

Abstract
SHH‐activated medulloblastomas (SHH‐MB) account for 25–30% of all medulloblastomas (MB) and occur with a bimodal age distribution, encompassing many infant and adult, but fewer childhood cases. Different age groups are characterized by distinct survival outcomes and age‐specific alterations of regulatory pathways. Here, we review SHH‐specific genetic aberrations and signaling pathways. Over 95% of SHH‐MBs contain at least one driver event – the activating mutations frequently affect sonic hedgehog signaling (PTCH1, SMO, SUFU), genome maintenance (TP53), and chromatin modulation (KMT2D, KMT2C, HAT complexes), while genes responsible for transcriptional regulation (MYCN) are recurrently amplified. SHH‐MBs have the highest prevalence of damaging germline mutations among all MBs. TP53‐mutant MBs are enriched among older children and have the worst prognosis among all SHH‐MBs. Numerous genetic aberrations, including mutations of TERT, DDX3X, and the PI3K/AKT/mTOR pathway are almost exclusive to adult patients. We elaborate on the newest development within the evolution of molecular subclassification, and compare proposed risk categories across emerging classification systems. We discuss discoveries based on preclinical models and elaborate on the applicability of potential new therapies, including BET bromodomain inhibitors, statins, inhibitors of SMO, AURK, PLK, cMET, targeting stem‐like cells, and emerging immunotherapeutic strategies. An enormous amount of data on the genetic background of SHH‐MB have accumulated, nevertheless, subgroup affiliation does not provide reliable prediction about response to therapy. Emerging subtypes within SHH‐MB offer more layered risk stratifications. Rational clinical trial designs with the incorporation of available molecular knowledge are inevitable. Improved collaboration across the scientific community will be imperative for therapeutic breakthroughs.
Introduction
Medulloblastoma (MB) is the most common pediatric brain malignancy, accounting for approximately 20% of childhood brain cancers and 10% of all childhood cancer deaths. Incidence culminates among children younger than 10 years of age, with about half of cases arising before the age of 5.1, 2 Up to 40% of patients are diagnosed with metastatic disease,3 with a grim outlook for survival.4 More than one‐third of patients die within 5 years after diagnosis, and survivors face treatment‐related long‐term adverse effects.5
MB treatment strategy involves maximal safe resection followed by craniospinal irradiation and cytotoxic chemotherapy, with specific type and intensity for high‐ or standard/average‐risk disease. Average‐risk patients are over 3 years of age with total or near‐total resection and no disease dissemination, while patients with suboptimal tumor resection, metastasis, and/or large cell/anaplastic (LCA) histology are treated for high‐risk disease.6 Infants under 3 years of age require delayed irradiation and are preferably treated by multiagent chemotherapy, with better results after gross total resection with the absence of dissemination compared to patients with residual or metastatic disease.7, 8, 9 Continuing advances in neuroimaging, neurosurgical techniques, radiation therapy, and combined chemotherapy have increased 5‐year survival rates to 70–80%,1, 5 although individual responses to treatment vary considerably and survival rates have reached a plateau.10 The highly toxic and invasive multimodal therapies frequently induce debilitating adverse effects on the long term.11 Evidently, interventions should be spared in patients likely to be cured and maximized in those with aggressive disease.
The molecular era lead to exciting transformations in patient stratifications with consequences for therapeutic approaches. Based on molecular alterations, four subgroups became widely accepted: sonic hedgehog‐activated (SHH‐MB), wingless‐activated (WNT‐MB), Group 3, and Group 4 MBs, each characterized by distinct patterns of somatic mutations, copy number alterations, transcriptional profiles, and clinical outcomes.12 WNT‐ and SHH‐activated MBs have abnormal activation of the WNT and SHH pathways, respectively, while no dominant signaling pathway alterations were identified in Group 3 and Group 4 MBs and appear as non‐WNT/non‐SHH in the revised WHO classification.13 Subgroup assignment is highly prognostic, with markedly different survival rates.14 The 5‐year overall survival is as high as 95% in WNT‐activated MBs. Group 3 patients face the worst 5‐year overall survival (45–60%), especially low among infants. Group 4 and SHH‐MBs are characterized by an intermediate (75–80%) 5‐year overall survival that also depends on disease dissemination, histology, and genetic aberrations, such as mutations and oncogene amplifications.15, 16, 17, 18 Within each primary MBs, additional subtypes are emerging with distinct biology and clinical outcomes,18, 19, 20 providing a constructive approach for therapy optimization.14 Here, we provide a comprehensive overview of SHH‐MBs with special focus on emerging prognostication schemes and novel therapeutic approaches.
Clinical Attributes
SHH‐MBs account for ~30% of all MBs and occur in a bimodal age distribution encompassing the majority of infant and adult, but relatively fewer childhood cases15, 21, 22 (Fig. 1A). Pediatric and adult tumors are molecularly and clinically distinct.12, 23 Approximately 21% of SHH‐MBs are enriched with TP53‐mutations, delineating a distinct subcategory – “SHH‐activated TP53‐wild‐type” is more frequent among adults and young children and confers a good prognosis with an 81% 5‐year overall survival (OS). In contrast, the “SHH‐activated TP53‐mutant” subtype occurs typically among older children between ages 5 and 18 and is associated with a dismal prognosis, including a 41% 5‐year OS. In children older than 5 years, tumors with TP53‐mutations account for two‐third of deaths.24
Figure 1.
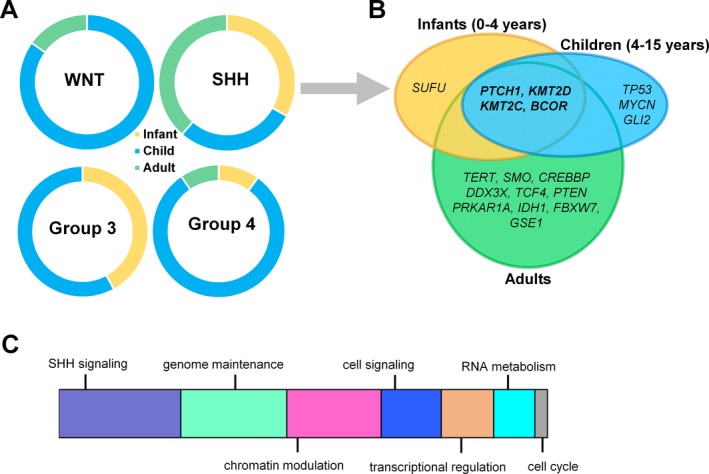
Age‐specific distribution of childhood medulloblastomas. (A) Infants, children, and adults are represented differently within each medulloblastoma subgroup. (B) Dominant mutations across three different age groups in SHH‐activated medulloblastomas (SHH‐MB). (C) Schematic representation of major mechanisms most frequently affected by somatic alterations contributing to SHH‐MB development.
The majority of SHH‐MBs are nodular/desmoplastic but can also have a classic or LCA histology, the latter especially frequent among children.25, 26 The nodular‐desmoplastic subtype predicts increased survival in infants, and in this young group of patients, radiation therapy may be successfully abolished.27, 28, 29 Radiation‐sparing treatments involve systemic chemotherapy with intraventricular therapy (HIT‐SKK'92 protocol),7 or high‐dose chemotherapy with stem cell rescue (CCG‐99703 protocol).30 Sequential high‐dose chemotherapy following the CCG‐99703 protocol resulted in excellent outcome for SHH‐MB patients with classical histology.31 Nevertheless, conventional chemotherapy, excluding intraventricular methotrexate, is not feasible as The Children's Oncology Group clinical trial (ACNS1221) for nonmetastatic infants under 4 years of age with nodular‐desmoplastic MB was closed prematurely due to an excessive number of relapses.32
Molecular Biology
SHH‐MBs frequently arise in the cerebellar hemisphere from cerebellar granule neuron precursors (CGNPs) or the cochlear nuclei of the brainstem.33 Constitutive SHH‐signaling leads to overproliferation,34, 35, 36 therefore SHH expressed during early postnatal development from Purkinje neurons promotes the rapid expansion of CGNPs in the external granule layer differentiating as granulate neurons migrate into the internal granule layer.
Distinct regulatory processes are altered in different age groups. Developmental processes and DNA/histone methylation include the most frequently disturbed pathways in infants, while chromatin organization and transcription regulation are most heavily involved in adults.26 Despite the age group‐specific molecular features, SHH tumors share common traits, such as high expression of SHH‐signaling target genes (e.g., Gli family of transcription factors) or CGNP specification genes (e.g., ATOH1) and relatively low expression levels of neuronal differentiation genes.26, 37, 38, 39
Genetic predispositions
SHH‐MBs have the highest, 14–20% prevalence of destructive germline mutations among all MBs, although less than half are projected based on family history and medical records. Genetic predispositions influence both progression‐free and overall survival.40 Gorlin syndrome is associated with mutations affecting PTCH1 and SUFU genes of the SHH pathway, and Li–Fraumeni syndrome is linked to TP53‐mutations predisposing carriers to multiple familial cancers.41 Germline PTCH1 and SUFU mutations are more common among infants with a median age of 2.0 years and are present in 21% of all infant SHH‐MBs. Hereditary TP53‐mutations are most frequent among children (median age of 9.8 years), present in ~21% of all SHH‐MBs patients aged 5–16, and are coupled with a low, 27% 5‐year survival.40 Li–Fraumeni syndrome‐related SHH‐MBs are associated with an increased incidence of chromothripsis, a massive genomic rearrangement during a single catastrophic event, resulting in gene fusions and/or highly amplified copy number states of recognized oncogenes.40 These findings suggest that TP53‐mutations predispose cells to catastrophic DNA rearrangements or facilitate cell survival after such events.42 Accordingly, an outstandingly high mutational rate characterizes Li–Fraumeni syndrome‐associated SHH‐MBs.37 Rare heterozygous germline mutations in BRCA2 and PALB2 genes were also identified among SHH‐MB patients, coupled with homologous recombination repair deficiency‐like mutation spectrum.40
Because of TP53′s crucial role in DNA repair, genome maintenance, and cell death, radiation therapy may accelerate tumor growth, which is particularly important for patients with Li–Fraumeni syndrome.43 Radiation also increases the likelihood of secondary malignancies, such as basal cell carcinomas and other tumors of the skin in patients with Gorlin syndrome (germline PTCH1 mutation).44 Genetic counseling is recommended for families in case of TP53‐mutations. Additionally, genetic testing is recommended for germline PTCH1 and SUFU mutations for children with MB, in children <3 years old, or those whose tumors show nodular or desmoplastic histologic features and/or somatic changes in the SHH pathway.45
Copy number variations
Losses of 9q, 10q, and 17p, frequently co‐occurring with TP53‐mutations, are the most frequent large‐scale chromosomal aberrations in SHH‐MB. Focal somatic copy number aberrations (SCNA) include MDM4 and PPM1D amplifications and focal deletions of TP53, all involved in TP53‐signaling; amplification of GLI2 and deletion of PTCH1, exclusive for SHH tumors; and amplifications of MYCN and CCND2. Some SCNAs (including IGF1R, PIK3C2G, PIK2C2B, KIT, MDM4, PDGFRA, and deletion of PTEN) are potentially targetable with already available small molecule inhibitors.38
Mutations
Over 95% of SHH‐MBs contain at least one driver event. However, the types of mutations are highly variable.22, 26 Activating mutations almost permanently involve the SHH‐signaling pathway,26 but alterations beyond canonical SHH‐signaling, such as mutations of the IDH1 gene with epigenetic regulatory function, have also been recently described.22
The most frequently mutated genes include PTCH1 (~43%), SUFU (~10%), and SMO (~9%), and the presence of these activating mutations is mutually exclusive and age group specific22, 26, 37, 39 (Fig. 1B). In a significant proportion of patients, where PTCH1, SMO, and SUFU mutations are absent, alternative mechanisms are responsible for SHH‐pathway activation.46 PTCH1 mutations are roughly equally numerous in adults, children, and infants, while SMO mutations are highly enriched in adults and SUFU mutations in infants (0–4 years), with both mutations barely present in children.26 Infants harboring germline PTCH1 mutations are diagnosed with Gorlin syndrome (nevoid basal cell carcinoma).41
SHH‐MBs enriched with TP53‐mutations (~20%) are coupled with the highest overall mutational rate of all MBs. Chromosome 17p loss is common in TP53‐mutant cases.26 Interestingly, WNT‐MBs are also enriched with TP53‐mutations, with the second highest mutation rate, but without the survival disadvantage observed in SHH‐MBs.37 In children, TP53‐mutations are mutually exclusive with PTCH1 mutations, but frequently co‐occur with GLI2 and MYCN amplifications, which are essential regulators of transcription22 (Fig. 1B): for example, GLI2 is the main transcription effector of SHH‐signaling in granule cell precursors.47
Chromatin modulation is frequently affected in SHH‐MBs (Fig. 1C). KMT2D and KMT2C methyltransferase mutations occur with relatively high frequency within both pediatric and adult samples.22 Mutations deregulating histone‐acetyltransferase (HAT) complexes involving genes such as CREBBP, KAT6B, EP300, BRPF1, and KANSL1 are present in approximately 19% of all SHH‐MB patients.22 Frequently mutated genes also include the nuclear receptor corepressor complex encoding BCOR, GPS2, LDB1, GABRG1, and NCOR2.39, 48
Mutations of various genes are almost exclusively specific to the adult subgroup, including alterations in BRPF1/3 associated with SMO mutations or CREBBP and KDM3B in PTCH1‐mutated tumors.26 Fifty‐four percent of adults and 7% of pediatric samples carry DDX3X mutations affecting RNA metabolism.26 Telomerase reverse transcriptase (TERT) promoter mutations that drive telomerase activity occur with high frequency in adult SHH‐MBs (83% of older patients),49 including the most‐frequent C228T and the less‐frequent C250T variants.26 TERT‐mutant SHH tumors carry very few previously described SCNAs and are mostly mutually exclusive with 10q loss, possibly underlying the comparatively favorable prognosis.49 Recurrent mutations of the PI3K/AKT/mTOR pathway involving PIK3CA, PTEN, and PIK3C2G mutations also occur mainly in adult patients, with a surprisingly high frequency: pathway activation based on p‐AKT and p‐S6 positivity assessed by IHC was identified in about 30% of adult SHH patients, with a strong association with poor outcome.26 We summarize the most common SHH‐MB‐specific genetic alterations in Table S1.
High expression of genes often cannot be traced back to specific mutations or chromosome aberrations, instead, might be regulated by epigenetic mechanisms, as methylation events are more common compared to sequence mutations. For example, high expression of cMET in SHH‐MBs is not linked to recurrent mutations or amplifications, but is among the most frequently hypo‐ or hypermethylated genes in MBs,50 also associated with prognostic significance.51
Molecular classification
According to the consensus of the International Medulloblastoma Working Group, two of four requirements are suggested to be met for verified SHH activation: (1) GAB1 immunoreactivity, (2) SHH‐signaling‐specific mutation, (3) methylation profiling, or (4) gene expression profiling consistent with SHH activation either based on genome‐wide arrays or focused gene expression panels.38, 52 Combined immunoreactivity for GAB1, YAP1, and filamin A distinguishes WNT‐ and SHH‐MBs from Group 3 and 4 MBs, but GAB1 immunoreactivity characterizes only SHH‐activated tumors.25 The NanoString 22 gene signature array determines SHH‐pathway activation based on the differential expression of five genes (PDLIM3, EYA1, HHIP, ATOH1, and SFRP1) optimized for FFPE samples.38 A second diagnostic multigene signature utilizes the expression of eight genes (BCHE, GLI1, ITIH2, MICAL1, PDLIM3, PTCH2, RAB33A, and SFRP1) for SHH‐subgroup identification.53 Another unique five‐gene signature (GLI1, SPHK1, SHROOM2, PDLIM3, and OTX2) identifies SHH‐MBs even in the absence of SHH‐pathway mutations.54 The signature developed by Shou et al. detects all SHH‐MBs, but unable to predict the response to SMO inhibitors26 (Table 1).
Table 1.
Identifying subgroup affiliation in childhood medulloblastomas
| WNT | SHH | Group 3 | Group 4 | Source | Comments | Ref. | |
|---|---|---|---|---|---|---|---|
| IHC | Nuclear β‐catenin | GAB1 | FFPE | Least robust: nuclear β‐catenin present in other subgroups | 25 | ||
| DKK1 | SFRP1 | NPR3 | KCNA1 | FFPE | 12 | ||
| Gene expression | WIF1, DKK2, TNC, CCDC46, PYGL | BCHE, GLI1, ITIH2, MICAL1, PDLIM3, PTCH2, RAB33A, SFRP1 | n/a | n/a | Frozen | 13‐gene multiplex mRNA expression assay | 53 |
| n/a | GLI1, SPHK1, SHROOM2, PDLIM3, OTX2 | n/a | n/a | Fresh frozen and FFPE | 54 | ||
| WIF1, DKK2, TNC, GAD1, EMX2 | PDLIM3, EYA1, HHIP, ATOH1, SFRP1 | IMPG2, GABRA5, EGFL11, NRL, MAB21L2, NPR3 | KCNA1, EOMES, KHDRBS2, RBM24, UNC5D, OAS1 | FFPE | 38 | ||
| Mutation | CTNNB1 exon 3 | 52 | |||||
| Structural aberrations | Chromosome 6 monosomy (FISH or DNA copy number array) | 52 | |||||
| Methylation | Illumina Infinium HumanMethylation 450 BeadChip array | Fresh frozen and FFPE | Also for copy number profiling | 121 | |||
Subtypes of SHH‐Activated MBs
Based on the presence of metastatic dissemination, TP53‐mutation, and MYCN amplification, the proposed consensus stratifies SHH‐MBs into three risk categories. Standard risk tumors are nonmetastatic and harbor wild‐type TP53 and no MYCN amplification. Metastatic and/or MYCN‐amplified tumors belong to the high‐risk category, while very‐high‐risk tumors contain either somatic or germline TP53‐mutations55 (Fig. 2). Amplification of MYCN is generally restricted to SHH and Group 4 patients but only associates with poor prognosis in SHH‐MBs and is frequently accompanied by TP53‐mutations.14, 15, 26, 56
Figure 2.
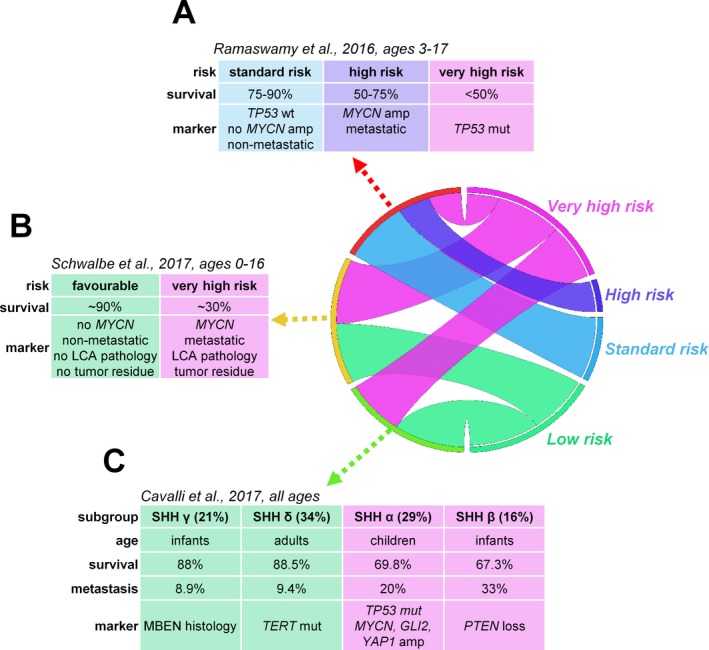
Risk stratification of SHH‐activated medulloblastomas including prognostic biomarker candidates across all age groups as defined by Ramaswamy et al. (2016) (A), Schwalbe et al. (2017) (B) and Cavalli et al. (2017) (C).
Additional prognostic biomarkers, such as GLI2 amplification and LCA histology, strongly affect outcomes in pediatric samples, but are extremely unusual in adult tumors.15, 26 A large‐scale cytogenetic study analyzing biomarkers in a subgroup‐specific manner identified GLI2 amplification, 14q loss, and leptomeningeal dissemination, but not anaplastic histology, as key biomarkers differentiating clinically high‐ and low‐risk SHH‐MBs.14
Integration of methylation‐ and gene expression‐based data suggests four subtypes: two in infants and one each in children and adults. SHH α appears primarily in children aged 3–16 years, has the worst prognosis of all SHH‐MBs, harbors TP53‐mutations, MYCN and GLI2 amplifications, 9q loss, 10q loss, 17p loss, and YAP1 amplifications. TP53‐ mutations are highly prognostic only within this subtype. SHH γ is present among infants and is enriched for MBEN histology, representing a low‐risk group potentially suitable for therapy de‐escalation. SHH β is also present in infants with high rates of metastasis and associates with focal PTEN deletions with worse survival compared to SHH γ. SHH δ is mainly present in adults and is associated with TERT promoter mutations and a favorable prognosis19 (Fig. 2). A methylomic profile‐based analysis divided SHH‐activated childhood MBs into infant and children subtypes with a cutoff at 4.3 years of age. In children, the presence of MYCN amplification, LCA pathology, metastasis, and incomplete resection separated very‐high‐risk disease from tumors with a favorable outcome20 (Fig. 2). Two methylation subtypes were identified in infants with markedly different molecular alterations and prognosis in another study: 5‐year progression‐free survival was 27.8% for the iSHH‐I‐subtype that harbored SUFU alterations and chromosome (chr) 2 gains versus 75.4% for the iSHH‐II‐subtype characterized by SMO mutations and chr9q deletions. Patients in the latter subtype profited from a radiation‐free therapy, supporting the validity of a molecularly driven treatment approach.57 However, inspecting subtype‐specific biomarkers reveals that iSHH‐I and iSHH‐II likely correspond to the already described SHH β and SHH γ subtypes. Additional discrepancy across subclassifications stems from diverse patient populations (only children in20) utilized data types, and clustering methods. Prospective clinical trials are in great demand to identify biomarkers suitable for effective patient stratification.
Proteomics reveal novel stratifications and translational opportunities
Genomic studies provide invaluable insight into differences in cancer biology and outcome across MB subgroups. Nevertheless, translation of the proposed findings to subgroup‐specific therapies remains difficult. Recent proteomic analyses recapitulated genomic subgrouping, what is more uncovered posttranscriptional MB heterogeneity not evident in the genome or the transcriptome.58, 59 Since proteomics are more representative of cellular biology, the approach is well suited to identify functionally relevant therapeutic targets.
Heterogeneous transcriptional patterns from untreated SHH, Group 3, and Group 4 MB samples converged only on two distinct protein‐signaling profiles that partially overlap with molecular subgroups. The profile with MYC‐like signaling encompasses all of SHH‐activated and the majority of Group 3 samples, and is associated with a rapid death postrecurrence. The rest of Group 3 and the bulk of Group 4 tumors show enrichment of DNA damage/apoptosis/neuronal signaling.60 Protein profiling uncovered putative targets for MBs with MYC‐like profile, including the inhibition of cell cycle progression and protein synthesis.60
Protein analysis resulted in an alternative subdivision of SHH‐MBs compared to the age‐based split according to expression‐ and methylation‐based studies.19, 20 The majority of adult samples clustered in the SHHa, while pediatric samples were split between SHHa and SHHb subtypes. SHHa signatures contained elevated SOX2, a regulator of neural progenitors, and mutations of PTCH1, TERT, and PRKAR1A consistent with canonical SHH‐pathway activation. Proteins in SHHb presented upregulation of calcium, glutamate, and RAS‐signaling pathways and elevated CD47, 58 more characteristic to Group 4 than to SHH‐MBs.19 CD47 is a membrane protein that blocks macrophages from destroying tumor cells,61 and the anti‐CD47 antibody, Hu5F9‐G4, demonstrated therapeutic efficacy in PDX models of Group 3 MBs.62 Hence, focusing at posttranscriptional alterations reveals functionally relevant novel mechanisms of tumorigenesis with translational potential. An integrative approach incorporating data from various “omics” – including protein profiling – would yield a more complete understanding of cancer biology, therefore collaborative initiatives facilitating data sharing are much desired.
Preclinical Models of SHH‐MB Reveal the Mechanisms of Tumorigenesis
Preclinical models provide invaluable tools to study biological mechanisms underlying MB development and for evaluating new therapies. Several SHH cell lines have been established, such as DAOY, UW228, UW426, ONS‐76, with confirmed subgroup identity based on transcriptional profiling.63 Out of the four SHH‐MB cell lines, two are TP53‐mutant, DAOY and UW228, reflecting that more aggressive subtypes are either easier to grow in cultures or more aggressive cells are selected and enriched in vitro.63
Cell cultures and culture‐derived allografts do not inevitably replicate the phenotypes of the original disease. Compared to allografts derived from original tumors with activated SHH‐signaling, dependence on SHH‐pathway activation is rapidly lost in cultured tumor cells and would not be restored when these cells are transplanted back to nude mice.64 The tumor microenvironment, particularly tumor‐associated astrocytes are starting to emerge as key components promoting SHH‐pathway activation in vivo.65 Altered in vitro signaling activity has therapeutic consequences, therefore preclinical models should be tested for how well they represent the original disease. Transplantation cancer models utilizing direct allografts maintain the genetic and histological profiles of original tumors, presumably better modeling patient responses to rational therapies.64
Several preclinical mouse models of MB recapitulate the development of SHH‐activated tumors, essentially through modeling SHH‐pathway dysregulation. SHH‐MBs arise from granule cells, yet they also develop in mice from granule neuron precursors of the cochlear nuclei.33 Approximately 10–20% of mice with a single‐allele knockout for the Ptch1 gene (Ptch1 +/−), a negative inhibitor of the Smo pathway (Fig. 3), develop cerebellar MBs.66 However the majority of CGNPs with Ptch1 loss will eventually differentiate into mature neurons,67 and the relatively low proportion of developing tumors indicates that, besides mutational activation of the SHH pathway, additional events are necessary for MB formation. For example, external beam irradiation considerably increases the tumor incidence rate (~80%).68, 69 Following this first attempt, various models emerged that crossed Ptch1 +/− with other aberrations, including disruption of DNA repair (DNA ligase IV loss) or cell cycle regulation (KIP1, INK4C, or INK4D inactivation) in conjunction with TP53 dysfunction.70, 71, 72, 73, 74 Nevertheless, simultaneous mutations in these pathways are rare in human SHH‐MBs. Constitutive activation of Smoothened (SmoA1) in mouse CGNPs resulted in highly penetrant tumors with leptomeningeal metastasis with 48% incidence rates, along with increased expression of both Sonic hedgehog (Gli1, NMyc) and Notch (Notch2 and Hes5) target genes.75, 76
Figure 3.
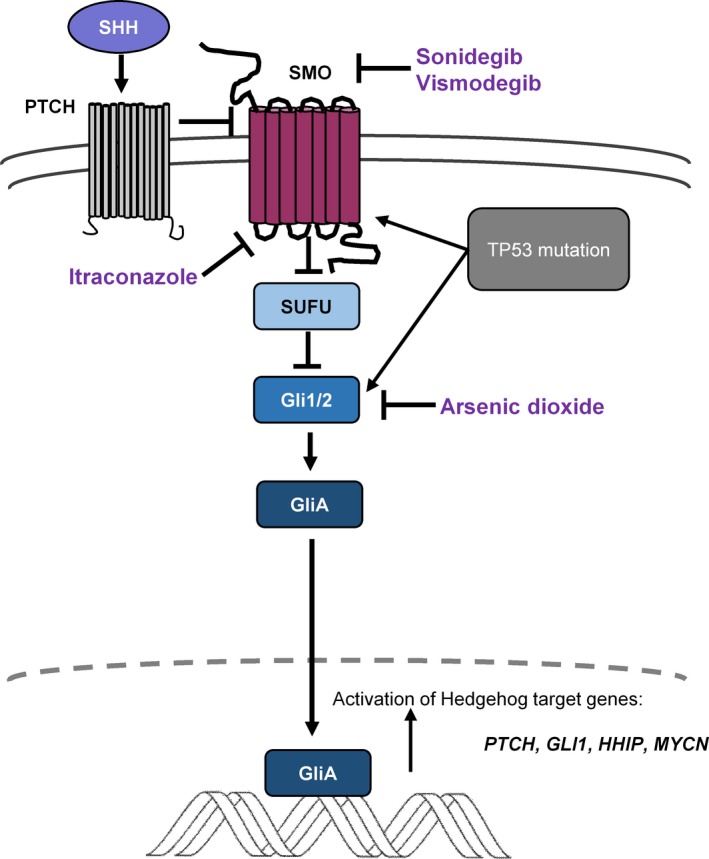
Sonic hedgehog signaling is crucial during normal development of cerebellum, and its dysregulation leads to medulloblastoma development. Mutations of PTCH1,SMO, and SUFU, or amplification of GLI2 contribute to downstream activation of Hedgehog signaling targets. Several small molecule inhibitors of SMO, such as sonidegib (LDE‐225) or vismodegib (GDC‐0449), are being investigated as potential targeted therapies in clinical trials. Mutations downstream of SMO render such inhibitors ineffective. Itraconazole has the ability to inhibit SMO activation including some SMO mutations that confer resistance to SMO inhibitors, and arsenic trioxide inhibits GLI2 ciliary accumulation.
Mouse models of MB utilizing the Sleeping Beauty (SB) transposon system provide excellent tools to discern driver events of tumorigenesis.77 An unbiased SB transposon‐based in vivo screen confirmed that a single‐allele loss of MyoD accelerated SHH‐MB formation, supporting the role of MyoD as a novel tumor suppressor in SHH‐MBs.78 A subsequent SB transposon‐mediated insertional mutagenesis screen in single‐allele Ptch1‐knockout mice identified transcription factor Nfia as a driver of SHH‐activated MB development, as reduced Nfia conjoined with SHH‐signaling perturbations.79 In the Ptch1 +/‐ tumor model, whole‐body SB mutagenesis activated a gene network of neuronal transcription factors associated with increased proliferation and decreased differentiation. Activity of this network was mostly driven by Pten and Mytil expression and associated with metastatic disease and poor survival outcome in human subjects, especially in SHH‐MBs. Network mutations converged on and increased the Igf2 expression critical for CGNP proliferation and tumor formation.80
Activation of SHH releases inhibition of Smo by the transmembrane receptor Ptch1 to activate Gli1/2 transcription activators (GLIA), Gli3, a transcriptional repressor, and downstream mitogenic genes such as Ccnd1 and Mycn (Fig. 3). In mice, Gli1 functions as an oncogene during MB development: inactivation of both Gli1 alleles significantly reduces the incidence of spontaneous MB in Ptch1 +/− mice.81 Hdac‐mediated deacetylation of Gli1 via Hdac1 enzyme promotes transcriptional activation and enhances SHH‐signaling. Members of the Kcash (Kctd containing, Cullin3 adaptor, suppressor of Hedgehog) gene family, coding for potassium channel tetramerization domains, are negative regulators of Hdac1 activity by promoting its ubiquitination, resulting in Gli1 acetylation. Kcash‐s are downregulated in human primary MBs, and their overexpression leads to growth suppression of MB cell lines. Kcash‐s may represent a novel endogenous agent capable of restraining SHH‐pathway activation.82
Aggressive MBs associated with poor prognosis express high levels of ATOH1, a critical transcriptional factor required for the differentiation of cerebellar granular cells during normal brain development. ATOH1 is usually absent after the first year of life.83 Deletion of Atoh1 prevents cerebellar neoplasia development in mice.84 In the Nd2:SmoA1 mouse MB model, Atoh1 protein reduction markedly decreases MB incidence and increases survival.85 Atoh1 has a central role in the regulation of Gli2, a main transcriptional effector with an important role in modulating proliferation.84 Jak2‐mediated phosphorylation increases Atoh1 transcriptional activity, thus inhibiting Jak2‐mediated tyrosine phosphorylation may provide a new mechanism to regulate MB formation.85
Low expression of GNAS has been strongly associated with decreased overall survival and appears as a potent tumor suppressor gene in SHH‐MBs.86 GNAS encodes the heterotrimeric Gs protein alpha‐subunit (Gsα) and contributes to signal transduction from the extracellular environment and controls motility, cell growth, and survival.87 Gnas‐knockout mice develop MB resembling human tumors in the cerebellum, associated with the upregulation of SHH‐pathway components, such as Gli1, Gli2, Ptch1, and CyclinD1 and widespread expression of granule precursor markers, such as Zic1 and Atoh1. Low GNAS levels due to inactivating mutations define a subset of SHH‐MBs with an aggressive phenotype and are suggested to be potential prognostic markers for treatment stratification.86
Increased expression of Gli1 is a hallmark of elevated SHH‐signaling, however not all hedgehog‐pathway activation requires Gli1. The tumor microenvironment is gaining increasing importance in SHH‐pathway activation. In a Ptch1‐deficient mouse model, the intermediate filament protein Nestin is required for MB formation via binding and inactivating Gli3.88 Nestin is expressed in mature tissues in pathological situations when developmental programs are recapitulated and is also a marker for neural stem cells.89 Gli3 is a transcriptional repressor that is proteolytically processed to its truncated form (Gli3R) and acts predominantly as a negative regulator of SHH‐signaling. Progressively increasing Nestin levels during MB development impair proteolytic processing of Gli3, abolishing its inhibitory function. The Nestin‐Gli3 interaction augments SHH‐signaling activation in the absence of Ptch1, leading to tumorigenesis.88 Tumor‐associated astrocytes (TAA) are multifunctional specialized glial cells abundant in MBs.65 TAAs secret a functional SHH ligand to the microenvironment that promotes cellular proliferation of MBs by inducing expression of Nestin. Targeting TAAs or the expressed Shh ligand may be an alternative treatment strategy against SHH‐dependent MBs.65
In summary, despite the large number of murine models approximating human SHHs, the field lacks adequate representation of the full spectrum of the disease. Infant and adult SHH‐MBs represent distinct subtypes with different gene expression patterns, genetic features, locations, and clinical outcomes.12 Based on genomic and transcriptional analyses, model systems utilizing Ptch1 and Smo mutations match to human MBs well,90 but are suggested to be more similar to adult tumors compared to infant SHH‐MBs.90 Mouse models of infant SHH‐MBs are needed for the development of more successful therapeutic approaches for this age group.
Prospective Therapeutic Opportunities
SMO inhibitors
Smoothened (SMO) regulates the suppression of SUFU91; thus, potential SMO inhibitors could prevent SUFU activation and translocation of GLI proteins to the nucleus (Fig. 3). Several small molecule inhibitors of SMO, such as sonidegib (LDE‐225) or vismodegib (GDC‐0449), are being investigated as potential agents in clinical trials,92, 93 including ongoing trial NCT01878617, and demonstrate particular efficacy in relapsed adult SHH‐MBs.93, 94 Approximately 80% of adult patients carry either PTCH1 or SMO mutations, rendering them to be likely responders to SMO inhibition. However, infants and children frequently harbor mutations downstream of SMO and thus may be resistant.26 GLI2 and MYCN amplification or SUFU‐driven SHH‐signaling have consequences for targeted therapy. SMO inhibitors act upstream of these genes; thus, GLI2‐ and MYCN‐amplified tumors might be refractory to agents targeting SMO.26, 95, 96 Resistance against SMO inhibitors rapidly develops, and its mechanisms are under intense research.97 De novo resistance is linked to SMO mutations (such as D473H or E518), but resistance can also develop downstream of SMO.94, 96 Finally, SMO inhibitions have serious side effects in pediatric patients because they inhibit bone and teeth development: vismodegib‐treated children developed widespread growth plate fusions that persisted long after therapy cessation.98 Drug‐resistant SMO mutations highlight the need for new therapies in SHH‐MB. Itraconazole has the ability to inhibit SMO activation, including some SMO mutations that confer resistance to SMO inhibitors. Arsenic trioxide inhibits GLI2 ciliary accumulation (Fig. 3). These two drugs alone or in combination inhibit in vitro cell growth and in vivo MB development in mice bearing wild‐type or SMO mutations and prolong the survival of mice with SMO inhibitor‐resistant medulloblastomas. Their combined administration seems to be effective against all known SMO mutations.99
Statins
Cholesterol is an essential component of plasma membranes, and its homeostasis is regulated by a tight network of proteins. Enhanced expression of genes related to cholesterol biosynthesis has been identified in SHH‐MBs.100 Statin treatment promotes differentiation and inhibited proliferation in MB cells isolated from Ptch1 +/− mice indicating that cholesterol is an essential component of MB progression. Cholesterol and vismodegib bind SMO at different binding sites, leading to synergistic effects on tumor cells: inhibition of cholesterol biosynthesis by statins alone or in combination with vismodegib repressed in vivo SHH‐MB proliferation and growths in subcutaneous allograft tumors of Ptch1 +/− mice.100 The efficacy of statins depends on SHH‐pathway mutations: MBs with mutations in SmoM2 or downstream of SMO, such as in SUFU or GLI2 are intrinsically resistant to SMO‐ and cholesterol inhibitors. Potential combination of statins with vismodegib may lead to reduced dosing of the latter to prevent serious side effects in pediatric populations. Targeting cholesterol biosynthesis characterizes a promising alternative treatment strategy for a subset of SHH‐MBs, nevertheless additional preclinical studies are required prior to their clinical use.100
BET bromodomain inhibitors
Genomic amplifications of zinc finger transcription factors GLI2 and GLI1 of the SHH‐signaling pathway have been associated with more aggressive disease.95, 101 BRD4 and other BET bromodomain proteins are critical regulators of GLI1 and GLI2 transcription downstream of SMO and SUFU. BET bromodomain inhibitors, such as JQ1, target BRD4 and significantly decrease tumor cell viability both in vitro and in vivo in genetically engineered mouse models, even when genetic lesions predispose tumors to resistance against SMO inhibitors.102
AURK or PLK inhibitors
A population of cells in the Ptch1 heterozygous mice model of MB that express surface carbohydrate antigen CD15/SSEA‐1 augment tumor propagation following transplantation. These cells display increased expression of genes responsible for G2 and M phase regulation throughout mitosis. CD15 is expressed in a subset of human MBs associated with poor prognosis. Inhibition of G2/M regulators, such as Aurora kinases (AURK) or Polo‐like kinases (PLK), reduces proliferation in vitro and tumor growth in vivo, and thus may represent a novel approach to treating CD15+SHH‐MBs.103 In addition, a class of Aurora kinase inhibitors may also disrupt the native conformation of Aurora A and, as a consequence, drive the proteolytic degradation of MYCN protein.104
cMET inhibitors
Aberrant MET‐signaling is involved in metastasis development in various solid tumors and is upregulated in primary SHH‐MBs,50 plus phosphorylated MET kinase‐activity correlates with MB recurrence and poor survival.51 A MET inhibitor, foretinib, induced apoptosis and inhibited migration and invasion in SHH‐MB cell lines, induced tumor regression, and prevented leptomeningeal metastases in mouse xenografts.51
Targeting stem‐like cells
Quiescent, therapy‐resistant cells serve as a reservoir for relapse. In a model of SHH‐MB, cells expressing the neural stem cell marker Sox2 comprised less than 5% of the tumor but drove tumor growth after antimitotic chemotherapy and SMO inhibition.68 Higher expression of stem cell genes is associated with worse outcome in numerous malignancies.105, 106 Likewise, high SOX2 immunoreactivity is linked to worse survival in SHH patients.68 Mithramycin, an agent against SOX2+ cells, reduced the proliferation of human‐derived primary SHH‐MB cell lines and growth of subcutaneous allografts.68
Transducing NMyc in embryonic cerebellar cells induced SHH‐activated MB formation with the contribution of transcription factor SOX9.107 SOX9 regulates stem cell properties, differentiation, proliferation, and survival and is commonly elevated in WNT‐ and SHH‐MBs, particularly during early tumor initiation in a WNT/β‐catenin‐dependent manner.108 FBW7, a protein participating in substrate recognition, regulates posttranslational regulation, particularly degradation of SOX9.109 FBW7 is frequently mutated in SHH‐MBs, and its expression is downregulated across all MB subtypes. Lower levels of FBW7 are associated with increased quantity of SOX9 protein, metastasis, and poor survival. SOX9 abundance reduced the efficacy of cisplatin treatment both in vitro and in vivo. Activation of PI3K/AKT/mTOR signaling is associated with poor survival in adult SHH‐MBs.26 Pharmacological inhibition of PI3K/AKT/mTOR pathway activity combined with cisplatin treatment stimulates FBW7 expression, degrades SOX9 protein levels, and increases apoptosis, providing a potential new opportunity to treat recurrent, chemoresistant MB patients.110 In an Nd2:SmoA1 transgenic mouse model of MB, tumor‐propagating CD15+ cells were regulated by Pten and PI3K signaling and displayed sensitivity to pan‐PI3K inhibitor both in vivo and in vitro but remained resistant to chemotherapy.111
The chemokine ligand CXCL12 and its receptor CXCR4 are expressed in brain tumors (including medulloblastomas), and their expression is associated with poor prognosis.112, 113, 114 CXCL12 and CXCR4 are potentially important coactivators of SHH‐signaling: SHH‐CXCR4 coactivated tumors express higher levels of ATOH1 and cyclin D1 and exhibit maximal tumor growth.114 Targeting Cxcr4 alone with the infusion of small molecule antagonist AMD 3100 (plerixafor) inhibits growth of intracranial MB xenografts by decreasing cellular proliferation and increasing apoptosis.115 Combined Shh and Cxcr4 antagonism by vismodegib and plerixafor results in a synergistic antitumoral effect in xenografts injected with SmoA1‐derived primary tumor cells by specifically targeting the MB stem‐like cell pool, revealed by decreased expression of stem cell markers Bmi1 and Sox2.116
Immunotherapy
Immunological differences within the tumor microenvironment across MB subtypes suggest different regulatory mechanisms and determine possible immunotherapeutic strategies. Increased expression of inflammation‐related genes and elevated infiltration of tumor‐associated macrophages (TAM) were described in SHH‐MBs compared to Group3 and Group 4 tumors.117 Likewise, increased frequencies of CD4+ and CD8+ T cells, myeloid cells, and dendritic cells were identified in Ptch1 +/− SHH‐MB‐bearing mice compared to another model of MYC‐amplified MBs, characterized by a higher proportion of CD8+ PD‐1+ double positive T cells.118 The higher percentage of infiltrating myeloid‐derived suppressor cells and TAMs in SHH‐MBs suggests an immunologically suppressive tumor microenvironment. Accordingly, treatment with either anti‐CTLA‐4 alone, anti‐PD‐1 alone, or in combination was not effective in Ptch1 +/− SHH‐MB‐bearing mice but showed survival benefits in MYC‐amplified tumor bearing animals.118
There is a pronounced urgency for alternative therapeutic modalities in pediatric brain malignancies. Identification of differentially expressed cell surface markers between tumor and normal cells may lead to novel immunotherapeutic strategies. The tumor‐associated antigen, PRAME, is detected in the majority of MB samples but not in normal tissue, and high PRAME expression correlates with worse survival.119 Genetically modified T cells directed toward the PRAME antigen both in vitro and in vivo were effective against multiple HLA‐A*02+ MB cell lines, including DAOY cells. Adoptive immunotherapy targeting PRAME represents a promising innovative approach for patients with HLA‐A*02+ MBs.119 Comparing high‐risk neuroblastomas and normal tissues identified differential expression of tumor‐specific cell surface molecule, Glypican 2 (GPC2), an extracellular proteoglycan signaling coreceptor, required for cellular proliferation. A GPC2‐directed antibody‐drug conjugate, D3‐GPC2‐PBD was effective against neuroblastoma cells in vitro in an antigen‐ and concentration‐dependent manner, and in vivo in murine PDX models.120 GPC2 is also highly expressed both in primary and metastatic MBs, and similarly to neuroblastomas, MBs express the GPC2 transcript. GPC2 expression is highest among Group 4 MBs, but the relatively high expression across SHH‐ and Group 3 MBs suggests its potential relevance in other subtypes as well.120
Conclusions
An enormous amount of data on the genetic background of SHH‐activated MBs have accumulated in the past decade. While subgroup affiliation still does not provide reliable prediction of therapy response, emerging models offer more layered patient stratification. A more collective approach could accelerate translation of new insights into clinical practice. Collaborative efforts with improved communication, material, and data sharing could permit the development of better tailored preclinical models for basic biology studies and therapeutic development, and facilitate the integration of multilayer (genomic, epigenetic, proteomic, and metabolomic) molecular data to uncover novel disease biomarkers. Rational clinical trial design with the incorporation of available molecular understanding, focusing especially on high‐risk patients are inevitable and may bring the much‐sought‐after breakthrough in the stagnant survival rates of the past decades.
Conflicts of Interest
The authors have declared no conflicts of interest.
Supporting information
Table S1. Frequent genetic alterations in SHH‐activated medulloblastomas according to 12, 22, 37, 38, 46, 48, 53, 54. % patient values refer to within subgroup percentages.
Funding Information
The study was supported by the NVKP_16‐1‐2016‐0037, 2018‐1.3.1‐VKE‐2018‐00032 and KH‐129581 grants of the National Research, Development and Innovation Office, Hungary.
Funding Statement
This work was funded by National Research, Development and Innovation Office, Hungary grants NVKP_16‐1‐2016‐0037, 2018‐1.3.1‐VKE‐2018‐00032, and KH‐129581.
References
- 1. Ward E, DeSantis C, Robbins A, et al. Childhood and adolescent cancer statistics, 2014. Cancer J Clin 2014;64:83–103. [DOI] [PubMed] [Google Scholar]
- 2. Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pizer BL, Clifford SC. The potential impact of tumour biology on improved clinical practice for medulloblastoma: progress towards biologically driven clinical trials. Br J Neurosurg 2009;23:364–375. [DOI] [PubMed] [Google Scholar]
- 4. Pui CH, Gajjar AJ, Kane JR, et al. Challenging issues in pediatric oncology. Nat Rev Clin Oncol 2011;8:540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smoll NR. Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs). Cancer 2012;118:1313–1322. [DOI] [PubMed] [Google Scholar]
- 6. Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children's Cancer Group 921 randomized Phase III Study. J Clin Oncol 1999;17:832–845. [DOI] [PubMed] [Google Scholar]
- 7. Rutkowski S, Bode U, Deinlein F, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 2005;10:978–986. [DOI] [PubMed] [Google Scholar]
- 8. Grill J, Sainte‐Rose C, Jouvet A, et al. Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol 2005;6:573–580. [DOI] [PubMed] [Google Scholar]
- 9. Geyer JR, Sposto R, Jennings M, et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol 2005;20:7621–7631. [DOI] [PubMed] [Google Scholar]
- 10. Leary SES, Olson JM. The molecular classification of medulloblastoma: driving the next generation clinical trials. Curr Opin Pediatr 2012;24:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Armstrong GT. Long‐term survivors of childhood central nervous system malignancies: the experience of the childhood cancer survivor study. Eur J Paediat Neurol 2010;14:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Northcott PA, Hielscher T, Dubuc A, et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol 2011;122:231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016;131:803–820. [DOI] [PubMed] [Google Scholar]
- 14. Shih DJ, Northcott PA, Remke M, et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 2014;32:886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: an international meta‐analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 2012;123:473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ellison DW, Kocak M, Dalton J, et al. Definition of disease‐risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol 2011;29:1400–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 2011;29:1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012;123:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 2017;31:737–754.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwalbe EC, Lindsey JC, Nakjang S, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a Cohort study. Lancet Oncol 2017;18:958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gajjar AJ, Robinson GW. Medulloblastoma‐translating discoveries from the bench to the bedside. Nat Rev Clin Oncol 2014;11:714–722. [DOI] [PubMed] [Google Scholar]
- 22. Northcott PA, Buchhalter I, Morrissy AS, et al. The whole‐genome landscape of medulloblastoma subtypes. Nature 2017;547:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Remke M, Hielscher T, Northcott PA, et al. Adult medulloblastoma comprises three major molecular variants. J Clin Oncol 2011;29:2717–2723. [DOI] [PubMed] [Google Scholar]
- 24. Zhukova N, Ramaswamy V, Remke M, et al. Subgroup‐specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 2013;31:2927–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non‐SHH/WNT molecular subgroups. Acta Neuropathol 2011;121:381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kool M, Jones DT, Jager N, et al. Genome sequencing of SHH medulloblastoma predicts genotype‐related response to smoothened inhibition. Cancer Cell 2014;25:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leary SE, Zhou T, Holmes E, et al. Histology predicts a favorable outcome in young children with desmoplastic medulloblastoma: a report from the children's oncology group. Cancer 2011;117:3262–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rutkowski S, Gerber NU, von Hoff K, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy and deferred radiotherapy. Neuro‐Oncol 2009;11:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rutkowski S, von Hoff K, Emser A, et al. Survival and prognostic factors of early childhood medulloblastoma: an international meta‐analysis. J Clin Oncol 2010;28:4961–4968. [DOI] [PubMed] [Google Scholar]
- 30. Cohen BH, Geyer JR, Miller DC, et al. Pilot study of intensive chemotherapy with peripheral hematopoietic cell support for children less than 3 years of age with malignant brain tumors, the CCG‐99703 Phase I/II Study. A report from the Children's Oncology Group. Pediat Neurol 2015;53:31–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lafay‐Cousin L, Smith A, Chi SN, et al. Clinical, pathological, and molecular characterization of infant medulloblastomas treated with sequential high‐dose chemotherapy. Pediatr Blood Cancer 2016;63:1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lafay‐Cousin L, Bouffet E, Onar‐Thomas A, et al. ACxml: a Phase II study for the treatment of non metastatic desmoplastic medulloblastoma in children less than 4 years of age—a report from the Children Oncology Group. J Clin Oncol 2017;35(15_Suppl):10505. [Google Scholar]
- 33. Grammel D, Warmuth‐Metz M, von Bueren AO, et al. Sonic hedgehog‐associated medulloblastoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol 2012;123:601–614. [DOI] [PubMed] [Google Scholar]
- 34. Gibson P, Tong Y, Robinson G, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010;468:1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wallace VA. Purkinje‐cell‐derived sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 1999;9:445–448. [DOI] [PubMed] [Google Scholar]
- 36. Dahmane N, Ruiz‐i‐Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 1999;126:3089–3100. [DOI] [PubMed] [Google Scholar]
- 37. Jones DT, Jager N, Kool M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012;2:100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Northcott PA, Shih DJ, Remke M, et al. Rapid, reliable, and reproducible molecular sub‐grouping of clinical medulloblastoma samples. Acta Neuropathol 2012;123:615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robinson G, Parker M, Kranenburg TA, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012;488:43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Waszak SM, Northcott PA, Buchhalter I, et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol 2018;19:785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smith MJ, Beetz C, Williams SG, et al. Germline mutations in SUFU cause Gorlin syndrome‐associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 2014;20:4155–4161. [DOI] [PubMed] [Google Scholar]
- 42. Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012;148:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tchelebi L, Ashamalla H, Graves PR. Mutant p53 and the response to chemotherapy and radiation. Sub‐Cell Biochem 2014;85:133–159. [DOI] [PubMed] [Google Scholar]
- 44. Thalakoti S, Geller T. Basal cell nevus syndrome or Gorlin syndrome. Handb Clin Neurol 2015;132:119–128. [DOI] [PubMed] [Google Scholar]
- 45. Foulkes WD, Kamihara J, Evans DGR, et al. Cancer surveillance in gorlin syndrome and rhabdoid tumor predisposition syndrome. Clin Cancer Res 2017;23:e62–e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 2006;24:1924–1931. [DOI] [PubMed] [Google Scholar]
- 47. Corrales JD, Rocco GL, Blaess S, et al. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development (Cambridge, England) 2004;131:5581–5590. [DOI] [PubMed] [Google Scholar]
- 48. Pugh TJ, Weeraratne SD, Archer TC, et al. Medulloblastoma exome sequencing uncovers subtype‐specific somatic mutations. Nature 2012;488:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Remke M, Ramaswamy V, Peacock J, et al. TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol 2013;126:917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schwalbe EC, Williamson D, Lindsey JC, et al. DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin‐fixed biopsies. Acta Neuropathol 2013;125:359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Faria CC, Golbourn BJ, Dubuc AM, et al. Foretinib is effective therapy for metastatic sonic hedgehog medulloblastoma. Can Res 2015;75:134–146. [DOI] [PubMed] [Google Scholar]
- 52. Gottardo NG, Hansford JR, McGlade JP, et al. Medulloblastoma Down Under 2013: a report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol 2014;127:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schwalbe EC, Lindsey JC, Straughton D, et al. Rapid diagnosis of medulloblastoma molecular subgroups. Clin Cancer Res 2011;17:1883–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shou Y, Robinson DM, Amakye DD, et al. A five‐gene hedgehog signature developed as a patient preselection tool for hedgehog inhibitor therapy in medulloblastoma. Clin Cancer Res 2015;21:585–593. [DOI] [PubMed] [Google Scholar]
- 55. Ramaswamy V, Remke M, Bouffet E, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol 2016;131:821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Korshunov A, Remke M, Kool M, et al. Biological and clinical heterogeneity of MYCN‐amplified medulloblastoma. Acta Neuropathol 2012;123:515–527. [DOI] [PubMed] [Google Scholar]
- 57. Robinson GW, Rudneva VA, Buchhalter I, et al. Risk‐adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 2018;19:768–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Archer TC, Ehrenberger T, Mundt F, et al. Proteomics, post‐translational modifications, and integrative analyses reveal molecular heterogeneity within medulloblastoma subgroups. Cancer Cell 2018;34:396–410.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rivero‐Hinojosa S, Lau LS, Stampar M, et al. Proteomic analysis of medulloblastoma reveals functional biology with translational potential. Acta Neuropathol Commun 2018;6:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zomerman WW, Plasschaert SLA, Conroy S, et al. Identification of two protein‐signaling states delineating transcriptionally heterogeneous human medulloblastoma. Cell Rep 2018;22:3206–3216. [DOI] [PubMed] [Google Scholar]
- 61. Jaiswal S, Jamieson CH, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009;138:271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gholamin S, Mitra SS, Feroze AH, et al. Disrupting the CD47‐SIRPα anti‐phagocytic axis by a humanized anti‐CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med 2017;9:eaaf2968. [DOI] [PubMed] [Google Scholar]
- 63. Ivanov DP, Coyle B, Walker DA, Grabowska AM. In vitro models of medulloblastoma: choosing the right tool for the job. J Biotechnol 2016;236:10–25. [DOI] [PubMed] [Google Scholar]
- 64. Sasai K, Romer JT, Lee Y, et al. Shh pathway activity is down‐regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res 2006;66:4215–4222. [DOI] [PubMed] [Google Scholar]
- 65. Liu Y, Yuelling LW, Wang Y, et al. Astrocytes promote medulloblastoma progression through hedgehog secretion. Can Res 2017;77:6692–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science (New York, NY) 1997;277:1109–1113. [DOI] [PubMed] [Google Scholar]
- 67. Yang ZJ, Ellis T, Markant SL, et al. Medulloblastoma can be initiated by deletion of Patched in lineage‐restricted progenitors or stem cells. Cancer Cell 2008;14:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vanner RJ, Remke M, Gallo M, et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014;26:33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pazzaglia S, Tanori M, Mancuso M, et al. Linking DNA damage to medulloblastoma tumorigenesis in patched heterozygous knockout mice. Oncogene 2006;25:1165–1173. [DOI] [PubMed] [Google Scholar]
- 70. Lee Y, Miller HL, Jensen P, et al. A molecular fingerprint for medulloblastoma. Can Res 2003;63:5428–5437. [PubMed] [Google Scholar]
- 71. Uziel T, Zindy F, Xie S, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev 2005;19:2656–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ayrault O, Zindy F, Rehg J, et al. Two tumor suppressors, p27Kip1 and patched‐1, collaborate to prevent medulloblastoma. Mol Cancer Res 2009;7:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Can Res 2001;61:513–516. [PubMed] [Google Scholar]
- 74. Romer JT, Kimura H, Magdaleno S, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/‐)p53(‐/‐) mice. Cancer Cell 2004;6:229–240. [DOI] [PubMed] [Google Scholar]
- 75. Hallahan AR, Pritchard JI, Hansen S, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog‐induced medulloblastomas. Can Res 2004;64:7794–7800. [DOI] [PubMed] [Google Scholar]
- 76. Hatton BA, Villavicencio EH, Tsuchiya KD, et al. The Smo/Smo model: hedgehog‐induced medulloblastoma with 90% incidence and leptomeningeal spread. Can Res 2008;68:1768–1776. [DOI] [PubMed] [Google Scholar]
- 77. Wu X, Northcott P, Dubuc A, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012;482:529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dey J, Dubuc AM, Pedro KD, et al. MyoD is a tumor suppressor gene in medulloblastoma. Cancer Res 2013;73:6828–6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Genovesi LA, Ng CG, Davis MJ, et al. Sleeping Beauty mutagenesis in a mouse medulloblastoma model defines networks that discriminate between human molecular subgroups. Proc Natl Acad Sci USA 2013;110:E4325–E4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Łastowska M, Al‐Afghani H, Al‐Balool HH, et al. Identification of a neuronal transcription factor network involved in medulloblastoma development. Acta Neuropathol Commun 2013;1:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kimura H, Stephen D, Joyner A, Curran T. Gli1 is important for medulloblastoma formation in Ptc1 + /‐ mice. Oncogene 2005;24:4026–4036. [DOI] [PubMed] [Google Scholar]
- 82. De Smaele E, Di Marcotullio L, Moretti M, et al. Identification and characterization of KCASH2 and KCASH3, 2 novel Cullin3 adaptors suppressing histone deacetylase and hedgehog activity in medulloblastoma. Neoplasia (New York, NY) 2011;13:374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Salsano E, Pollo B, Eoli M, et al. Expression of MATH1, a marker of cerebellar granule cell progenitors, identifies different medulloblastoma sub‐types. Neurosci Lett 2004;370:180–185. [DOI] [PubMed] [Google Scholar]
- 84. Flora A, Klisch TJ, Schuster G, Zoghbi HY. Deletion of Atoh1 disrupts sonic hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science (New York, NY) 2009;326:1424–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Klisch TJ, Vainshtein A, Patel AJ, Zoghbi HY. Jak2‐mediated phosphorylation of Atoh1 is critical for medulloblastoma growth. eLife 2017;6:e31181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. He X, Zhang L, Chen Y, et al. The G‐protein alpha subunit Gsα is a tumor suppressor in sonic hedgehog‐driven medulloblastoma. Nat Med 2014;20:1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Neves SR, Ram PT, Iyengar R. G protein pathways. Science 2002;296:1636. [DOI] [PubMed] [Google Scholar]
- 88. Li P, Lee EH, Du F, et al. Nestin mediates hedgehog pathway tumorigenesis. Can Res 2016;76:5573–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Michalczyk K, Ziman M. Nestin structure and predicted function in cellular cytoskeletal organisation. Histol Histopathol 2005;20:665–671. [DOI] [PubMed] [Google Scholar]
- 90. Poschl J, Stark S, Neumann P, et al. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol 2014;128:123–136. [DOI] [PubMed] [Google Scholar]
- 91. Svard J, Heby‐Henricson K, Persson‐Lek M, et al. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian hedgehog signaling pathway. Dev Cell 2006;10:187–197. [DOI] [PubMed] [Google Scholar]
- 92. Rodon J, Tawbi HA, Thomas AL, et al. A phase I, multicenter, open‐label, first‐in‐human, dose‐escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res 2014;20:1900–1909. [DOI] [PubMed] [Google Scholar]
- 93. Robinson GW, Orr BA, Wu G, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog–subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC‐025B and PBTC‐032. J Clin Oncol 2015;33:2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC‐0449. N Engl J Med 2009;361:1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cho YJ, Tsherniak A, Tamayo P, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 2011;29:1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Dijkgraaf GJ, Alicke B, Weinmann L, et al. Small molecule inhibition of GDC‐0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Can Res 2011;71:435–444. [DOI] [PubMed] [Google Scholar]
- 97. Samkari A, White J, Packer R. SHH inhibitors for the treatment of medulloblastoma. Expert Rev Neurother 2015;15:763–770. [DOI] [PubMed] [Google Scholar]
- 98. Robinson GW, Kaste SC, Chemaitilly W, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017;8:69295–69302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kim J, Aftab BT, Tang JY, et al. Itraconazole and arsenic trioxide inhibit hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013;23:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gordon RE, Zhang L, Peri S, et al. Statins synergize with hedgehog pathway inhibitors for treatment of medulloblastoma. Clin Cancer Res 2018;24:1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Buczkowicz P, Ma J, Hawkins C. GLI2 is a potential therapeutic target in pediatric medulloblastoma. J Neuropathol Exp Neurol 2011;70:430–437. [DOI] [PubMed] [Google Scholar]
- 102. Tang Y, Gholamin S, Schubert S, et al. Epigenetic targeting of hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med 2014;20:732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Markant SL, Esparza LA, Sun J, et al. Targeting sonic hedgehog‐associated medulloblastoma through inhibition of Aurora and Polo‐like kinases. Cancer Res 2013;73:6310–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gustafson WC, Meyerowitz JG, Nekritz EA, et al. Drugging MYCN through an allosteric transition in Aurora Kinase A. Cancer Cell 2014;26:414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 2011;17:1086–1093. [DOI] [PubMed] [Google Scholar]
- 106. Merlos‐Suarez A, Barriga FM, Jung P, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011;8:511–524. [DOI] [PubMed] [Google Scholar]
- 107. Swartling FJ, Savov V, Persson AI, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N‐MYC. Cancer Cell 2012;21:601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Larsimont JC, Youssef KK, Sanchez‐Danes A, et al. Sox9 controls self‐renewal of oncogene targeted cells and links tumor initiation and invasion. Cell Stem Cell 2015;17:60–73. [DOI] [PubMed] [Google Scholar]
- 109. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008;8:83–93. [DOI] [PubMed] [Google Scholar]
- 110. Suryo Rahmanto A, Savov V, Brunner A, et al. FBW7 suppression leads to SOX9 stabilization and increased malignancy in medulloblastoma. EMBO J 2016;35:2192–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Singh AR, Joshi S, Zulcic M, et al. PI‐3K inhibitors preferentially target CD15 + cancer stem cell population in SHH driven medulloblastoma. PLoS ONE 2016;11:e0150836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bian XW, Yang SX, Chen JH, et al. Preferential expression of chemokine receptor CXCR1 by highly malignant human gliomas and its association with poor patient survival. Neurosurgery 2007;61:570–578; discussion 8–9. [DOI] [PubMed] [Google Scholar]
- 113. Calatozzolo C, Maderna E, Pollo B, et al. Prognostic value of CXCL12 expression in 40 low‐grade oligodendrogliomas and oligoastrocytomas. Cancer Biol Ther 2006;5:827–832. [DOI] [PubMed] [Google Scholar]
- 114. Sengupta R, Dubuc A, Ward S, et al. CXCR1 activation defines a new subgroup of sonic hedgehog–driven medulloblastoma. Cancer Res 2012;72:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Rubin JB, Kung AL, Klein RS, et al. A small‐molecule antagonist of CXCR117 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci 2003;100:13513–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ward SA, Warrington NM, Taylor S, et al. Reprogramming medulloblastoma‐propagating cells by a combined antagonism of Sonic Hedgehog and CXCR118. Can Res 2017;77:1416–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Margol AS, Robison NJ, Gnanachandran J, et al. Tumor‐associated macrophages in SHH subgroup of medulloblastomas. Clin Cancer Res 2015;21:1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Pham CD, Flores C, Yang C, et al. Differential Immune microenvironments and response to immune checkpoint blockade among molecular subtypes of murine medulloblastoma. Clin Cancer Res 2016;22:582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Orlando D, Miele E, De Angelis B, et al. Adoptive immunotherapy using PRAME‐specific T cells in medulloblastoma. Can Res 2018;78:3337–3349. [DOI] [PubMed] [Google Scholar]
- 120. Bosse KR, Raman P, Zhu Z, et al. Identification of GPC2 as an oncoprotein and candidate immunotherapeutic target in high‐risk neuroblastoma. Cancer Cell 2017;11:295–309.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hovestadt V, Remke M, Kool M, et al. Robust molecular subgrouping and copy‐number profiling of medulloblastoma from small amounts of archival tumour material using high‐density DNA methylation arrays. Acta Neuropathol 2013;125:913–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Frequent genetic alterations in SHH‐activated medulloblastomas according to 12, 22, 37, 38, 46, 48, 53, 54. % patient values refer to within subgroup percentages.