

Abstract
Treatment strategies for metastatic renal cell carcinoma (RCC) have been limited due to chemotherapy and radiotherapy resistance. The development of targeted drugs has now opened novel therapeutic options. In the present study, anti‐tumoral properties of the histone deacetylase inhibitor valproic acid (VPA) were tested in vitro and in vivo on pre‐clinical RCC models. RCC cell lines Caki‐1, KTC‐26 or A498 were treated with various concentrations of VPA to evaluate tumour cell adhesion to vascular endothelial cells or to immobilized extracellular matrix proteins. In vivo tumour growth was conducted in subcutaneous xenograft mouse models. VPA was also combined with low dosed interferon‐α (IFN‐α) and the efficacy of the combination therapy, as opposed to VPA monotherapy, was compared. VPA significantly and dose‐dependently prevented tumour cell attachment to endothelium or matrix proteins, accompanied by elevated histones H3 and H4 acetylation. VPA altered integrin‐α and ‐β subtype expression, in particular α3, α5 and β3, and blocked integrin‐dependent signalling. In vivo, VPA significantly inhibited the growth of Caki‐1 in subcutaneous xenografts with the 200 mg/kg being superior to the 400 mg/kg dosing schedule. VPA‐IFN‐α combination markedly enhanced the effects of VPA on RCC adhesion, and in vivo tumour growth was further reduced by the 400 mg/kg but not by the 200 mg/kg VPA dosing schedule. VPA profoundly blocked the interaction of RCC cells with endothelium and extracellular matrix and reduced tumour growth in vivo. Therefore, VPA should be considered an attractive candidate for clinical trials.
Keywords: HDAC, valproic acid, renal cell carcinoma, adhesion, integrin receptors
Introduction
Renal cell carcinoma (RCC) accounts for 2–3% of adult cancers worldwide, with the highest rates observed in the United States, Australia and Europe. Nearly 40,000 new cases are diagnosed each year in the Western countries, leading to an estimated 20,000 deaths [1]. 30% of RCC patients have metastatic disease at the time of diagnosis. An additional 20% to 30% with clinically localized disease at the time of nephrectomy will subsequently develop metastatic disease [2]. The 5‐year survival for those patients with distant metastases is less than 10%[3].
Metastatic RCC is generally resistant to standard chemotherapy, radiotherapy and hormonal therapy. Cytokine‐based immunotherapy has until recently been considered standard care for first‐line treatment of metastatic RCC. However, this approach is associated with significant toxicity, only 10–20% of patients experienced objective disease response, and life was prolonged only in selected patients [4].
A novel concept to manage RCC is based on the development of potent anticancer compounds directed against specific and relevant biological targets. Histone deacetylases (HDACs) represent one of the most important intracellular molecules, as they modulate a wide variety of cellular functions. Abnormal histone acetylation status can result in undesirable phenotypic changes, including developmental disorders and cancer [5]. Hence, HDAC inhibitors may be useful in cancer prevention, due to their ability to ‘reactivate’ the expression of epigenetically silenced genes, including those involved in differentiation, invasion and metastasis. Among the growing list of HDAC‐inhibitors, the branched‐chain fatty acid valproic acid (VPA) has been shown to possess distinct HDAC inhibitory properties and to affect the growth and survival of several tumour cells in vitro and in vivo[6, 7]. VPA is an established drug in the long‐term therapy of epilepsy. It can be applied orally, negative side effects are rare and it demonstrates expedient pharmacokinetic properties.
Very recently, administration of VPA has been reported to result in a marked decrease in proliferation of RCC cells in vitro and a significant reduction in tumour volume in vivo[8]. Nevertheless, close communication of RCC cells with vascular endothelium and underlying extracellular matrix proteins is required to allow tumour transmigration and metastatic spread. Since invasion and metastasis are the critical events of malignant tumour progression and the main cause of treatment failure, a potent anticancer compound should not only prevent RCC proliferation but – more importantly – prevent transit of circulating tumour cells from the blood vessel into the target tissue.
To explore whether VPA fulfils this essential criterion, the potential of VPA to block adhesion properties of RCC cells was investigated and the underlying mode of action explored. Also, VPA was combined with low‐dosed interferon‐α (IFN‐α) and the effects of the combination regimen compared to the single drug application. The experimental strategy was based on earlier reports demonstrating that IFN‐α may enhance VPA’s potency both in vivo and in vitro[9, 10, 11].
VPA was shown to potently block RCC tumour cell adhesion in vitro and prevent RCC tumour growth in vivo. VPA’s activity was associated with reduction of HDAC and elevated acetylation of histones H3 and H4. VPA altered integrin‐α and ‐β subtype expression and blocked integrin‐dependent signalling. It is of particular interest that VPA‐IFN‐α combination induced stronger effects on RCC cell adhesion than VPA alone. Based on these results, VPA provides potent anti‐tumour activity and, therefore, may reveal significant therapeutic benefit in treating advanced RCC.
Materials and methods
Antibodies
Integrins: anti‐α1 (IgG1; clone SR84, dilution 1:1000), anti‐α2 (IgG2a; clone 12F1‐H6, dilution 1:250), anti‐α3 (IgG1; clone C3II.1, dilution 1:1000), anti‐α4 (IgG1; clone 9F10, dilution 1:200), anti‐α5 (IgG1; clone IIA1, dilution 1:5000), anti‐α6 (IgG2a; clone GoH3, dilution 1:200), anti‐β1 (IgG1; clone MAR4, dilution 1:2500), anti‐β3 (IgG1; clone VI‐PL2, dilution 1:2500) or anti‐β4 (IgG2a; clone 439–9B, dilution 1:250) integrins were all from BD Biosciences (Heidelberg, Germany). Anti‐integrin‐linked kinase (ILK; clone 3, dilution 1:1000), anti‐focal adhesion kinase (FAK; clone 77, dilution 1:1000) and anti‐phospho‐specific FAK (pY397; clone 18, dilution 1:1000) were also derived from BD Biosciences.
Histones: Anti‐histone H3 (IgG, clone Y173, dilution 1:5000), anti‐acetylated H3 (IgG, clone Y28, dilution 1:500), anti‐histone H4 (polyclonal IgG, dilution 1:250), anti‐acetylated H4 (Lys8, polyclonal IgG, dilution 1:500) and anti‐HDAC3 (polyclonal IgG, dilution 1:2000) were all from Biomol GmbH (Hamburg, Germany).
Anti‐β‐actin monoclonal antibody was obtained from Sigma (Taufenkirchen, Germany).
Cell cultures
Kidney carcinoma Caki‐1 and KTC‐26 cells were purchased from LGC Promochem (Wesel, Germany). A498 were derived from CLS (Heidelberg, Germany). Tumour cells were grown and subcultured in RPMI 1640 medium (Seromed, Berlin, Germany) supplemented with 10% foetal calf serum (FCS), 100 IU/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified, 5% CO2 incubator.
Endothelial cells (human umbilical vein endothelial cells) were isolated from human umbilical veins and harvested by enzymatic treatment with chymotrypsin. HUVEC were grown in Medium 199 (Biozol, Munich, Germany), 10% FCS (Gibco, Karlsruhe, Germany), 10% pooled human serum (Blood Bank of The German Red Cross, Frankfurt am Main, Germany), 20 μg/ml endothelial cell growth factor (Boehringer, Mannheim, Germany), 0.1% heparin (Roche, Basel, Switzerland), 100 ng/ml gentamycin (Gibco) and 20 mM N‐2‐Hydroxyethylpiperazine‐N′‐2‐ethanesulfonic acid buffer (Seromed, Berlin, Germany). To control the purity of HUVEC cultures, cells were stained with fluorescein isothiocyanate labelled monoclonal antibody against factor VIII‐associated antigen (Von Willebrand factor; clone F8/86; Dako, Hamburg, Germany) and analysed microscopically or by FACscan (Becton Dickinson, Heidelberg, Germany; FL‐1H (log) channel histogram analysis; 1 × 104 cells/scan). Cell cultures with a purity >95% were serially passaged. Subcultures from passages 2–4 were selected for experimental use.
Drug treatment
Tumour cells were treated with VPA (gift from G. L. Pharma GmbH, Lannach, Austria) at a final concentration of 0.25, 0.5 or 1 mM for 3 or 5 days (if not otherwise indicated). Controls remained untreated (i.e. treated with cell culture medium alone). In a further experiment, tumour cells were incubated simultaneously with VPA and IFN‐α (Roferon A; Roche Pharma AG, Grenzach‐Wyhlen, Germany; 200 U/ml), and compared to cells treated with VPA or IFN‐α, or to those which remained untreated.
Tumour cell adhesion
To analyse tumour cell adhesion, HUVEC were transferred to six‐well multiplates (Falcon Primaria; BD Biosciences) in complete HUVEC medium. When 80–100% confluency was reached, Caki‐1, KTC‐26 or A498 cells were detached from the culture flasks by accutase treatment (PAA Laboratories, Cölbe, Germany) and 0.5 × 106 cells were then added to the HUVEC monolayer for 60 min. Subsequently, non‐adherent tumour cells were washed off using warmed (37°C) Medium 199. The remaining cells were fixed with 1% glutaraldehyde. Adherent tumour cells were counted in five different fields of a defined size (5 × 0.25 mm2) using a phase contrast microscope and the mean cellular adhesion rate was calculated.
Attachment to extracellular matrix components
Six‐well plates were coated with collagen G (extracted from calfskin, consisting of 90% collagen type I and 10% collagen type III; Seromed; diluted to 100 μg/ml in (phosphate buffered saline), laminin (derived from the Engelbreth–Holm–Swarm mouse tumour; BD Biosciences; diluted to 50 μg/ml in PBS), or fibronectin (derived from human plasma; BD Biosciences; diluted to 50 μg/ml in PBS) overnight. Plastic dishes served as the background control. Plates were washed with 1% BSA (bovine serum albumin) in PBS to block nonspecific cell adhesion. Thereafter, 0.5 × 106 tumour cells were added to each well for 60 min. Subsequently, non‐adherent tumour cells were washed off, the remaining adherent cells were fixed with 1% glutaraldehyde and counted microscopically. The mean cellular adhesion rate, defined by adherent cellscoated well− adherent cellsbackground, was calculated from five different observation fields.
Evaluation of integrin surface expression
Tumour cells were washed in blocking solution (PBS, 0.5% BSA) and then incubated for 60 min. at 4°C with phycoerythrin (PE)‐conjugated monoclonal antibodies directed against integrin subtypes indicated above. Integrin expression of tumour cells was then measured using a FACscan (Becton Dickinson; FL‐1H or FL‐2H (log) channel histogram analysis; 1 × 104 cells/scan) and expressed as mean fluorescence units (MFU). A mouse IgG1‐PE (MOPC‐21) or IgG2a‐PE (G155–178; all: BD Biosciences) was used as an isotype control.
Western blotting
Acetylated histones H3 and H4 were evaluated in Caki‐1 tumour cells by Western blot analysis. Intracellular integrin subtype level and integrin related signalling were also explored in treated versus non‐treated cell populations. tumour cell Lysates were applied to a 7% polyacrylamide gel and electrophoresed for 90 min. at 100 V. The protein was then transferred to nitrocellulose membranes. After blocking with non‐fat dry milk for 1 hr, the membranes were incubated overnight with the antibodies, diluted as listed above. HRP‐conjugated goat‐antimouse IgG (Upstate Biotechnology, Lake Placid, NY, USA; dilution 1:5000) served as the secondary antibody. The membranes were briefly incubated with enhanced chemiluminescence detection reagent (ECL™, Amersham/GE Healthcare, München, Germany) to visualize the proteins and exposed to an x‐ray‐film (Hyperfilm™ EC™, Amersham/GE Healthcare). The β‐actin (1:1000) served as the internal control.
HDAC activity
For determining the inhibitory activity of VPA and/or IFN‐α for HDACs, a cell‐free assay (Color de Lys, Biomol GmbH) detecting HDAC 1 and 2 was used according to the manufacturer’s protocol. A nuclear extract of HeLa cells containing HDAC 1 and 2 was incubated for 10 min. at 37°C with trichostatin A (TSA, 1μM final concentration) as a positive control of inhibition. The HDAC reaction was initiated by adding a substrate of acetylated peptides, incubated for 15–30 min., and followed by adding a colour developer. The absorbance of triplicate analyses was assayed at 405 nm with a Bio‐Tec microtitre‐plate reader. To determine the HDAC activity of Caki‐1 cells, the cell extracts (50 μg) which had been exposed to VPA, IFN‐α, to both VPA + IFN‐α, or to control medium, were added to the substrate.
Tumour growth in vivo
For in vivo testing, 107 Caki‐1 cells were injected subcutaneously into male NMRI: nu/nu mice (EPO GmbH, Berlin, Germany). Treatment was initiated when tumours had grown to a palpable size (5–6 mm diameter). VPA was dissolved in 10% PEG 400/saline. It was injected intraperitoneally in doses of 100, 200 or 400 mg/kg/day once daily (n= 8). A second group received IFN‐α 5 × 105 IU/kg/day once daily (n= 8) and a third group both VPA and IFN‐α (n= 8). The control group of mice was treated with the solvent (n= 10). Tumour size was measured with callipers. Tumour volumes, relative tumour volumes (relative to the first treatment day) and treated/control (T/C) values were calculated. Body weight and mortality were recorded continuously to estimate tolerability.
Statistics
All in vitro experiments were performed three to six times, in vivo experiments were done 8–10 times. Statistical significance was investigated by the Wilcoxon–Mann‐Whitney‐U‐test. Differences were considered statistically significant at a P‐value less than 0.05.
Results
Inhibition of RCC cell adhesion by VPA
RCC cell–HUVEC interaction was explored in a co‐culture model. Short‐term pre‐treatment (1 hr) of tumour cells with VPA did not alter their adhesion behaviour (data not shown). Rather, a 3‐day pre‐incubation with 0.5 or 1 mM VPA was necessary to significantly prevent tumour cell attachment to endothelium (1 mM VPA > 0.5 mM VPA). A 3‐day application of 0.25 mM VPA did not prevent tumour cell attachment to HUVEC. However, extension of the 0.25 mM VPA incubation period to 5 days significantly diminished RCC cell adhesion, compared to the control values (Fig. 1).
Figure 1.
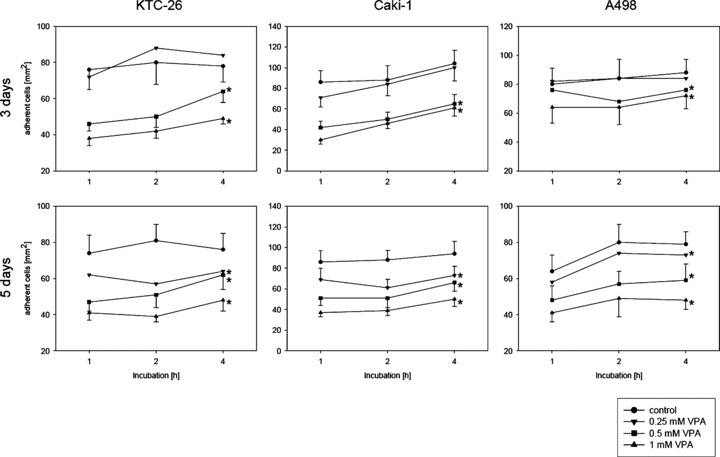
Adhesion of kidney cancer cells to HUVEC is down‐regulated by VPA. KTC‐26, Caki‐1 or A498 cells were treated with various concentrations of VPA for three or five days, and then added at a density of 0.5 × 106 cells/well to HUVEC monolayers for different time periods. Non‐adherent tumour cells were washed off in each sample, the remaining cells were fixed and counted in five different fields (5 × 0.25 mm2) using a phase contrast microscope. Mean values were calculated from five counts. Mean adhesion capacity is depicted as counted cells/mm2. * indicates significant difference to controls.
Further studies concentrated on VPA dosages which induced minimum (0.25 mM) or maximum (1 mM) effects. Since all RCC cell lines showed identical adhesion characteristics, ongoing experiments were restricted to Caki‐1 as the representative cell line.
IFN‐α enhances the adhesion blocking properties of VPA
A 3‐day pre‐incubation of Caki‐1 with 1 mM VPA significantly prevented tumour cell adhesion to endothelial cells. More cells detached from HUVEC when VPA and IFN‐α were applied in combination, although IFN‐α alone did not influence the adhesion behaviour of Caki‐1 (Fig. 2, left). With respect to a 5‐day pre‐treatment, both 0.25 and 1 mM VPA altered HUVEC–Caki‐1 interaction (1 mM > 0.25 mM). The effects observed were drastically enhanced by the VPA‐IFN‐α combination therapy. Application of IFN‐α alone did not result in any adhesion differences, compared to non‐treated controls (Fig. 2, right).
Figure 2.

Time dependent adhesion of Caki‐1 to HUVEC. Caki‐1 cells were treated with low (0.25 mM) or high (1 mM) concentrations of VPA, applied alone or in combination with IFN‐α. After a three or five day pre‐incubation, tumour cells were added at a density of 0.5 × 106 cells/well to HUVEC monolayers for different time periods. Non‐adherent tumour cells were washed off in each sample, the remaining cells were fixed and counted in five different fields (5 × 0.25 mm2) using a phase contrast microscope. Mean values were calculated from five counts. Mean adhesion capacity is depicted as counted cells/mm2. * indicates significant difference to controls. # indicates significant differences between VPA monotherapy and VPA‐IFN‐α combination therapy. If VPA‐IFN‐α combination therapy was not superior to the VPA monotherapy but evoked significant differences to the untreated controls, respective figure symbols were also marked with *.
The attachment of Caki‐1 cells to immobilized laminin, collagen, or fibronectin was investigated next, since RCC transmigration includes both adhesion to endothelial cells and to sub‐endothelial matrix components. Attachment rates of treated tumour cells were compared to the attachment rate of non‐treated controls which were set to 100%. In doing so, 0.25 mM VPA, given with or without IFN‐α for 3 days, did not change tumour cell binding to collagen or laminin. Attachment to fibronectin was diminished by nearly 20%. IFN‐α did not further enhance this effect. The 1 mM VPA strongly prevented Caki‐1 binding to all matrix proteins. A combination regimen based on 1 mM VPA and IFN‐α further diminished the amount of tumour cells which bound to laminin and collagen. However, this was not true with respect to the fibronectin matrix (Fig. 3). Pre‐incubation of Caki‐1 cells with 0.25 mM VPA for 5 days significantly reduced tumour binding to fibronectin. VPA (0.25 mM)‐IFN‐α combination therapy further enhanced this effect and even induced down‐regulation of Caki‐1 attachment to collagen. Interaction with laminin was not influenced. When 1 mM VPA was applied for 5 days, binding of Caki‐1 to collagen, laminin or fibronectin was blocked significantly. The therapeutic response was even stronger when IFN‐α was included, although IFN‐α alone did not influence tumour cell binding (Fig. 3).
Figure 3.
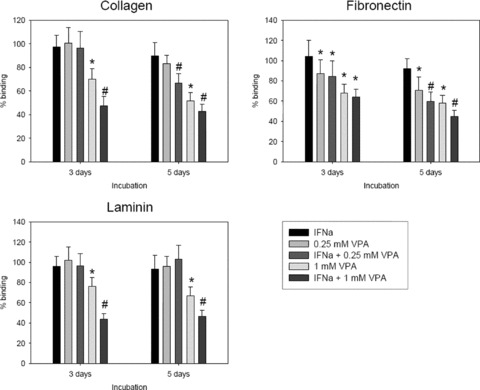
Adhesion of RCC cells to extracellular matrix proteins is down‐regulated by VPA or VPA‐IFN‐α. Caki‐1 cells were pre‐treated with low (0.25 mM) or high (1 mM) concentrations of VPA, applied alone or in combination with IFN‐α. Non‐treated cells served as the controls. Cells were then added to immobilized fibronectin, laminin or collagen at a density of 0.5 × 106 cells/well for 60 min. Plastic dishes were used to evaluate unspecific binding (background control). Non‐adherent tumour cells were washed off in each sample, the remaining cells were fixed and counted in five different fields (5 × 0.25 mm2) using a phase contrast microscope. Mean values were calculated from the five counts. Specific adhesion capacity (background adhesion on plastic surface was subtracted from adhesion to matrix proteins) is depicted as% binding and related to non‐treated controls which were set to 100%. * indicates significant difference to controls. # indicates significant differences between VPA monotherapy and VPA‐IFN‐α combination therapy. If VPA‐IFN‐α combination therapy was not superior to the VPA monotherapy but evoked significant differences to the untreated controls, figure symbols were also marked with *.
VPA and VPA‐IFN‐α combination increase histones H3 and H4 acetylation
Caki‐1 cells were treated with VPA (1 and 5 mM) [12] or VPA‐IFN‐α combination for 12 hrs, and histone acetylation was assessed by Western blotting. Caki‐1 showed distinct increase of acetylated H3 and H4 under VPA treatment. Interestingly, VPA’s effects were more prominent in the presence of IFN‐α, although IFN‐α alone did not modify histones (Fig. 4, up). Determination of HDAC activity revealed strong inhibition under VPA, the effect of which became much more prominent in the presence of IFN‐α (Fig. 4, down).
Figure 4.
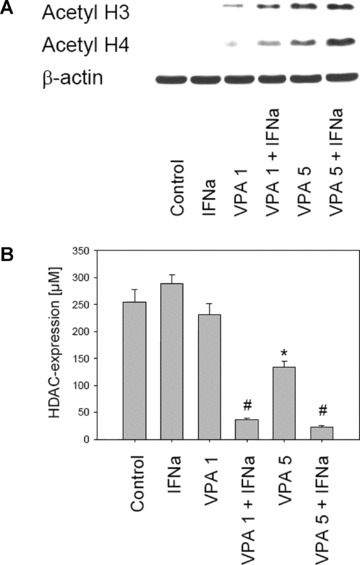
Figure 4A: Western blot analysis of H3 and H4 acetylation in Caki‐1, treated with 1 mM or 5 mM VPA or with VPA‐IFN‐α combination. Caki‐1 were incubated with VPA/VPA‐IFN‐α for 12 or 24 hrs. Cell lysates were then analysed by specific antibodies as listed in materials and methods. The β‐actin served as the internal control. One representative experiment of three is shown. 4B: For determining the inhibitory activity of VPA or VPA‐IFN‐α combination on HDAC activity, the Color de Lys cell‐free assay was used. Amount of HDAC is given in μM. * indicates significant difference to controls. # indicates significant differences between VPA monotherapy and VPA‐IFN‐α combination therapy. One of three experiments is shown here.
VPA modifies integrin expression pattern and blocks integrin activation
The α‐ and β‐integrin subtypes were analysed next, since integrins are deeply involved in tumour cell adhesion and transmigration [13]. With respect to the receptor surface expression, explored by flow cytometry, α3 was detected most extensively on Caki‐1, whereas α1, α2 and α5 were expressed to a lower extent (Fig. 5). The α4 and α6 subtypes were not significantly elevated over the background values (data not shown). The β1 and β3 subtypes were found to be highly expressed, β4 to be moderately expressed on Caki‐1. Figure 5 is related to a 1 mM VPA concentration, given for 5 days, and demonstrates enhanced receptor levels, in particular of the α3, α5 and β3 subtype, which were caused by VPA alone. Comparison between VPA and the VPA‐IFN‐α combination triggered effects revealed no differences with respect to integrin β1, β3, β4 and α3 surface expression. However, a VPA‐IFN‐α regimen induced a stronger α1, α2 and α5 expression than VPA alone.
Figure 5.

FACS analysis of integrin surface expression on Caki‐1 cells. Caki‐1 cells were incubated with 1 mM VPA (VPA), with 1 mM VPA‐IFN‐α (V‐IFN) combination, or remained untreated (control). Cells were then washed in blocking solution and stained with specific monoclonal antibodies as listed in materials and methods. A mouse IgG1‐PE or IgG2a‐PE was used as the isotype control. Fluorescence was analysed using a FACScan flow cytometer, and a histogram plot was generated to show PE‐fluorescence. MFU (mean fluorescence units) values are given below each histogram. One of three independent experiments is shown here.
Intracellular integrin levels were analysed by Western blotting (Fig. 6). The α1 protein expression increased in the presence of VPA, independently from the VPA concentration. When VPA‐IFN‐α combination was applied for 5 days, α1 further increased, whereas α1 was slightly reduced by the drug combination after 3 days (each compared to VPA alone). IFN‐α given alone did not alter α1 expression. The α2 became slightly elevated by VPA or VPA‐IFN‐α after a 5‐day pre‐incubation. The α3 was down‐regulated equally well by VPA or VPA‐IFN‐α combination after 3 days. However, IFN‐α did not act on α3 when given for 5 days. In contrast, the α5 protein level was altered particularly by IFN‐α and VPA‐IFN‐α combination, but not by VPA alone. With respect to the β3 and β4 integrin subunits, 5‐day pre‐incubation with VPA or VPA‐IFN‐α was necessary to evoke a distinct (and similar) reduction of these proteins. IFN‐α alone did not act on Caki‐1 cells in this matter. The influence of the compounds on β1 was ambiguous. The β1 was diminished after 3 days by 0.25 mM VPA exclusively. With respect to the 5‐day application, β1 was reduced by IFN‐α, 0.25 mM VPA and 1mM VPA‐IFN‐α combination, but not by 1 mM VPA alone.
Figure 6.
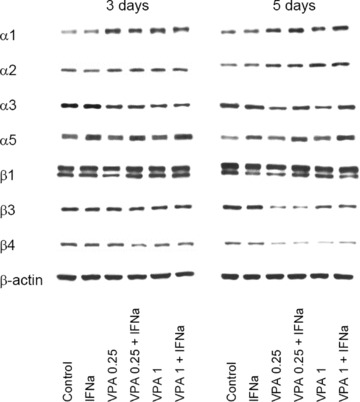
VPA or VPA‐IFN‐α modifies intracellular integrin proteins. Caki‐1 cells were incubated with IFN‐α, with low‐ (0.25 mM) or high‐dosed (1 mM) VPA, with VPA‐IFN‐α combination or remained untreated (control). Incubation lasted for three or five days. Cell lysates were then analysed by specific antibodies against integrin subtypes as listed in materials and methods. The β‐actin served as the internal control. One representative experiment of three is shown.
Quantitative alterations of integrins may not necessarily be coupled with reduced receptor activity. We, therefore, explored ILK, FAK and phosphorylation of FAK (pFAK) which are involved in the regulation of integrin function [14]. Figure 7 demonstrates that both FAK and pFAK were markedly down‐regulated in Caki‐1 cells when the combination regimen, but not the VPA monotherapy, was applied for 3 days. Moderate effects were also seen on ILK protein expression. When the incubation time was extended to 5 days, pFAK was nearly lost, this effect being caused by both VPA monotherapy as well as VPA‐IFN‐α combination.
Figure 7.
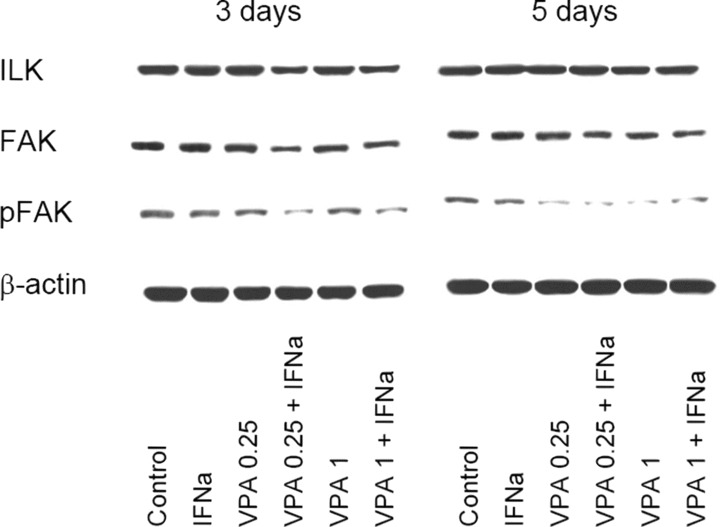
Influence of VPA or VPA‐IFN‐α on integrin‐dependent signalling. Caki‐1 cells were incubated with IFN‐α, with low‐ (0.25 mM) or high‐dosed (1 mM) VPA, with VPA‐IFN‐α combination or remained untreated (control). Incubation lasted for three or five days. Intracellular signalling was evaluated using the appropriate monoclonal antibodies recognizing ILK, FAK or phosphorylated FAK (pFAK). The β‐actin served as the internal control. One representative experiment of three is shown.
VPA treatment inhibits progression of tumour xenografts
Tumour xenografts were established in athymic nu/nu mice using Caki‐1 cells to evaluate the effects of VPA or VPA‐IFN‐α combination on RCC cell growth in vivo. Compared to the untreated animals, application of 200 mg/kg VPA significantly diminished the tumour volume, with reduction of 70% at day 46, compared to the control (Fig. 8). 400 mg/kg VPA induced minor effects with a reduction of 40% at day 46, whereas 100 mg/kg VPA did not influence tumour growth. Simultaneous application of both 400 mg/kg VPA and IFN‐α led to a further reduction of the tumour volume, compared to 400 mg/kg VPA injected alone (mean tumour volume 0.60 + 0.08 versus 0.47 + 0.07 cm3, day 46). However, VPA (200 mg/kg schedule) – IFN‐α combination treatment was not superior to the 200 mg/kg VPA monotherapy (data not shown).
Figure 8.

Effect of VPA on RCC xenografts. Caki‐1 xenografts were established in male athymic mice. Animals in the treatment arm received 100, 200 or 400 mg/kg VPA (n= 8). The control group of mice was treated with the solvent (n= 10). *indicates significant difference to the control animals.
Discussion
The results presented here provide evidence that the HDAC inhibitor VPA potently blocks RCC cell adhesion to endothelial cells and to extracellular matrix proteins. The attachment of circulating RCC cells to vascular endothelium and subsequent disruption of the basement membrane are crucial steps in haematogenous metastasis [15, 16]. Thus, the blocking characteristics of VPA could serve to optimize metastatic RCC treatment by suppressing tumour transmigration, thereby slowing RCC progression.
Earlier data have already demonstrated that VPA alters RCC cell growth dynamics [8]. Furthermore, VPA inhibited hypoxia‐inducible factor 1α in RCC cells which plays a critical role in transcriptional gene activation involved in tumour angiogenesis [17]. Obviously, VPA exerts multi‐targeted effects on cancer cells and thus may represent an attractive candidate for therapeutic intervention.
In the in vitro system, pre‐treatment of RCC cells with VPA for several days was necessary to induce a significant adhesion blockade. Effects of 0.25 mM VPA were not seen before a 5‐day pre‐incubation, whereas 1 mM VPA modified adhesion of RCC cells already after a 3‐day pre‐treatment period. This observation concurs with earlier studies dealing with the influence of VPA on RCC cell growth in vitro and in vivo[8]. Prolonged VPA exposure was also necessary to modify neuroectodermal tumour cells [18, 19], and Xia et al. has suggested that chronic administration of VPA is required to achieve therapeutic benefits with prostate carcinoma [12]. In our RCC xenograft model significant tumour reduction was not seen until 10 days after starting chronic VPA application. We, therefore, propose that long‐term application of VPA is necessary to delay tumour cell growth and block metastatic processes.
To analyse the mechanistic background responsible for VPA’s adhesion blocking properties, integrin receptor expression was explored, since these molecules play key roles in cancer metastasis by controlling tumour cell targeting, arrest, adhesion and migration [20, 21]. Incubation of Caki‐1 with VPA evoked a distinct receptor increase on the cell surface, notably of the α integrin subtypes and β3 integrins. It is still not clear which subtypes are involved in RCC transmigration and overall malignancy and how they contribute to these processes. RCC specimens taken from higher grade tumours showed decreased α3, α5 and α6 expression [22]. It has recently been demonstrated that down‐regulation of α2, α3 and α5 surface levels is necessary to allow transendothelial RCC migration [23]. Presumably, enhanced presentation of integrins at the cell surface, caused by VPA, impairs RCC motile behaviour. In line with this assumption, novel findings suggest that integrin internalization contributes to tumour metastasis and, conversely, integrin translocation from the cytoplasm to the cell membrane may prevent tumour cells from crossing the endothelial barrier [24, 25, 26]. Indeed, up‐regulation of integrin surface expression by VPA was paralleled by a down‐regulation of intracellular integrin proteins of the α3, β2 and β3 subtypes in the RCC in vitro model. Nevertheless, whether α3, β2 and β3 subtype translocation or improper localization of all integrins contributes to the diminished adhesion capacity of RCC cells requires further investigation.
Beside quantitative integrin alterations, phosphorylated FAK became strongly reduced in Caki‐1 by VPA, pointing to a specific de‐activation of the integrin receptors. It is generally accepted that FAK promotes cell adhesion, and in fact, integrin‐stimulated cell adhesion requires FAK and FAK activity [27, 28]. Satoh and coworkers demonstrated that FAK constitutes a functional unit which may be essential in determining malignant properties of RCC cells. [29]. We reported recently that FAK phosphorylation modifies RCC migration [30]. Consequently, loss of FAK activity seen under VPA therapy may be (at least in part) responsible for the diminished adhesion capacity of RCC cells. It has not been elucidated yet how VPA contributed to FAK de‐activation. Lee and coworkers demonstrated loss of FAK and FAK activity in colon cancer cells following exposure to the HDAC‐inhibitor butyric acid. They hypothesized that beside transcriptional regulation of HDAC‐mediated pathways down‐regulation of FAK might also be achieved by events other than histone acetylation and deacetylation since HDAC inhibitors could also acetylate non‐histone targets as well [31]. A similar scenario may also hold true in our RCC model.
Xenograft studies have been included in our experimental design. Although they may not be fully predictive of the therapeutic efficacy of cancer therapies in clinical trials, they may offer additional information to cell culture studies. 200 mg/kg body weight VPA significantly reduced the growth of xenografted RCC cells. An altered expression of proteins related to the malignant phenotype, including a massive increase of p21 and bax in this model has been reported [8]. The 200 mg/kg dosing schedule has been recommended by others to diminish prostate cancer xenografts [32], and to suppress tumour angiogenesis in vivo[33]. However, a different VPA regimen may be required to treat other tumour types. Daily i.p. injections of 366 mg/kg VPA were necessary to inhibit gastrointestinal tumour growth in nu/nu mice [34], and neuroblastoma xenograft studies were based on 400 mg/kg VPA [9, 35]. On the other hand, the 400 mg/kg VPA dosage induced only minor effects in our RCC model. The reason for the lower response in the 400 mg/kg protocol compared to the 200 mg/kg application is not clear. Speculatively, tumours may activate feedback or compensatory pathways to promote RCC growth and survival. Further studies are necessary to concentrate on this issue.
Finally, whether IFN‐α added to VPA in low concentrations could offer an advantage over VPA monotherapy was investigated. It has recently been shown that IFN‐α, when used together with VPA, significantly potentiates the anti‐tumoral activity of VPA on the human N‐myc amplified cell line BE(2)‐C, whereas IFN‐α on its own has little or no effect [10]. Most strikingly, VPA plus IFN‐α synergistically inhibited growth of UKF‐NB‐3 xenograft tumours in nude mice and induced complete cures in two out of six animals, while single treatment merely inhibited tumour growth [9]. Furthermore, IFN‐α has been documented to enhance the anti‐angiogenic action of HDAC‐inhibitors in neuroblastoma bearing transgenic mice [10], and to potentiate the influence of HDAC‐inhibitors on growth and invasion of lung and liver cancer cells [36, 37].
A combination VPA‐IFN‐α regimen was more effective than a VPA monotherapy in vitro. IFN‐α alone did not act on RCC adhesion, showing that IFN‐α boosts VPA’s anti‐tumoral properties. The conclusion is corroborated by histone analysis, since VPA‐IFN‐α combination induced much stronger alterations on HDAC activity and histone acetylation than VPA alone, whereas IFN‐α given separately was without any effect. A similar phenomenon has been observed in neuroblastoma cell cultures. Treatment with VPA alone decreased the ability of BE(2)‐C cells to adhere to and penetrate human endothelium. All these effects of VPA were significantly enhanced when combined with IFN‐α which on its own had little or no effect [10].
The mode of action of IFN‐α is not clear regarding this matter. However, a similar behaviour has recently been noticed on melanoma cell lines, where IFN‐α alone did not induce apoptosis but drastically enhanced the pro‐apoptotic effect of VPA [38]. Intriguingly and consistent with further reports, treatment of these cells with IFN‐α evoked strong increase of the Stat1 protein level which optimized the response of the melanoma cell lines to VPA [38, 39]. Although purely speculative, enhancement of Stat1 by IFN‐α might also render RCC cells to become more susceptible to VPA treatment. Nevertheless, it may not be logical to exclude any specific activity of IFN‐α. Indeed, IFN‐α but not VPA triggered alterations of α5 integrin expression in the experiments presented here. Recently, IFN‐α but not VPA was shown to reduce bcl‐2 expression in vivo[8], and Kaneko et al. reported down‐regulation of matrix metalloproteinase activity in a hepatocellular carcinoma invasion model which was caused by IFN‐α[37].
Surprisingly, the in vitro RCC data were not always confirmed by the in vivo model, since the 200 mg/kg VPA‐IFN‐α combination was not superior to the 200 mg/kg VPA monotherapy. However, when interpreting the results, we should be aware that different experimental protocols have been used. VPA and IFN‐α were administered once in the in vitro system whereas animals were treated chronically over a prolonged time period. Therefore, drug concentrations reaching the target cells may vary in vitro and in vivo and, consequently, different tumour responses may be evoked. It should also be considered that optimum therapeutic response may have already been achieved with 200 mg/kg VPA and, therefore, additional drugs may provide no further benefit. The latter hypothesis might explain why additive effects became obvious when the sub‐optimal concentration of 400 mg/kg VPA was used in combination with IFN‐α. Nevertheless, the molecular background of VPA‐IFN‐α interaction in vivo has not been evaluated in detail. Thus, this assumption remains speculative.
In summary, administration of VPA resulted in a marked decrease in adhesion of RCC cells in vitro and significant reduction in tumour volume in vivo. We postulate that VPA’s effects are (partially) based on the alterations of integrin expression and signalling. For the future, primary tumour cells should be tested, because they more closely reflect the clinical situation than the established cell lines we used. Since VPA has been approved by the U.S. Food and Drug Administration, with an established safety profile, and since drug concentrations used in the present study are within the therapeutic range, it can be considered an attractive candidate for clinical trials.
Acknowledgements
We thank Karen Nelson for critically reading the manuscript. This work was supported by the ‘Horst Müggenburg‐Stiftung’, ‘Jung‐Stiftung’, ‘Walter Schulz‐Stiftung’, ‘Ebert‐Stiftung’ and ‘Held‐Hecker‐Stiftung’.
References
- 1. Parkin DM, Bray F, Ferlay J, et al . Global cancer statistics, 2002. CA Cancer J Clin. 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Jacobsohn KM, Wood CG. Adjuvant therapy for renal cell carcinoma. Semin Oncol. 2006; 33: 576–82. [DOI] [PubMed] [Google Scholar]
- 3. Oudard S, George D, Medioni J, et al . Treatment options in renal cell carcinoma: past, present and future. Ann Oncol. 2007; 18S: x25–x31. [DOI] [PubMed] [Google Scholar]
- 4. McDermott DF, Atkins MB. Application of IL‐2 and other cytokines in renal cancer. Expert Opin Biol Ther. 2004; 4: 455–68. [DOI] [PubMed] [Google Scholar]
- 5. Mottet D, Castronovo V. Histone deacetylases: target enzymes for cancer therapy. Clin Exp Metastasis. 2008; 25: 183–9. [DOI] [PubMed] [Google Scholar]
- 6. Cinatl J Jr, Cinatl J, Scholz M, et al . Antitumor activity of sodium valproate in cultures of human neuroblastoma cells. Anticancer Drugs. 1996; 7: 766–73. [DOI] [PubMed] [Google Scholar]
- 7. Blaheta RA, Michaelis M, Driever PH, et al . Evolving anticancer drug valproic acid: insights into the mechanism and clinical studies. Med Res Rev. 2005; 25: 383–97. [DOI] [PubMed] [Google Scholar]
- 8. Jones J, Juengel E, Mickuckyte A, et al . The histone deacetylase inhibitor valproic acid alters growth properties of renal cell carcinoma in vitro and in vivo J Cell Mol Med. 2009; 8A: 2376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michaelis M, Suhan T, Cinatl J, et al . Valproic acid and interferon‐alpha synergistically inhibit neuroblastoma cell growth in vitro and in vivo . Int J Oncol. 2004; 25: 1795–9. [PubMed] [Google Scholar]
- 10. Cinatl J Jr, Kotchetkov R, Blaheta R, et al . Induction of differentiation and suppression of malignant phenotype of human neuroblastoma BE(2)‐C cells by valproic acid: enhancement by combination with interferon‐alpha. Int J Oncol. 2002; 20: 97–106. [PubMed] [Google Scholar]
- 11. Kuljaca S, Liu T, Tee AE, et al . Enhancing the anti‐angiogenic action of histone deacetylase inhibitors. Mol Cancer. 2007; 6: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xia Q, Sung J, Chowdhury W, et al . Chronic administration of valproic acid inhibits prostate cancer cell growth in vitro and in vivo . Cancer Res. 2006; 66: 7237–44. [DOI] [PubMed] [Google Scholar]
- 13. Moschos SJ, Drogowski LM, Reppert SL, et al . Integrins and cancer. Oncology. 2007; 21: 13–20. [PubMed] [Google Scholar]
- 14. Hehlgans S, Haase M, Cordes N. Signalling via integrins: implications for cell survival and anticancer strategies. Biochim Biophys Acta. 2007; 1775: 163–80. [DOI] [PubMed] [Google Scholar]
- 15. Steinbach F, Tanabe K, Alexander J, et al . The influence of cytokines on the adhesion of renal cancer cells to endothelium. J Urol. 1996; 155: 743–8. [PubMed] [Google Scholar]
- 16. Jin JS, Hsieh DS, Lin YF, et al . Increasing expression of extracellular matrix metalloprotease inducer in renal cell carcinoma: tissue microarray analysis of immunostaining score with clinicopathological parameters. Int J Urol. 2006; 13: 573–80. [DOI] [PubMed] [Google Scholar]
- 17. Qian DZ, Kachhap SK, Collis SJ, et al . Class II histone deacetylases are associated with VHL‐independent regulation of hypoxia‐inducible factor 1 alpha. Cancer Res. 2006; 66: 8814–21. [DOI] [PubMed] [Google Scholar]
- 18. Blaheta RA, Michaelis M, Natsheh I, et al . Valproic acid inhibits adhesion of vincristine‐ and cisplatin‐resistant neuroblastoma tumour cells to endothelium. Br J Cancer. 2007; 96: 1699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beecken WD, Engl T, Ogbomo H, et al . Valproic acid modulates NCAM polysialylation and polysialyltransferase mRNA expression in human tumor cells. Int Immunopharmacol. 2005; 5: 757–69. [DOI] [PubMed] [Google Scholar]
- 20. Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004; 5: 816–26. [DOI] [PubMed] [Google Scholar]
- 21. Moschos SJ, Drogowski LM, Reppert SL, et al . Integrins and cancer. Oncology. 2007; 21S: 13–20. [PubMed] [Google Scholar]
- 22. Markovic‐Lipkovski J, Brasanac D, Muller GA, et al . Cadherins and integrins in renal cell carcinoma: an immunohistochemical study. Tumori. 2001; 87: 173–8. [DOI] [PubMed] [Google Scholar]
- 23. Jones J, Berkhoff S, Weich E, et al . Transient down‐regulation of beta1 integrin subtypes on kidney carcinoma cells is induced by mechanical contact with endothelial cell membranes. J Cell Mol Med. 2007; 11: 826–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winterwood NE, Varzavand A, Meland MN, et al . A critical role for tetraspanin CD151 in alpha3beta1 and alpha6beta4 integrin‐dependent tumor cell functions on laminin‐5. Mol Biol Cell. 2006; 17: 2707–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Skalski M, Coppolino MG. SNARE‐mediated trafficking of alpha5beta1 integrin is required for spreading in CHO cells. Biochem Biophys Res Commun. 2005; 335: 1199–210. [DOI] [PubMed] [Google Scholar]
- 26. Powelka AM, Sun J, Li J, et al . Stimulation‐dependent recycling of integrin beta1 regulated by ARF6 and Rab11. Traffic. 2004; 5: 20–36. [DOI] [PubMed] [Google Scholar]
- 27. Hauck CR, Sieg DJ, Hsia DA, et al . Inhibition of focal adhesion kinase expression or activity disrupts epidermal growth factor‐stimulated signaling promoting the migration of invasive human carcinoma cells. Cancer Res. 2001; 61: 7079–90. [PubMed] [Google Scholar]
- 28. Sieg DJ, Hauck CR, Ilic D, et al . FAK integrates growth‐factor and integrin signals to promote cell migration. Nat Cell Biol. 2000; 2: 249–56. [DOI] [PubMed] [Google Scholar]
- 29. Satoh M, Nejad FM, Ohtani H, et al . Association of renal cell carcinoma antigen, disialylgalactosylgloboside, with c‐Src and Rho A in clustered domains at the surface membrane. Int J Oncol. 2000; 16: 529–36. [DOI] [PubMed] [Google Scholar]
- 30. Jones J, Marian D, Weich E, et al . CXCR4 chemokine receptor engagement modifies integrin dependent adhesion of renal carcinoma cells. Exp Cell Res. 2007; 313: 4051–65. [DOI] [PubMed] [Google Scholar]
- 31. Lee JC, Maa MC, Yu HS, et al . Butyrate regulates the expression of c‐Src and focal adhesion kinase and inhibits cell invasion of human colon cancer cells. Mol Carcinog. 2005; 43: 207–14. [DOI] [PubMed] [Google Scholar]
- 32. Shabbeer S, Kortenhorst MS, Kachhap S, et al . Multiple Molecular pathways explain the anti‐proliferative effect of valproic acid on prostate cancer cells in vitro and in vivo . Prostate. 2007; 67: 1099–110. [DOI] [PubMed] [Google Scholar]
- 33. Gao D, Xia Q, Lv J, et al . Chronic administration of valproic acid inhibits PC3 cell growth by suppressing tumor angiogenesis in vivo . Int J Urol. 2007; 14: 838–45. [DOI] [PubMed] [Google Scholar]
- 34. Greenblatt DY, Vaccaro AM, Jaskula‐Sztul R, et al . Valproic acid activates notch‐1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells. Oncologist. 2007; 12: 942–51. [DOI] [PubMed] [Google Scholar]
- 35. Yang Q, Tian Y, Liu S, et al . Thrombospondin‐1 peptide ABT‐510 combined with valproic acid is an effective antiangiogenesis strategy in neuroblastoma. Cancer Res. 2007; 67: 1716–24. [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto‐Yamaguchi Y, Okabe‐Kado J, Kasukabe T, et al . Induction of apoptosis by combined treatment with differentiation‐inducing agents and interferon‐alpha in human lung cancer cells. Anticancer Res. 2003; 23: 2537–47. [PubMed] [Google Scholar]
- 37. Kaneko F, Saito H, Saito Y, et al . Down‐regulation of matrix‐invasive potential of human liver cancer cells by type I interferon and a histone deacetylase inhibitor sodium butyrate. Int J Oncol. 2004; 24: 837–45. [PubMed] [Google Scholar]
- 38. Krämer OH, Baus D, Knauer SK, et al . Acetylation of Stat1 modulates NF‐kappaB activity. Genes Dev. 2006; 20: 473–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wong LH, Sim H, Chatterjee‐Kishore M, et al . Isolation and characterization of a human STAT1 gene regulatory element. Inducibility by interferon (IFN) types I and II and role of IFN regulatory factor‐1. J Biol Chem. 2002; 277: 19408–17. [DOI] [PubMed] [Google Scholar]