

Abstract
The Z variant of α1‐antitrypsin (AT) polymerizes within the liver and gives rise to liver cirrhosis and the associated plasma deficiency leads to emphysema. In this work, a combinatorial approach based on the inhibitory mechanism of α1‐AT was developed to arrest its pathogenic polymerization. One peptide, Ac‐TTAI‐NH2, emerged as the most tight‐binding ligand for Z α1‐AT. Characterization of this tetrapeptide by gel electrophoresis and biosensor analysis revealed its markedly improved binding specificity and affinity compared with all previously reported peptide inhibitors. In addition, the peptide is not cytotoxic to lung cell lines. A model of the peptide‐protein complex suggests that the peptide interacts with nearby residues by hydrogen bonds, hydrophobic interactions, and cavity‐filling stabilization. The combinatorially selected peptide not only effectively blocks the polymerization but also promotes dissociation of the oligomerized α1‐AT. These results are a significant step towards the potential treatment of Z α1‐AT related diseases.
Keywords: antitrypsin, polymerization, cirrhosis, emphysema, peptide, combinatorial chemistry, surface plasmon resonance, urea gel
Introduction
The α1‐antitrypsin (AT) is the most abundant circulating protease inhibitor in plasma (1–2 mg/ml) and a prototypical member of the serpin (serine protease inhibitor) superfamily [1, 2]. It is primarily synthesized by the hepatocyte and enters the lung by passive diffusion to protect the alveolar matrix from proteolysis, particularly by neutrophil elastase [1, 2, 3, 4, 5, 6]. The secondary structure of active AT is composed of three β‐sheets (A, B and C), nine α‐helices (A–I), and a reactive centre loop (RCL) which tightly entraps neutrophil elastase and other target proteases [6, 7, 8]. The majority of people carry the normal M form of AT (M‐AT), but the Z variant of AT (E342K; Z‐AT) is present in approximately 4% of the Caucasians of Northern European descent [9]. This variant renders the conformation of the protein more susceptible to loop‐sheet polymerization; i.e. the RCL of one AT is inserted into the A‐sheet of another AT as s4A and polymerizes to form pathogenic inclusion bodies [10, 11, 12]. The accumulation of Z‐AT polymers within the hepatocyte causes liver cirrhosis and the resultant plasma deficiency can give rise to early‐onset emphysema [13, 14, 15, 16, 17]. Polymerization of other serpin variants such as those of antithrombin III, α1‐antichymotrypsin, C1‐inhibitor and neuroserpin have also been described and related to thrombosis, emphysema, angioedema and dementia, respectively [18, 19].
Given that protein aggregation is the origin for the pathogenesis of the liver and lung diseases associated with Z‐AT, the crux of the matter is to inhibit the oligomerization process and thus prevent the intracellular accumulation of Z‐AT polymers. Previous studies have shown that 11‐ to 13‐mer synthetic peptides with homology to the RCL could anneal to the A‐sheet of AT [18, 20, 21, 22, 23, 24]. However, the molecular size and lack of specificity preclude these peptides as potential therapeutic options. Recently, a benchmark hexapeptide (Ac‐FLEAIG) as a RCL segment of AT was reported to preferentially block the polymerization of Z‐AT, yet this peptide remains too large to be clinically relevant [25]. A more recent attempt to identify AT binding ligands was performed by screening some 40 small RCL peptides and analogues primarily derived from antithrombin and AT [26]. A tetrapeptide (WMDF‐NH2) was found to block the polymerization of papaya proteinase IV treated M‐AT, but it impractically required 100‐fold molar excess of peptide and a long 24 hrs of incubation. Another tetrapeptide (VIKF‐NH2) was reported to require concentration at 200‐fold molar excess and a long incubation of more than 3 weeks to dissociate AT polymers [27]. Therefore, an efficient and comprehensive screening protocol of greatly expanded molecular diversity is imperative to identify novel and potent peptide inhibitors of Z‐AT polymerization.
Combinatorial chemistry is a powerful technique to harness molecular diversity for lead discovery and offers an unparalleled opportunity for accelerating the development of new drugs and materials [28, 29]. The expanded breadth and scope of molecular structures in combinatorial libraries have brought the screening power for the identification of potent enzyme inhibitors and protein ligands [30]. In this work, combinatorial chemistry was employed for the first time to tackle the biological problem of Z‐AT polymerization. Prior to the construction of libraries, we had set out to perform a systematic study on the benchmark Ac‐FLEAIG with a specific aim to define the minimal peptide length necessary for binding to AT [31]. An acetylated tetrapeptide amide Ac‐FLAA‐NH2 was identified to be the shortest and most potent inhibitor of Z‐AT polymerization. This finding together with the analysis of various human serpin RCL sequences were used to construct a series of β‐strand‐directed libraries. Through the combinatorial screening approach (iterative deconvolution), the optimal peptide sequences for binding to M‐ and Z‐AT were sequentially determined by cycles of re‐synthesis and screening. The newly identified Z‐AT ligand (Ac‐TTAI‐NH2) was not cytotoxic to lung cell lines and has significantly improved binding affinity and target specificity. It therefore can serve as an excellent template to develop mimetic drugs for treating Z‐AT‐related disorders. Moreover, the combinatorial approach presented in this work should be applicable for the discovery of new inhibitors of other pathogenic serpins.
Materials and methods
Isolation and purification of M‐ and Z‐AT
M‐ and Z‐AT proteins were purified from human plasma of corresponding M‐AT and Z‐AT homozygotes as described previously [31]. The purity and authenticity of isolated proteins were verified by SDS‐PAGE, native‐PAGE and Western blotting. M‐ and Z‐AT proteins were stored in Tris buffer (50 mM Tris, 50 mM KCl, pH 7.4) and snap frozen in liquid nitrogen and stored at –80°C until use.
Chemical synthesis of combinatorial peptide libraries
The soluble tetrapeptide libraries were constructed by solid‐phase peptide synthesis with standard Fmoc chemistry using peptide amide linker (PAL) resin (Advanced ChemTech, Inc., Louisville KY, USA) as solid support on a homemade peptide synthesis apparatus. The first generation of tetrapeptide libraries with diversity in the last three residues represented by formulas of Ac‐AX2X3X4‐NH2, Ac‐FX2X3X4‐NH2, Ac‐HX2X3X4‐NH2, Ac‐IX2X3X4‐NH2, Ac‐LX2X3X4‐NH2, Ac‐MX2X3X4‐NH2, Ac‐TX2X3X4‐NH2, Ac‐VX2X3X4‐NH2, Ac‐WX2X3X4‐NH2 and Ac‐YX2X3X4‐NH2 were constructed. Each randomized position (Xi) was a composition of 10 amino acids (A, F, H, I, L, M, T, V, W and Y). Therefore, the anticipated diversity of these directed libraries was 1 × 104 different sequences (1 × 103 in each sub‐library). The randomized positions were generated using a split‐and‐mix method to construct the peptide libraries [32]. In brief, after deprotection of the N‐Fmoc group on a solid support with 20% (v/v) piperidine in dimethylformamide (DMF), the resin was evenly divided into 10 portions and placed into separate reaction vessels. The selected amino acids were coupled to the resin in each reaction vessel. All coupling reactions were performed in DMF at ambient temperature for 2 hrs using a 4‐fold excess of Fmoc‐ protected amino acids relative to resin loading. Each coupling step was monitored by a ninhydrin test to ensure that the amino acids had completely incorporated into the forming peptides. After completion of coupling, the resin from all vessels was thoroughly washed, mixed, deprotected and redistributed into the reaction vessels. These processes were repeated to prepare the 10 sub‐libraries. For the acetylation on N terminus, the resin was soaked in acetic anhydride/diisopropylethylamine/DMF for 2 hrs. Final release of the peptides from the resin and side chain deprotection was concomitantly achieved with 95% (v/v) trifluoroacetic acid in water for 2 hrs. All peptides were lyophilized three times prior to use.
Iterative deconvolution of peptide libraries
Both M‐ and Z‐AT (0.5 mg/ml) were incubated with a 10‐fold molar excess of each synthetic N α‐acetyl tetrapeptide in sub‐library in Tris buffer (50 mM Tris, 50 mM KCl, pH 7.4) at 37°C. Assessment of peptide annealing to AT was achieved by an established protocol of 8 M urea PAGE from one of us (R. M.) and performed on 8% (w/v) gels [25]. Gel patterns were digitized with a scanner. The amounts of binary complex (BC) were measured by Gel‐Pro analyzer computer software (Media Cybernetic, Silver Spring, MD, USA), and densitometric values were normalized to obtain the percentage of BC formation in each sub‐library. The most potent sub‐library was judged by the percentage of the BC formation. The sub‐libraries were deconvoluted by cycles of re‐synthesis and densitometric analysis to sequentially determine the optimal peptide sequences. Finally, the identified peptides were manually re‐synthesized and their purity was verified by a Hitachi high‐pressure liquid chromatography system (L7420/L7100, Hitachi High‐Technologies, Tokyo, Japan) using a Mightysil C18 column (25 cm × 4.6 mm, 5 μm; Kanto Chemical Co., Ltd., Tokyo, Japan). Products were assessed at 220 nm using a 30‐min. linear gradient of 0–100% acetonitrile [0.1% trifluoroacetic acid (TFA)] in H2O (0.1% TFA) at a flow rate of 1 ml/min. Molecular weight was determined on a LCT Premier™ XE oa‐TOF (orthogonal acceleration time‐of‐flight) mass spectrometer (Waters Corp., Manchester, UK). High‐resolution mass spectrometry (HRMS) calcd for the Ac‐TTAI‐NH2 445.25, found 468.24 [M+Na]+; Ac‐TTAF‐NH2 479.24, found 502.23 [M+Na]+; Ac‐FLAA‐NH2 461.26, found 484.25 [M+Na]+.
Comparison of peptide binding to Z‐AT
Four AT binding peptides Ac‐TTAI‐NH2, Ac‐FLAA‐NH2, WMDF‐NH2 and Ac‐FLEAIG were mixed with Z‐AT (0.5 mg/ml) in 50 mM Tris, 50 mM NaCl, pH 7.4 at 37°C for a short incubation time of 2 hrs. Peptides were incubated in 10, 50 or 100‐fold molar excess of Z‐AT. The formation of a BC between AT and peptide was assessed on 8 M urea PAGE.
The inhibition of Z‐AT polymerization by Ac‐TTAI‐NH2
To assess the inhibition of AT polymerization, Z‐AT (0.5 mg/ml) was incubated with the tetrapeptide Ac‐TTAI‐NH2 (in 20‐fold molar excess) and the hexapeptide Ac‐FLEAIG (in 100‐fold molar excess) in 50 mM Tris, 50 mM NaCl, pH 7.4, at 37°C. After a 4‐hr incubation time, samples were treated with a heat pulse of 30 min. at 62°C before assay by 8% (w/v) native PAGE.
Assessment of the reversal of Z‐AT oligomers by Ac‐TTAI‐NH2
To assess the reversal of Z‐AT oligomers, Z‐AT (0.5 mg/ml) was first heated to 62°C for 15 min. to produce oligomers, and then incubated with the peptide Ac‐TTAI‐NH2 (in 100‐fold molar excess) in 50 mM Tris, 50 mM NaCl, pH 7.4, at 37°C for up to 72 hrs. The reversal of Z‐AT oligomers was assessed on 8% (w/v) native PAGE.
Surface plasmon resonance (SPR) experiments
The SPR experiments were recorded with BIAcore 3000 optical biosensor (Biacore, Inc., Uppsala, Sweden). Z‐AT (50 μg/ml) was suspended in 10 mM acetate (pH 4.5) and immobilized to a CM5 sensor chip by a standard amine coupling procedure as recommended by the manufacturer. The identified peptide inhibitor Ac‐TTAI‐NH2, and a negative control peptide Ac‐WWWH‐NH2 were chosen as analytes for the SPR analysis. All the binding experiments were carried out at 37°C using phosphate buffer (20 mM Na2HPO4, 150 mM NaCl, pH 7.4), and different concentrations of peptides were injected over the Z‐AT chip to generate the sensorgrams. The sensorgram was recorded as a plot of binding response (resonance unit) versus time. All experiments were performed at a flow rate of 2 μl/min. and multiple washes with 10 mM glycine buffer (pH 3.0) were performed between the injection cycles.
Trypsin inhibitory activity
The measurement of antitryptic activity was a modification of a reported protocol [33]. The inhibitory activity was measured as residual trypsin activity using a chromogenic substrate N‐α‐benzoyl‐L‐arginine‐4‐nitroanilide. Trypsin and its substrate were purchased from Sigma (St. Louis, MO, USA). Z‐AT (5 μg), partially oligomerized Z‐AT (5 μg; produced by incubation of 37°C for 20 hrs) and BC (produced by incubation of 5 μg Z‐AT with 100‐fold molar excess of the peptide Ac‐TTAI‐NH2 at 37°C for 20 hrs) were pre‐incubated with 1 μg trypsin at room temperature for 15 min., and then the substrate was added for additional 10 min. The absorbance at 405 nm was determined on a UV‐visible spectrometer (JASCO V‐570, Nihon Bunko Co. Ltd, Tokyo, Japan). Results were calculated as the percentage of residual trypsin activity. All experiments were carried out in 5% dimethyl sulfoxide (DMSO) in Tris buffer (100 mM, pH 7.36).
Cell culture
Human BEAS‐2B normal lung epithelial cells (Bioresource Collection and Research Center, Hsinchu, Taiwan) were grown in Ham’s F12 Medium (Gibco‐Invitrogen, Carlsbad, CA, USA) supplemented with 5 μg/ml insulin (Gibco‐Invitrogen), 10 μg/ml transferrin (Gibco‐Invitrogen), 0.5 μg/ml hydrocortisone (Sigma‐Aldrich, Inc., St. Louis, MO, USA), 2.5 ng/ml recombinant human epidermal growth factor (PeproTech, Rocky Hill, NJ, USA), and 12 μg/ml endothelial cell growth supplement (Millipore, Billerica, MA, USA). Human NL20 normal lung epithelial cells were cultured in Ham’s F12 medium supplemented with 2.7 g/l glucose (Gibco‐Invitrogen), 2 mM L‐glutamine (Gibco‐Invitrogen), 0.1 mM non‐essential amino acids (Biological Industries, Kibbutz Beit Haemek, Israel), 5 μg/ml insulin, 1 μg/ml transferrin, 0.5 μg/ml hydrocortisone, 10 ng/ml recombinant human epidermal growth factor, and 4% (v/v) fetal bovine serum (FBS) (Biological Industries). Human normal lung fibroblast WI‐38 and IMR‐90 cell lines were grown in Eagle’s minimum essential medium (Gibco‐Invitrogen) supplemented with 2 mM L‐glutamine, and 10% (v/v) FBS. Human A2058 melanoma cells were cultivated in Dulbecco’s modified Eagle’s medium (Gibco‐Invitrogen) supplemented with 4.5 mM L‐glutamine, 4.5 g/l glucose, 1 mM sodium pyruvate (Sigma‐Aldrich) and 10% (v/v) FBS. All cells were maintained at 37°C in a humidified atmosphere containing 5% (v/v) CO2 in air.
MTT cell proliferation assay
In order to evaluate the cytotoxicity of the synthetic Ac‐TTAI‐NH2 peptide, we performed MTT [3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide] cell proliferation assays using various human cell lines in particular normal lung epithelial and fibroblast cells. Briefly, an optimal number of cells (6 × 103 for all normal cell lines tested and 4 × 103 cells for highly proliferated A2058 cancer cell line) containing 100 μl of appropriate culture medium with 1% (v/v) FBS was seeded in each well of 96‐well microplates (Corning, Lowell, MA, USA) and allowed to attach overnight. The synthetic Ac‐TTAI‐NH2 peptide dissolved in sterile filtered 5% (v/v) DMSO (Sigma‐Aldrich) was then added into each well to a final concentration of 10 μM. On the second and fourth day of the 4‐day incubation, 10 μl of 0.5 mg/ml MTT (Sigma‐Aldrich) in phosphate buffer saline was added into each well and cells were incubated at 37°C for another 4 hrs. After the reaction, the yellow MTT was reduced to purple formazan in the mitochondria of living cells. A 100 μl DMSO was then added to dissolve the insoluble purple formazan product into a coloured solution. Finally, the absorbance of this coloured solution was quantified by measuring at an absorbance of 550 nm by a UV‐visible spectrophotometer.
Results and discussion
Design and synthesis of combinatorial peptide library
A key consideration in the construction of combinatorial library is to achieve the optimal level of molecular diversity. As many design criteria can be accommodated in a library, prior in‐depth characterization of lead compounds should be advantageous to reduce redundancy and therefore improve the screening efficiency. Recently, we reported a systematic study on the benchmark Ac‐FLEAIG as a lead compound to investigate structural requirements necessary for efficient peptide annealing to AT, and most importantly, with a specific goal to define the minimal peptide length required for binding [31]. An acetylated tetrapeptide amide (Ac‐FLAA‐NH2) was identified to bind much more tightly to AT than its N‐ or/and C‐terminally unprotected analogues, a result implying the contribution of additional interactions to stronger affinity. Altogether, our result clearly suggests that N‐acetylated tetrapeptide amide can serve as the optimum library scaffold.
While constructing combinatorial peptide libraries, all 20 proteinogenic amino acids should in principle be incorporated as building blocks to reach the maximal molecular diversity. However, a potential problem in our library design is that high molar excess of peptides are necessary to recognize AT. As library screening was to perform in solution, tetrapeptide libraries containing all 20 amino acids may risk impractical or futile screening due to poor solubility of libraries to achieve the required screening concentration. We had estimated that, to meet the needed 100:1 molar ratio, 4.04 g of tetrapeptides (average molecular weight 528 Da) per millilitre for each library were required. To provide a suitable number of peptides in these soluble libraries, a selection criterion of building blocks was therefore necessary not only to control the library size, but also to guide the synthesis of a tailor‐made library.
The AT inhibitory mechanism provides an excellent base to select building blocks for library construction. The RCL of AT plays a pivotal role and accounts for effective inhibition of neutrophil elastase [33, 34]. The irreversible suicide substrate mechanism is mediated by the insertion of the RCL as a central strand (s4A) and renders the original five‐strand β‐sheet A into a fully antiparallel six‐strand β‐sheet to become more energetically stable. Therefore, we reasoned that residues within the RCL of inhibitory serpins are expected to be β‐sheet preferred amino acids. To confirm the hypothesis, we performed an analysis of the RCL sequences of human serpins with Chou–Fasman helix and sheet propensities in the prediction of secondary structure (Table 1) [35]. Our simple analysis showed that 60.1% (69.0% if restricted to P12‐P3’) of the RCL residues of inhibitory serpins are composed of the top 10 β‐sheet forming amino acids (only 38.3% for those of non‐inhibitory serpins). Consequently, the 10 amino acids (A, F, H, I, L, M, T, V, W and Y) found in higher frequency in β‐sheet formation were selected to incorporate as the building blocks for library construction. The method of split‐and‐mix synthesis was employed to construct a series of β‐strand‐directed libraries in this study [32]. The major consideration of using this established protocol was to ensure the equimolar representation of all the individual peptides in the library for subsequent screening.
Table 1.
Reactive centre loop sequences for human serpins*
| Serpin | P15 | P14 | P13 | P12 | P11 | P10 | P9 | P8 | P7 | P6 | P5 | P4 | P3 | P2 | P1 | P1’ | P2’ | P3’ | P4’ | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inhibitory | A1 | G | T | E | A | A | G | A | M | F | L | E | A | I | P | M | S | I | P | P |
| A2 | G | T | E | A | T | G | A | P | H | L | E | E | K | A | W | S | K | Y | Q | |
| A3 | G | T | E | A | S | A | A | T | A | V | K | I | T | L | L | S | A | L | V | |
| A4 | G | T | E | A | A | A | A | T | T | F | A | I | K | F | F | S | A | Q | T | |
| A5 | G | T | R | A | A | A | A | T | G | T | I | F | T | F | R | S | A | R | L | |
| A9 | G | T | E | A | T | A | A | T | T | T | K | F | I | V | R | S | K | D | G | |
| A10 | G | T | E | A | V | A | G | I | L | S | E | I | T | A | Y | S | M | P | P | |
| B1 | G | T | E | A | A | A | A | T | A | G | I | A | T | F | C | M | L | M | P | |
| B2 | G | T | E | A | A | A | G | T | G | G | V | M | T | G | R | T | G | H | G | |
| B3 | G | A | E | A | A | A | A | T | A | V | V | G | F | G | S | S | P | A | S | |
| B4 | G | V | E | A | A | A | A | T | A | V | V | V | V | E | L | S | S | P | S | |
| B6 | G | T | E | A | A | A | A | T | A | A | I | M | M | M | R | C | A | R | F | |
| B7 | G | T | E | A | T | A | A | T | G | S | N | I | V | E | K | Q | L | P | Q | |
| B8 | G | T | E | A | A | A | A | T | A | V | V | R | N | S | R | C | S | R | M | |
| B9 | G | T | E | A | A | A | A | S | S | C | F | V | V | A | E | C | C | M | E | |
| B10 | G | T | E | A | A | A | G | S | G | S | E | I | D | I | R | I | R | V | P | |
| B11 | G | T | E | A | A | A | A | T | G | D | S | I | A | V | K | S | L | P | M | |
| B12 | G | T | Q | A | A | A | A | T | G | A | V | V | S | E | R | S | L | R | S | |
| B13 | G | T | E | A | A | A | A | T | G | I | G | F | T | V | T | S | A | P | G | |
| C1 | G | S | E | A | A | A | S | T | A | V | V | I | A | G | R | S | L | N | P | |
| D1 | G | T | Q | A | T | T | V | T | T | V | G | F | M | P | L | S | T | Q | V | |
| E1 | G | T | V | A | S | S | S | T | A | V | I | V | S | A | R | M | A | P | E | |
| E2 | G | T | K | A | S | A | A | T | T | A | I | L | I | A | R | S | S | P | P | |
| F2 | G | V | E | A | A | A | A | T | S | I | A | M | S | R | M | S | L | S | ||
| G1 | G | V | E | A | A | A | A | S | A | I | S | V | A | R | T | L | L | V | ||
| I1 | G | S | E | A | A | A | V | S | G | M | I | A | I | S | R | M | A | V | L | |
| I2 | G | S | E | A | A | T | S | T | G | I | H | I | P | V | I | M | S | L | A | |
| Non‐inhibitory | A6 | G | V | D | T | A | G | S | T | G | V | T | L | N | L | T | S | K | P | I |
| A7 | G | T | E | A | A | A | V | P | E | V | E | L | S | D | Q | P | E | N | T | |
| A8 | G | R | E | P | T | E | S | T | Q | Q | L | N | K | P | E | V | L | E | V | |
| B5 | G | G | D | S | I | E | V | P | G | A | R | I | L | Q | H | K | D | E | L | |
| F1 | G | A | G | T | T | P | S | P | G | L | Q | P | A | H | L | T | F | P | L | |
| H1 | G | N | P | F | D | Q | D | I | Y | G | R | E | E | L | R | S | P | K | L | |
| H2 | G | N | P | F | D | Q | D | I | Y | G | R | E | E | L | R | S | P | K | L |
*Based on Chou–Fasman helix and sheet propensities, the RCL residues were dichotomized to give the top 10 β‐sheet prone amino acids (A, F, H, I, L, M, T, V, W and Y) and shaded in the table. A gap has been introduced at P6 for F2 and G1 to maintain the alignment of residues.
Combinatorial library screening
Library screening was achieved by the method of iterative deconvolution [36, 37]. The deconvolution is a strategy in which the efficacy of each predefined sub‐library defines the synthesis of the next generation of libraries for screening. The stepwise cycles of re‐synthesis and screening were repeated until the optimal peptide sequences for binding to M‐ and Z‐AT were identified. A series of β‐strand‐directed libraries in conjunction with analysis on 8 M urea PAGE were used to identify the preference of amino acids at each randomized position. This established gel‐based assay is conformation sensitive and requires no labelling chromophores or encoding tags [25]. For results from the first‐round library screening, a striking preference of Thr (Ac‐TX2X3X4‐NH2) at the X1 position was identified for both M‐ (Fig. 1A, panel I) and Z‐AT (Fig. 1B, panel I). All other residues in X1 were distant second without ambiguity. This clear consensus of Thr is intriguing since the RCL sequences of human inhibitory serpins exhibit a conserved Thr residue at both the P14 (74.1%) and P8 (77.8%) sites. Moreover, Thr at the P8 site has been reported as a ‘peptide anchor’ in a recent study by Carrell and coworkers [26]. The concordance of these findings suggests that Thr is a key element for effective peptide annealing to AT.
Figure 1.

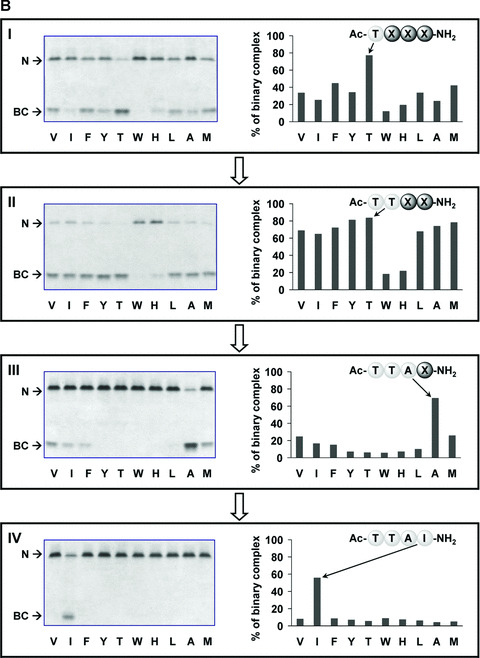
Iterative deconvolution of β‐strand‐directed tetrapeptide library binding to M and Z‐AT. The potency of each sub‐library was determined by the ability to form binary complex (BC) with native M‐ (A) and Z‐AT (B) as screened by 8 M urea gels (left side), and further quantitatively measured to give the percentage of BC formation by densitometric analysis (right side). The unbound M‐ and Z‐AT were designated as N in the figures. Urea was an additive and served as a chaotropic reagent to distinguish the native AT and its peptide‐inserted BC. More anodal migration of the BC most likely was due to the acquisition of additional stability of a fully antiparallel β‐sheet A from the incorporated peptide, leading to greater resistance to unfolding and hence faster migration in electrophoresis. The letters below each graph were standard amino acid symbols. The most reactive sub‐library in each screening cycle was indicated on the right side (X represented the randomized position), which defined the synthesis of next round sub‐libraries for subsequent screening. Panels I, II, III and IV showed the screening results of rounds 1, 2, 3 and 4, respectively. All screening were performed with a calculated 10‐fold molar excess of each individual peptide in each sub‐library compared with AT, apart from panel V which was performed with a 100‐fold molar excess of peptide compared with AT. Incubations were performed at 37°C for 2 hrs, apart from panels IV and V which were 1‐hr incubations.
The second‐round library screening revealed that all 10 amino acids except Trp and His were candidates for the X2 position (Fig. 1A and B, panel II). Thr was further identified as the best X2 residue (Ac‐TX2X3X4‐NH2) by a quantitative densitometric analysis. As a result, Thr was subsequently incorporated to refine the third generation libraries. In the third round of library screening, Ala emerged clearly as the best residue at the X3 position (Ac‐TTAX4‐NH2) (Fig. 1A and B, panel III). The first three rounds of library screening produced the identical Ac‐TTAX4‐NH2 for both M‐ and Z‐AT under the same experimental condition of 10:1 molar ratio of each peptide to AT. The final round of screening for M‐AT was initially ineffective (Fig. 1A, panel IV), hence the molar ratio of library was increased to 100:1 and observable BCs were identified (Fig. 1A, panel V). This indicated that the effectiveness of the preceding sub‐library (Ac‐TTAX4‐NH2) was a summation of nearly all peptides within. Further quantitative densitometric analysis revealed that Ac‐TTAF‐NH2 was the most potent peptide for binding to M‐AT, yet other peptides with aliphatic or non‐polar residues at the X4 position (Ac‐TTAA‐NH2, Ac‐TTAI‐NH2, Ac‐TTAL‐NH2, Ac‐TTAM‐NH2 and Ac‐TTAV‐NH2) exhibited comparable potency. On the other hand, Ac‐TTAI‐NH2 was identified exclusively as the best Z‐AT binder even under the stringent conditions of 10‐fold molar excess of peptide at 37°C for 1 hr (Fig. 1B, panel IV).
At the early stage of our combinatorial library screening, Thr at the X1 position was preferentially identified when each peptide was used in 100‐fold molar excess. However, under identical condition, the X2 residue could not be determined unambiguously, as 8 out of 10 sub‐libraries were found to be nearly equally potent (for result, see Fig. S1). A more stringent 10‐fold molar excess of peptides was thus chosen to aim at identifying the best X2 residue, yet the same trend was observed under this rigorous condition. This result reflected the strategy of using β‐strand prone amino acids rendering these peptides exhibited stronger affinity and therefore prompted us to decide to use the more strict 10‐fold molar excess of individual peptides as the final screening condition throughout this work.
Binding comparison of the previously reported peptides and the identified Ac‐TTAI‐NH2 to Z‐AT
To demonstrate the potency of the identified Z‐AT ligand, we set out to evaluate and compare Ac‐TTAI‐NH2 with the previously reported peptides Ac‐FLEAIG, WMDF‐NH2 and Ac‐FLAA‐NH2 in a short 2‐hr incubation time (Fig. 2) [25, 26, 31]. As these three peptides were known to require a considerably long incubation time (>1 day) and high molar ratios (>50‐fold) to effectively bind to Z‐AT, no significant binding to Z‐AT was observed under such rigorous conditions (Fig. 2A [lane 5] and B [lanes 2 and 3]). The result showed that, with only a 2‐hr incubation time and a 10‐fold molar excess, Ac‐TTAI‐NH2 was sufficient to form BC with Z‐AT (Fig. 2A [lane 6] and B [lane 4]). In addition, no observable BC was formed by incubating this peptide with M‐AT under the same experimental condition, which clearly demonstrated its preferential specificity to Z‐AT (Fig 2A, lane 3). Together these results indicate that the identified Ac‐TTAI‐NH2 tetrapeptide has a much higher binding affinity and specificity than all previously reported peptides found by other approaches.
Figure 2.

The comparison of the binding of the reported peptides Ac‐FLEAIG, WMDF‐NH2, Ac‐FLAA‐NH2 and the combinatorially selected Ac‐TTAI‐NH2 to AT by 8 M urea gels. (A) M‐ (lanes 1–3) and Z‐AT (lanes 4–6) were incubated with the peptides Ac‐FLEAIG (lanes 2 and 5; AT/peptide = 1:100) and Ac‐TTAI‐NH2 (lanes 3 and 6; AT/peptide = 1:10) at 37°C for 2 hrs. Each lane contained 2.5 μg of protein. The stronger affinity of the peptide Ac‐TTAI‐NH2 with Z‐AT was clearly distinguished by its lower molar ratio and short incubation required to form BC, whereas as predicted, no BC was formed with Ac‐FLEAIG. In addition, the specificity of the peptide Ac‐TTAI‐NH2 was also demonstrated as no BC was formed with M‐AT. (B) Z‐AT (lane 1) was incubated with the peptides WMDF‐NH2 (lane 2; AT/peptide = 1:100), Ac‐FLAA‐NH2 (lane 3; AT/peptide = 1:50) and Ac‐TTAI‐NH2 (lane 4; AT/peptide = 1:10) at 37°C for 2 hrs. As anticipated, no BC was formed with WMDF‐NH2 and Ac‐FLAA‐NH2 under this strict condition. The potency of the peptide Ac‐TTAI‐NH2 was demonstrated by the fact that a low molar ratio and short incubation time were required to form BC. Each lane contained 2.5 μg of protein.
Assessment of the inhibition of AT polymerization and the reversal of AT oligomers
One major goal of this work was to identify new inhibitors that not only prevent the pathogenic polymerization of Z‐AT, but also facilitate the dissociation of existing oligomers. Therefore, for the assessment of Ac‐TTAI‐NH2, the heat‐induced Z‐AT polymers were produced following a previously reported protocol [31]. As shown in Fig. 3, Ac‐TTAI‐NH2 was able to effectively inhibit the formation of Z‐AT polymers under these stringent conditions. The increased stability of Z‐AT protein was likely due to the formation of the fully antiparallel six‐strand β‐sheet upon the insertion of Ac‐TTAI‐NH2. In addition, the ability of the peptide Ac‐TTAI‐NH2 to dissociate Z‐AT polymers was also evaluated. As shown in Fig. 4, observable BC was formed only after a short 6‐hr of incubation time of the peptide Ac‐TTAI‐NH2 with Z‐AT polymers at 37°C. Not only was the amount of oligomers reduced gradually, BC was also increased considerably over time (up to 72 hrs). Despite the fact that the robust polymers remained tightly aggregated, Ac‐TTAI‐NH2 nevertheless facilitated the dissociation of oligomers to monomeric Z‐AT likely through BC formation, which represents a significant step from a therapeutic perspective. Previous study has shown that the polymerized Z‐AT is stable for several weeks in the absence of synthetic peptides [10, 25]. The property of Ac‐TTAI‐NH2 to prevent polymers formation and promote oligomers reversal is promising for the potential treatment of Z‐AT related diseases.
Figure 3.

The 4‐mer peptide Ac‐TTAI‐NH2 effectively blocked the polymerization of Z‐AT. Z‐AT was incubated with the previous identified peptide Ac‐FLEAIG (lane 3; 100‐fold molar excess) and the combinatorially selected peptide Ac‐TTAI‐NH2 (lane 4; 20‐fold molar excess) at 37°C for 4 hrs, and then heated to 62°C for 30 min. to intentionally accelerate the polymerization process before assay. Lane 2 showed the effect of heat pulse on Z‐AT without peptide. While Ac‐FLEAIG was able to inhibit polymerization under physiological conditions [25], it was unable to prevent polymerization under such harsh conditions. In contrast, polymerization of Z‐AT was completely inhibited by the peptide Ac‐TTAI‐NH2 at a low molar ratio. Each lane contained 2.5 μg of protein.
Figure 4.
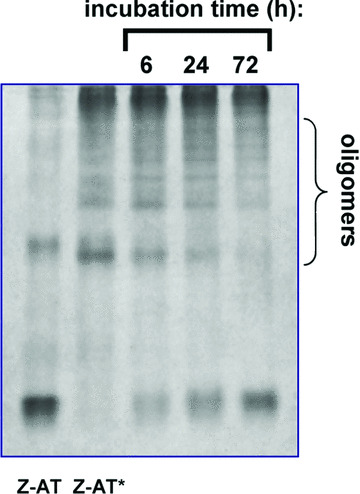
Reversal of Z‐AT oligomers by the identified peptide Ac‐TTAI‐NH2. Oligomers of Z‐AT were prepared by heating Z‐AT (0.5 mg/ml) at 58°C for 1 hr and labelled as Z‐AT* in the figure. The oligomerized Z‐AT was then incubated with the peptide Ac‐TTAI‐NH2 (100‐fold molar excess) for 6, 24 and 72 hrs at 37°C. After 6 hrs of incubation, BC was observable and increased with time, and as indicated there was the gradual resolution of oligomers of Z‐AT. Note that the native protein and BC bands appeared at almost the same position on a native PAGE. Each lane contained 3.5 μg of protein.
Sequence alignment of Ac‐TTAI‐NH2 with human inhibitory serpin RCLs
Upon sequence alignment with the RCL regions of human inhibitory serpins (Table 1), the Ac‐TTAI‐NH2 peptide was surprisingly found to be identical to that of the P8‐P5 segment of protease nexin 1 (PN‐1). We thus anticipated that the juxtaposed strands 3 and 5 of PN‐1 and AT might have consensus sequences interacting with the peptide Ac‐TTAI‐NH2. Using the ClustalW program [38], sequence alignment revealed that the residues of PN‐1 and AT in strand 3 (VLVN in PN‐1 versus ALVN in AT), strand 5 (HILQ versus KAVH) and F‐helix connecting loop (LLSP versus LVKE) were phylogenically conserved if not identical. Based on the conservation of the deduced binding sites of PN‐1 and AT, the identified AT ligand Ac‐TTAI‐NH2 might also be a PN‐1 ligand. On the other hand, the peptide Ac‐MFLE‐NH2 derived from the corresponding P8‐P5 sequence of AT might also be considered as another possible ligand of AT. Unpredictably however, neither the synthetic peptide Ac‐MFLE‐NH2 was recognized by AT (both M and Z), nor Ac‐TTAI‐NH2 was a PN‐1 ligand even with a 100‐fold molar excess of peptides and lengthy incubation time (2 days for AT and 7 days for PN‐1). This unpredictability of serpin ligands may reflect the natural selection of RCL sequences in protein phylogenies, and also underscores the usefulness of our combinatorial approach for the discovery of new serpin ligands.
Confirmation of specific binding of Ac‐TTAI‐NH2 to Z‐AT by SPR
To further validate the library screening result, we set out to investigate the binding of the combinatorially selected Ac‐TTAI‐NH2 to Z‐AT by SPR. The chip‐based SPR technology has proved to be valuable for biomolecular interaction analyses in real time without the need for tags or labels [39]. Our previous separate work has also demonstrated that SPR is a powerful tool for the study of binding interactions of proteins with small molecules [40, 41]. In our SPR experiment, the macromolecular Z‐AT protein was intentionally immobilized onto the sensor chip to avoid any hindrance of protein‐peptide interactions that might result from the modification of peptide structure as a consequence of immobilization chemistry. In addition, the protein chip is reusable to the assay of different peptides. The best Z‐AT binder Ac‐TTAI‐NH2 (125 and 250 μM) and a negative control peptide Ac‐WWWH‐NH2 (250 μM) were injected over the immobilized Z‐AT to give the binding sensorgrams. The peptide Ac‐WWWH‐NH2 was selected from the least active amino acid in each library screening cycle. As shown in Fig. 5, even while monitoring 2 hrs of dissociation time, Ac‐TTAI‐NH2 was not washed out completely from the chip, which reflected its strong binding affinity with Z‐AT. In contrast, the peptide Ac‐WWWH‐NH2 was hardly recognized by Z‐AT under the identical condition. This SPR result confirmed the specificity and tight binding of the combinatorially selected peptide Ac‐TTAI‐NH2 to Z‐AT.
Figure 5.
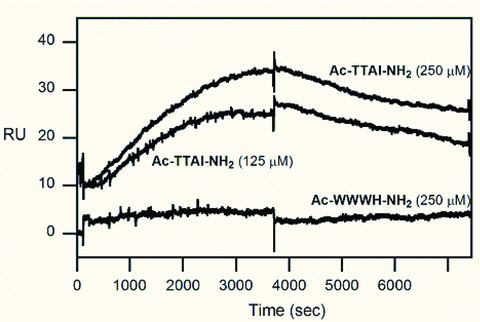
Binding of the combinatorially selected peptide Ac‐TTAI‐NH2 to a Z‐AT chip by SPR. Overlaid plot demonstrated the specific binding of the peptide Ac‐TTAI‐NH2 (125 and 250 μM), and the control peptide Ac‐WWWH‐NH2 (250 μM) was not recognized by the immobilized Z‐AT. The sensorgrams of the peptide Ac‐TTAI‐NH2 were shifted (10 resonance units) for clarity. The resonance unit was converted from the refractive index changes (measured by resonance angle shift in reflected light) upon binding on the sensor chip surface. The peptides and injection concentrations were as indicated in the figure.
Assessment of the antitryptic activity of the binary complex
A small peptide identified by the new approach described herein represents a significant step. The inhibitory activity of the BC was assessed by measuring the residual trypsin activity. The assay revealed that BC of Ac‐TTAI‐NH2 and Z‐AT is inactive against trypsin. As its measured residual trypsin activity was 0%, while the activity of Z‐AT was 39.7%. These data supports the premise that the peptide binds to β‐sheet A thus subverting the inhibitory mechanism. Therefore it is likely that although Ac‐TTAI‐NH2 may be effective at inhibiting polymerization and thus preventing the liver cirrhosis associated with Z‐AT polyemrization, α1‐AT replacement therapy would continue to be required to prevent Z‐AT associated lung disease. An effort targeted at finding small molecules that bind to positions apart from the central strand (s4A) site has been reported recently. However, the complex was extraordinarily stable and reported to be inactive as a protease inhibitor [42]. In terms of chemotactic activity, the effect of BC of Z‐AT and the peptide Ac‐TTAI‐NH2 is anticipated to be abrogated. As a previous study has shown that cleaved AT is not chemotactic while both the BC and the cleaved form of AT contain a peptide or loop insertion into the A‐sheet [9].
Assessment of the cytotoxicity of Ac‐TTAI‐NH2
To determine whether the synthetic Ac‐TTAI‐NH2 peptide has any cytotoxic effect on cells, we performed MTT cell proliferation assays using different type of human cells including two normal lung epithelial cell lines (BEAS‐2B and NL20), two normal lung fibroblast cell lines (WI‐38 and IMR‐90) and one cancer cell line (A2058). As shown in the representative experiment in Fig. 6, even at a concentration as high as 10 μM, the cell viability was not affected by the addition of the Ac‐TTAI‐NH2 peptide, indicating that this small‐size tetramer peptide is not cytotoxic and may therefore possess clinical potential as an effective and safe substance for treating α1‐AT polymerization.
Figure 6.
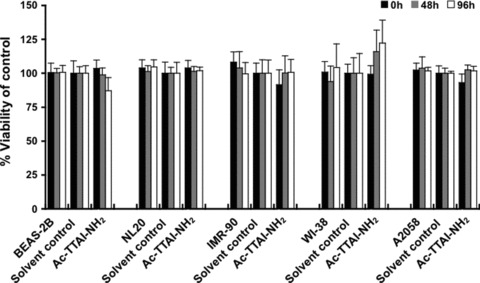
MTT assay of various human cell lines treated with the Ac‐TTAI‐NH2 peptide. Five human cell lines including BEAS‐2B and NL20 normal lung epithelial cell lines, WI‐38 and IMR‐90 normal lung fibroblast cell lines, and A2058 melanoma cell line were treated for 0, 48 or 96 hrs with DMSO solvent control or 10 μM of the synthetic Ac‐TTAI‐NH2 peptide. The percentage of viable cells, relative to the solvent control‐treated cells, was measured by MTT cell proliferation assay. The results showed that no obvious cytotoxicity was detected for the Ac‐TTAI‐NH2 tetramer peptide. All experiments were performed in octuplicate in 96‐well microplates as described in the Materials and Methods section. Data are presented as mean ± S.D.
Proposed model for the combinatorially selected ligand Ac‐TTAI‐NH2 binding to AT
The proposed structure for the identified peptide (Ac‐TTAI‐NH2) interacting with AT was modelled and illustrated in Fig. 7. Besides the canonical β‐sheet hydrogen bonds between the peptide backbone and nearby protein residues on both strands 3 (Ala183, Leu184, Val185 and Asn186) and 5 (Lys331, Ala332, Val333 and His334), additional hydrogen bonds formed by the acetyl group on Ac‐TTAI‐NH2 with the side chain of His334 and backbone NH of Lys335 were also recognized (Fig. 7). Moreover, the interactions of Ac‐TTAI‐NH2 peptide side chains with AT may play a pivotal role in the contribution of tight binding. Our model of the BC structure suggested a unique hydrogen bonding between Ser56 and the N‐terminal Thr (P8) on Ac‐TTAI‐NH2. This Thr at the P8 site has been reported to be a critical junction to link a strong hydrogen bond network with residues Ser53, Ser56 and His334 [26]. The subsequent P7 site showed considerable tolerance of the β‐strand prone amino acids, as 8 out of the 10 residues exhibited similar activity (panel II in Fig. 1A and B). Therefore, the identification of the P7 site as Thr was emerged and determined using densitometric analysis. The following P6 site resided in a small hydrophobic pocket (Leu64, Leu184 and Ala332), and Ala was experimentally identified as the best residue to fill in this site (panel III in Fig. 1A and B). Interestingly, another small hydrophobic amino acid Val was identified as the second active residue, which also correlated with the hydrophobic pocket at the P6.
Figure 7.

Proposed structure of the identified peptide Ac‐TTAI‐NH2 binding to AT. The peptide inserted into the lower part of A‐sheet (labelled in blue) of AT and interacted with its nearby residues. The NH and CO groups from the backbone of the incorporated peptide were hydrogen bonded to the backbones of adjacent strands 3 and 5 (residues in yellow) of AT (square panel on the left) and rendered the A‐sheet into a six‐stranded antiparallel β‐sheet. The circle on the upper right revealed the hydrogen bond (light green dashed line) between the N‐terminal Thr (P8) of peptide Ac‐TTAI‐NH2 and Ser56. Additional hydrogen bonds derived from the acetyl group of peptide with the side chain of His334 (light green dashed line) and the backbone NH group of Lys335 (light green dashed line) were also illustrated. The circle on the lower right showed the hydrophobic side chain of Ile occupies and interacted with a pocket surrounding by residues of Val173, Glu175, Leu176, Ala183 and Lys331. The complex orientations in the two circle panels on the right were changed to better present the peptide‐protein interactions. Carbon, nitrogen and oxygen atoms were shown in white, light blue and red, respectively.
Analysis of the first three identified residues on Ac‐TTAI‐NH2 using the Venn diagram of amino acids revealed that these residues are all small amino acids (Ala, Cys, Gly, Ser and Thr) [43]. Furthermore, by comparing and examining the P8 and P7 sites of human inhibitory serpin RCL sequences (Table 1), it was evident that 88.9% of both sites were Ala, Gly, Ser or Thr (four of the five defined small amino acids). The percentage of the small amino acids on the P6 site was also calculated but decreased to 44.0%. However, 72.0% were found to be hydrophobic residues and this explained that small hydrophobic Ala and Val were experimentally identified at the P6 site. Therefore, we reasoned that smaller side chains at P8‐P6 would be advantageous to efficiently incorporate into the A‐sheet by evading the connecting loop of the F‐helix, which was reported to undergo a reversible conformational change during the inhibitory mechanism [44]. Finally, the last round of screening of individual peptides showed that non‐polar amino acids (Ala, Ile, Leu, Met, Phe, and Val) at the C‐terminal of peptides were more potent. Among them, Phe was densitometrically determined as the P5 residue for M‐AT (panel V in Fig. 1A) and, most strikingly, Z‐AT accommodated Ile as the only residue at the P5 site (panel IV in Fig. 1B). This finding correlates with a study on cavity‐filling mutations of AT, which demonstrated that substitution of bulky and hydrophobic residues such as Ile and Phe at Val173 or Ala183 stabilized the metastable nature of protein structure [45]. As shown in Fig. 7, the bulky and hydrophobic Ile on Ac‐TTAI‐NH2 resided well in the cavity surrounding by Val173, Leu176, Ala183, Glu175 and Lys331. The specificity of different ligands identified for M‐ and Z‐AT is likely to originate from the slight fluctuation of binding interfaces of proteins. These results indicate that the formation and stabilization of the protein/peptide complex are facilitated by hydrogen bonds, hydrophobic interactions and cavity‐filling from the side chains of the combinatorially selected peptidyl ligands.
Conclusion
Abnormal protein aggregation is responsible for numerous diseases such as Alzheimer’s disease, Parkinson’s disease and α1‐AT deficiency. The Z variant of α1‐AT aggregates in the liver leading to plasma deficiency, and the consequence is a predisposition to cirrhosis and emphysema. Reviewing a database of 373 control cohorts in 58 countries, it was estimated that 3.4 millions of individuals worldwide carry the deficiency allele combinations [46]. Currently, there is no cure for patients with Z‐AT related diseases. Although augmentation of plasma AT concentration is available, its efficacy for treatment of the lung disease is under detailed evaluation [47, 48]. Furthermore, it would not ameliorate the development of liver disease which underscores the need to develop new treatments. As Z‐AT polymerization underlies the pathogenesis of diseases, blocking its aggregation is a promising strategy to ameliorate Z‐AT related diseases. In this work, combinatorial chemistry was employed for the first time to expand the molecular diversity with the aim to discover potent Z‐AT polymerization inhibitors. The most potent Z‐AT ligand to date, Ac‐TTAI‐NH2 was identified in a library of 10,000 peptides. It has been revealed that the peptide Ac‐TTAI‐NH2 was able to bind to Z‐AT at a considerably reduced molar ratio (10:1) and with significantly shorter incubation times (1–2 hrs) compared with previously identified peptides. The ability to block polymerization and the potential to dissociate oligomers of Z‐AT were also clearly demonstrated. In addition, it is not cytotoxic to lung cell lines. In terms of drug discovery, this low molecular weight ligand should be beneficial for designing peptidomimetics or small organic molecules. Finally, the screening platform developed here is applicable to other polymerizing pathogenic serpins with the ultimate goal of identifying novel drugs for currently incurable conditions.
Supporting information
Fig. S1 Iterative deconvolution of the first two rounds of libraries binding to M‐AT. The library screening was performed with the 8 M urea PAGE and potency of sub‐libraries was judged by the formation of binary complex (BC). (A) and (B) show the first and second round of screening, respectively. The initial screening condition was performed with a calculated 100‐fold molar excess of each individual peptide of AT. Incubations were performed at 37°C for 2 hrs, and then given with a heat‐pulse of 20 min. at 62°C before gel electrophoresis. The letters below each graph are standard amino acid symbols.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgements
This work was supported by the National Science Council of Taiwan, R.O.C. (to Y.‐H.C.), and the Department of Health and National Health Research Institutes of Taiwan, R.O.C. (DOH95‐TD‐G‐111–014, PA‐094‐PP‐10, and CA‐095‐PP‐36 to W.‐S.W.C.), Wellcome Trust (to R.M.). We thank Ms. Hui‐Chin Wen (Institute of Cancer Research, National Health Research Institutes, Taiwan, R.O.C.) for helpful discussions.
References
- 1. Brantly M, Nukiwa T, Crystal RG. Molecular basis of alpha‐1‐antitrypsin deficiency. Am J Med. 1988; 84: 13–31. [DOI] [PubMed] [Google Scholar]
- 2. Mahadeva R, Lomas DA. Alpha1‐antitrypsin deficiency, cirrhosis and emphysema. Thorax. 1998; 53: 501–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Elliott PR, Lomas DA, Carrell RW, et al . Inhibitory conformation of the reactive loop of α1‐antitrypsin. Nat Struct Biol. 1996; 3: 676–81. [DOI] [PubMed] [Google Scholar]
- 4. Loebermann H, Tokuoka R, Deisenhofer J, et al . Human α1‐proteinase inhibitor: crystal structure analysis of two crystal modifications, molecular model, and preliminary analysis of the implications for function. J Mol Biol. 1984; 177: 531–57. [PubMed] [Google Scholar]
- 5. Stratikos E, Gettins PWG. Major proteinase movement upon stable serpin‐pro teinase complex formation. Proc Natl Acad Sci USA. 1997; 94: 453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stratikos E, Gettins PWG. Formation of the covalent serpin‐proteinase complex involves translocation of the proteinase by more than 70 Å and full insertion of the reactive center loop into β‐Sheet A. Proc Natl Acad Sci USA. 1999; 96: 4808–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huntington JA, Read RJ, Carrell RW. Structure of a serpin‐protease complex shows inhibition by deformation. Nature. 2000; 407: 923–6. [DOI] [PubMed] [Google Scholar]
- 8. Lomas DA, Carrell RW. α1‐Antitrypsin deficiency – A model for conformational diseases. N Eng J Med. 2002; 346: 45–53. [DOI] [PubMed] [Google Scholar]
- 9. Parfrey H, Dafforn TR, Belorgey D, et al . New therapeutic strategies for Z α1‐antitrypsin‐related emphysema. Am J Respir Cell Mol Biol. 2004; 31: 133–9. [DOI] [PubMed] [Google Scholar]
- 10. Lomas DA, Evans DL, Finch JT, et al . The mechanism of Z alpha1‐antitrypsin accumulation in the liver. Nature. 1992; 357: 605–7. [DOI] [PubMed] [Google Scholar]
- 11. Teckman JH, An JK, Blomenkamp K, et al . Mitochondrial autophagy and injury in the liver in α1‐antitrypsin deficiency. Am J Physiol. 2004; 286: G851–62. [DOI] [PubMed] [Google Scholar]
- 12. Lomas DA, Evans DL, Stone SR, et al . Effect of the Z mutation on the physical and inhibitory properties of α1‐antitrypsin. Biochemistry. 1993; 32: 500–8. [DOI] [PubMed] [Google Scholar]
- 13. Lieberman J, Mittman C, Gordon HW. Alpha1 antitrypsin in the livers of patients with emphysema. Science. 1972; 175: 63–5. [DOI] [PubMed] [Google Scholar]
- 14. Janciauskiene S, Dominaitiene R, Sternby NH, et al . Detection of circulating and endothelial cell polymers of Z and wild type α1‐antitrypsin by a monoclonal antibody. J Biol Chem. 2002; 277: 26540–6. [DOI] [PubMed] [Google Scholar]
- 15. Mulgrew AT, Taggart CC, Lawless MW, et al . α1‐Antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractant. Chest. 2004; 125: 1952–7. [DOI] [PubMed] [Google Scholar]
- 16. Mahadeva R, Atkinson C, Li Z, et al . Polymers of Z α1‐antitrypsin co‐localize with neutrophils in emphysematous alveoli and are chemotactic in vivo . Am J Pathol. 2005; 166: 377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stoller JK, Aboussouan LS. Alpha1‐antitrypsin deficiency. Lancet. 2005; 365: 2225–36. [DOI] [PubMed] [Google Scholar]
- 18. Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. 2002; 3: 759–68. [DOI] [PubMed] [Google Scholar]
- 19. Sivasothy P, Dafforn TR, Gettins PGW, et al . Pathogenic α1‐antitrypsin polymers are formed by reactive loop beta‐sheet A linkage. J Biol Chem. 2000; 275: 33663–8. [DOI] [PubMed] [Google Scholar]
- 20. Schulze AJ, Baumann U, Knof S, et al . Structural transition of alpha1‐antitrypsin by a peptide sequentially similar to beta‐strand s4A. Eur J Biochem. 1990; 194: 51–6. [DOI] [PubMed] [Google Scholar]
- 21. Björk I, Nordling K, Larsson I, et al . Kinetic characterization of the substrate reaction between a complex of antithrombin with a synthetic reactive‐bond loop tetradecapeptide and four target proteinases of the inhibitor. J Biol Chem. 1992; 267: 19047–50. [PubMed] [Google Scholar]
- 22. Chang WSW, Wardell MR, Lomas DA, et al . Probing serpin reactive loop conformations by proteolytic cleavage. Biochem J. 1996; 314: 647–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fitton HL, Pike RN, Carrell RW, et al . Mechanisms of antithrombin polymerisation and heparin activation probed by insertion of synthetic reactive loop peptides. Biol Chem. 1997; 378: 1059–63. [DOI] [PubMed] [Google Scholar]
- 24. Skinner R, Chang WSW, Jin L, et al . Implications for function and therapy of a 2.9 Å structure of binary‐complexed antithrombin. J Mol Biol. 1998; 283: 9–14. [DOI] [PubMed] [Google Scholar]
- 25. Mahadeva R, Dafforn T, Carrell RW, et al . 6‐Mer peptide selectively anneals to a pathogenic serpin conformation and blocks polymerisation: implications for the prevention of Z α1‐antitrypsin related cirrhosis. J Biol Chem. 2002; 277: 6771–4. [DOI] [PubMed] [Google Scholar]
- 26. Zhou A, Stein PE, Huntington JA, et al . How small peptides block and reverse serpin polymerization. J Mol Biol. 2004; 342: 931–41. [DOI] [PubMed] [Google Scholar]
- 27. Chowdhury P, Wang W, Lavender S, et al . Fluorescence correlation spectroscopic study of serpin depolymerization by computationally designed peptides. J Mol Biol. 2007; 369: 462–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Horton DA, Bourne GT, Smythe ML. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem Rev. 2003; 103: 893–930. [DOI] [PubMed] [Google Scholar]
- 29. McFarland EW, Weinberg WH. Combinatorial approaches to materials design. TIBTECH. 1999; 17: 107–15. [DOI] [PubMed] [Google Scholar]
- 30. Kim W, Kim Y, Min J, et al . A high‐throughput screen for compounds that inhibit aggregation of the Alzheimer’s peptide. ACS Chem Biol. 2006; 1: 461–9. [DOI] [PubMed] [Google Scholar]
- 31. Chang YP, Mahadeva R, Chang WSW, et al . Identification of a 4‐mer peptide inhibitor that effectively blocks the polymerization of pathogenic Z α1‐antitrypsin. Am J Respir Cell Mol Biol. 2006; 35: 540–8. [DOI] [PubMed] [Google Scholar]
- 32. Furka Á. Combinatorial chemistry: 20 years on…. Drug Discov Today. 2002; 7: 1–7. [DOI] [PubMed] [Google Scholar]
- 33. Schulze AJ, Frohnert PW, Engh RA, et al . Evidence for the extent of insertion of the active site loop of intact α1‐proteinase inhibitor in β‐Sheet A. Biochemistry. 1992; 31: 7560–5. [DOI] [PubMed] [Google Scholar]
- 34. Mast AE, Enghild JJ, Salvesen G. Conformation of the reactive site loop of α1‐proteinase inhibitor probed by limited proteolysis. Biochemistry. 1992; 31: 2720–8. [DOI] [PubMed] [Google Scholar]
- 35. Chou PY, Fasman GD. Empirical predictions of protein conformation. Annu Rev Biochem. 1978; 47: 251–76. [DOI] [PubMed] [Google Scholar]
- 36. Dooley CT, Chung NN, Wilkes BC, et al . An all D‐amino acid opioid peptide with central analgesic activity from a combinatorial library. Science. 1994; 266: 2019–22. [DOI] [PubMed] [Google Scholar]
- 37. Yu Z, Tu J, Chu YH. Tight‐binding streptavidin ligands from a cyclic peptide library. Anal Chem. 1997; 69: 4515–18. [DOI] [PubMed] [Google Scholar]
- 38. Chenna R, Sugawara H, Koike T, et al . Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003; 31: 3497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rich RL, Myszka DG. Survey of the year 2005 commercial optical biosensor literature. J Mol Recognit. 2006; 19: 478–534. [DOI] [PubMed] [Google Scholar]
- 40. Chang YP, Chu YH. Using surface plasmon resonance to directly determine binding affinities of combinatorially selected cyclopeptides and their linear analogs to a streptavidin chip. Anal Biochem. 2005; 340: 74–9. [DOI] [PubMed] [Google Scholar]
- 41. Chang YP, Tseng MJ, Chu YH. Using surface plasmon resonance to directly measure slow binding of low‐molecular mass inhibitors to a VanX chip. Anal Biochem. 2006; 359: 63–71. [DOI] [PubMed] [Google Scholar]
- 42. Mallya M, Phillips RL, Saldanha SA, et al . Small molecules block the polymerization of Z α1‐antitrypsin and increase the clearance of intracellular aggregates. J Med Chem. 2007; 50: 5357–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Taylor WR. The classification of amino acid conservation. J Theor Biol. 1986; 119: 205–18. [DOI] [PubMed] [Google Scholar]
- 44. Cabrita LD, Dai W, Bottomley SP. Different conformational changes within the F‐Helix occur during serpin folding, polymerization, and proteinase inhibition. Biochemistry. 2004; 43: 9834–39. [DOI] [PubMed] [Google Scholar]
- 45. Lee C, Maeng JS, Kocher JP, et al . Cavities of α1‐antitrypsin that play structural and functional roles. Protein Sci. 2001; 10: 1446–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. de Serres FJ. Worldwide racial and ethnic distribution of α1‐antitrypsin deficiency. Chest. 2002; 122: 1818–29. [DOI] [PubMed] [Google Scholar]
- 47. Stoller JK, Fallat R, Schluchter MD, et al . Augmentation therapy with alpha1‐antitrypsin: patterns of use and adverse events. Chest. 2003; 123: 1425–34. [DOI] [PubMed] [Google Scholar]
- 48. Gildea TR, Shermock KM, Singer ME, et al . Cost‐effectiveness analysis of augmentation therapy for severe alpha1‐antitrypsin deficiency. Am J Respir Crit Care Med. 2003; 167: 1387–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Iterative deconvolution of the first two rounds of libraries binding to M‐AT. The library screening was performed with the 8 M urea PAGE and potency of sub‐libraries was judged by the formation of binary complex (BC). (A) and (B) show the first and second round of screening, respectively. The initial screening condition was performed with a calculated 100‐fold molar excess of each individual peptide of AT. Incubations were performed at 37°C for 2 hrs, and then given with a heat‐pulse of 20 min. at 62°C before gel electrophoresis. The letters below each graph are standard amino acid symbols.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item