

Abstract
Current bone disease therapy with bone marrow‐derived mesenchymal stromal cells (MSC) is hampered by low efficiency. Advanced allogeneic studies on well‐established mouse genetic and disease models are hindered by difficulties in isolating murine MSC (mMSC). And mMSC prepared from different laboratories exhibit significant heterogeneity. Hence, this study aimed to identify and isolate a sub‐population of mMSC at an early passage number with high osteogenic potential. Enrichment of mMSC was achieved by 1‐hr silica incubation and negative selection. Approximately 96% of these cells synthesized osteocalcin after 28 days of osteogenic induction in vitro, and displayed a complete dynamic alteration of alkaline phosphatase (ALP) activity with increasing osteogenic maturation and strong mineralization. Moreover, the cells displayed uniform and stable surface molecular profile, long‐term survival, fast proliferation in vitro with maintenance of normal karyotype and distinct immunological properties. CD73 was found to be expressed exclusively in osteogenesis but not in adipogenesis. These cells also retained high osteogenic potential upon allogeneic transplantation in an ectopic site by the detection of bone‐specific ALP, osteopontin, osteocalcin and local mineralization as early as 12 days after implantation. Hence, these cells may provide a useful source for improving current strategies in bone regenerative therapy, and for characterizing markers defining the putative MSC population.
Keywords: mesenchymal stromal cells, differentiation, osteogenesis, biology, efficiency, CD73, silica
Introduction
With a predicted doubling of the aged population from 1990 to 2020, bone‐related diseases are predicted to be a major healthcare challenge where effective treatment strategies are lacking [1]. This has spurred the WHO supported global organization, ‘The Bone and Joint Decade’, to encourage research in the prevention, diagnosis and treatment of musculoskeletal disorders [2]. Bone marrow derived mesenchymal stromal cells (MSC), being a readily available cell source with high proliferative capacity and multilineage differentiation potential, have been intensively studied as a potential therapeutic cell source in bone diseases. Although the positive therapeutic efficacy of MSC in patients has been widely reported [2, 3], it must be noted that the efficacy of MSC therapy in both animals and patients is in fact extremely low [4, 5, 6]. Compared to the 1–2% successful engraftment of MSC in vivo[2], the low efficiency of MSC osteogenesis in vitro is seldom reported. The low efficiency of MSC osteogenesis in vitro may reflect the poor osteogenic performance of these cell grafts in vivo. Additionally, MSC of various species showed limited proliferative capacities, displaying senescence after approximately 12 passages (40 population doublings) for human MSC [6, 7], 20 passages for rhesus monkey MSC [8], 4 passages (22 population doubling) for rat MSC [9], and varying lifespan for mouse MSC, ranging from 10 passages [10] to more than 80 population doublings [11, 12, 13]. This increasing senescence of MSC during serial passage will limit the available number of cells upon ex vivo expansion and confine their therapeutic efficacy within a limited time window. Besides increasing senescence, the physiology of MSC changes with serial passage and their multi‐lineage differentiation capacity also diminishes [8, 14]. Additionally, it is recognized that MSC are composed of highly heterogeneous sub‐populations that display variable differentiation capability and proliferation rates [15, 16]. The application of MSC sub‐population with poor osteogenic potential and short lifespan will undoubtedly decrease the therapeutic efficacy. Hence, we suggested that the efficacy of MSC therapy could be enhanced by the purification of a MSC sub‐population with high osteogenic potential and prolonged lifespan within in vitro culture. In this study, we attempted to isolate such an MSC sub‐population from mice rather than human, because of the high inter‐batch variability and inconsistent availability of donated human material. The osteogenic potential of the isolated mouse MSC (mMSC) sub‐population was well characterized both in vitro and in vivo.
Materials and methods
Mice
Female C57BL/6 (C57) and Balb/c (Bc) mice, 6–8 weeks old, were purchased from the Laboratory Animals Center and housed in the satellite Animal Holding Unit of the National University of Singapore. All experimental protocols involving animals were approved by the Institutional Animal Care & Use Committee of the National University of Singapore.
Isolation and enrichment of mMSC
After C57 mice were killed by CO2 asphyxiation, both femurs and tibiae were dissected out. The ends of each femur and tibiae were removed to expose the bone marrow cavity. Bone marrow from the femurs and tibiae of the same mouse were cultured in 5 ml of complete media (CM): mesencult basal medium (Catalogue# 05501, Lot# 07B21265, StemCell Technologies, Inc., Vancouver, BC, Canada) containing mesenchymal stem cell stimulatory supplements (Catalogue# 05502, Lot# 06M20759, StemCell Technologies, Inc.) and 1% penicillin–streptomycin solution (PS, Sigma‐Aldrich, St. Louis, MO, USA) in T25 flasks at 37°C, 5% CO2 and 95% humidity for 3 days. Non‐adherent cells were discarded, and adherent cells attained confluence after 3–5 days. The confluent cells were detached with 0.05% Trypsin/ethylenediaminetetraacetic acid (EDTA) (Invitrogen, San Diego, CA, USA) at 37°C for 2 min., for subsequent serial passage.
Cells without any enrichment (M1, n= 10) were grown up to passage (P) 5 with a split ratio of 1:2, and a seeding density of 5 × 103 cells/cm2 after P5. Cells enriched by silica depletion were grown up to P2 at a split ratio of 1:2 and detached at 100% confluence. Two different methods were used to enrich mMSC at P2. One method involved incubating these cells (M2, n= 7) at 1 × 106 cells/ml within a 15 ml conical tube with 100 μg/ml of silica microspheres (diameter range from 1.00–2.49 μm, Catalogue# SS04N, Bangs Laboratories, Inc., Fishers, IN, USA) in RPMI‐1640 media (Sigma‐Aldrich) without heat‐inactivated foetal bovine serum (FBS, HyClone, Logan, UT, USA) at 37°C for 1 hr. The other method involved incubating these cells (M4, n= 4) at 1 × 106 cells/ml with 100 μg/ml of silica microspheres in RPMI‐1640 media and 5% of FBS at 37°C for 4.5 hrs. Cells and silica microspheres were mixed every 20 min. Subsequently, the cells were resuspended in 4 ml of phosphate‐buffered saline (PBS) and layered onto Ficoll‐Paque PREMIUM (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Interlayer cells were collected from Ficoll after centrifugation at 300 ×g for 30 min. at room temperature and washed with PBS twice. The cells were cultured in CM at 1 × 104 cells/cm2. M1, M2 and M4 were derived from bone marrow explants of different mice.
To remove the small amount of remaining haematopoietic cells after silica depletion, cells were negatively selected at P3 with a Lineage Cell Depletion Kit (130‐090‐858, Miltenyi Biotec, Inc., Bergisch Gladbach, Germany) following the manufacturer’s instructions. The negatively selected cells were then seeded in CM at 1 × 104 cells/cm2. Upon reatimesng confluence, the cells were detached and characterized by flow cytometric analysis. After P5, cells were seeded at 5 × 103 cells/cm2 and attained confluence around 3–4 days. Cell surface molecular profile was characterized every other passage after P2. Cell growth dynamics from P5 onwards were graphed as the logarithm of total cell numbers from P5 to the testing day with 2 as the base (y‐axis) against the accumulated days up to the testing day (x‐axis).
Colony‐forming units assay
M2 cells at P8 (M2P8), M4 cells at P8 (M4P8) and M2 cells at P39 (M2P39) were seeded into 6‐well plates at 10 cells/cm2, 50 cells/cm2 and 100 cells/cm2 in duplicates. Culture media was changed every 3–4 days. On day 10, cells were stained with 0.25% crystal violet (Sigma) for 10 min. After photomicrographs were taken under an inverted fluorescence microscope (Olympus IX70, Tokyo, Japan), the dye was extracted with 1 ml of 10% acetic acid per well. When the colour was uniform, every 200 μl aliquot was transferred into each well of a 96‐well flat bottom plate and the absorbance was read at 600 nm using the TECAN INFINITE M200 microplate reader (Tecan Group Ltd., Männedorf, Switzerland).
Differentiation of mMSC
Osteogenic differentiation
M1 at P9, M2 and M4 at P10, M2 at P32 (abbreviated as M1P9, M2P10, M4P10 and M2P32, respectively) were induced in Dulbecco’s minimum essential medium (DMEM) containing 10% FBS, 10 mM sodium β‐glycerophosphate (Sigma), 50 μg/ml ascorbate‐2‐phosphate (Sigma), 10−8 M dexamethasone (Sigma) and 1% PS for 28 days. During differentiation, MSC were separately cultured in T75 flasks for flow cytometric analysis, and in 6‐well culture plates (2 × 103 cells/cm2) for characterization. Cells were cultured in CM as controls. Medium was collected from the 6‐well culture plates on day 14 and day 28, respectively, for alkaline phosphatase (ALP) quantitative assay [17, 18]. On day 28, cells were stained with alizarin red (pH4.1, Sigma) for 30 sec.
Chondrogenic differentiation
M1P9, M2P10, M4P10 and M2P32 were induced in DMEM containing 1% insulin‐transferrin‐selenium (ITS) Premix (BD Biosciences, San Jose, CA, USA), 10 ng/ml transforming growth factor (TGF‐)β1 (R&D Systems, Minneapolis, MN, USA), 50 μg/ml ascorbate‐2‐phosphate, 10−7 M dexamethasone, 40 μg/ml L‐proline (Sigma), 1% sodium pyruvate (Sigma), 1% nonessential amino acids (Invitrogen), 1% Glutamax (Invitrogen) and 1% PS at 2.5 × 105 cells/tube in 3‐D pellet culture for 28 days. Pellets cultured in media without TGF‐β1 served as the controls. The differentiation was confirmed by toluidine blue staining and sulphated glycosaminoglycan (s‐GAG) quantitation using Blyscan Glycosaminoglycan Assay kit (Biocolor Ltd., Newtownabbey, Northern Ireland, UK), as previously described [17, 18].
Adipogenic differentiation
M1P9, M2P10, M4P10 and M2P32 were induced in DMEM containing 10% FBS, 5 μg/ml Insulin (Sigma), 60 μM Indomethacin (Sigma), 50 μM 3‐isobutyl‐1‐methylxanthine (Sigma), 10−7 M dexamethasone and 1% PS at 2 × 105 cells/well within 6‐well plates for 7–10 days. Cells were stained with 0.36% of oil‐red‐O (Sigma) for 1 hr. After being imaged, the dye was extracted by 100% isopropanol at room temperature for 10 min. Every 200 μl of dye extraction was transferred into one well of a 96‐well flat bottom plate and the absorbance was read at 500 nm using the microplate reader.
Flow cytometry analysis
For flow cytometry analysis, 2 × 105 cells were suspended in 10 μl of PBS for binding with each specific antibody. Briefly, cells were incubated with rat anti‐mouse CD16/CD32 (mouse FcR blocking reagent, Pharmingen, BD Biosciences San Diego, CA) antibody at 4°C for 5 min., followed by the addition of fluorescence conjugated monoclonal antibodies of Sca‐1 (553335, Pharmingen), CD11b (130‐081‐201, Miltenyi Biotec, Inc.), CD29 (555005, Pharmingen), CD34 (11‐0341, eBioscience, San Diego, CA, USA), CD44 (11‐0441, eBioscience), CD45 (130‐091‐609, Miltenyi Biotec, Inc.), CD73 (550741, Pharmingen), CD90.2 (553003, Phamingen), CD105 (12‐1051, eBioscience), CD117 (130‐091‐730, Miltenyi Biotec, Inc.), MHC‐Ib (11‐5999, eBioscience) and MHC‐II (11‐5322, eBioscience), respectively, at 4°C for 30 min. Cells were then washed with PBS once and fixed in 1% paraformaldehyde for analysis by using the CyAn™ ADP Analyzer (Beckman Coulter, Fullerton, CA, USA) and Summit v4.3.
In the case of cells stained for intracellular osteocalcin (OC), the cells were fixed in 4% paraformaldehyde at room temperature for 5 min. and washed with PBS once, prior to incubation in perm buffer (PBS containing 1% FBS, 10% rabbit serum, 5 mM EDTA [6381‐92‐6, Duchefa Biochemie, Netherlands] and 0.1% Saponin [Sigma]) at room temperature for 30 min., and incubation with anti‐mouse CD16/CD32 antibody at 4°C for 5 min. and Goat anti‐mouse OC antibody (BT‐592, 1:250, Biomedical Technologies, Inc., Stoughton, MA, USA) [19] at 4°C for 45 min. Cells were then washed with perm buffer once and incubated with Cy5 Rabbit anti‐goat IgG (81‐1616, 1:500, Zymed Laboratories, Invitrogen) at 4°C for 15 min. Cells were subsequently washed with PBS and fixed in 1% paraformaldehyde for analysis.
Karyotyping of mMSC chromosomes
Metaphase spreads preparation
Subconfluent M2 at P25, P35, P45 and P55 were incubated in Colcemid (0.25 μg/ml, Invitrogen) at 37°C for 6 hrs and detached. The cell pellet was suspended in hypotonic KCl (0.075 M) at 37°C for 15 min. and was fixed in chilled Carnoy’s fixative (glacial acetic acid: absolute methanol = 1: 3). Metaphase spreads were prepared by dripping the fixed cell suspension from 20 cm above the pre‐cleaned slides (Fisher Scientific, Inc., Pittsburgh, PA, USA) and drying on a 65°C hotplate. The short hypertonic treatment time and low height of dripping cell suspension were meant to prevent the formation of broken metaphases. The slides were aged at room temperature for 10 days before G‐banding.
Giemsa banding
Slides with murine metaphase spreads were immersed for 30 sec. in a coplin jar containing 2.5% trypsin/0.9% NaCl mixture, followed by washing with 0.9% NaCl solution twice. The slides were then stained with Gurr’s Giemsa working solution for 5 min., followed by washing in Gurr’s 6.8 buffer (Invitrogen) twice. The slides were air‐dried and cover slipped with Cytoseal 60 (Cole‐parmer, USA Vernon Hills, IL). Metaphase analysis was performed with a Zeiss Axioplan microscope (Carl Zeiss GmbH, Oberkochen, Germany).
Allo‐immunogenicity testing of mMSC
One‐way mixed lymphocyte culture in vitro
C57 and Bc mononuclear cells (MNC) were obtained by Ficoll fractionation at 300 ×g for 20 min. For stimulators, M2P5, M2P14, M2P5DOC (differentiated osteogenic cells) and M2P10DOC as well as C57 MNC were inactivated by 25 μg/ml of mitomycin C in darkness at 37°C for 20 min. prior to being suspended in RPMI‐1640 containing 5% FBS, 50 μM 2‐mercaptoethanol (Sigma) and 1% PS. C57 MNC and Bc MNC were suspended at 1 × 106 cells/ml, while M2P5, M2P14, M2P5DOC and M2P10DOC were suspended at 105 cells/ml. Ten thousand M2P5/M2P14/ M2P5DOC/ M2P10DOC were co‐cultured with 105 of Bc MNC or were added into the co‐culture of 105 of Bc MNC and 105 of C57 MNC in triplicates within a 96‐well U Bottom Plate in 0.2 ml/well of medium for 6 days. Sixteen hours before harvesting, tritium‐labelled thymidine (0.5 μCi/well, Bio‐Rad Laboratories, USA) was added into the co‐culture. Counts per minute (CPM) of tritium was recorded with a liquid scintillation counter (PerkinElmer, Waltham, MA, USA).
Allogeneic transplantation and osteogenic differentiation in vivo
After anaesthesia, Bc mice were injected subcutaneously with 5 × 106 of M1P14 and M2P14 in 200 μl of PBS in both sides of the abdominal wall. The successful injection showed protrusion of skin at the injection sites were labelled with gentian violet. At 12 days after implantation, the whole skin around the injection area were cut, fixed and embedded in OCT compound media (Jung, Leica Microsystems, Wetzlar, Germany). Specimens were sectioned at 6 μm in thickness. The slides were permeabilized and blocked in perm buffer (the same as that for flow cytometric analysis) at 37°C for 30 min. The slides were stained with primary antibodies (MHC‐Ib[1:12.5], and antibodies to osteogenic markers, such as anti‐mouse ALP antibody [B4‐78, bone and liver specific, DSHB, Iowa City, IA, USA; diluted to 5 μg/ml], antimouse osteopontin [OPN] antibody [MPIIIB10, DSHB, diluted to 5 μg/ml], Goat anti‐mouse OC antibody [diluted at 1:50]) in perm buffer at room temperature for 1 hr and subsequently with secondary antibody (Qdot655 Goat anti‐mouse IgG [diluted at 1:200] for ALP and OPN detection, Cy5 Rabbit anti‐goat IgG [diluted at 1:100] for OC detection) at room temperature for 1 hr. Slides were mounted in VECTASHIELD Mounting Medium (H‐1200, Vector Laboratories, Burlingame, CA, USA) and observed under fluorescence microscope. Besides fluorescence staining, haematoxylin and eosin staining was performed to demonstrate tissue morphology as well as alizarin red staining to investigate the existence of mineral deposition.
Statistical analysis
Quantitative data were analysed by SPSS 11.5 for windows (SPSS, Inc., Chicago, IL, USA) using ANOVA. A value of P < 0.05 was considered significantly different.
Results
mMSC enrichment and expansion
With normal passaging, CD11b+, CD45+, CD117+ and lineage+ cells would be limited to 5% or less of the total cell population after eight passages (data not shown). When duration of trypsinization was shortened to 2 min., the detached cells would be free of haematopoietic cell contamination with 99% purity at P6 (Fig. 1). With silica incubation, monocytes and macrophages that engulf silica became heavy and consequently sediment to the bottom of the tube after centrifugation in Ficoll. After 4.5 hrs incubation with silica, the contamination of CD11b+ cells decreased from 81.86 ± 10.96% (mean ± S.E.) at P2 to 13.96 ± 3.03% upon reaching confluence at P3. However, when bone marrow cells were incubated with silica for 1 hr, the contamination by CD11b+ cells decreased to only 46.11 ± 10.54% upon reaching confluence at P3. Subsequently, these cells were then subjected to negative selection at P3. Upon reaching confluence at P5, the contamination by haematopoietic cells was reduced to within 1% (Fig. 1).
Figure 1.
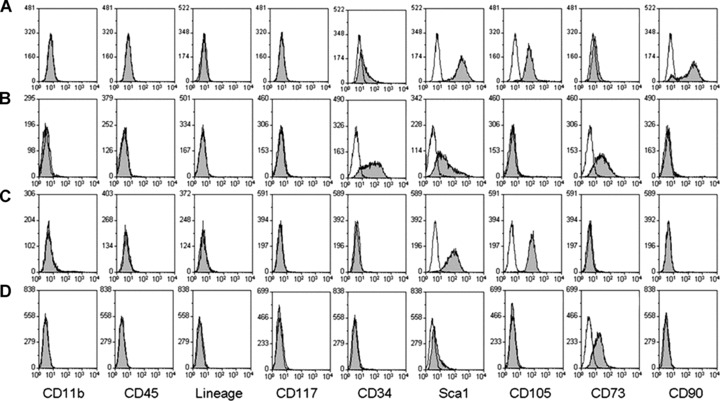
Surface molecule profile of M1P6, M2P5, M4P5 and M2P36. (A) The surface molecule profile of M1 at P6 which were not treated with silica. (B) The surface molecule profile of M2 at P5 which were incubated with silica for 1 hr and were negatively selected. (C) The surface molecule profile of M4 at P5 which were incubated with silica for 4.5 hrs and were negatively selected. (D) The surface molecule profile of M2 at P36. Open peaks were unstained cells as negative control, solid peaks were cells stained with corresponding antibodies after FcR blocking.
As in previous studies, mMSC displayed a much slower rate of proliferation compared to MSC isolated from other species. With our protocol, it took approximately 5–7 days to attain confluence in the first two passages, at a split ratio of 1:2. Higher passage splitting ratio of mMSC caused a delay or stoppage of growth. After silica treatment, the time duration to attain confluence would range from 5 to 16 days. However at a seeding density of 5 × 103 cells/cm2, the proliferation rate would speed up from the fifth passage onwards, with the time duration required to attain confluence being shortened to 3–4 days.
Phenotypic analysis
All cells showed strong expression of CD29 and CD44 (data not shown) regardless of passage number or treatment with silica. When bone marrow cells were not incubated with silica (M1, Fig. 1A), they did not express CD11b, CD45, CD117 and other haematopoietic lineage markers at P6, while displaying strong expression of Sca‐1, CD105 and CD90, as well as low level expression of CD34 and CD73. After 1 hr incubation with silica followed by Ficoll fractionation, M2 (Fig. 1B) at P5 were negative for expression of CD11b, CD45, CD90, CD105, CD117 and other haematopoietic lineage markers, but displayed positive expression of CD34, Sca‐1 and CD73. At an identical passage number, M4 (Fig. 1C), which were incubated with silica for 4.5 hrs, displayed strong expression of Sca‐1 and CD105, but were negative for other tested markers. During prolonged culture of M2, the expression of CD34, Sca‐1, CD73 and CD105 was analysed at even passage numbers. The results show that the expression of CD34 completely diminished by passage 36 (Fig. 1D) and that of Sca‐1 completely diminished by passage 40 (data not shown). However, M2 did not express CD105 but did express CD73 at the same level until the last passage in culture.
Cell morphology and tri‐lineage differentiation capability
The cultured mMSC displayed heterogeneous morphology, with mixture of predominantly spindle‐shaped fibroblastic cells and small rounded cells, together with a minority of giant flat cells. M2P39 appeared to have the smallest cell size (Fig. 2M), while M1P9, M2P10 and M4P10 were similar (Fig. 2A, E and I). M1P9, M2P10, M4P10 and M2P32 displayed tri‐lineage differentiation potential on day 28 of osteogenesis (Fig. 2C, G, K and O) and chondrogenesis (Fig. 2B, F, J and N), as well as on day 10 of adipogenesis (Fig. 2D, H, L and P) after induction. However, the differentiation potential was different in each lineage. M1 osteogenic nodules were small and disperse, whereas M2P10, M4P10 and M2P32 osteogenic nodules were dense. As observed macroscopically, a higher percentage of M2 entered osteogenesis (Fig. 3A and B) as compared to M4 (Fig. 3C). Under microscopic observation (Fig. 2), more chondrogenic cells of typical morphology with dense cartilaginous matrix were displayed by M1P9, M2P10 and M2P32 compared to M4P10. A much lower percentage of mature fat cells were observed in M1P9, M2P10 and M2P32 than in M4P10. The lipid droplets in M2P32 had a small and immature morphology.
Figure 2.
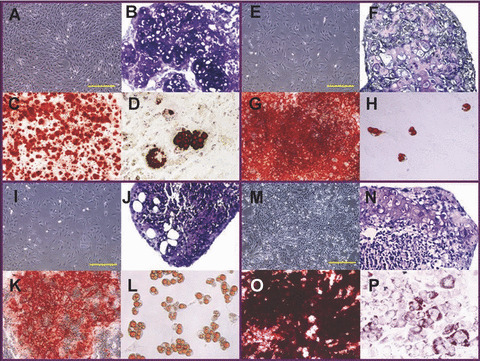
M1P9, M2P10, M4P10 and M2P32 morphology and tri‐lineage differentiation. (A) Morphology of M1P9 (4×). (B) Chondrogenesis of M1P9 for 28 days (20×). (C) Osteogenesis of M1P9 for 28 days (10×). (D) Adipogenesis of M1P9 for 10 days (20×). (E) Morphology of M2P10 (4×). (F) Chondrogenesis of M2P10 for 28 days (20×). (G) Osteogenesis of M2P10 for 28 days (10×). (H) Adipogenesis of M2P10 for 10 days (20×). (I) Morphology of M4P10 (4×). (J) Chondrogenesis of M4P10 for 28 days (20×). (K) Osteogenesis of M4P10 for 28 days (10×). (L) Adipogenesis of M4P10 for 10 days (20×). (M) Morphology of M2P32 (4×). (N) Chondrogenesis of M2P32 for 28 days (20×). (O) Osteogenesis of M2P32 for 28 days (10×). (P) Adipogenesis of M2P32 for 10 days (20×). Yellow scale bars represent 500 μm.
Figure 3.
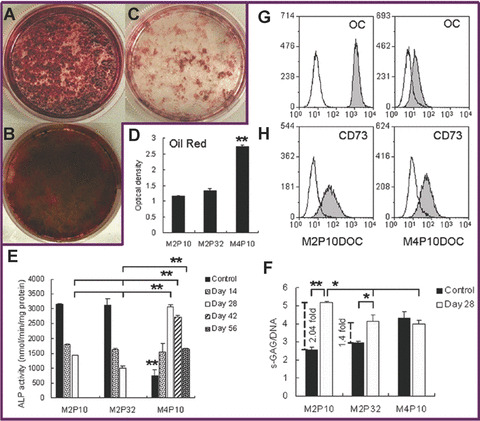
Comparison of tri‐lineage differentiation capability and CD73 expression on M2 and M4. (A) Osteogenesis of M2P10 for 28 days. (B) Osteogenesis of M2P32 for 28 days. (C) Osteogenesis of M4P10 for 28 days. (D) Quantity of oil red extracted from adipogenic progenitors of M2P10, M2P32 and M4P10. (E) ALP activity of osteogenic progenitors of M2P10, M2P32 and M4P10. Dark bars are controls, representing the ALP activity of the same cells cultured in mMSC complete medium. (F) Quantity of s‐GAG synthesis per cell of chondrogenic progenitors of M2P10, M2P32 and M4P10. Dark bars are controls, representing the same cell pellet cultured in chondrogenic differentiation media without TGF‐β1. (G) Synthesis of osteocalcin by day‐28 osteogenic progenitors of M2P10 (left) and M4P10 (right) detected by intracellular fluorescent staining. Open peaks were controls, representing cells stained with the secondary antibody. (H) Expression of CD73 by day‐28 osteogenic progenitors of M2P10 (left) and M4P10 (right). Open peaks were controls, representing unstained cells. All bars in D, E and F represent mean ± S.E. of duplicates of two independent experiments (*, P < 0.05; **, P < 0.01).
To further characterize the difference between M2 and M4, quantitative assays of osteogenesis, chondrogenesis and adipogenesis were performed on M2P10, M2P32 and M4P10. On day 28 of osteogenic differentiation, there was much more intracellular synthesis of OC in M2P10 compared to M4P10 (Fig. 3G). The ALP activity of both M2P10 and M2P32 decreased with further extension of osteogenesis (Fig. 3E). Low ALP activity was displayed by M4P10 at the starting point of differentiation, but the ALP activity gradually increased up to day 28 and then decreased thereafter. On day 28 of differentiation, the ALP activity of M4 was significantly higher than that of both M2P10 and M2P32 (P < 0.01). After an extension of 14 days and 28 days of differentiation, the ALP activity of M4 remained significantly higher than that of M2 (P < 0.01). Interestingly, the expression of CD73 was maintained during the osteogenesis of M2P10, which was also manifested in M4P10 during osteogenic differentiation (Fig. 3H). Upon extraction of oil red from adipogenic progenitors of M2 and M4, M4P10 showed significantly higher absorbance than M2P10 and M2P32 (P < 0.01) (Fig. 3D). In chondrogenic differentiation, M2 demonstrated significantly higher level of s‐GAG/DNA, when compared to the control media (*, P<0.05; **, P<0.01), while M4 did not (Fig. 3F). Chondro‐differentiated M2 also secreted significantly higher s‐GAG synthesis than chondro‐differentiated M4 in a single cell level (P < 0.05). Upon comparison of M2P10 and M2P32, the level of s‐GAG/DNA decreased with longer passage cells (P < 0.05).
Colony‐forming unit assay
Both M2P8 and M2P39 displayed dense and compact colonies with clear boundaries, while M4P8 displayed less compact, more diffuse colonies (Fig. 4). Upon extraction of crystal violet dye, M2P39 showed the highest concentration of dye, while M4P8 showed the lowest. The difference between them was significant (*, P < 0.05; **, P < 0.01).
Figure 4.
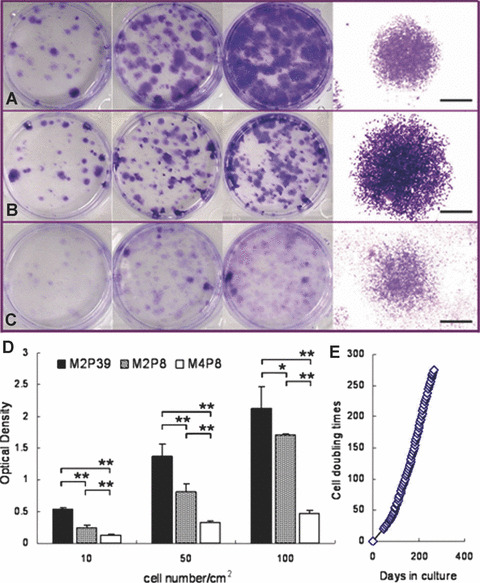
M2 showed rapid proliferation and long‐term survival in vitro. (A) M2P8 colonies at seeding density of 10, 50, 100 cells/cm2 (from left to right) and morphology of a typical colony (1.25×). (B) M2P39 colonies at seeding density of 10, 50, 100 cells/cm2 (from left to right) and morphology of a typical colony (1.25×). (C) M4P8 colonies at seeding density of 10, 50, 100 cells/cm2 (from left to right) and morphology of a typical colony (1.25×). Black scale bars represent 2 mm. (D) Quantity of crystal violet extracted from M2P39, M2P8 and M4P8. All bars represent mean ± S.E. of duplicates of two independent experiments (*, P < 0.05; **, P < 0.01). (E) Cell doubling times against culture days in vitro of M2 after P5.
Long‐term survival and karyotyping
M2 could be cultured for more than 70 passages in vitro (Fig. 4), which approximates to about 275 doublings. Cell doubling was calculated after P5. The maximal number of mMSC in one T75 flask was approximately 6.48 ± 1.71 × 106 cells from P17 to P34. One extra day of culture would not significantly increase the cell number at confluence. After 67 passages, the proliferation rate slowed down. By Giemsa banding, majority of M2 revealed normal diploid karyotype (Fig. 5A) in all tested passage numbers of cells. Less than 5 % of cells were aneuploid in all tested cells (Fig. 5B). No time‐dependent increase in number of aneuploid cells was observed. Additionally, various euploid M2 were detected in different copy numbers, such as 3N, 4N and 5N. No cells with >5N ploidy were seen. Polyploidy cells were less than 8% of total cells at P25 and P35, while they increased up to approximate 20% of total cells at P45 and P55.
Figure 5.
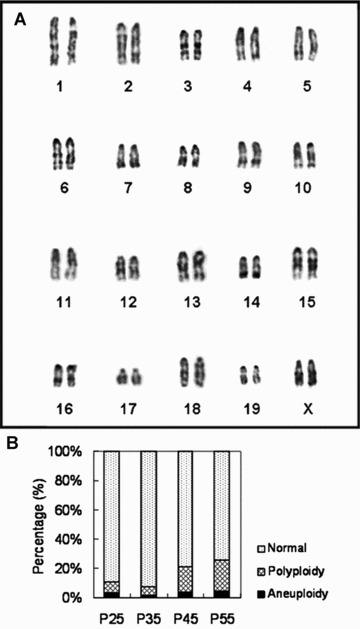
Chromosomal analysis of M2 cells in different passage numbers. (A) A typical karyogram of M2. (B) The percentage of aneuploid cells, polyploid cells and normal cells in passage 25, 35, 45 and 55 of M2.
Immunological properties
M2 had low expression of MHC‐Ib and negative expression of MHC‐II at P5 (Fig. 6). With continuous passage (M2P36) and osteogenic differentiation from M2P5, the expression of MHC‐Ib was hardly detected.
Figure 6.
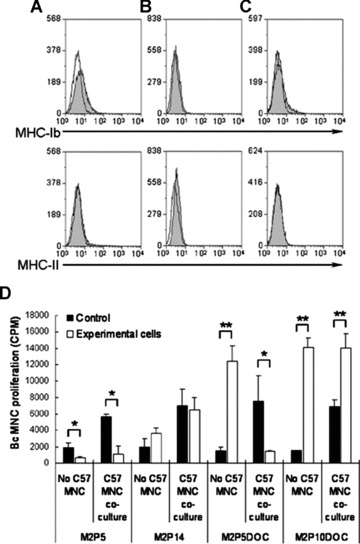
Immune properties of M2 with prolonged culture and differentiation. (A) Expression of MHC‐Ib (Top) and MHC‐II (bottom) of M2P5. (B) Expression of MHC‐Ib (Top) and MHC‐II (bottom) of M2P36. (C) Expression of MHC‐Ib (Top) and MHC‐II (bottom) of M2P5 differentiated osteogenic cells (DOC). Open peaks were controls, representing unstained cells. (D) Two combinations of mixed lymphocyte culture. One was the co‐culture of naïve Balb/c (Bc) mononuclear cells (MNC) with M2P5 or M2P14 or M2P5DOC or M2P10DOC. For this group, x‐axis labels are ‘No C57 MNC’. Dark bars were controls, representing the count per minute of Bc MNC alone without the addition of any other cells in each test. The other combination was the co‐culture of Bc MNC and inactivated C57BL/6 (C57) MNC in the presence of M2P5 or M2P14 or M2P5DOC or M2P10DOC. For this group, x‐axis labels are ‘C57 MNC co‐culture’. The dark bars are controls, representing the count per minute of Bc MNC under the stimulation of inactivated C57 MNC in the same testing. Bars represent mean ± S.E. of triplicates of two independent experiments (*, P < 0.05; **, P < 0.01).
In mixed lymphocyte culture (Fig. 6), M2P5, M2P14, M2P5DOC and M2P10DOC exerted distinct effects on the proliferation of Bc MNC. In the absence of C57 MNC, M2P5 did not induce proliferation of naïve Bc MNC but instead significantly inhibited proliferation (P < 0.05). However, M2P14 had no inhibitory effect on naïve Bc MNC. After differentiation, both M2P5DOC and M2P10DOC showed significant stimulatory effect on Bc MNC proliferation (P < 0.01). In the co‐culture of Bc MNC with inactivated C57 MNC, M2 at P5 significantly inhibited the proliferation of Bc MNC (P < 0.05), but had no significant inhibitory effect at P14. Interestingly, M2P5DOC significantly inhibited the proliferation of Bc MNC (P < 0.05), whereas M2P10DOC significantly enhanced its proliferation (P < 0.01).
Osteogenic differentiation in vivo
Although M1 displayed similar high proliferative capacity and osteogenic potential as M2 in vitro, the skin protuberance, which was caused by M1 subcutaneous injection, disappeared after 12 days after injection. By staining with MHC‐Ib antibody, only a small group of M1 cell grafts were detected at injection sites of abdominal skin specimens (Fig. 7A). In contrast to M1, the skin protuberance caused by M2 injection could be observed after 12 days. After cutting through the centre of the M2 protuberance, a white area could be observed (Fig. 7B). Upon cryosectioning and immunofluoresence staining, low levels of ALP could be detected in M1 graft specimens (Fig. 7E), while low level of ALP as well as moderate levels of OPN and OC could be detected in M2 graft specimens (Fig. 7F, H and J). By haematoxylin and eosin staining, M2 graft area (Fig. 7D) showed much more plentiful matrix and higher cell density than M1 graft area (Fig. 7C). There were red blood cells filling in blood vessel‐like tissue gaps in M2 graft area. And leucocytes infiltration could be observed in both M1 and M2 graft areas. With further alizarin red staining, stained areas indicative of mineral deposition could be observed scattering in M2 graft specimens (Fig. 7L).
Figure 7.
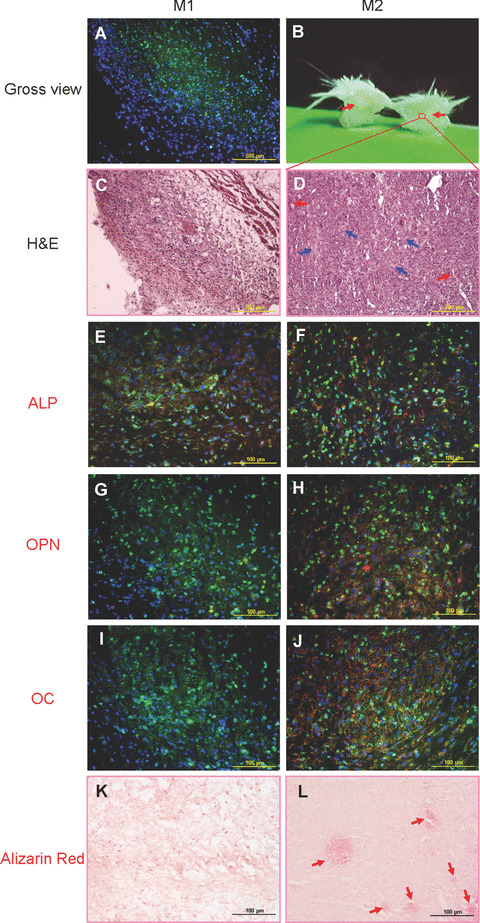
In vivo osteogenic differentiation of M1P14 and M2P14. (A) Twelve days after subcutaneous injection of M1P14, only a small area of abdominal skin specimens were stained with MHC‐Ib antibody (green) under microscopic observation (10×), which showed the existence of cell grafts. (B) After cutting across the protuberance of abdominal skin injected with M2P14, a white area (as pointed by red arrows) could be seen in the skin layers subcutaneously. (C) Haematoxylin and eosin staining view of the M1P14 graft specimens (10×). (D) Haematoxylin and eosin staining view of the white area of M2P14 graft specimens (10×). Blood vessel examples filled with red blood cells are pointed by red arrows. And some matrix‐rich areas are indicated by blue arrows. (E) ALP staining (red) could be detected in M1P14 cell graft area (green) (20×). (F) ALP staining (red) could be detected in M2P14 cell graft area (green) (20×). (G) No OPN staining could be detected in M1P14 cell graft area (green) (20×). (H) OPN staining (red) could be detected in M2P14 cell graft area (green) (20×). (I) No OC staining could be detected in M1P14 cell graft area (green) (20×). (J) OC staining (red) could be detected in M2P14 cell graft area (green) (20×). (K) Alizarin red staining showed absence of mineral deposition in M1P14 graft specimens (20×). (L) Alizarin red staining showed some positively stained areas of mineral deposition (as indicated by red arrows) in M2P14 graft specimens (20×). Cell nuclei in all pictures were stained with DAPI (blue).
Discussion
Our attempt to isolate a sub‐population of MSC with high osteogenic potential was carried out in the mouse, which is an important mammalian model system for both scientific and pre‐clinical investigations before proceeding to human systems. To date, several publications have reported a variety of different methods for isolating and expanding mMSC, including the use of different isolation and expansion media containing serum from different species [20], varying seeding densities and various enzymes for detachment [21], suspension culture [22], and seeding on fibronectin substratum [12, 23] as well as the use of culture media containing different growth factor combinations [12, 13, 23, 24, 25]. In enriching mMSC from bone marrow, the greatest technical challenge is the removal of haematopoietic cells, in particular monocytes and macrophages. Previously, it was reported that bone marrow monocyte‐macrophages play an important role in the proliferation and differentiation of haematopoietic cells in vitro[26]. Besides shortening trypsinization time, this study utilized silica for achieving this goal. Distinguished from previous published methods, the principle of using dense silica particles here is the rapid silica uptake by cultured macrophages which consequently caused cellular apoptosis upon internalization [27]. Additionally, the silica‐loaded cells can be separated from the rest of the cell population by Ficoll fractionation. Results showed abrupt decrease of CD11b+ cells after silica depletion. And followed by further negative selection, a highly purified population of CD11b−CD45−CD117−lineage− bone marrow cells with 99% purity can be attained as early as P5.
Another major goal of this study was to isolate mMSC with a high capacity for differentiation into the osteogenic lineage. In this study, M1, M2 and M4 were plastic adherent and could be successfully induced into the tri‐lineages. By fulfilling these criteria [28], all of them can thus be defined as MSC. However, M2 was much more efficient in osteogenic and chondrogenic differentiation compared to M4 in vitro, both qualitatively and quantitatively. Furthermore, high efficiency of osteogenic and chondrogenic differentiation was maintained up to late passage number (P32) of M2. Moreover, ALP, OPN and OC synthesis could readily be detected in M2 grafts as early as 12 days after implantation even by ectopic subcutaneous injection. Although M1 showed high osteogenic potential in vitro, most of them were lost after 12 days of implantation, and those remaining cell grafts demonstrated little deposition of osteogenic proteins and matrix. This implies that the M2 cell population is easily localized and is innately committed to the osteogenic lineage. It is well known that OC is expressed only at the terminal stage of osteogenesis, playing a role in bone mineralization [29, 30]. By alizarin red staining, small areas of mineral deposit had been detected in M2 graft. This further suggests that some M2 graft cells might undergo mineralization as early as 12 days after engraftment. Thus, the properties of better localization and enhanced osteogenic potential would make M2 a better candidate in cellular therapy of bone diseases.
In this study, a key difference was that M2 was derived after 1 hr incubation with silica, whereas M4 was derived after 4.5 hrs incubation with silica. This suggests that the bone marrow subpopulation with the greatest osteogenic potential might have the tendency to engulf silica particles after 1 hr incubation. Another implication is that within a heterogeneous bone marrow population, cells with high capacity for osteogenesis might proliferate much faster than cells with high capacity for adipogenesis. Recent studies reported that the osteogenic differentiation of MSC could be enhanced by internalization of silica nanoparticles and that the survival of MSC was not adversely affected by the internalization [31, 32]. Nevertheless, this observed stimulation of osteogenesis was limited. It is unclear whether the silica utilized in this study had any influence on the osteogenic differentiation of M2. In the tri‐lineage differentiation, we observed that CD73 expression by M2 was maintained to the terminal stage of osteogenesis. CD73 was also expressed by osteogenic progenitors of M4 but not by undifferentiated M4 and its differentiated adipocytes. This would imply that CD73 expression might be a candidate marker for osteogenesis by mMSC. Indeed, this phenomenon was also similarly observed in human and rat MSC [33, 34].
Another goal of this study was to isolate an mMSC subpopulation with high proliferative capacity in vitro. In previous studies, mMSC exhibited a limited duration of survival ex vivo[10], and their survival was dependent upon supplementation of the culture medium with appropriate combination of growth factors [11, 12, 13]. In our protocol, M2 could proliferate steadily for more than 70 passages without the need for any growth factor supplementation. However, there may be a caveat to this in that spontaneous transformation of mMSC had been observed in culture by other groups. Zhou et al. reported increased chromosome number and multiple Robertsonian translocations at P3 that were coincident with the loss of contact inhibition [35], while Miura et al. observed mMSC transformation around P15 [36]. In our study, we also observed aneuploidity and polyploidity in M2, but these were at low frequency in cells up to P35. At the same time, contact inhibition was maintained.
Because of the quality of MSC being dependent on the age and health status of the donor, autologous MSC may not be optimal for the treatment of aged patients [37, 38]. It is important to identify a subpopulation of MSC suitable for allogeneic transplantation. In this study, the immune properties of M2 were thoroughly evaluated at early and late passage numbers, as well as before and after osteogenic differentiation. From our results, M2 displayed immuno‐privileged and immuno‐inhibitory properties at P5. It is well established that the immuno‐inhibitory effect is a fundamental property of all stromal cells [39]. However, on further analysis, the immuno‐stimulatory effect was manifested at P14 and after osteogenic differentiation; whereas the expression of MHC‐I and ‐II molecules did not change along with serial passage and differentiation in vitro. This would imply that the observed alteration in immunogenicity was not initiated by MHC molecules and that the immune rejection of MSC could not be predicted by changes in expression of classical MHC molecules in vitro. Indeed, the same phenomenon had also been observed with rabbit MSC [40]. However, the expression of MHC molecules on rabbit MSC was induced after implantation [40], so did mMSC. As a consequence, leucocytes infiltration was observed at allogeneic mMSC graft area. Interestingly, the in vitro immuno‐inhibitory effect of M2P5 on Bc MNC proliferation by C57 MNC stimulation was maintained by M2P5DOC but weakened at P14 and was reversed by M2P10DOC. This suggests that although the immunogenicity of mMSC increased with serial passage and terminal differentiation, the immuno‐inhibitory effect of osteogenic cells differentiated from mMSC at early passage numbers could be maintained at the same level as that of their undifferentiated mMSC progenitors, until perhaps the tenth passage. However, the survival of M2P14 cell grafts 12 days after subcutaneous injection in Bc mice suggests that these cells might display additional mechanisms to protect themselves from the attack of allogeneic immune cells. This protective effect could not be detected by in vitro mixed lymphocyte culture. On day 12 after implantation, M2 cell grafts were matrix embedded, with high deposition of OPN and OC. It is possible that bone‐associated extracellular matrix produced rapidly after implantation might hinder physical interaction between the implanted allogeneic M2 cells and host immune cells, thereby conferring a protective effect. Indeed, less leucocytes infiltration was observed in the matrix‐rich area of M2 graft. Another possibility is that mMSC exerted a balance of inhibitory and stimulatory effect on the growth of immune cells. This balance might skew towards stimulation with serial passage and differentiation in vitro, while it might skew towards inhibition upon encountering some unspecified factors in vivo. A third possibility is that the implanted mMSC selectively support the activity of those regulatory haematopoietic cells by means of cell‐to‐cell contact or secreted cytokines.
To conclude our findings, a subpopulation of plastic‐adherent mMSC with high capacity for osteogenic differentiation was isolated by silica depletion and negative selection. These cells could proliferate extensively in vitro with normal karyotype and could survive at an ectopic site within a competent allogeneic immune system without compromise to their osteogenic potential. Additionally, the property of easy localization makes these cells an ideal candidate cell source for improving the local integration of MSC grafts. Their therapeutic efficacy and efficiency could be further testified in mouse bone‐related disease and transgenic models, which are important research tools to provide critical solutions to similar problems in human beings. In addition, the methodology used to isolate osteogenic sub‐populations of mMSC might also be applicable for isolating osteogenic subpopulations of human MSC.
Based on our data, it is obvious that none of tested surface molecules is a specific marker of mMSC, since tri‐lineage differentiation was observed with mMSC populations deficient in some of these surface molecules. And each population had different capacity of tri‐lineage differentiation. Nevertheless, the results would still imply that MSC expressing the combination of a number of these molecules might have higher capacity to differentiate into certain lineages. Thus, M2 may provide a good model system for tissue engineering of bone and regenerative medicine, as well as the discovery of new osteogenic surface markers. After implantation, mineral accumulation was only observed in some areas of M2 graft tissue. This result further certifies the contemporary understanding that MSC are a heterogeneous cell population which contains cells at different developmental stages. Areas that demonstrate mineralization may represent a group of cells which proceed to bone formation faster. Therefore, M2 may also provide a good model system to identify bona fide MSC and osteogenic progenitors. Although silica can be used effectively to remove macrophages and monocytes with high affinity for internalizing silica particles, it is still necessary to use negative selection to remove the remaining contaminating cells. Further optimization of silica particle size and incubation time for mMSC isolation may improve the efficiency of removing the unwanted haematopoietic subpopulation.
Acknowledgements
The authors thank Dr. Boon Chin Heng for his help in manuscript preparation, Mr. Yen Leong Chua (Life Sciences Institute, Immunology Programme, NUS) for ordering and taking care of mice, Ms. Fei Chuin Lew (OLS Immunology Programme, NUS) for her assistance in flow cytometry, as well as Mr. Gen Lin (OLS Immunology Programme, NUS) for his technical support. The ALP monoclonal antibody developed by Katzmann J.A. and the OPN monoclonal antibody developed by Termine J.D. were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA, USA.
References
- 1. Weinstein SL. 2000‐2010: the bone and joint decade. J Bone Joint Surg Am . 2000; 82: 1–3. [PubMed] [Google Scholar]
- 2. Horwitz EM, Prockop DJ, Gordon PL, et al . Clinical responses to bone marrow transplantation in children with severe osteogenesis imperfecta. Blood . 2001; 97: 1227–31. [DOI] [PubMed] [Google Scholar]
- 3. Adachi N, Ochi M, Deie M, et al . Transplant of mesenchymal stem cells and hydroxyapatite ceramics to treat severe osteochondral damage after septic arthritis of the knee. J Rheumatol . 2005; 32: 1615–8. [PubMed] [Google Scholar]
- 4. Brooke G, Cook M, Blair C, et al . Therapeutic applications of mesenchymal stromal cells. Semin Cell Dev Biol . 2007; 18: 846–58. [DOI] [PubMed] [Google Scholar]
- 5. Granero‐Molto F, Weis JA, Longobardi L, et al . Role of mesenchymal stem cells in regenerative medicine: application to bone and cartilage repair. Expert Opin Biol Ther . 2008; 8: 255–68. [DOI] [PubMed] [Google Scholar]
- 6. Nakahara H, Bruder SP, Haynesworth SE, et al . Bone and cartilage formation in diffusion chambers by subcultured cells derived from the periosteum. Bone . 1990; 11: 181–8. [DOI] [PubMed] [Google Scholar]
- 7. Wagner W, Horn P, Castoldi M, et al . Replicative senescence of mesenchymal stem cells: a continuous and organized process. PLoS One. 2008; 3: e2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Izadpanah R, Kaushal D, Kriedt C, et al . Long‐term in vitro expansion alters the biology of adult mesenchymal stem cells. Cancer Res . 2008; 68: 4229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Y, Song J, Liu W, et al . Growth and differentiation of rat bone marrow stromal cells: does 5‐azacytidine trigger their cardiomyogenic differentiation Cardiovasc Res . 2003; 58: 460–8. [DOI] [PubMed] [Google Scholar]
- 10. Nadri S, Soleimani M, Hosseni RH, et al . An efficient method for isolation of murine bone marrow mesenchymal stem cells. Int J Dev Biol . 2007; 51: 723–9. [DOI] [PubMed] [Google Scholar]
- 11. Fehrer C, Laschober G, Lepperdinger G. Aging of murine mesenchymal stem cells. Ann N Y Acad Sci . 2006; 1067: 235–42. [DOI] [PubMed] [Google Scholar]
- 12. Tropel P, Noël D, Platet N, et al . Isolation and characterisation of mesenchymal stem cells from adult mouse bone marrow. Exp Cell Res . 2004; 295: 395–406. [DOI] [PubMed] [Google Scholar]
- 13. Jiang Y, Jahagirdar BN, Reinhardt RL et al . Pluripotency of mesenchymal stem cells derived from adult marrow. Nature . 2002; 418: 41–9. [DOI] [PubMed] [Google Scholar]
- 14. Roobrouck VD, Ulloa‐Montoya F, Verfaillie CM. Self‐renewal and differentiation capacity of young and aged stem cells. Exp Cell Res . 2008; 314: 1937–44. [DOI] [PubMed] [Google Scholar]
- 15. Bianco P, Gehron Robey P. Marrow stromal stem cells. J Clin Invest . 2000; 105: 1663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lodie TA, Blickarz CE, Devarakonda TJ, et al . Systematic analysis of reportedly distinct populations of multipotent bone marrow‐derived stem cells reveals a lack of distinction. Tissue Eng . 2002; 8: 739–51. [DOI] [PubMed] [Google Scholar]
- 17. Toh WS, Yang Z, Liu H, et al . Effects of culture conditions and bone morphogenetic protein 2 on extent of chondrogenesis from human embryonic stem cells. Stem Cells . 2007; 25: 950–60. [DOI] [PubMed] [Google Scholar]
- 18. Toh WS, Liu H, Heng BC, et al . Combined effects of TGFbeta1 and BMP2 in serum‐free chondrogenic differentiation of mesenchymal stem cells induced hyaline‐like cartilage formation. Growth Factors . 2005; 23: 313–21. [DOI] [PubMed] [Google Scholar]
- 19. Olmsted‐Davis EA, Gugala Z, Camargo F, et al . Primitive adult hematopoietic stem cells can function as osteoblast precursors. Proc Natl Acad Sci USA . 2003; 100: 15877–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peister A, Mellad JA, Larson BL, et al . Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood . 2004; 103: 1662–8. [DOI] [PubMed] [Google Scholar]
- 21. Meirelles Lda S, Nardi NB. Murine marrow‐derived mesenchymal stem cell: isolation, in vitro expansion, and characterization. Br J Haematol . 2003; 123: 702–11. [DOI] [PubMed] [Google Scholar]
- 22. Shiota M, Heike T, Haruyama M, et al . Isolation and characterization of bone marrow‐derived mesenchymal progenitor cells with myogenic and neuronal properties. Exp Cell Res . 2007; 313: 1008–23. [DOI] [PubMed] [Google Scholar]
- 23. Breyer A, Estharabadi N, Oki M, et al . Multipotent adult progenitor cell isolation and culture procedures. Exp Hematol . 2006; 34: 1596–601. [DOI] [PubMed] [Google Scholar]
- 24. Sun S, Guo Z, Xiao X, et al . Isolation of mouse marrow mesenchymal progenitors by a novel and reliable method. Stem Cells . 2003; 21: 527–35. [DOI] [PubMed] [Google Scholar]
- 25. Baddoo M, Hill K, Wilkinson R, et al . Characterization of mesenchymal stem cells isolated from murine bone marrow by negative selection. J Cell Biochem . 2003; 89: 1235–49. [DOI] [PubMed] [Google Scholar]
- 26. Mori M, Sadahira Y, Kawasaki S, et al . Macrophage heterogeneity in bone marrow culture in vitro . J Cell Sci . 1990; 95: 481–5. [DOI] [PubMed] [Google Scholar]
- 27. Gilberti RM, Joshi GN, Knecht DA. The phagocytosis of crystalline silica particles by macrophages. Am J Respir Cell Mol Biol . 2008; 39: 619–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dominici M, Le Blanc K, Mueller I, et al . Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy . 2006; 8: 315–7. [DOI] [PubMed] [Google Scholar]
- 29. Marie PJ. Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys . 2008; 473: 98–105. [DOI] [PubMed] [Google Scholar]
- 30. Young MF. Bone matrix proteins: their function, regulation, and relationship to osteoporosis. Osteoporos Int . 2003; 14: S35–42. [DOI] [PubMed] [Google Scholar]
- 31. Huang DM, Chung TH, Hung Y, et al . Internalization of mesoporous silica nanoparticles induces transient but not sufficient osteogenic signals in human mesenchymal stem cells. Toxicol Appl Pharmacol . 2008; 231: 208–15. [DOI] [PubMed] [Google Scholar]
- 32. Lipski AM, Pino CJ, Haselton FR, et al . The effect of silica nanoparticle‐modified surfaces on cell morphology, cytoskeletal organization and function. Biomaterials . 2008; 29: 3836–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gharibi B, Lewis BM, Elford C, et al . Adenosine is an important regulator of mesenchymal stem cell differentiation into osteoblasts. Endocrine Abstracts. 2008; 15: 16. [Google Scholar]
- 34. Siddappa R, Martens A, Doorn J, et al . cAMP/PKA pathway activation in human mesenchymal stem cells in vitro results in robust bone formation in vivo . Proc Natl Acad Sci USA . 2008; 105: 7281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou YF, Bosch‐Marce M, Okuyama H, et al . Spontaneous transformation of cultured mouse bone marrow‐derived stromal cells. Cancer Res . 2006; 66: 10849–54. [DOI] [PubMed] [Google Scholar]
- 36. Miura M, Miura Y, Padilla‐Nash HM, et al . Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem Cells . 2006; 24: 1095–103. [DOI] [PubMed] [Google Scholar]
- 37. Muschler GF, Nitto H, Boehm CA, et al . Age‐ and gender‐related changes in the cellularity of human bone marrow and the prevalence of osteoblastic progenitors. J Orthop Res . 2001; 19: 117–25. [DOI] [PubMed] [Google Scholar]
- 38. Quarto R, Thomas D, Liang CT. Bone progenitor cell deficits and the age‐associated decline in bone repair capacity. Calcif Tissue Int . 1995; 56: 123–9. [DOI] [PubMed] [Google Scholar]
- 39. Jones S, Horwood N, Cope A, et al . The antiproliferative effect of mesenchymal stem cells is a fundamental property shared by all stromal cells. J Immunol . 2007; 179: 2824–31. [DOI] [PubMed] [Google Scholar]
- 40. Liu H, Kemeny DM, Heng BC, et al . The immunogenicity and immunomodulatory function of osteogenic cells differentiated from mesenchymal stem cells. J Immunol . 2006; 176: 2864–71. [DOI] [PubMed] [Google Scholar]