by which DUSP11 expression is regulated by p53, we looked for putative p53‐binding sites in its genomic regions. In silico analysis (Genomatix, MatInspector http://www.genomatix.de/) identified two putative p53‐binding sites (A and B) in Intron 1, as shown in Fig. 4A.
Figure 4.
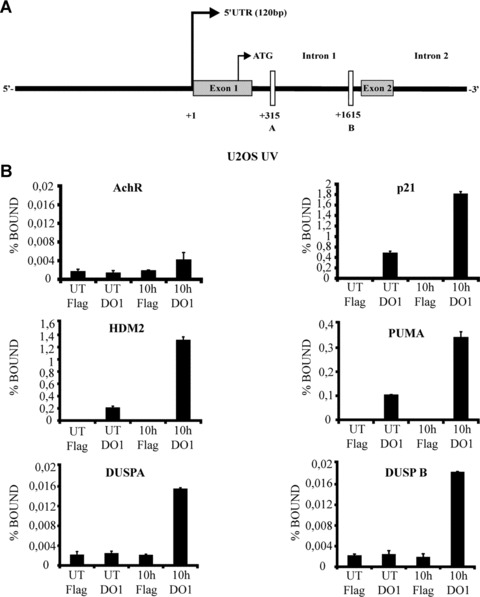
Genotoxic stress induced binding of p53 to the first intron of DUSP11. (A) Schematic presentation of DUSP11 genomic region: the two putative p53‐binding sites, located in Intron 1, are indicated as A and B. (B) ChIP analysis of U2OS cells treated with 15 J/m2 UV for the indicated times. UT: untreated cells. ChIP assays were performed using an antibody specific for p53 (DO1) and the anti‐Flag was used as a negative control. The resulting DNA was amplified by qPCR with primers corresponding to the genes shown. p21 (CDKN1A), HDM2 and PUMA promoters were used as positive controls, while Acetylcholine muscarinic receptor (AchR) promoter functioned as a negative control.
Site A: (5’ half site) ccatcgcaagcttgaCTTGtccg; (3’ half site): aaagcaagcggcggaCAAGtcaa; site B: (3’ half site) agagaggaga‐gaggaCATGttgt; (5’ half site): tcatagcccgcacaaCATGtcct.
Where the DNA‐binding consensus sites for p53 are shown in bold. The p53 consensus half site is RRRCWWGYYY (R is A or G, W is A or T, and Y is C or T) [23, 24]. To confirm the functionality of these putative p53‐binding sites, chromatin IP (ChIP) experiments were performed using specific primers for site A and B of DUSP11 regulatory regions, respectively. Moreover, specific primers for known p53‐dependent transactivated genes p21 (CDKN1A), HDM2 and PUMA were used as positive controls while enrichment of p53 on the acetylcholine muscarinic receptor promoter (AchR) was used as negative control. ChIP experiments were performed using chromatin DNA prepared from U2OS cells that were untreated (UT), or treated for 10 hrs with 15 J/m2 UV. The negative control of the ChIP did not show any significant enrichment for p53. In contrast the three positive controls showed a specific increase in the binding of p53 to the respective promoters (p21, HDM2 and PUMA). As expected, p53 was also bound to sites A and B of DUSP11 regulatory regions. This experiment established the binding of p53 to DUSP11 regulatory regions in vivo upon DNA damaging treatment (Fig. 4B). Taken together, these results demonstrate that DUSP11 is a direct physiological target of p53.
To assess the effect of increased expression of DUSP11, we transfected proliferating U2OS cells with expression plasmids for DUSP11, catalytically inactive DUSP11 mutant, p21 as a control for a p53‐induced gene that inhibits cell proliferation and an empty plasmid as a negative control. As shown in Fig. S2, the expression of DUSP11, but not the catalytic inactive mutant, led to a strong decrease in the colony formation number of U2OS. Quantification of these results showed that DUSP11 expression led to a four‐ to sixfold decrease in colony formation as compared to control, whereas the overexpression of p21 almost eliminated cell proliferation. Importantly, the expression of a catalytic inactive mutant of DUSP11 (C/S) does not affect growth (Fig. 5A), demonstrating that the growth suppressive function of DUSP1 requires its phosphatase activity. The strong decrease in cell proliferation induced by DUSP11, and not the catalytic inactive mutant is not due to different levels of the proteins expression (Fig. 5B), and the transfection efficiency of the different constructs used were also very similar (Fig. S3). Consistent with this, mild overexpression of DUSP11, but not the catalytic inactive mutant, led to a decrease in proliferation of MEFs infected with the indicated constructs (Fig. 5C and D). E2F1 served as a positive control in these assays (25; 26).
Figure 5.
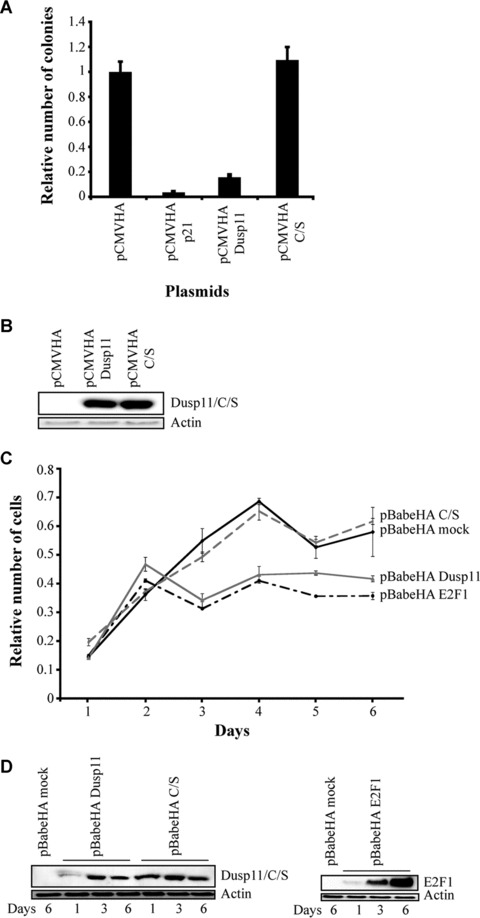
Ectopic expression of DUSP11 inhibits cell proliferation. (A) U2OS cells were stably transfected with the indicated plasmids (pCMV: negative control, pCMV p21: control for the inhibition of cellular proliferation). The relative number of colonies is plotted in a graph (see ‘Materials and methods’). Error bars indicate standard deviations based on three independent transfections. (B) Western blot analysis showing the levels of overexpressed Dusp11 and C/S proteins. The blots were probed with anti‐HA antibody. (C) Mouse embryonic fibrolasts were infected with the indicated plasmids and selected in puromycin 2.5 μg/ml for 5 days. Once selected, they were plated in triplicate and their growth was analysed performing an MTT assay at the indicated time‐points. Error bars indicate standard deviations based on three independent experiments. (D) Western blot analysis of Dusp11, C/S and E2F1 overexpressed proteins at days 1, 3 and 6 after selection. The blots were probed with anti‐HA antibody.
To understand if DUSP11 expressed at physiological levels is a growth suppressor, we designed different shRNA oligonucleotides to inhibit its expression. Two different shRNA constructs were used (namely shDUSP11‐a and shDUSP11‐b) that inhibit DUSP11 expression at both RNA and protein levels (Fig. 6A). U2OS cells were infected, selected with puromycin and plated at low density in six‐well plates. As shown in Fig. 6B and Fig. S4B the inhibition of DUSP11, as well as p21, confers a growth advantage when cells are grown at low density. Moreover, the inhibition of DUSP11 expression confers a significant growth advantage to cells treated with DNA damaging agents such as UV and Doxorubicin (Fig. 6C and D and Fig. S4B and C). Thus, these results suggest that DUSP11 contributes significantly to the p53‐dependent growth arrest.
Figure 6.
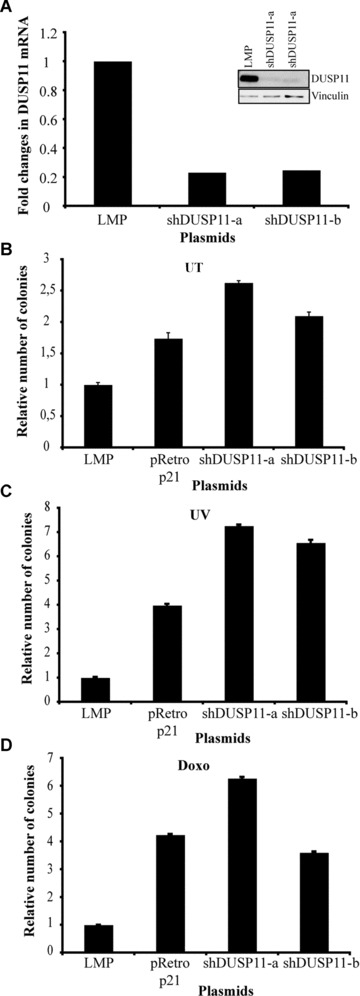
Inhibition of DUSP11 expression by shRNA stimulates cell proliferation and protects from DNA damage. (A) Left panel: qPCR analysis of DUSP11 expression upon infection with the indicated plasmids. Specific primers were used to analyse the expression of DUSP11. β‐ACTIN levels were used for normalization. Right panel: Western blot analysis of DUSP11 protein levels upon infection with the indicated plasmids. The blots were probed with anti‐DUSP11 antibody. U2OS cells were infected with the indicated plasmids (LMP: negative control, pRetroSuper p21: positive control for the stimulation of cell proliferation). Once selected, cells were plated at low density (B), or treated with 15 J/m2 UV (C), or 0.3 μg/ml doxorubicin (D) and then plated at low density. The relative number of colonies is plotted in a graph (see ‘Materials and methods’). Error bars indicate standard deviations based on three independent infections.
To obtain information regarding the biological and biochemical functions of DUSP11, we decided to find potential interactors of the protein by performing a yeast two‐hybrid assay. In this assay, we identified SAM68, which is a nuclear RNA‐binding protein whose function has been proposed to depend on protein modification in response to extracellular cues. SAM68 was shown to regulate the alternative splicing of CD44 variant exon v5 upon phosphorylation by ERK1/2 [27]. The alternative spliced form of CD44 has been suggested to play a role in metastatic cancers [28, 29].
To confirm that DUSP11 can bind to SAM68 in other assays, we tested its ability to bind to the mouse and human forms of SAM68, in vitro. To this extent, DUSP11 was in vitro translated and incubated with the bacterially expressed mouse GST‐Sam68. As shown in the left panel of Fig. 7A, it specifically bound to the mouse Sam68 protein. The same result was obtained performing the in vitro binding with the human GST‐SAM68 protein (Fig. 7A, right panel). To test whether DUSP11 could bind to SAM68 also in vivo, 293 cells were transfected with plasmids expressing HA‐epitope tagged DUSP11. Forty‐eight hours after transfection the cells were lysed and proteins were immunoprecipitated with an antibody recognizing the HA‐tag or a non‐related antibody recognizing RAS. The antibodies to exogenous DUSP11 specifically precipitated endogenous human SAM68 (Fig. 7B, left panel). Similarly, immunoprecipitation with a SAM68‐specific antibody showed that endogenous human SAM68 was bound to overexpressed DUSP11 HA‐tagged proteins (Fig. 7B, right panel). Moreover, endogenous DUSP11 was also found in a complex with endogenous SAM68 (Fig. 7C). These results show that DUSP11 binds to SAM68 in vivo and in vitro and that DUSP11 in this way may provide a link between DNA damage, p53 and the function of this protein.
Figure 7.
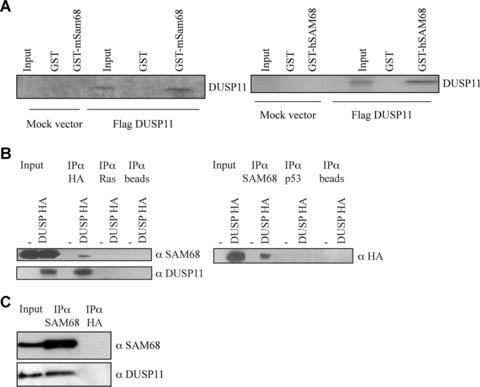
DUSP11 binds to SAM68 both in vitro and in vivo. (A) In vitro translated human DUSP11 specifically binds to mouse Sam68 (left panel). In vitro translated human DUSP11 specifically binds to human SAM68 (right panel). (B) DUSP11 binds endogenous SAM68. A total of 293 cells were transfected with mock (pCMV) or an expression plasmid containing HA‐tagged DUSP11. The indicated antibodies were used for immunoprecipitation. The immunoprecipitated material was separated by SDS‐PAGE and immunoblotted with the indicated antibodies. (C) Co‐immunoprecipitation of endogenous DUSP11 and SAM68. The indicated antibodies were used to perform immunoprecipitation in 293 cells. The immunoprecipitated material was separated by SDS‐PAGE and immunoblotted with the indicated antibodies. A total of 40 μg lysates were used in the input lanes, whereas 800 μg was used for the immunoprecipitations.
Conclusions
In this report we have shown that DUSP11 is a novel p53 target gene. Specifically we show that p53 associates with the promoter region of DUSP11, and although this binding appears to be weaker than for the positive controls, a similar low affinity binding has been observed for other known p53 target genes such as BAX, AIP1 and PIG3[13][30]. Taken together with our results showing that DNA damaging agents lead to an increase in DUSP11 levels in a p53‐dependent manner this establishes DUSP11 as a novel p53 target gene.
Interestingly, consistent with being a p53 target gene working as a growth suppressor, increased expression of DUSP11 inhibits cell proliferation. Furthermore, overexpression of its catalytic inactive mutant does not change growth, demonstrating that DUSP11 phosphatase activity is required to inhibit cellular growth. Significantly, our results also show that endogenous DUSP11 contributes to DNA damage and p53‐dependent growth arrest.
In order to understand the molecular mechanisms involved in the regulation of this process, and to be able to place DUSP11 in a specific cellular pathway, DUSP11 putative protein interactors were searched and SAM68 was isolated. DUSP11 and SAM68 were shown to associate both in vitro and in vivo. SAM68 is a nuclear RNA‐binding protein that regulates the alternative splicing of CD44 variant exon v5, upon phosphorylation by ERK1/2 [27]. Interestingly, this form of CD44 has been proposed to stimulate metastatic cancers [28, 29]. Since DUSP11 is a phosphatase, we speculated that it could act as a negative regulator of SAM68, counteracting the phosphorylation by ERK1/2 and thus preventing the growth factor‐induced alternative splicing of CD44, acting as a tumour suppressor gene. However, to date we have not been able to demonstrate any role of DUSP11 in phorbol ester (TPA) induced stimulation of CD44 splicing, nor the activity of SAM68 in these specific assays.
Interestingly, basal levels of p53 was demonstrated in a recent paper [31] to repress expression of CD44, via binding to a non‐canonical p53‐binding sequence in the CD44 promoter. The p53‐mediated repression of CD44 was shown to be important for the stress‐induced apoptosis in normal cells, whereas CD44 contributes to tumourigenesis in transformed cells lacking p53 function. Although, we have so far been unable to demonstrate a specific role for DUSP11 in regulating CD44 expression and/or splicing, we speculate that, upon stress stimuli, p53 can inhibit CD44 activity also through the action of its novel target gene DUSP11.
p53 has also been proposed to have a role in the regulation of splicing processes [6]. Loss of fidelity of the splicing process occurs during tumour progression and can have a deleterious effect on tumour suppressor genes. It has been reported that the presence of aberrant transcripts of the TSG101 gene in breast cancer cells is associated with a mutation of the TP53 gene. In addition, it has been demonstrated that the induction of p53 in several cellular models leads to a significant decrease of aberrant transcripts levels, and that stress‐activated p53 is able to modulate the TSG101 splicing [7]. Since ionizing radiation triggers a decrease of aberrant transcript levels of TSG101 both in p21 (CDNK1A)‐proficient and ‐deficient HCT116 cells, the activation of p21, the best‐characterized p53 target gene, does not seem to be required for the p53‐dependent modulation of splicing process.
Still, the underlying mechanism remains to be elucidated [7]. Moreover, a very recent paper confirmed the involvement of p53 in regulating the splicing process in tumour cells. In 162 breast carcinomas, p53 mutations were found to be significantly associated with an increased expression of survivin and, in particular, its anti‐apoptotic splice variants. These results provide for the first time in vivo evidence that, in human breast cancer, the survivin expression as well as its splicing depends on the p53 status [32]. Very interestingly RNPC1, an RNA‐binding protein and a target of the p53 family, was shown to be required for maintaining the stability of the basal and stress‐induced p21 transcript [33].
Due to the previously described role of DUSP11 in RNA processing, this newly identified p53 target gene could provide an interesting link between p53 and the suppression of abnormal mRNA splicing forms. We are currently investigating this intriguing possibility.
In summary, the results presented here demonstrate that DUSP11 is a novel p53 target gene that works as growth suppressor dependent on its phosphatase activity. Moreover, the demonstration that DUSP11 can bind to SAM68 provides a number of testable hypotheses for how p53 through the regulation of DUSP11 expression could counteract the function of SAM68. Such studies may reveal the mechanism by which DNA damaging signals and p53 can regulate RNA splicing.
Supporting information
Fig. S1 FACS analysis of RKO cells or RKO cells expressing E6 treated with actinomycin D or doxorubicin. (A) Cells were treated with 5 nM actinomycin D (ActD) for 48 hrs, before fixation. +: treated cells; ‐: untreated cells. Percentage of apoptotic and G1 arrested cells, are indicated. (B) Cells were treated with 0.3 μg/ml doxorubicin (Doxo) for 48 hrs, before fixation. +: treated cells; ‐: untreated cells. Percentage of apoptotic and G1 arrested cells are indicated.
Fig. S2 Ectopic expression of DUSP11 inhibits cell proliferation. U2OS cells were stably transfected as described in Fig. 5A and plated at low density. Cells were then fixed and stained with crystal violet.
Fig. S3 Estimation of transfection efficiency for the cells used to perform the Colony formation assay, described in Fig. 5. Cells were transfected with the indicated plasmids plus one tenth of the pCMV EGFP expression plasmid. The percentage of cells expressing EGFP was analysed by FACS.
Fig. S4 Inhibition of DUSP11 expression by shRNA stimulates cell proliferation and protects from DNA damage. (A) U2OS cells were infected as described in Fig. 6B. Once selected, cells were plated at low density. Cells were then fixed and stained with crystal violet. (B) U2OS cells were infected as described in Fig. 6C. Once selected, cells were treated with UV 15 J/m2 and then plated at low density. Cells were then fixed and stained with crystal violet. (C) U2OS cells were infected as described in Fig. 6D. Once selected, cells were treated with doxorubicin 0.3 μg/ml and then plated at low density. Cells were then fixed and stained with crystal violet.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgements
We thank Heiko Müller for the high‐density oligonucleotide microarray analysis; Emma S. Hickman for U2OSp53ER cells; Karen Vousden for p53 expression constructs and RKO cells; M. Cristina Moroni and Elena Colli for U2OSpRetroSuper and U2OSpRetroSuperp53 cells; Bruno Amati for AchR primers; Dr. Harald Koënig for SAM68 constructs; Elena Prosperini for technical assistance and Esther Hulleman for critical review of the manuscript. This work was supported by grants from AIRC, FIRC and the Italian Health Ministry.
References
- 1. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature . 2000; 408: 307–10. [DOI] [PubMed] [Google Scholar]
- 2. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell . 2001; 7: 683–94. [DOI] [PubMed] [Google Scholar]
- 3. Dulic V, Kaufmann WK, Wilson SJ, et al . p53‐dependent inhibition of cyclin‐dependent kinase activities in human fibroblasts during radiation‐induced G1 arrest. Cell . 1994; 76: 1013–23. [DOI] [PubMed] [Google Scholar]
- 4. Sax JK, El‐Deiry WS. p53 downstream targets and chemosensitivity. Cell Death Differ . 2003; 10: 413–7. [DOI] [PubMed] [Google Scholar]
- 5. Kim KT, Ongusaha PP, Hong YK, et al . Function of Drg1/Rit42 in p53‐dependent mitotic spindle checkpoint. J Biol Chem . 2004; 279: 38597–602. [DOI] [PubMed] [Google Scholar]
- 6. Turpin E, Dalle B, de Roquancourt A, et al . Stress‐induced aberrant splicing of TSG101: association to high tumor grade and p53 status in breast cancers. Oncogene . 1999; 18: 7834–7. [DOI] [PubMed] [Google Scholar]
- 7. Moyret‐Lalle C, Duriez C, Van Kerckhove J, et al . p53 induction prevents accumulation of aberrant transcripts in cancer cells. Cancer Res . 2001; 61: 486–8. [PubMed] [Google Scholar]
- 8. Deshpande T, Takagi T, Hao L, et al . Human PIR1 of the protein‐tyrosine phosphatase superfamily has RNA 5’‐triphosphatase and diphosphatase activities. J Biol Chem . 1999; 274: 16590–4. [DOI] [PubMed] [Google Scholar]
- 9. Yuan Y, Li DM, Sun H. PIR1, a novel phosphatase that exhibits high affinity to RNA. ribonucleoprotein complexes. J Biol Chem . 1998; 273: 20347–53. [DOI] [PubMed] [Google Scholar]
- 10. Dickins RA, Hemann MT, Zilfou JT, et al . Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet . 2005; 37: 1289–95. [DOI] [PubMed] [Google Scholar]
- 11. Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus‐mediated RNA interference. Cancer Cell . 2002; 2: 243–7. [DOI] [PubMed] [Google Scholar]
- 12. Ausubel FM, Brent R, Kingston RE, et al . Current protocols in molecular biology. New York : Green Publishing Associates & Wiley‐Interscience; 1988. [Google Scholar]
- 13. Kaeser MD, Iggo RD. Chromatin immunoprecipitation analysis fails to support the latency model for regulation of p53 DNA binding activity in vivo . Proc Natl Acad Sci USA . 2002; 99: 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carnero A, Hudson JD, Price CM, et al . p16INK4A and p19ARF act in overlapping pathways in cellular immortalization. Nat Cell Biol . 2000; 2: 148–55. [DOI] [PubMed] [Google Scholar]
- 15. Harlow E, Lane D. Antibodies: a laboratory manual. Cold Spring Harbor, New York : Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- 16. Smith DB, Johnson KS. Single‐step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S‐transferase. Gene . 1988; 67: 31–40. [DOI] [PubMed] [Google Scholar]
- 17. Moroni MC, Hickman ES, Denchi EL, et al . Apaf‐1 is a transcriptional target for E2F and p53. Nat Cell Biol . 2001; 3: 552–8. [DOI] [PubMed] [Google Scholar]
- 18. Yin Y, Terauchi Y, Solomon GG, et al . Involvement of p85 in p53‐dependent apoptotic response to oxidative stress. Nature . 1998; 391: 707–10. [DOI] [PubMed] [Google Scholar]
- 19. Christophorou MA, Martin‐Zanca D, Soucek L, et al . Temporal dissection of p53 function in vitro and in vivo . Nat Genet . 2005; 37: 718–26. [DOI] [PubMed] [Google Scholar]
- 20. Hou MH, Robinson H, Gao YG, et al . Crystal structure of actinomycin D bound to the CTG triplet repeat sequences linked to neurological diseases. Nucleic Acids Res . 2002; 30: 4910–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Beer EL, Bottone AE, Voest EE. Doxorubicin and mechanical performance of cardiac trabeculae after acute and chronic treatment: a review. Eur J Pharmacol . 2001; 415: 1–11. [DOI] [PubMed] [Google Scholar]
- 22. Crook T, Tidy JA, Vousden KH. Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans‐activation. Cell . 1991; 67: 547–56. [DOI] [PubMed] [Google Scholar]
- 23. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell . 1997; 88: 323–31. [DOI] [PubMed] [Google Scholar]
- 24. Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ . 2006; 13: 951–61. [DOI] [PubMed] [Google Scholar]
- 25. Denchi EL, Helin K. E2F1 is crucial for E2F‐dependent apoptosis. EMBO Rep . 2005; 6: 661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salon C, Eymin B, Micheau O, et al . E2F1 induces apoptosis and sensitizes human lung adenocarcinoma cells to death‐receptor‐mediated apoptosis through specific downregulation of c‐FLIP(short). Cell Death Differ . 2006; 13: 260–72. [DOI] [PubMed] [Google Scholar]
- 27. Matter N, Herrlich P, Konig H. Signal‐dependent regulation of splicing via phosphorylation of Sam68. Nature . 2002; 420: 691–5. [DOI] [PubMed] [Google Scholar]
- 28. Arch R, Wirth K, Hofmann M, et al . Participation in normal immune responses of a metastasis‐inducing splice variant of CD44. Science . 1992; 257: 682–5. [DOI] [PubMed] [Google Scholar]
- 29. Cooper DL, Dougherty GJ. To metastasize or not? Selection of CD44 splice sites. Nat Med . 1995; 1: 635–7. [DOI] [PubMed] [Google Scholar]
- 30. Jackson JG, Pereira‐Smith OM. p53 is preferentially recruited to the promoters of growth arrest genes p21 and GADD45 during replicative senescence of normal human fibroblasts. Cancer Res . 2006; 66: 8356–60. [DOI] [PubMed] [Google Scholar]
- 31. Godar S, Ince TA, Bell GW, et al . Growth‐inhibitory and tumor‐ suppressive functions of p53 depend on its repression of CD44 expression. Cell . 2008; 134: 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vegran F, Boidot R, Oudin C, et al . Association of p53 gene alterations with the expression of antiapoptotic survivin splice variants in breast cancer. Oncogene . 2007; 26: 290–7. [DOI] [PubMed] [Google Scholar]
- 33. Shu L, Yan W, Chen X. RNPC1, an RNA‐binding protein and a target of the p53 family, is required for maintaining the stability of the basal and stress‐induced p21 transcript. Genes Dev . 2006; 20: 2961–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 FACS analysis of RKO cells or RKO cells expressing E6 treated with actinomycin D or doxorubicin. (A) Cells were treated with 5 nM actinomycin D (ActD) for 48 hrs, before fixation. +: treated cells; ‐: untreated cells. Percentage of apoptotic and G1 arrested cells, are indicated. (B) Cells were treated with 0.3 μg/ml doxorubicin (Doxo) for 48 hrs, before fixation. +: treated cells; ‐: untreated cells. Percentage of apoptotic and G1 arrested cells are indicated.
Fig. S2 Ectopic expression of DUSP11 inhibits cell proliferation. U2OS cells were stably transfected as described in Fig. 5A and plated at low density. Cells were then fixed and stained with crystal violet.
Fig. S3 Estimation of transfection efficiency for the cells used to perform the Colony formation assay, described in Fig. 5. Cells were transfected with the indicated plasmids plus one tenth of the pCMV EGFP expression plasmid. The percentage of cells expressing EGFP was analysed by FACS.
Fig. S4 Inhibition of DUSP11 expression by shRNA stimulates cell proliferation and protects from DNA damage. (A) U2OS cells were infected as described in Fig. 6B. Once selected, cells were plated at low density. Cells were then fixed and stained with crystal violet. (B) U2OS cells were infected as described in Fig. 6C. Once selected, cells were treated with UV 15 J/m2 and then plated at low density. Cells were then fixed and stained with crystal violet. (C) U2OS cells were infected as described in Fig. 6D. Once selected, cells were treated with doxorubicin 0.3 μg/ml and then plated at low density. Cells were then fixed and stained with crystal violet.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item