

Abstract
The anti‐inflammatory properties of transforming growth factor‐β1 (TGF‐β1) account for its protection against atherosclerotic plaque rupture. This study investigates whether activation of the Nrf2 (nuclear factor erythroid 2 [NF‐E2]‐related factor 2) transcription pathway is involved in TGF‐β1 mediated induction of the antioxidant enzyme heme oxygenase‐1 (HO‐1) in smooth muscle cells (SMC). Human aortic smooth muscle cells (HAoSMC) or wild‐type and Nrf2‐deficient mouse (MAoSMC) aortic SMC were treated with TGF‐β1 (2.5–10 ng/ml, 0–24 hrs). We report the first evidence that TGF‐β1 induces Nrf2 mediated HO‐1 expression and antioxidant response element activity, which was paralleled by enhanced superoxide production and expression of the NAD(P)H oxidase subunit p22phox. TGF‐β1 failed to induce HO‐1 expression in MAoSMC derived from Nrf2‐deficient mice, and HO‐1 induction by TGF‐β1 in HAoSMC was attenuated by inhibition of extracellular signal regulated kinase or c‐jun‐N‐terminal kinase but not p38 mitogen activated protein kinase. Inhibition of NAD(P)H oxidase or scavenging of superoxide diminished HO‐1 induction in response to TGF‐β1. The oxidative stress agents glucose oxidase (GOx) and diethylmaleate enhanced TGF‐β1 generation and HO‐1 expression in HAoSMC, while antagonism of TGF‐β1 signalling by adenoviral Smad7 overexpression attenuated their induction of HO‐1. Pre‐treatment of HAoSMC with TGF‐β1 reduced nuclear translocation of the pro‐apoptotic mediator p53 elicited by GOx. Our findings demonstrate that Nrf2 is a new target of TGF‐β1 signalling in the vasculature which may contribute to the atheroprotective properties attributed to this growth factor.
Keywords: vascular smooth muscle cells, transforming growth factor‐β1, Nrf2, antioxidant response element, heme oxygenase‐1, reactive oxygen species, NADPH oxidase, p53, apoptosis, Smad, mitogen activated protein kinases
Introduction
Transforming growth factor‐β1 (TGF‐β1) is one of the most important and extensively studied modulators of the inflammatory balance within atherosclerotic and restenotic lesions [1, 2]. The anti‐proliferative and anti‐apoptotic effects of TGF‐β1 in cultured vascular smooth muscle cells (SMC) have been well documented [3], although atheroprotective actions of TGF‐β1 have only recently been highlighted in vivo in experimental models of aortic aneurysm and in human atherosclerotic plaques [3, 4, 5, 6]. Reactive oxygen species (ROS) play a key role in the progression of atherosclerosis leading to plaque rupture [7, 8]. Superoxide anions (O2 −.) are generated during atherogenesis primarily by NAD(P)H oxidase isoforms expressed in endothelial cells, SMC and macrophages [9]. Moreover, TGF‐β1 has been shown to mediate phenotypic changes in human cardiac fibroblasts [10] and umbilical vein endothelial cells [11] through enhanced O2 −. generation by NAD(P)H oxidase.
Endogenous cellular antioxidant defence proteins, such as heme oxygenase‐1 (HO‐1), are induced in vascular cells in response to oxidative stress [12, 13]. HO‐1 is a microsomal enzyme that catalyses the rate‐limiting step in the degradation of the pro‐oxidant heme to biliverdin, iron and carbon monoxide (CO) [14]. Biliverdin is subsequently converted by biliverdin reductase to bilirubin, an antioxidant which can scavenge lipid peroxyl radicals [15]. CO exhibits both anti‐apoptotic and anti‐inflammatory properties [16], and is a vasodilator of potential importance in atherogenesis when the regulation of vascular tone by the endothelial nitric oxide synthase is impaired [17]. HO‐1 is present in human atherosclerotic lesions [18], and its overexpression in rodent models of vascular disease protects against atherogenesis and restenosis [19, 20].
We have recently established that induction of HO‐1 in human and murine aortic SMC and murine macrophages by oxidatively modified low‐density lipoproteins (LDL) is mediated by activation of nuclear factor erythroid 2 (NF‐E2)‐related factor 2 (Nrf2) [21, 22] the predominant transcription factor that binds to antioxidant response elements (ARE) in the promoter region of target genes [23]. Extracellular signal‐regulated kinase (ERK1/2), c‐Jun NH2‐terminal kinase (JNK) and p38 mitogen activated protein kinase (MAPK) are involved in phosphorylation and stabilization of Nrf2, nuclear translocation and binding of Nrf2 to ARE sequences [24]. Although activation of the classical TGF‐β1 mediated Smad [25] and MAPK cascades are involved in the phenotypic modulation of hepatic stellate cells [26] and HO‐1 expression in pulmonary epithelial cells [27], there is currently a paucity of information on the involvement of these pathways in TGF‐β1 mediated Nrf2‐ARE signalling in human or murine vascular SMC.
The present study has thus investigated whether TGF‐β1 enhances HO‐1 expression, nuclear translocation of Nrf2 and ARE activation. Our findings demonstrate that the Nrf2‐ARE pathway represents a novel target for TGF‐β1 in human vascular SMC which has not been previously described in any vascular cell type. Moreover, TGF‐β1 mediated signalling through superoxide generation via NAD(P)H oxidase involves activation of Smad and MAPK pathways in the induction of HO‐1. TGF‐β1 also attenuated the nuclear accumulation of p53, a key mediator of apoptosis and senescence [28], elicited by oxidative stress. Therefore, induction of antioxidant genes via Nrf2 activation by TGF‐β1 provides further mechanistic insights for the ‘protective cytokine hypothesis’[3] in relation to its role in modulating restenosis, atherogenesis and atherosclerotic plaque stability.
Material and methods
Cell culture
Human aortic smooth muscle cells (HAoSMC) derived from primary explant cultures or murine smooth muscle cells (MAoSMC) isolated from aortic explants of C57BL/6 wild‐type or Nrf2‐deficient mice were cultured in phenol red‐free Dulbecco’s modified Eagle’s medium (Sigma‐Aldrich, Poole, UK) containing 1/gl D‐glucose and supplemented with 20% foetal calf serum (FCS) [21, 22]. For experimental treatments, confluent cultures were equilibrated in medium containing 1% FCS for 24 hrs prior to treatments with recombinant TGF‐β1 (2–24 hrs, 2.5–10 ng/ml, R&D Systems, Abingdon, UK), diethylmaleate (DEM, 50–100 μM, 24 hrs, Sigma) or the hydrogen peroxide generator glucose oxidase (GOx, 24 hrs, 5–10 mU/ml, Merck, Nottingham, UK).
Experiments designed to investigate inhibition of MAPK pathways were performed in cells equilibrated in medium containing 1% FCS for 24 hrs before treatment with the specific MAPK inhibitors [21], U0126 (1 μM, MEK inhibitor), SP600125 (10 μM, JNK inhibitor) or SB203580 (2 μM, p38MAPK inhibitor) or vehicle controls for 30 min. prior to TGF‐β1 exposure. To determine the effects of scavenging superoxide or inhibiting NADPH oxidase activity on HO‐1 induction, cells were treated concomitantly with TGF‐β1 (2.5–5 ng/ml, 12 hrs) and either superoxide dismutase (SOD, 200 U/ml) or the NADPH oxidase flavoprotein inhibitor diphenylene iodonium (DPI, 2 μM) [29], respectively. In some experiments cells were pre‐treated with TGF‐β1 (5 ng/ml) for 4 hrs prior to exposure to GOx (0–20 mU/ml) in the continued presence of TGF‐β1.
Antagonism of TGF‐β1 signalling by Smad7 adenoviral transfection
To antagonize Smad mediated TGF‐β1 signalling in SMC, cells were transfected with an adenoviral vector co‐ordinating expression of Smad7 (generous gift of Dr. A. Nakao, Juntendo University, Japan), which inhibits phosphorylation of Smad2 [30] an early event in the classical TGF‐β signalling cascade [25]. Replication defective adenoviral vectors coordinating expression of β‐galactosidase (AdLacZ), or Smad7 (AdSmad7), under regulation by the CMV promoter, were propagated and purified as previously described [31]. Confluent HAoSMC were transfected (100 MOI, 24 hrs) with AdLacZ (control) or AdSmad7 and, following gene transfer, cells were equilibrated (24 hrs) in fresh medium containing 20% FCS prior to initiation of experimental protocols.
Western blot analyses
Activation of MAPK and Smad2 phosphorylation and changes in HO‐1, p22phox and nuclear Nrf2 expression were determined by western blot analysis in cells treated with TGF‐β1. Incubations were terminated by washing cells with ice‐cold PBS. Cells were lysed in buffer (pH 6.8, 2% w/v SDS, 10% v/v glycerol, 50 mM Tris‐HCl) containing and protease and phosphatase inhibitor cocktails (Sigma). Nuclear extracts were prepared following cell lysis in 0.5% Nonidet P‐40 containing buffer as described previously [21, 22]. Protein content was determined using the bicinchoninic acid assay (BCA, Thermo Scientific, Cramlington, UK), and lysates were subjected to gel electrophoresis, electroblotting and then probed with a monoclonal antibody against HO‐1 (BD Biosciences, Oxford, UK) or polyclonal antibodies against p22phox, Nrf2, p53 or phosphorylated isoforms of ERK1/2, JNK and p38MAPK (Santa Cruz Biotechnology). Monoclonal antibodies against either α‐tubulin or lamin A (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used as loading controls. Enhanced chemiluminescence was used to visualize bands on autoradiographic film, which were quantified using by scanning densitometry.
Nrf2 localization by immunofluorescence
HAoSMC were grown on Lab‐Tek II (Fisher Scientific, Loughborough, UK) chamber slides and treated with TGF‐β1 (0–10 ng/ml, 2 hrs). Cells were then washed, fixed using phosphate buffered saline containing paraformaldehyde (4%), permeabilized with triton X‐100 (0.1%) and examined by immunofluorescence using an anti‐Nrf2 primary antibody (Santa Cruz Biotechnology) and Alexa Fluor 488‐labelled secondary antibody (Invitrogen, Paisley, UK). Nuclei were co‐labelled by staining cells with propidium iodide (500 ng/ml, 15 min.) prior to visualizing the cells. An inverted microscope (Nikon, Eclipse TE2000‐U, Kingston‐upon‐Thames, UK) fitted with appropriate fluorescence filters and a CCD digital camera (Nikon, DXM1200F) was used to capture images [21].
Detection of superoxide generation
Superoxide production was assessed by lucigenin [32] or L‐012 [33] enhanced chemiluminescence in live HAoSMC cultures and cell homogenates. Briefly, confluent cell cultures were pre‐treated with TGF‐β1 (5 ng/ml, 0–60 min. or 12 hrs) in medium prior to measurement of superoxide (O2 −.) generation by chemiluminescence in cells incubated with Krebs–Henseleit buffer in the presence of lucigenin (5 μM) and NADPH (100 μM) or L‐012 (50 μM) without exogenous NADPH, and luminescence detected in a microtitre plate reader (Chameleon V, Hidex) over 40 min. at 37°C. Cells were also treated with TGF‐β1 (5 ng/ml, 4 hrs) and treatments terminated by washing monolayers twice with cold PBS. Cells were detached using trypsin/ethylenediaminetetraacetic acid (0.05% v/v), and cell pellets disrupted using a polytron homogenizer. Superoxide production in cell homogenates (10 μg protein) following 4‐hr treatment with TGF‐β1 was assessed in the presence of NADPH (100 μM) and lucigenin (5 μM) in the luminescence plate reader over 30 min. at 37°C. The specificity of the luminescence signals for O2 −. was assessed by co‐incubation of cells with the membrane permeable polyethylene glycol‐superoxide dismutase (PEG‐SOD, 200 U/ml). Cells were also preincubated with DPI (2 μM), or apocynin (100 μM), an inhibitor of NAD(P)H oxidase subunit assembly [34], for 30 min. prior to and during TGF‐β1 treatments.
Functional ARE‐luciferase reporter assay
An ARE‐luciferase reporter construct, consisting of the 5’‐flanking region of the mouse cystine/glutamate membrane transporter (xCT) gene containing four ARE sequences [35], was used to monitor TGF‐β1 mediated Nrf2‐ARE activation in mouse aortic SMC. Constructs containing the xCT‐Luc reporter gene or the pGL3 empty vector were transfected into MAoSMC using jetPEI (MP Biomedicals, Cambridge, UK) in culture medium containing 10% FCS. Cells were then recovered and equilibrated in medium containing 1% FCS for 12 hrs. Cells were harvested following treatments with TGF‐β1 (0–10 ng/ml, 12 hrs) and luciferase reporter activity determined using a commercially available reporter gene assay kit (LucLite, Perkin Elmer, Waltham, MA, USA) and a luminescence plate reader (Hidex Chameleon V, Hidex, Turku, Finland). Cell lysates were analysed for protein content using the BCA assay method and luminescence units normalized for total protein content.
TGF‐β1 enzyme linked immunosorbant assay
A commercially available TGF‐β1‐specific solid phase sandwich ELISA (Quantikine, R&D Systems) was used to determine total TGF‐β1 levels in the conditioned medium from HAoSMC treated with the oxidative stress agents DEM and GOx over 24 hrs. Values were normalized to total cellular protein content measured using the BCA assay.
Statistical analysis
Statistical variance from the mean was determined using the normal distribution and denoted as means ± S.E.M. (n= 3–6 different cell cultures). Confidence limits were established using an unpaired Student’s t‐test and two‐way ANOVA were used to compare multiple groups. Statistical significance between data sets was established at P < 0.05 to P < 0.001.
Results
Induction of HO‐1 protein expression by TGF‐β1 in HAoSMC
Under basal conditions, expression of HO‐1 protein was low in HAoSMC; however, TGF‐β1 (2.5 ng/ml, 4–24 hrs) significantly increased HO‐1 expression within 4 hrs, with a 9.4‐fold induction above basal levels measured at 8–12 hrs. Following 24 hrs treatment with TGF‐β1 (2.5 ng/ml), HO‐1 protein expression returned to basal levels (Fig. 1A, B). Induction of HO‐1 by TGF‐β1 was found to be also dose dependent with significant increases in protein expression induction observed following treatments at 2.5–10 ng/ml for 8 hrs (Fig. 1C).
Figure 1.
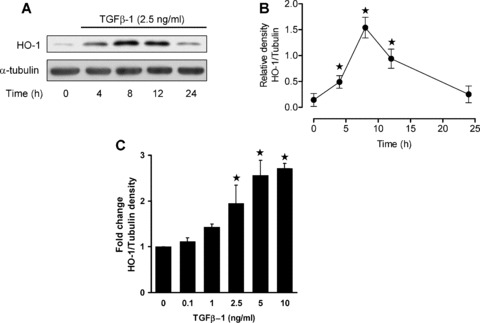
Time‐ and dose‐dependent induction of HO‐1 by TGF‐β1 in HAoSMC. Confluent cultures were equilibrated in medium containing 1% FCS prior to treatment with TGF‐β1 (0.1–10 ng/ml, 0–24 hrs). (A) Immunoblot analyses of HO‐1 protein expression in cells treated with TGF‐β1 (2.5 ng/ml) relative to α‐tubulin used as a loading control and (B) denstometric quantification of HO‐1 expression. (C) Denstometric quantification of immunoblot analyses of HO‐1 expression in cells treated with TGF‐β1 for 12 hrs. Immunoblots are representative of results obtained in n= 3–8 different cell cultures, data are means ± S.E.M., ★P < 0.05 relative to untreated cells.
Role of MAPK in TGF‐β1 signalling and HO‐1 induction in HAoSMC
To investigate whether MAPK pathways are activated by TGF‐β1 in HAoSMC, cells were treated with TGF‐β1 (2.5 or 5 ng/ml) for 5– 30 min. or 2–4 hrs and phosphorylation of p38MAPK, ERK1/2 and JNK determined by immunoblotting (Fig. 2A, C). TGF‐β1 acutely increased phosphorylation of p38MAPK and ERK1/2 after 5–20 min. (data not shown). p38MAPK and JNK phosphorylation were also induced after 2‐hr treatments, with negligible changes detected after 4 hrs. Basal levels of phospho‐ERK1 were higher than phospho‐ERK2; however, phosphorylation of ERK2 was markedly enhanced above basal levels following treatment of cells with TGF‐β1 for 2 hrs (5 ng/ml) or at 4 hrs (2.5 ng/ml). To determine whether MAPK signalling pathways are involved in TGF‐β1 mediated HO‐1 induction, HAoSMC were pre‐treated for 30 min. with either SB203580 (2 μM, p38MAPK inhibitor), SP600125 (10 μM, JNK inhibitor) or U0126 (1 μM, MEK inhibitor), prior to co‐treatment with TGF‐β1 (2.5 ng/ml) for a further 12 hrs. Inhibition of either JNK or MEK significantly (P < 0.05) attenuated HO‐1 induction by TGF‐β1; however, inhibition of p38MAPK had negligible effects on TGF‐β1 stimulated HO‐1 induction (Fig. 2B, C).
Figure 2.
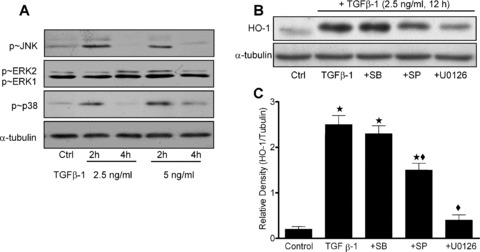
Activation of JNK, ERK1/2 and p38MAPK by TGF‐β1 and effects of MAPK inhibition on HO‐1 induction in HAoSMC. Confluent cultures were equilibrated in medium containing 1% FCS prior to treatment with TGF‐β1 (2.5– 5 ng/ml, 2–4 hrs). (A) Phosphorylation of p38MAPK, ERK1/2 and JNK were determined by immunoblot analyses. (B) Immunoblots and (C) denstometric quantification showing effects of MAPK inhibitors on TGF‐β1 (2.5 ng/ml, 12 hrs) stimulated HO‐1 induction. HAoSMC were pre‐treated for 30 min. with U0126 (1 μM, MEK inhibitor), SB203580 (2 μM, p38MAPK inhibitor) or SP600125 (10 μM, JNK inhibitor) prior to TGF‐β1 treatments. Representative immunoblots shown are representative of results obtained in n= 3–8 different cell cultures, data are means ± S.E.M., ★P < 0.01 relative to untreated cells, ♦P < 0.01 relative to cells treated with TGF‐β1.
Role of ROS generation in TGF‐β1 mediated HO‐1 induction in HAoSMC
To determine whether TGF‐β1 modulates the generation of superoxide (O2 −.) in HAoSMC, expression of p22phox, an essential component in the assembly and activation of the NADPH oxidase complex [9], and NAD(P)H‐dependent superoxide generation were measured. Pre‐treatment of HAoSMC acutely with TGF‐β1 (1– 60 min., 5 ng/ml) significantly (P < 0.05) enhanced NAD(P)H‐dependent generation of O2 −. in live cells (Fig. 3A). Treatment of cells with the membrane permeable PEG‐SOD (200 U/ml) or the NAD(P)H oxidase inhibitors DPI (2 μM) or apocynin (100 μM), in combination with TGF‐β1 (5 ng/ml, 10 min.) significantly abrogated the enhanced O2 −. generation (Fig. 3B). Treatment with TGF‐β1 for 4 hrs (2.5–5 ng/ml) also significantly (P < 0.05) enhanced superoxide production in HAoSMC cell homogenates (data not shown), and inhibited by co‐treatment with apocynin (100 μM). Generation of superoxide was sustained for at 12 hrs following treatment with TGF‐β1 (5 ng/ml), as assessed by L‐012 enhanced‐chemiluminescence in live cell cultures (Fig 3C), and significantly attenuated by co‐treatment of cells with apocynin or SOD. Expression of p22phox protein in HAoSMC was markedly enhanced by treatment with 2.5–10 ng/ml TGF‐β1 for 12 hrs (Fig. 4A, B) and TGF‐β1 (5 ng/ml, 12 hrs) mediated HO‐1 protein expression was also found to be reduced by co‐treatments with the superoxide scavenger SOD or the NADPH oxidase flavoprotein inhibitor DPI (Fig. 4C, D).
Figure 3.
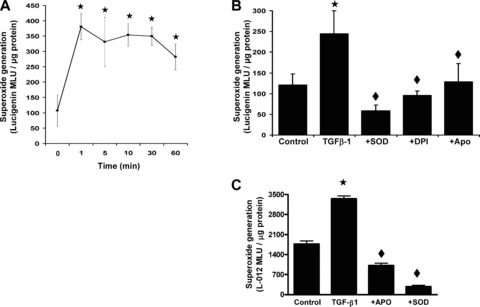
TGF‐β1 enhanced generation of superoxide in HAoSMC. Confluent cultures were equilibrated in medium containing 1% FCS prior to treatments (0–60 min.) with TGF‐β1 (5 ng/ml). (A) NAD(P)H‐dependent superoxide generation in live HAoSMC was determined by lucigenin‐enhanced chemiluminescence. (B) Inhibition of NAD(P)H‐dependent superoxide detection by TGF‐β1 treatment (10 min.) in the absence (control) or presence of SOD (200 U/ml), DPI (2 μM) or apocynin (100 μM). (C) Superoxide generation determined by L‐012 enhanced chemiluminescence in live HAoSMC treated with TGF‐β1 (5 ng/ml) for 12 hrs in the presence or absence of apocynin (100 μM) or SOD (200 U/ml). Values denote the means ± S.E.M. of measurements in n= 3–6 different cell cultures, ★P < 0.01 relative to control untreated cells, ♦P < 0.01 relative to cells treated with TGF‐β1.
Figure 4.
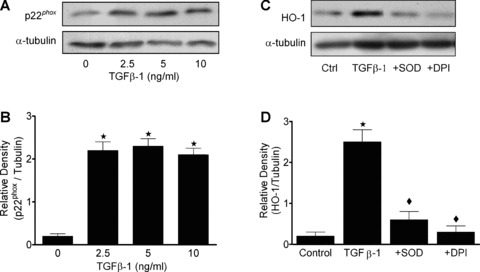
TGF‐β1 enhances expression of p22phox and HO‐1 in HAoSMC. Confluent cultures were equilibrated in medium containing 1% FCS prior to treatment with TGF‐β1 for 12 hrs. (A) Expression of p22phox was determined by western blot analyses relative to α‐tubulin and (B) quantification by densitometry. (C) Immunoblot analyses of HO‐1 protein expression in cells treated with TGF‐β1 (5 ng/ml) in the absence or presence of SOD (200 U/ml) or DPI (2 μM) and (D) quantification by denstometry. Immunoblots are representative of results obtained in n= 3–8 different cell cultures, data are means ± S.E.M., ★P < 0.01 relative to untreated cells, ♦P < 0.01 relative to cells treated with TGF‐β1.
Activation of Nrf2 nuclear translocation by TGF‐β1 in HAoSMC
As nuclear translocation of the Nrf2 is associated with transcriptional activation of HO‐1 [23], we analysed HAoSMC nuclear lysates for Nrf2 protein expression following treatment TGF‐β1. As shown in Fig. 5(A), treatment of cells with TGF‐β1 for 2 hrs resulted in enhanced nuclear accumulation of Nrf2. Immunofluorescent analysis revealed that Nrf2 was predominantly cytosolic in control untreated cells (Fig. 5B–D); however, nuclear translocation of Nrf2 following treatment of HAoSMC with TGF‐β1 was demonstrated by the enhanced co‐localization with nuclear propidium iodide (Fig. 5E–G).
Figure 5.
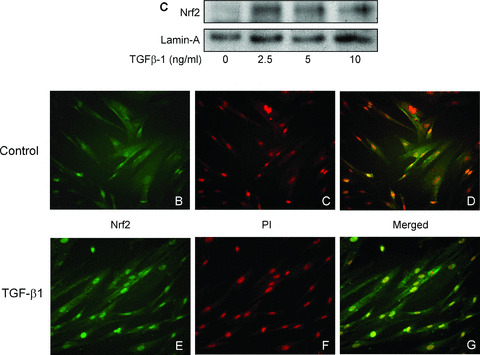
Nuclear translocation of Nrf2 in HAoSMC treated with TGF‐β1. Confluent cultures were equilibrated in medium containing 1% FCS prior to treatment with TGF‐β1 (2.5–10 ng/ml, 2 hrs). (A) Immunoblot analysis of Nrf2 protein expression in nuclear extracts relative to the loading control lamin‐A. Cellular localization of Nrf2 was determined by immunofluorescent microscopy in untreated cells (B–D) and cells treated with TGF‐β1 (2.5 ng/ml, 2 hrs, E–G) and stained using a specific antibody against Nrf2 and an Alexa Fluor‐488 conjugated secondary antibody (B, E). Nuclei were counterstained with propidium iodide (C, F) and images merged using Photoshop software (D, G). Data are representative of similar results in three different cell cultures.
Nrf2 dependent transcriptional activation of HO‐1 and ARE by TGF‐β1 in MAoSMC
To further confirm that HO‐1 induction by TGF‐β1 was dependent on the transcriptional activity of Nrf2, MAoSMC from wild‐type or Nrf2‐deficient mice were treated for 12 hrs with TGF‐β1 (2.5– 10 ng/ml). HO‐1 protein expression was increased by TGF‐β1 only in SMC from wild‐type Nrf2+/+ mice, while basal HO‐1 expression was not detectable in SMC from Nrf2−/– mice (Fig. 6A). In these same experiments, treatment with the electrophilic stress agent DEM (100 μM) resulted in a significant induction of HO‐1 in Nrf2+/+ cells but only limited induction in Nrf2−/– MAoSMC (not shown), confirming our previous findings in this cell type [22]. Lysates from wild‐type MAoSMC transfected with an ARE‐luciferase reporter construct containing four ARE‐like sequences from the mouse xCT cystine transporter gene [34] exhibited a dose‐dependent increase in luciferase mediated luminescence following treatment of cells for 12 hrs with TGF‐β1 (2.5–10 ng/ml), confirming enhanced ARE‐mediated transcriptional activity (Fig. 6B). No luciferase activity was detectable in TGF‐β1 treated cells transfected with the control empty pGL3 vector (data not shown).
Figure 6.
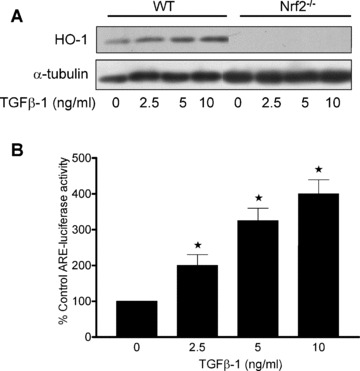
Nrf2 dependent induction of HO‐1 and ARE‐luciferase reporter activity in MAoSMC treated with TGF‐β1. Confluent cultures of aortic SMC from wild‐type or Nrf2‐deficient (Nrf2−/–) mice were equilibrated in medium containing 1% FCS prior to treatment with TGF‐β1 (2.5– 10 ng/ml, 12 hrs). (A) HO‐1 protein expression in whole cell lysates from Nrf2+/+ and Nrf2−/– MAoSMC using α‐tubulin as an internal loading control. (B) Confluent cultures of Nrf2+/+ MAoSMC were transfected with the mouse ARE‐luciferase reporter construct (xCT‐Luc) and then equilibrated in medium containing 1% FCS prior to stimulation with TGF‐β1 (2.5–10 ng/ml, 12 hrs). Cells were then lysed and luciferase activity expressed as a percentage change from control untreated cells. Values denote means ± S.E.M. of measurements in three different cell cultures, ★P < 0.05 relative to values in control untreated cells.
Role of Smad signalling and TGF‐β1 generation in HO‐1 induction by stress agents in HAoSMC
To establish whether induction of HO‐1 in HAoSMC by the stress agents DEM or H2O2, generated by GOx, involves activation of the classical TGF‐β1 mediated Smad signalling pathway [25], HAoSMC were transfected with either an adenoviral vector (AdSmad7) coordinating expression of Smad7, an endogenous inhibitor of Smad signalling [29], or a control vector (AdLacZ), coordinating expression of β‐galactosidase. Transfection of cells with AdSmad7 markedly enhanced Smad7 levels (Fig. 7A) and antagonized TGF‐β1 signalling as shown by the diminished Smad2 phosphorylation in response to treatments with exogenous TGF‐β1 (Fig. 7B). Control AdLacZ transfected cells did not exhibit changes in basal HO‐1 levels, whereas, HO‐1 induction was evident in response to DEM (50–100 μM, 24 hrs) or GOx (5–10 mU/ml, 24 hrs). Cells transfected with Smad7 exhibited a marked attenuation in stress agent induced HO‐1 induction (Fig. 7C). Since TGF‐β1 signalling pathways appear to be involved in stress mediated HO‐1 induction, we determined TGF‐β1 release into the supernatant of control or Smad7 transfected HAoSMC in response to DEM or GOx. TGF‐β1 levels were elevated above basal in both control AdLacZ and AdSmad7 transfected HAoSMC (Fig. 7D) and the lack of a difference in TGF‐β1 levels between control and AdSmad7 transfected cells indicates that Smad7 overexpression did not alter TGF‐β1 generation in HAoSMC challenged with either DEM or GOx.
Figure 7.
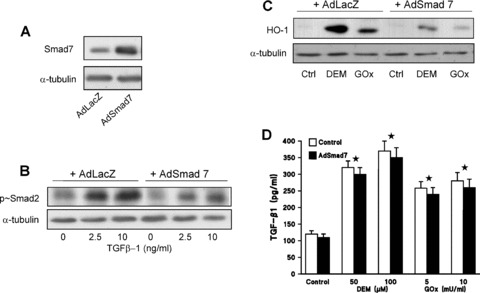
Stress agents enhance TGF‐β1 release from HAoSMC and induce HO‐1 via a Smad mediated pathway. Confluent cultures of HAoSMC were transfected with either AdLacZ (control) or AdSmad7 prior to equilibration in medium containing 1% FCS and treatment for 24 hrs with DEM (50–100 μM) or GOx (5–10 mU/ml). Immunoblot of Smad7 (A) or phospho‐Smad2 (B) protein expression in whole cell lysates of AdLacZ or AdSmad7 transfected cells treated with TGF‐β1 (4 hrs) relative to α‐tubulin loading control. (C) HO‐1 protein expression in whole cell lysates of AdLacZ or AdSmad7 transfected HAoSMC treated for 24 hrs with DEM (100 μM) or GOx (10 mU/ml) relative to α‐tubulin loading control. (D) Total TGF‐β1 secreted into the culture medium was assessed by ELISA, and levels corrected for total protein content in LacZ (open bars) or Smad7 (solid bars) transfected cells. Values denote the means ± S.E.M. of measurements in three different cell cultures, ★P < 0.01 relative to control untreated cells in values obtained from both LacZ and AdSmad7 transfected cells.
TGF‐β1 attenuates p53 nuclear accumulation induced by GOx
As nuclear accumulation of the transcription factor p53 is associated with activation of pro‐apoptotic genes [28], we analysed p53 protein expression in nuclear lysates from HAoSMC following treatment of cells with the oxidative stress agent GOx (0–20 mU/ml, 8 hrs) in the absence or presence of pre‐treatments with TGF‐β1 (5 ng/ml, 4 hrs). As shown in Fig. 8, treatment of HAoSMC with GOx resulted in a dose‐dependent increase in nuclear p53 expression which was significantly attenuated by prior pre‐treatment of cells with TGF‐β1.
Figure 8.
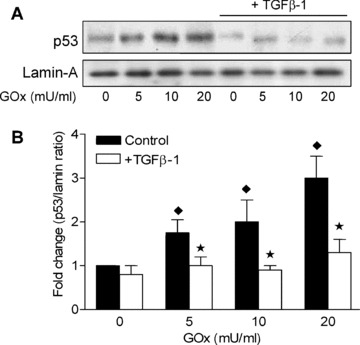
TGF‐β1 attenuates nuclear accumulation of p53 induced by GOx in HAoSMC. Confluent cultures of HAoSMC were treated in medium containing 1% FCS the absence or presence of TGF‐β1 (5 ng/ml, 4 hrs) prior to exposure to GOx (0–20 mU/ml, 8 hrs). (A) Representative immunoblot of p53 protein expression in nuclear lysates relative to the loading control lamin‐A. (B) Denstometric quantification of fold changes in p53 expression compared to levels in control untreated cells. Immunoblots are representative of results obtained in n= 3–4 different cell cultures, data are means ± S.E.M., ★P < 0.01 relative to cells treated with GOx in the absence of TGF‐β1, ♦P < 0.05 relative to control untreated cells.
Discussion
The present study provides novel evidence in HAoSMC that TGF‐β1 activates nuclear translocation of Nrf2, leading to expression of the vasculoprotective gene HO‐1 [12, 13, 19, 20]via transcriptional activation of ARE, generation of ROS and activation of MAPK and Smad signalling pathways. TGF‐β1 is regarded as a protective cytokine that limits atherosclerotic plaque rupture and stabilizes the diseased vessel wall [1, 2, 3, 4, 5, 6]. However, the mechanisms underlying the protective properties of TGF‐β1 in vascular cells remain to be fully elucidated. TGF‐β1 can elicit generation of ROS in human cardiac fibroblasts [10], umbilical vein endothelial [11] and pulmonary artery SMC [36]via activation of NAD(P)H oxidase to mediate phenotypic changes, cytoskeletal alterations and enhance cell proliferation, respectively. Our study provides an important insight into the mechanisms underlying adaptive redox responses to TGF‐β1 in HAoSMC, a cell type of direct relevance to atherogenesis and plaque stability [37].
As enhanced ROS generation and redox imbalance are primary stimuli for the activation of endogenous antioxidant defence mechanisms [23], we sought to elucidate whether TGF‐β1 induced ROS generation leads to transcriptional activation of HO‐1 via Nrf2‐ARE signalling. Nrf2 is now established to be a key transcription factor involved in cytoprotection in pulmonary disorders [38] and cancer chemoprotection [39], but to our knowledge there have been no reports addressing whether the Nrf2/ARE signalling and HO‐1 expression are modulated by TGF‐β1 in vascular SMC. Our findings provide the first evidence that Nrf2 mediates HO‐1 induction in response to TGF‐β1, highlighting a novel mechanism by which this cytokine confers protection against vascular cell dysfunction and plaque rupture. We previously reported that elevated HO‐1 expression in HAoSMC is associated with an increase in its capacity to generate the antioxidant bilirubin, reflecting increased HO‐1 activity [21].
We have demonstrated that induction of HO‐1 expression by TGF‐β1 is associated with an acute and sustained (2–4 hrs) increase in phosphorylation of p38MAPK, ERK2 and JNK (Fig. 2A). This finding is consistent with the increasing number of reports documenting that TGF‐β1 can activate MAPK pathways in addition to classical Smad signalling [40, 41, 42, 43]. Moreover, inhibition of ERK1/2 or JNK partially attenuated TGF‐β1 mediated HO‐1 induction; however, inhibition of p38MAPK had no effect on TGF‐β1 stimulated HO‐1 expression (Fig. 2B, C). The involvement of MAPK cascades in HO‐1 induction in vascular smooth muscle appears to be agonist and cell type specific, since induction of HO‐1 by α‐lipoic acid in the A10 rat aortic SMC line involves ERK1/2 [44], whereas the anti‐tumour and anti‐restenosis drug paclitaxel induces HO‐1 in primary rat aortic SMC via JNK alone [45]. Nevertheless, our findings establish that HO‐1 induction in response to TGF‐β1 is mediated in via activation of ERK1/2 and JNK in addition to Smad signalling pathways.
Changes in ROS generation in vascular cells or exposure to oxidative stress agents are often associated with a parallel induction of endogenous antioxidant defence genes such as HO‐1 [12, 13, 14]. In our study, TGF‐β1 also enhanced p22phox expression in HAoSMC (Fig. 4A), which was associated with an increase in NAD(P)H oxidase‐dependent O2 −. generation (Fig. 3A, B). The p22phox subunit of NAD(P)H oxidase has a central stabilizing and regulatory role in the enzyme complex and is essential for O2 −. generation in aortic SMC [9, 46]. Scavenging of O2 −. by SOD or inhibition of NAD(P)H oxidase complex activity and assembly by co‐treatment of cells with DPI or apocynin [34] abolished the increases in NAD(P)H‐dependent O2 −. generation, suggesting that NAD(P)H oxidase was the major source of superoxide generated in response to TGF‐β1 treatments. This is consistent with previous findings that NAD(P)H oxidase is activated by TGF‐β1 in fibroblasts [10, 47]. It has been previously reported in the literature that specific knock‐down of p22phox in human aortic SMC using antisense oligonucleotides substantially decreases (i) ROS generation, activation of the transcription factor HIF‐1α and mRNA levels of VEGF and PAI‐1 in response to thrombin stimulation [48], (ii) ROS generation and tissue factor gene activation in response to platelet‐derived products, including TGF‐β1[49] and (iii) ROS generation and DNA damage following arsenite treatment [50]. Moreover, we have shown for the first time that scavenging of O2 −. by co‐treatment of HAoSMC with SOD or inhibition of NAD(P)H oxidase flavoprotein activity with DPI markedly attenuated HO‐1 induction in response to TGF‐β1, suggesting that enhanced NAD(P)H oxidase dependent ROS generation was partly responsible for antioxidant gene induction (Fig. 4C, D). Given that p22phox knock‐down has been demonstrated to significantly reduce ROS generation [48, 48, 50], it is likely that this would also result in diminished HO‐1 induction by TGF‐β1. It is also likely that other subunits of the NADPH oxidase enzyme complex such as Nox4 are involved in the observed enhanced ROS generation [9, 10].
We have previously reported that Nrf2 mediates induction of HO‐1 by oxidized LDL in human HAoSMC [21], and now report the first evidence that TGF‐β1 acutely enhances nuclear translocation of Nrf2 (Fig. 5). Moreover, TGF‐β1 failed to induce HO‐1 in MAoSMC from Nrf2‐deficient mice (Fig. 6A), suggesting that transcriptional regulation by Nrf2 is essential for induction of HO‐1 expression by TGF‐β1. In addition, TGF‐β1 significantly enhanced ARE‐mediated gene transcription in MAoSMC transfected with a murine xCT ARE‐luciferase reporter construct (Fig. 5B). Induction of the cystine membrane transporter xCT has been shown to be regulated by Nrf2/ARE signalling [35] and is essential for the maintenance of glutathione (GSH) levels and redox balance in cultured cells [51].
As TGF‐β1 induced ROS generation was paralleled by Nrf2 activation and HO‐1 expression, we further investigated the involvement of the TGF‐β/Smad signalling pathway in HO‐1 induction in HAoSMC treated with the oxidative stress agents DEM and GOx. The electrophile DEM rapidly depletes intracellular GSH levels, leading to activation of Nrf2‐dependent gene transcription in SMC [22, 52], while GOx continuously generates physiological low micromolar levels of H2O2[53]. Overexpression of Smad7, an endogenous inhibitory modulator of the TGF‐β1 mediated Smad signalling pathway [25, 30], significantly attenuated HO‐1 induction in response to DEM and GOx compared to levels in cells transfected with the control LacZ vector (Fig. 7A). Interestingly, these stress agents also augmented generation of TGF‐β1 (Fig. 7B), which is likely to have acted in an autocrine manner to further increase ROS generation and activation of both MAPK and Nrf2/ARE signalling pathways in SMC. Our findings are consistent with previous reports that H2O2 enhances TGF‐β1 synthesis in human mesangial cells [54], and that overexpression of Smad7 inhibits TGF‐β1‐mediated induction of HO‐1 gene expression in human renal epithelial cells [55]. There is a paucity of information at present on the interactions between Smad and Nrf2 signalling pathways in the regulation of HO‐1 in vascular cells. However, it seems likely that cross talk between Smad, TGF‐β activated kinases (TAK) and MAPK signalling pathways contribute to Nrf2/ARE activation and subsequent induction of protective antioxidant defence genes such as HO‐1 [13, 23, 24, 40].
Although the complex interactions between Smad and MAPK signalling pathways remain to be fully elucidated and are likely to be cell type and stimulus specific, our findings provide a novel link between TGF‐β1 signalling and activation of Nrf2 and induction of cytoprotective antioxidant genes such as HO‐1. Our observation that pre‐treatment of HAoSMC with TGF‐β1 attenuates GOx‐induced nuclear accumulation of the pro‐apoptotic mediator p53 (Fig. 8) provides further evidence that TGF‐β1 can protect against ROS‐mediated SMC dysfunction. Induction of cell death by hydrogen peroxide has been previously characterized in vascular SMC [56, 57] through activation of p53‐mediated signalling events leading to apoptosis and senescence [28]. Our novel data suggest that activation of Nrf2 and subsequent HO‐1 expression may constitute an additional pathway to account for the anti‐inflammatory properties of TGF‐β1. It is likely that antagonism of TGF‐β1 signalling by Smad7 overexpression will enhance apoptosis as previously reported in non‐vascular cells [58, 59] perhaps due to down‐regulation of HO‐1 expression [60, 61]. A complex relationship has been recently described between the p53 and Smad signalling pathways [62] and the mechanisms by which TGF‐β1 ameliorates ROS‐mediated p53 activation remain to be elucidated in SMC. As apoptosis is commonly observed within human unstable atherosclerotic plaques [63], it is therefore possible that activation of Nrf2 signalling by TGF‐β1 in vascular SMC ameliorates oxidative stress through induction of HO‐1 and other antioxidant genes to attenuate ROS generation and activation of p53 and pro‐ apoptotic signalling mediators [64].
Although repression of ARE‐dependent phase II genes in a murine mammary epithelial cell line was recently attributed to carcinogenic properties of TGF‐β1[65], our present study in primary vascular SMC has demonstrated that TGF‐β1 alone induces Nrf2/ARE activity and HO‐1 expression. Moreover, oxidative stress agents elicited a compensatory increase in endogenous TGF‐β1 generation in SMC, which would augment nuclear translocation of Nrf2 and induction of antioxidant genes. Identification of Nrf2 as a target of TGF‐β1 in the vasculature further highlights the ‘multi‐organ’ cytoprotective potential of Nrf2 [66] and provides a basis for further examining the ‘protective cytokine’ hypothesis that has been ascribed to TGF‐β1 in vascular diseases [3, 4, 5, 6]. Activation of Nrf2 may represent a novel vasculoprotective mechanism of relevance to current therapeutic strategies used to modulate TGF‐β1 activity within the vessel wall in atherogenesis and restenosis.
Acknowledgements
The authors thank Dr. Atsuhito Nakao, Juntendo University, Tokyo, Japan for the generous gift of the AdSmad7 adenoviral vector and Mr. Abdulaziz Albarhi and Dr Meis Moukayed for assistance in performing preliminary experiments. This study was supported by the British Heart Foundation (PG/04/067) and the Japanese Ministry of Education. Anila Anwar was supported by the Guy’s & St. Thomas’ Charity and Francois Li is supported by Heart Research UK and Wing Yip & Brothers Charitable Trust.
References
- 1. Lutgens E, Gijbels M, Smook M, et al. Transforming growth factor‐β mediates balance between inflammation and fibrosis during plaque progression. Arterioscler Thromb Vasc Biol. 2002; 22: 975–82. [DOI] [PubMed] [Google Scholar]
- 2. McCaffrey TA. TGF‐β and TGF‐β receptors in atherosclerosis. Cytokine Growth Factor Rev. 2000; 11: 103–14. [DOI] [PubMed] [Google Scholar]
- 3. Grainger DJ. Transforming growth factor‐β and atherosclerosis: so far, so good for the protective cytokine hypothesis. Arterioscler Thromb Vasc Biol. 2004; 24: 399–404. [DOI] [PubMed] [Google Scholar]
- 4. Dai J, Losy F, Guinault AM, et al. Overexpression of transforming growth factor‐β1 stabilizes already‐formed aortic aneurysms: a first approach to induction of functional healing by endovascular gene therapy. Circulation. 2005; 112: 1008–15. [DOI] [PubMed] [Google Scholar]
- 5. Cipollone F, Fazia M, Mincione G, et al. Increased expression of transforming growth factor‐β1 as a stabilizing factor in human atherosclerotic plaques. Stroke. 2004; 35: 2253–7. [DOI] [PubMed] [Google Scholar]
- 6. Mallat Z, Tedgui A. The role of transforming growth factor‐β in atherosclerosis: novel insights and future perspectives. Curr Opin Lipidol. 2002; 13: 523–9. [DOI] [PubMed] [Google Scholar]
- 7. Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury – part I: basic mechanisms and in vivo monitoring of ROS. Circulation. 2003; 108: 1912–6. [DOI] [PubMed] [Google Scholar]
- 8. Channon KM. Oxidative stress and coronary plaque stability. Arterioscler Thromb Vasc Biol. 2002; 22: 1751–2. [DOI] [PubMed] [Google Scholar]
- 9. Griendling KK, Sorescu D, Ushio‐Fukai M. NAD(P)H oxidase – role in cardiovascular biology and disease. Circ Res. 2000; 86: 494–501. [DOI] [PubMed] [Google Scholar]
- 10. Cucoranu I, Clempus R, Dikalova A, et al. NAD(P)H oxidase 4 mediates transforming growth factor‐β 1‐induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005; 97: 900–7. [DOI] [PubMed] [Google Scholar]
- 11. Hu TS, RamachandraRao SP, Siva S, et al. Reactive oxygen species production via NADPH oxidase mediates TGF‐β induced cytoskeletal alterations in endothelial cells. Am J Physiol. 2005; 289: F816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Siow RC, Sato H, Mann GE. Heme oxygenase carbon monoxide signalling pathway in atherosclerosis: anti‐atherogenic actions of bilirubin and carbon monoxide? Cardiovasc Res. 1999; 41: 385–94. [DOI] [PubMed] [Google Scholar]
- 13. Bach FH. Heme oxygenase‐1: a therapeutic amplification funnel. FASEB J. 2005; 19: 1216–9. [DOI] [PubMed] [Google Scholar]
- 14. Maines MD. The heme oxygenase system: a regulator of second messenger gases. Ann Rev Pharmacol Toxicol. 1997; 37: 517–54. [DOI] [PubMed] [Google Scholar]
- 15. Baranano DE, Rao M, Ferris CD, et al. Biliverdin reductase: A major physiologic cytoprotectant. Proc Natl Acad Sci. 2002; 99: 16093–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Otterbein LE, Bach FH, Alam J, et al. Carbon monoxide has anti‐inflammatory effects involving the mitogen‐activated protein kinase pathway. Nature Med. 2000; 6: 422–8. [DOI] [PubMed] [Google Scholar]
- 17. Sammut IA, Foresti R, Clark JE, et al. Carbon monoxide is a major contributor to the regulation of vascular tone in aortas expressing high levels of haeme oxygenase‐1. Br J Pharmacol. 1998; 125: 1437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang LJ, Lee TS, Lee FY, et al. Expression of heme oxygenase‐1 in atherosclerotic lesions. Am J Pathol. 1998; 152: 711–20. [PMC free article] [PubMed] [Google Scholar]
- 19. Juan SH, Lee TS, Tseng KW, et al. Adenovirus‐mediated heme oxygenase‐1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E‐deficient mice. Circulation. 2001; 104: 1519–25. [DOI] [PubMed] [Google Scholar]
- 20. Otterbein LE, Zuckerbraun BS, Haga M, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nature Med. 2003; 9: 183–90. [DOI] [PubMed] [Google Scholar]
- 21. Anwar AA, Li FYL, Leake DS, et al. Induction of heme oxygenase 1 by moderately oxidized low‐density lipoproteins in human vascular smooth muscle cells: role of mitogen‐activated protein kinases and Nrf2. Free Radic Biol Med. 2005; 39: 227–36. [DOI] [PubMed] [Google Scholar]
- 22. Ishii T, Itoh K, Ruiz E, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages – activation by oxidatively modified LDL and 4‐hydroxynonenal. Circ Res. 2004; 94: 609–16. [DOI] [PubMed] [Google Scholar]
- 23. Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Ann Rev Pharmacol Toxicol. 2003; 43: 233–60. [DOI] [PubMed] [Google Scholar]
- 24. Yu R, Chen C, Mo YY, et al. Activation of mitogen‐activated protein kinase pathways induces antioxidant response element‐mediated gene expression via a Nrf2‐dependent mechanism. J Biol Chem. 2000; 275: 39907–13. [DOI] [PubMed] [Google Scholar]
- 25. Attisano L, Wrana JL. Signal transduction by the TGF‐β superfamily. Science. 2002; 296: 1646–7. [DOI] [PubMed] [Google Scholar]
- 26. Tsukada S, Westwick JK, Ikejima K, et al. SMAD and p38 MAPK signaling pathways independently regulate alpha 1(I) collagen gene expression in unstimulated and transforming growth factor‐alpha‐stimulated hepatic stellate cells. J Biol Chem. 2005; 280: 10055–64. [DOI] [PubMed] [Google Scholar]
- 27. Ning W, Song RP, Li CJ, et al. TGF‐β1 stimulates HO‐1 via the p38 mitogen‐activated protein kinase in A549 pulmonary epithelial cells. Am J Physiol. 2002; 283: L1094–102. [DOI] [PubMed] [Google Scholar]
- 28. Jimenez GS, Khan SH, Stommel JM, et al. p53 regulation by post‐translational modification and nuclear retention in response to diverse stresses. Oncogene. 1999; 18: 7656–65. [DOI] [PubMed] [Google Scholar]
- 29. Chakraborty S, Massey V. Reaction of reduced flavins and flavoproteins with diphenyliodonium chloride. J Biol Chem. 2002; 277: 41507–16. [DOI] [PubMed] [Google Scholar]
- 30. Nakao A, Afrakhte M, Moren A, et al. Identification of Smad7, a TGF‐β‐inducible antagonist of TGF‐β signalling. Nature. 1997; 389: 631–5. [DOI] [PubMed] [Google Scholar]
- 31. Mallawaarachchi CM, Weissberg PL, Siow RC. Smad7 gene transfer attenuates adventitial cell migration and vascular remodeling after balloon injury. Arterioscler Thromb Vasc Biol. 2005; 25: 1383–7. [DOI] [PubMed] [Google Scholar]
- 32. Sorescu D, Somers MJ, Lassegue B, et al. Electron spin resonance characterization of the NAD(P)H oxidase in vascular smooth muscle cells. Free Radic Biol Med. 2001; 30: 603–12. [DOI] [PubMed] [Google Scholar]
- 33. Daiber A, August M, Baldus S, et al. Measurement of NAD(P)H oxidase‐derived superoxide with the luminol analogue L‐012. Free Radic Biol Med. 2004; 36: 101–11. [DOI] [PubMed] [Google Scholar]
- 34. Yu J, Weïwer M, Linhardt RJ, et al. The role of the methoxyphenol apocynin, a vascular NADPH oxidase inhibitor, as a chemopreventative agent in the potential treatment of cardiovascular diseases. Curr Vasc Pharmacol. 2008; 6: 204–17. [DOI] [PubMed] [Google Scholar]
- 35. Sasaki H, Sato H, Kuriyama‐Matsumura K, et al. Electrophile response element‐mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem. 2002; 277: 44765–71. [DOI] [PubMed] [Google Scholar]
- 36. Sturrock A, Cahill B, Norman K, et al. Transforming growth factor‐β 1 induces Nox4 NAD(P)H oxidase and reactive oxygen species‐dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol. 2006; 290: L661–73. [DOI] [PubMed] [Google Scholar]
- 37. Schwartz SM, Virmani R, Rosenfeld ME. The good smooth muscle cells in atherosclerosis. Curr Atheroscler Rep. 2000; 2: 422–9. [DOI] [PubMed] [Google Scholar]
- 38. Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006; 8: 76–87. [DOI] [PubMed] [Google Scholar]
- 39. Jeong WS, Jun M, Kong AN. Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxid Redox Signal. 2006; 8: 99–106. [DOI] [PubMed] [Google Scholar]
- 40. Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen‐activated protein kinase pathways and Smad signaling downstream of TGF‐β: implications for carcinogenesis. Oncogene. 2005; 24: 5742–50. [DOI] [PubMed] [Google Scholar]
- 41. Deaton RA, Su C, Valencia TG, et al. Transforming growth factor‐β1‐induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J Biol Chem. 2005; 280: 31172–81. [DOI] [PubMed] [Google Scholar]
- 42. Rhyu DY, Yang Y, Ha H, et al. Role of reactive oxygen species in TGF‐β1‐induced mitogen‐activated protein kinase activation and epithelial‐mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005; 16: 667–75. [DOI] [PubMed] [Google Scholar]
- 43. Undevia NS, Dorscheid DR, Marroquin BA, et al. Smad and p38‐MAPK signaling mediates apoptotic effects of transforming growth factor‐β1 in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004; 287: L515–24. [DOI] [PubMed] [Google Scholar]
- 44. Cheng PY, Lee YM, Shih NL, et al. Heme oxygenase‐1 contributes to the cytoprotection of alpha‐lipoic acid via activation of p44/42 mitogen‐activated protein kinase in vascular smooth muscle cells. Free Radic Biol Med. 2006; 40: 1313–22. [DOI] [PubMed] [Google Scholar]
- 45. Choi BM, Kim YM, Jeong YR, et al. Induction of heme oxygenase‐1 is involved in anti‐proliferative effects of paclitaxel on rat vascular smooth muscle cells. Biochem Biophys Res Commun. 2004; 321: 132–7. [DOI] [PubMed] [Google Scholar]
- 46. Hanna IR, Hilenski LL, Dikalova A, et al. Functional association of nox1 with p22phox in vascular smooth muscle cells. Free Radic Biol Med. 2004; 37: 1542–9. [DOI] [PubMed] [Google Scholar]
- 47. Sato M, Kawai‐Kowase K, Sato H, et al. c‐Src and hydrogen peroxide mediate transforming growth factor‐β1‐induced smooth muscle cell‐gene expression in 10T1/2 cells. Arterioscler Thromb Vasc Biol. 2005; 25: 341–7. [DOI] [PubMed] [Google Scholar]
- 48. Herkert O, Diebold I, Brandes RP, et al. NADPH oxidase mediates tissue factor‐dependent surface procoagulant activity by thrombin in human vascular smooth muscle cells. Circulation. 2002; 105: 2030–6. [DOI] [PubMed] [Google Scholar]
- 48. Gorlach A, Brandes RP, Bassus S, et al. Oxidative stress and expression of p22phox are involved in the up‐regulation of tissue factor in vascular smooth muscle cells in response to activated platelets. FASEB J. 2000; 14: 1518–28. [PubMed] [Google Scholar]
- 50. Lynn S, Gurr JR, Lai HT, et al. NADH oxidase activation is involved in arsenite‐induced oxidative DNA damage in human vascular smooth muscle cells. Circ Res. 2000; 86: 514–19. [DOI] [PubMed] [Google Scholar]
- 51. Sato H, Shiiya A, Kimata M, et al. Redox imbalance in cystine/glutamate transporter‐deficient mice. J Biol Chem. 2005; 280: 37423–9. [DOI] [PubMed] [Google Scholar]
- 52. Ruiz E, Siow RC, Bartlett SR, et al. Vitamin C inhibits diethylmaleate‐induced L‐cystine transport in human vascular smooth muscle cells. Free Radic Biol Med. 2003; 34: 103–10. [DOI] [PubMed] [Google Scholar]
- 53. Miura K, Ishii T, SugitaY , et al. Cystine uptake and glutathione level in endothelial cells exposed to oxidative stress. Am J Physiol. 1992; 262: C50–8. [DOI] [PubMed] [Google Scholar]
- 54. Iglesias‐de la Cruz MC, Ruiz‐Torres P, Alcami J, et al. Hydrogen peroxide increases extracellular matrix mRNA through TGF‐β in human mesangial cells. Kidney Int. 2001; 59: 87–95. [DOI] [PubMed] [Google Scholar]
- 55. Hill‐Kapturczak N, Truong L, Thamilselvan V, et al. Smad7‐dependent regulation of heme oxygenase‐1 by transforming growth factor‐β in human renal epithelial cells. J Biol Chem. 2000; 275: 40904–9. [DOI] [PubMed] [Google Scholar]
- 56. Li PF, Dietz R, von Harsdorf R. Differential effect of hydrogen peroxide and superoxide anion on apoptosis and proliferation of vascular smooth muscle cells. Circulation. 1997; 96: 3602–9. [DOI] [PubMed] [Google Scholar]
- 57. Deshpande NN, Sorescu D, Seshiah P et al . Mechanism of hydrogen peroxide‐induced cell cycle arrest in vascular smooth muscle. Antioxid Redox Signal. 2002; 4: 845–4. [DOI] [PubMed] [Google Scholar]
- 58. Hong S, Lee C, Kim SJ. Smad7 sensitizes tumor necrosis factor induced apoptosis through the inhibition of antiapoptotic gene expression by suppressing activation of the nuclear factor‐kappaB pathway. Cancer Res. 2007; 67: 9577–83. [DOI] [PubMed] [Google Scholar]
- 59. Lallemand F, Mazars A, Prunier C, et al. Smad7 inhibits the survival nuclear factor kappaB and potentiates apoptosis in epithelial cells. Oncogene. 2001; 20: 879–84. [DOI] [PubMed] [Google Scholar]
- 60. Liu XM, Peyton KJ, Ensenat D, et al. Nitric oxide stimulates heme oxygenase‐1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc Res. 2007; 75: 381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brunt KR, Fenrich KK, Kiani G, et al. Protection of human vascular smooth muscle cells from H2O2‐induced apoptosis through functional codependence between HO‐1 and AKT. Arterioscler Thromb Vasc Biol. 2006; 26: 2027–34. [DOI] [PubMed] [Google Scholar]
- 62. Atfi A, Baron R. p53 brings a new twist to the Smad signaling network. Sci Signal . 2008; 1: pe33. [DOI] [PubMed] [Google Scholar]
- 63. Rossi ML, Marziliano N, Merlini PA, et al. Different quantitative apoptotic traits in coronary atherosclerotic plaques from patients with stable angina pectoris and acute coronary syndromes. Circulation. 2004; 110: 1767–73. [DOI] [PubMed] [Google Scholar]
- 64. Li PF, Dietz R, von Harsdorf R. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c‐independent apoptosis blocked by Bcl‐2. EMBO J. 1999; 18: 6027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bakin AV, Stourman NV, SekharKR , et al. Smad3‐ATF3 signaling mediates TGF‐β suppression of genes encoding Phase II detoxifying proteins. Free Radic Biol Med. 2005; 38: 375–87. [DOI] [PubMed] [Google Scholar]
- 66. Lee JM, Li J, Johnson DA, et al. Nrf2, a multi‐organ protector FASEB J. 2005; 19: 1061–6. [DOI] [PubMed] [Google Scholar]