

Abstract
Objective
Diagnosis of frontotemporal dementia (FTD) is complicated by the overlap of clinical symptoms with other dementia disorders. Development of robust fluid biomarkers is critical to improve the diagnostic work‐up of FTD.
Methods
CSF concentrations of placental growth factor (PlGF) were measured in the discovery cohort including patients with FTD (n = 27), Alzheimer disease (AD) dementia (n = 75), DLB or PDD (n = 47), subcortical vascular dementia (VaD, n = 33), mild cognitive impairment that later converted to AD (MCI‐AD, n = 34), stable MCI (sMCI, n = 62), and 50 cognitively healthy controls from the Swedish BioFINDER study. For validation, CSF PlGF was measured in additional independent cohort of FTD patients (n = 22) and controls (n = 18) from the Netherlands.
Results
In the discovery cohort, MCI, MCI‐AD, AD dementia, DLB‐PDD, VaD, and FTD patients all showed increased CSF levels of PlGF compared with controls (sMCI P = 0.019; MCI‐AD P = 0.005; AD dementia, DLB‐PDD, VaD, and FTD all P < 0.001). PlGF levels were 1.8–2.1‐fold higher in FTD than in AD, DLB‐PDD and VaD (all P < 0.001). PlGF distinguished with high accuracy FTD from controls and sMCI performing better than tau/Aβ42 (AUC 0.954–0.996 versus 0.564–0.754, P < 0.001). A combination of PlGF, tau, and Aβ42 (tau/Aβ42/PlGF) was more accurate than tau/Aβ42 when differentiating FTD from a group of other dementias (AUC 0.972 vs. 0.932, P < 0.01). Increased CSF levels of PlGF in FTD compared with controls were corroborated in the validation cohort.
Interpretation
CSF PlGF is increased in FTD compared with other dementia disorders, MCI, and healthy controls and might be useful as a diagnostic biomarker of FTD.
Introduction
Frontotemporal dementia (FTD) is one of the most common early‐onset dementia with a reported prevalence rate of 3–26% in demented people with disease onset before 65 years of age.1 The core features of FTD are progressive deterioration in behavior, executive function or language caused by neuronal loss in frontal and anterior temporal cortices.2, 3 Based on clinical presentation FTD is broadly classified into behavioral‐variant FTD (bvFTD), semantic‐variant primary progressive aphasia (svPPA or semantic dementia (SD)) and nonfluent variant primary progressive aphasia (nfvPPA or PNFA).4, 5 Neuropathologically, FTD is characterized by either intraneuronal inclusions containing tau, TAR DNA‐binding protein with molecular weight 43 kDa (TDP‐43), or fused‐in‐sarcoma (FUS) proteins.1, 6 Approximately 10–20% of all FTD cases show autosomal dominant inheritance.7 The majority of genetic FTD is due to mutations in MAPT, 8, 9, 10 GRN, 11, 12 or C9orf72 13, 14, 15 genes, which are associated with accumulation of tau (in MAPT mutation carriers) or TDP‐43 (in GRN and C9orf72 mutation carriers) inclusions. Diagnosis of FTD is challenging because of the heterogeneity of clinical presentations, symptomatic overlap with other neurodegenerative disorders and difficulties to distinguish bvFTD, particularly in early stages, from primary psychiatric conditions which leads to long periods of diagnostic delay.16 Although progression of symptoms and imaging biomarkers may provide important diagnostic clues, there is a need for more cost‐efficient and less time‐consuming fluid biomarkers that could improve differential diagnosis of FTD.17 In this study, we identified placental growth factor (PlGF) as a new candidate biomarker of FTD. PlGF is a member of the vascular endothelial growth factor (VEGF) family, originally described in placenta but later found to be expressed in other organs.18 In addition to its regulatory role in pregnancy, accumulating evidence point to the biological effects of PlGF in pathological inflammation and angiogenesis associated with ischemia, hematologic diseases, and cancer.19 Several studies have implicated PlGF in central nervous system disorders. Upregulation of PlGF mRNA and protein in the brain has been shown in mouse models of ischemia.20, 21 Furthermore, we have demonstrated elevated CSF levels of PlGF in Parkinson's disease (PD), Parkinson's disease dementia (PDD) and dementia with Lewy bodies (DLB).41 Here, we measured cerebrospinal fluid (CSF) levels of PlGF in FTD and four major forms of neurodegenerative disorders with dementia. The discovery cohort included a total of 278 patients with FTD, AD, DLB, PDD, and VaD as well as stable MCI (sMCI), MCI that progressed to AD (MCI‐AD) and 50 cognitively healthy controls. We validated findings in the discovery cohort in additional independent cohort of FTD patients (n = 22) and controls (n = 18) from the Netherlands. Finally, in the discovery cohort, we assessed the performance of PlGF as a biomarker distinguishing FTD from controls or patients with other dementias.
Subjects and Methods
Study participants
Discovery cohort: Seventy‐five patients with AD, 47 patients with DLB‐PDD, 33 patients with VaD, 27 patients with FTD (25 bvFTD, 2 SD) and 50 healthy controls were recruited at the Memory Clinic of Skåne University Hospital in Malmö, Sweden. This cohort also included 96 individuals (recruited from the same clinic) with a baseline diagnosis of MCI. After an average clinical follow‐up period of 5.7 years (3.0–9.6), 34 of those had converted to AD (MCI‐AD), whereas 62 remained cognitively stable (sMCI). All study participants were assessed by medical doctors with extensive experience in cognitive disorders. All patients with a clinical syndrome of dementia met the DSM‐IIIR criteria for dementia22 combined with the NINCDS‐ADRDA criteria for AD,23 the NINDS‐AIREN criteria for VaD24 or criteria of probable DLB according to the 2005 consensus criteria.25 FTD patients were diagnosed according to Rascovsky (bvFTD)26 or Neary (SD) criteria.27 The FTD patients were recruited either from clinical practice or from a longitudinal FTD research study.28 All patients had minimum cerebral computed tomography (most often MRI) as imaging modality, and CSF analysis of AD biomarkers were used as exclusion criteria with in‐house cutoffs for clinical routine practice established at the Clinical Neurochemistry Laboratory, University of Gothenburg, Sweden following strict procedures for quality control to assure long‐term stability of biomarker levels.29 Of the 27 bvFTD patients, 18 were probable bvFTD, and 3 possible bvFTD, 4 definite bvFTD (3 by confirmation of TDP‐43 pathology postmortem and one C9orf72 repeat expansion carrier), and 2 SD. Patients with MCI at baseline had to fulfill the criteria advocated by Petersen.30 The healthy participants were not allowed to have any cognitive complaints or any significant neurological or psychiatric illness and they needed to have a well‐preserved general cognitive functioning. A careful clinical interview, together with an assessment of global function (Mini‐Mental State Examination, MMSE), delayed recall (Alzheimer's Disease Assessment Scale Cognitive Subscale, ADAS Cog, 10 word list delayed recall), attention (a quick test of cognitive speed, AQT) and visuospatial and executive function (cube‐drawing test and clock test), was done to rule out mild cognitive impairment. AD biomarkers were not considered in the diagnostic process. The characteristics of the cohort are given in Table 1.
Table 1.
Discovery cohort, demographic data, clinical characteristics, and CSF levels of PlGF
| Control (n = 50) | sMCI (n = 62) | MCI‐AD (n = 34) | AD (n = 75) | DLB‐PDD (n = 47) | VaD (n = 33) | FTD* (n = 27) | |
|---|---|---|---|---|---|---|---|
| Age | 74.2 (5.1) | 69.2 (7.5)a | 74.9 (7.7)b | 76.4 (7.4)b | 74.5 (6.3)b | 75.9 (7.9)b | 70.1 (6.6)a , c , d , e , f |
| Sex, (% female) | 72% | 56% | 65% | 68% | 40%a , c , d | 46%a | 44%a , d |
|
APOE 1 or 2 ε4 alleles |
27% | 47%a | 82%a , b | 65%a , b | 54%a , c | 25%b , c , d , e | 27%c , d |
| MMSE | 29.0 (1.0) | 28.2 (1.2) | 26.4 (1.7)a , b | 19.5 (3.3)a , b , c | 21.9 (5.1)a , b , c , d | 21.7 (4.4)a , b , c , d | 22.8 (6.3)a , b , c , d |
| Aβ42, pg/mL | 695 (282) | 486 (201)a , b | 317 (78)a , b | 260 (105)a , b | 340 (173)a , d | 396 (190)a , b , d | 709 (295)b , c , d , e , f |
| Aβ40, pg/mL | 5206 (1545) | 3821 (1377)a | 4232 (1345)a | 3899 (1376)a | 3170 (1137)a , b , c , d | 3238 (1285)a , c , d | 4509 (1660)a , b , e , f |
| tau, pg/mL | 443 (165) | 437 (175) | 645 (227)a , b | 766 (266)a , b , c | 472 (171)c , d | 441 (192)c , d | 385 (214)c , d |
| PlGF, pg/mL | 54.8 (15.8) | 64.1 (31.8)a | 70.5 (20.8)a | 79.5 (33.6)a | 89.5 (41.4)a , b | 94.2 (40.5)a , b , c | 166.7 (63.4)a , c , d , e , f |
AD, Alzheimer disease; DLB‐PDD, dementia with Lewy bodies or Parkinson's disease with dementia; F, female; FTD, frontotemporal dementia; sMCI, mild cognitive impairment; MCI‐AD, MCI that progressed to AD; MMSE, Mini Mental State Examinations; PlGF, placental growth factor; VaD, vascular dementia.
FTD group included 25 bvFTD (1 patient with C9orf72 mutations and 3 patients with TDP‐43 positivity neuropathologically) and 2 SD cases. APOE data were only available from 11 FTD patients.
Data are shown as mean (SD, n) unless otherwise specified. Demographic factors and clinical characteristics were compared using one‐way ANOVA and chi‐square tests. PlGF was analyzed with univariate general linear models controlling for age and sex. a P < 0.05 compared with controls, b P < 0.05 compared with sMCI, c P < 0.05 compared with MCI‐AD, d P < 0.05 compared with AD, e P < 0.05 compared with DLB‐PDD, f P < 0.05 compared with VaD.
Validation cohort: This independent cohort included 18 cognitively healthy controls, 22 patients with FTD (14 bvFTD, 6 SD, 2 PNFA) and 5 presymptomatic individuals with a GRN mutation that were recruited at the memory clinic of the Erasmus Medical Center. FTD patients were diagnosed according to Rascovsky (bvFTD)26 or Gorno‐Tempini (SD and PNFA) criteria.31 Healthy controls and presymptomatic GRN mutation carriers were ascertained in our longitudinal FTD‐RisC cohort in which asymptomatic first‐degree relatives (at‐risk individuals) of patients with autosomal dominant FTD are followed.32 Screening of the familial mutation is performed to divide at‐risk individuals into presymptomatic mutation carriers and healthy controls, and investigators remain blinded to individual mutation status. The characteristics of the cohort are given in Table 2.
Table 2.
Validation cohort, demographic data, clinical characteristics, and CSF levels of PlGF
| Control (n = 18) | FTD* (n = 22) | |
|---|---|---|
| Age | 54.0 (9.2) | 62.4 (7.6)a |
| Sex, (% female) | 61% | 54% |
|
APOE 1 or 2 ε4 alleles |
N/A | N/A |
| MMSE** | 29.6 (0.7) | 23.5 (5.2)a |
| PlGF, pg/mL | 42.2 (19.9) | 59.7 (23.9)a |
F, female; FTD, frontotemporal dementia; MMSE, Mini Mental State Examinations; PlGF, placental growth factor.
FTD group included 14 bvFTD, 6 SD, and 2 PNFA cases (2 patients with GRN mutation and 1 patient FTD with motor neuron disease).
MMSE was available from 18 controls and 15 FTD patients.
Data are shown as mean (SD, n) unless otherwise specified. Differences between the groups were compared using Student's t‐ and chi‐square tests; a P < 0.05 compared with controls.
The design of this study has been approved by the Local Ethics Committee of Lund University, Sweden and by the Local Ethics Committee of Erasmus Medical Center, the Netherlands and the study procedure was conducted in accordance with the Helsinki Declaration. All study participants (or legal representatives) gave their written informed consent to research.
CSF sampling and biological assays
For all patients and controls, CSF samples were drawn with the patients nonfasting. CSF was collected in polypropylene tubes and mixed gently to avoid gradient effects. All samples were centrifuged within 30 min at +4°C at 2000g for 10 min to remove cells and debris. Samples were stored in aliquots at −80°C pending biochemical analysis. CSF PlGF was measured using electrochemiluminescence immunoassay as per the manufacturer's protocol (Meso Scale Discovery, Gaithersburg, MA) with some modifications. Briefly, 10% bovine serum albumin was added to the blocking buffer and the samples were incubated overnight at +4°C. All samples were measured in duplicates. Samples from the validation and discovery cohorts were analyzed on separate occasions using different PlGF assay lots. Detection limits in the validation and discovery cohorts were 2.7 pg/mL and 3.1 pg/mL, respectively. Mean intraplate and interplate coefficients of variance (CV) were 4.5% and 7.5% in the discovery cohort and 5.2% and 3.3% in the validation cohort. Intraplate CV was below 20% for all samples except one with CV of 23%. This sample did not affect the results and was therefore included in statistical analysis. Samples were randomized according to diagnosis across plates/runs to minimize the effects of run‐to‐run variation. Our previous study has shown that PlGF levels do not correlated with CSF storage time (unpublished data). CSF amyloid β (Aβ) 42, Aβ40, and tau (total) were analyzed with Euroimmun immunoassay (EUROIMMUN AG, Lübeck, Germany). CSF neurofilament light chain (NfL) was analyzed as previously described.33
Statistical analysis
SPSS version 22 (IBM, Armonk, NY) and R version 3.3.134 was used for statistical analysis. CSF PlGF levels were not normally distributed and therefore ln‐transformed before the analysis. The effects of age, sex, and APOE genotype were tested with Pearson's correlation analysis and Student's t‐tests. Group differences were assessed using Student's t‐tests, one‐way ANOVA and univariate general linear models (GLM). Linear regressions were used to investigate associations with CSF Aβ and tau and clinical characteristics. Age and sex were included in all regression models to control the confounding effects of these factors. Because of the relatively small sample size we did not adjust statistical analysis for age and sex in the validation cohort. Diagnostic accuracies of CSF biomarkers were assessed using receiver operating characteristic (ROC) curve analysis. Area under the curve (AUC) of two ROC curves were compared using a bootstrap procedure (n = 2000 iterations). Alpha‐level of significance was set at P < 0.05.
Results
Discovery cohort
Associations with demographic and clinical characteristics
CSF levels of PlGF correlated positively with age in controls (r = 0.284, P = 0.045) and in sMCI (r = 0.529, P < 0.001), AD (r = 0.231, P = 0.046), and FTD (r = 0.550, P = 0.003) patients and were higher in men than women in controls (P = 0.004) and in patients with sMCI (P = 0.011), DLB‐PDD (P = 0.032), and VaD (P = 0.011). We did not find any differences in CSF PlGF concentrations between APOE ε4 allele carriers and noncarriers. CSF PlGF did not correlate with MMSE scores in any of the diagnostic groups or with disease duration in FTD group.
CSF levels of PlGF in diagnostic groups
We next compared PlGF levels between different diagnostic groups using GLM adjusted for age and sex. CSF levels of PlGF were elevated in sMCI (P = 0.019), MCI‐AD (P = 0.005), AD dementia (P < 0.001), DLB‐PDD (P < 0.001), VaD (P < 0.001), and FTD (P < 0.001) compared with cognitively healthy controls (Fig. 1A and Table 1). Notably, FTD patients showed 1.8‐ to 2.1‐fold higher PlGF levels compared to other dementias: AD, DLB‐PDD, and VaD (all P < 0.001, Fig. 1A and Table 1). PlGF concentrations were also increased in FTD compared to sMCI and MCI‐AD (both P < 0.001, Fig. 1A and Table 1). The results were very similar when two patients with SD were excluded from the analysis (data not shown).
Figure 1.
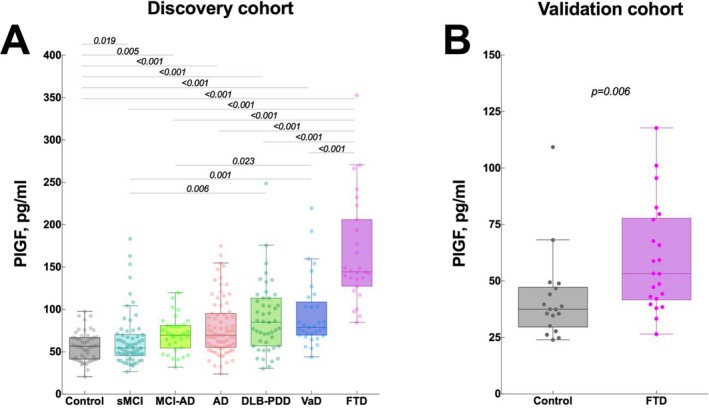
CSF levels of PlGF in dementia disorders. (A) Discovery cohort, CSF levels of PlGF in patients with AD, sMCI, MCI‐AD, AD, DLB‐PDD, VaD, FTD (25 bvFTD and 2 SD) and cognitively healthy controls. (B) Validation cohort, CSF levels of PlGF in patients with FTD (14 bvFTD, 6 SD, 2 PNFA) and cognitively healthy controls. AD, Alzheimer's disease; DLB‐PDD dementia with Lewy bodies or Parkinson's disease with dementia; FTD, frontotemporal dementia; bvFTD, behavioral variant FTD; sMCI, stable mild cognitive impairment; MCI‐AD, MCI that progressed to AD; SD, semantic dementia; VaD, vascular dementia.
In addition, we measured CSF levels of PlGF in another group of 14 cognitively healthy controls and 8 patients with FTD (Supplementary Methods and Table S1) on a separate occasion and using different PlGF assay lot. Similarly, we found increased levels of PlGF in FTD patients compared to controls (P < 0.001, Fig. S1).
CSF PlGF as biomarkers of FTD
Previous studies have suggested that the CSF tau/Aβ42 ratio can accurately distinguish FTD from AD dementia (AUC 0.86‐0.93).35, 36, 37 Here we compared the accuracy of tau/Aβ42, PlGF, tau/PlGF, and tau/Aβ42/PlGF in separating FTD patients from other diagnostic groups (Table 3 and Table S2). PlGF alone showed very high accuracies, sensitivities, and specificities when differentiating FTD from both controls (AUC 0.996, sensitivity 100%, specificity 96%; Fig. 2A) and sMCI (AUC 0.954, sensitivity 100%, specificity 84%) performing significantly better than tau/Aβ42 (AUC 0.954–0.996 vs. 0.564–0.754, P < 0.01). We did not observe any further improvement in AUCs for tau/PlGF and tau/Aβ42/PlGF.
Table 3.
Discovery cohort, Receiver Operating Characteristic (ROC) analysis of PlGF as a biomarker of FTD
| tau/Aβ42 | PlGF | tau/PlGF | tau/Aβ42/PlGF | |
|---|---|---|---|---|
| FTD versus controls* | 0.564 (0.425–0.704) | 0.996a (0.988–1.000) | 0.984a (0.962–1.000) | 0.946a (0.889–1.000) |
| FTD versus sMCI | 0.754 (0.638–0.870) | 0.954b (0.913–0.995) | 0.962a (0.927–0.996) | 0.967a (0.934–1.000) |
| FTD versus other dementia | 0.932 (0.892–0.972) | 0.905 (0.856–0.955) | 0.934 (0.894–0.975) | 0.972b , c , d (0.949–0.996) |
| FTD versus MCI‐AD | 0.983 (0.957–1.000) | 0.981 (0.955–1.000) | 0.991 (0.976–1.000) | 0.999 (0.995–1.000) |
| FTD versus AD | 0.990 (0.977–1.000) | 0.925e (0.875–0.975) | 0.991c (0.978–1.000) | 0.997c (0.992–1.000) |
| FTD versus DLB‐PDD | 0.897 (0.828–0.966) | 0.895 (0.822–0.969) | 0.882 (0.806–0.958) | 0.954d , e , f (0.912–0.996) |
| FTD versus VaD | 0.850 (0.754–0.945) | 0.875 (0.786–0.964) | 0.881 (0.795–0.967) | 0.941e (0.883–0.998) |
AD, Alzheimer disease; AUC, area under the ROC curve; CI, confidence interval; DLB‐PDD, dementia with Lewy bodies or Parkinson's disease with dementia; FTD, frontotemporal dementia; sMCI, mild cognitive impairment; MCI‐AD, MCI that progressed to AD; PlGF, placental growth factor; VaD, vascular dementia.
Data are shown as AUC (95%CI). *tau data were missing from three individuals (1 control, 1 sMCI, and 1 FTD) and these individuals were excluded from all ROC analysis.
a P < 0.001 compared with tau/Aβ42; b P < 0.01 compared with tau/Aβ42; c P < 0.01 compared with PlGF; d P < 0.05 compared with tau/PlGF; e P < 0.05 compared with tau/Aβ42; f P < 0.05 compared with PlGF.
Figure 2.
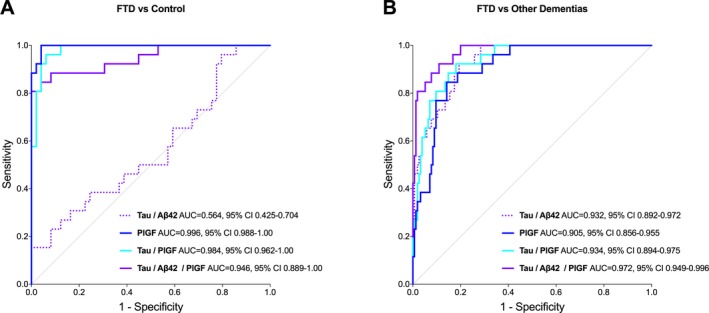
ROC curve analysis in the discovery cohort. ROC curve analysis of CSF biomarkers for distinguishing FTD from controls (A) and FDT from other dementias (B). AUC, area under the ROC curve; CI, confidence interval.
We then studied whether PlGF could improve the differential diagnosis of FTD versus prodromal AD (MCI‐AD), AD dementia, and other dementia types, that is, DLB‐PDD and VaD. The performance of tau/Aβ42/PlGF was significantly better compared to tau/Aβ42 when distinguishing FTD from the group of other dementias (AUC 0.972 vs. 0.932, P < 0.01, Fig. 2B), FTD from DLB‐PDD (AUC 0.954 vs. 0.897, P < 0.05), and FTD from VaD (AUC 0.941 vs. 0.850, P < 0.05). In addition, tau/Aβ42/PlGF showed higher sensitivities and/or specificities compared with tau/Aβ42 for differentiating FTD from other dementias (Table S2).
We also compared PlGF with neurofilament light chain (NfL), another promising biomarker of neuronal damage in FTD.38, 39, 40 In a subcohort of 267 individuals, PlGF, tau/PlGF and/or tau/Aβ42/PlGF showed higher accuracies than NfL when differentiating FTD from other dementia groups including AD (Table S3).
Associations with CSF Aβ and tau
CSF PlGF was positively associated with Aβ40 in FTD patients (β = 0.501, P = 0.020; adjusted for age and sex). In contrast, we found a negative correlation between PlGF and Aβ42 in the controls (β = −0.354, P = 0.034; adjusted for age and sex) but not in other groups. There were no significant associations between CSF PlGF and tau.
Validation cohort
To confirm our findings in the discovery cohort, we measured CSF levels of PlGF in the validation cohort from the Netherlands. Similar to the discovery cohort, we found increased levels of PlGF in FTD patients (not including the 5 presymptomatic individuals with GRN mutations) compared to controls (P = 0.006, Fig. 1B, Table 2). Notably, the differences in PlGF levels between controls and FTD were more pronounced in the discovery cohort. The range of CSF concentration of PlGF also differed between the two cohorts. Possible explanations for these results might be differences in preanalytical sample handling and lot‐to‐lot variation in the performance of the PlGF kits.
The validation cohort comprised 14 bvFTD and 6 SD patients. We found that CSF PlGF levels were increased in bvFTD but not in SD (P = 0.006 and P = 0.200, Fig. S2).
Finally, we measured PlGF in five presymptomatic individuals with GRN mutation. The mean PlGF concentration in this presymptomatic GRN group was almost as high as in bvFTD (60.3 ± 37.9 pg/mL and 63.4 ± 25.4 pg/mL), however, the difference in the levels between the controls and presymptomatic GRN did not reach statistical significance (P = 0.156) most likely due to the small sample size.
Discussion
In the discovery cohort, we demonstrated that CSF levels of PlGF were increased in different dementia subtypes and particularly in FTD compared to cognitively healthy controls, with FTD patients showing 1.8‐ to 2.1‐fold higher PlGF concentration than individuals with AD, DLB‐PDD, and VaD. We corroborated our findings of elevated CSF PlGF in another group of controls and FTD patients from the same clinical center in Sweden and in the validation cohort from the Netherlands. Furthermore, we report that when combined with tau and Aβ42, PlGF performed better than tau/Aβ42 alone in distinguishing FTD from DLB‐PDD, VaD, and all other dementias grouped together. Finally, PlGF showed higher accuracy than tau/Aβ42 in differentiating FTD from controls and sMCI.
These findings are in agreement with, and extend, our previous data on increased CSF levels of PlGF in PDD, DLB, and PD patients compared to control individuals.41 Studies investigating the role of PlGF in neurodegenerative diseases are sparse and it is at present unclear how PlGF could be linked to the core pathological features of the FTD or other dementia disorders. Hypoxia and reactive oxygen species are strong inducers of VEGF family members, including PlGF.42, 43, 44 Expression of PlGF is increased in mouse astrocytes and endothelial cells following cerebral ischemia.20, 21 Interestingly, astrogliosis in frontal and temporal regions is one of the core histopathological hallmarks of FTD.6 Furthermore, frontotemporal lobar degeneration has been shown to be accompanied by oxidative damage that targets primarily astrocytes.45 Thus, it is possible that in FTD, PlGF is increased in response to astrogliosis and oxidative stress.
In contrast to CSF Aβ42 and tau, PlGF showed very high accuracy when discriminating FTD patients from controls and even sMCI patients (AUCs > 0.95) with the performance similar to CSF NfL (Table S3).38, 39, 40 Although CSF NfL is a promising biomarker of neuronal damage in neurodegenerative disorders and disease severity in FTD,39, 40, 46, 47 it does not provide significant added value to CSF Aβ42 and tau for differential diagnosis of FTD because CSF levels of NfL are also elevated in many other dementias, for example, progressive supranuclear palsy (PSP) and VaD.48, 49 Postmortem investigations previously indicated that 10‐30% of patients clinically diagnosed with FTD, had Alzheimer disease (AD).50, 51, 52, 53 However, it was later found that FTD and AD dementia differ in CSF levels of the core AD biomarkers, Aβ42 and tau: FTD patients have consistently shown higher Aβ42 and lower tau levels compared to AD dementia patients.54, 55, 56 Furthermore, several studies including one in an autopsy‐proven cohort, have reported that the tau/Aβ42 (or Aβ42/tau) ratio discriminated with high sensitivity (70‐86%) and specificity (82‐94%) between FTD and AD cases.35, 36, 37 Nevertheless, there is a lack of biomarkers that could differentiate FTD from other forms of dementia such as, for example, DLB‐PDD or VaD both of which may share clinical symptoms with FTD.57, 58 In this study, we demonstrated that PlGF combined with tau and Aβ42 (tau/Aβ42/PlGF) distinguished with high accuracy (AUCs > 0.94) FTD from DLB‐PDD, VaD, and from all other types of dementia (DLB‐PDD, VaD, and AD) grouped together performing significantly better than tau/Aβ42. While PlGF did not differentiate FTD from AD any better than tau/Aβ42, its accuracy was very high with AUC over 0.92. Furthermore, PlGF and/or its ratios, performed better than NfL when distinguishing FTD from other dementia groups including AD. Of note, although the diagnosis of FTD was in the first hand based on assessment of clinical symptoms and neuroimaging findings, treating physicians had access to CSF Aβ42 and tau data. Consequently, it is possible that the diagnostic performance of PlGF in comparison with Aβ42 and tau was underestimated given the availability of CSF AD biomarkers (but not PlGF) in the diagnostic process.
One limitation of this study is that we did not measure p‐tau. Recent data have indicated that p‐tau/Aβ42 preforms better than t‐tau/Aβ42 when differentiating autopsy‐confirmed frontotemporal lobar degeneration from AD.59 Thus, future studies are needed to establish if PlGF could further improve the accuracy of p‐tau/Aβ42 in distinguishing FTD from other dementias. Another limitation is that because FTD is a rare disease the samples size was small with only few cases had autopsy‐confirmed diagnosis. Future studies in larger cohorts of neuropathologically confirmed cases should investigate PlGF levels across different dementia disorders and different clinical, pathological, and genetic FTD subtypes.
We demonstrate that CSF PlGF is increased in FTD compared with sMCI, MCI‐AD, DLB‐PDD, VaD, and control groups and that PlGF in combination with Aβ42 and tau accurately differentiates FTD from other dementia disorders, stable MCI patients, and cognitively healthy controls. These results suggest that PlGF offers significant promise as diagnostic biomarker of FTD and merit further studies in larger clinical cohorts.
Author Contributions
OH, AFS, LHM, KN, MLW, CN, KB, JCS, and SJ collected the data and reviewed the manuscript for intellectual content. OH and SJ designed the study, analyzed, and interpreted the data, prepared figures, and cowrote the manuscript. All authors read and approved the final manuscript.
Conflict of Interest
Santillo, Meeter, Landqvist Waldö, Nilsson, van Swieten, Janelidze report no disclosures. Blennow has served as a consultant or at advisory boards for Alector, Alzheon, CogRx, Biogen, Lilly, Novartis, and Roche Diagnostics, and is a cofounder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture‐based platform company at the University of Gothenburg, all unrelated to the work presented in this paper. Dr Hansson has acquired research support (for the institution) from Roche, GE Healthcare, Biogen, AVID Radiopharmaceuticals, Fujirebio, and Euroimmun. In the past 2 years, he has received consultancy/speaker fees (paid to the institution) from Lilly, Roche, and Fujirebio.
Supporting information
Table S1. Demographic data, clinical characteristics, and CSF levels of PlGF in another group of 14 cognitively healthy controls and 8 patients with FTD where CSF levels of PlGF were measured on a separate occasion and using different PlGF assay.
Table S2. Sensitivities, specificities, and maximized Youden index for PlGF as FTD biomarker in the discovery cohort.
Table S3. Receiver Operating Characteristic (ROC) analysis of PlGF and NfL as biomarkers of FTD in the discovery cohort.
Figure S1. CSF levels of PlGF in cognitively healthy controls and FTD patients in another group of 14 cognitively healthy controls and eight patients with FTD where CSF levels of PlGF were measured on a separate occasion and using different PlGF assay.
Figure S2. CSF levels of PlGF in FTD subtypes.
Acknowledgments
The authors thank the collaborators of this study and the entire BioFINDER Study group (www.biofinder.se), including Susanna Vestberg for classifying the MCI‐AD patients into MCI subgroups. Work in the authors’ laboratory was supported by the European Research Council, the Swedish Research Council, the Strategic Research Area MultiPark (Multidisciplinary Research in Parkinson's disease) at Lund University, the Crafoord Foundation, the Swedish Brain Foundation, the Swedish Alzheimer Foundation, the Torsten Söderberg Foundation, Skåne Research Hospital research funds, the Greta and Johan Kock Foundation, the Koch's Foundation, the Swedish Society for Medical Research, the Bente Rexed Gersteds Foundation for Brain Research and the Swedish federal government under the ALF agreement. This study was also funded by European Joint Programme ‐ Neurodegenerative Disease Research, the Netherlands Organisation for Health Research and Development, Alzheimer Nederland. and the Dioraphte Foundation (grant numbers: RiMod‐FTD 733051024, Memorabel 733050103, WE.09‐2014‐04).
Funding Information
Work in the authors’ laboratory was supported by the European Research Council, the Swedish Research Council, the Strategic Research Area MultiPark (Multidisciplinary Research in Parkinson's disease) at Lund University, the Crafoord Foundation, the Swedish Brain Foundation, the Swedish Alzheimer Foundation, the Torsten Söderberg Foundation, Skåne Research Hospital research funds, the Greta and Johan Kock Foundation, the Koch's Foundation, the Swedish Society for Medical Research, the Bente Rexed Gersteds Foundation for Brain Research, and the Swedish federal government under the ALF agreement. This study was also funded by European Joint Programme – Neurodegenerative Disease Research, the Netherlands Organisation for Health Research and Development, Alzheimer Nederland and the Dioraphte Foundation (grant numbers: RiMod‐FTD 733051024, Memorabel 733050103, WE.09‐2014‐04).
Funding Statement
This work was funded by Skåne Research Hospital research funds grant ; Alzheimer Nederland grant ; European Research Council grant ; Swedish Research Council grant ; Strategic Research Area MultiPark (Multidisciplinary Research in Parkinson's disease) at Lund University grant ; Crafoord Foundation grant ; Swedish Brain Foundation grant ; Swedish Alzheimer Foundation grant ; Torsten Söderberg Foundation grant ; Greta and Johan Kock Foundation grant ; Koch's Foundation grant ; Swedish Society for Medical Research grant ; Bente Rexed Gersteds Foundation for Brain Research grant ; Swedish federal government under the ALF agreement grant ; European Joint Programme – Neurodegenerative Disease Research grant ; Netherlands Organisation for Health Research and Development grant ; Dioraphte Foundation grants RiMod‐FTD 733051024, Memorabel 733050103, and WE.09‐2014‐04.
Contributor Information
Oskar Hansson, Email: oskar.hansson@med.lu.se.
Shorena Janelidze, Email: shorena.janelidze@med.lu.se.
References
- 1. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet 2015;386:1672–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hodges JR, Davies RR, Xuereb JH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol 2004;56:399–406. [DOI] [PubMed] [Google Scholar]
- 3. Warren JD, Rohrer JD, Rossor MN. Clinical review. Frontotemporal dementia. BMJ 2013;347:f4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seelaar H, Rohrer JD, Pijnenburg YA, et al. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry 2011;82:476–486. [DOI] [PubMed] [Google Scholar]
- 5. Woollacott IO, Rohrer JD. The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J Neurochem 2016;138(Suppl 1):6–31. [DOI] [PubMed] [Google Scholar]
- 6. Sieben A, Van Langenhove T, Engelborghs S, et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 2012;124:353–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldman JS, Farmer JM, Wood EM, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 2005;65:1817–1819. [DOI] [PubMed] [Google Scholar]
- 8. Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5’‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 1998;393:702–705. [DOI] [PubMed] [Google Scholar]
- 9. Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 1998;43:815–825. [DOI] [PubMed] [Google Scholar]
- 10. Spillantini MG, Murrell JR, Goedert M, et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 1998;95:7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baker M, Mackenzie IR, Pickering‐Brown SM, et al. Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature 2006;442:916–919. [DOI] [PubMed] [Google Scholar]
- 12. Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin‐positive frontotemporal dementia linked to chromosome 17q21. Nature 2006;442:920–924. [DOI] [PubMed] [Google Scholar]
- 13. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gijselinck I, Van Langenhove T, van der Zee J, et al. A C9orf72 promoter repeat expansion in a Flanders‐Belgian cohort with disorders of the frontotemporal lobar degeneration‐amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 2012;11:54–65. [DOI] [PubMed] [Google Scholar]
- 15. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Passant U, Elfgren C, Englund E, Gustafson L. Psychiatric symptoms and their psychosocial consequences in frontotemporal dementia. Alzheimer Dis Assoc Disord 2005;19(Suppl 1):S15–S18. [DOI] [PubMed] [Google Scholar]
- 17. Meeter LH, Kaat LD, Rohrer JD, van Swieten JC. Imaging and fluid biomarkers in frontotemporal dementia. Nat Rev Neurol 2017;13:406–419. [DOI] [PubMed] [Google Scholar]
- 18. Ribatti D. The discovery of the placental growth factor and its role in angiogenesis: a historical review. Angiogenesis 2008;11:215–221. [DOI] [PubMed] [Google Scholar]
- 19. Newell LF, Holtan SG. Placental growth factor: what hematologists need to know. Blood Rev 2017;31:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beck H, Acker T, Puschel AW, et al. Cell type‐specific expression of neuropilins in an MCA‐occlusion model in mice suggests a potential role in post‐ischemic brain remodeling. J Neuropathol Exp Neurol 2002;61:339–350. [DOI] [PubMed] [Google Scholar]
- 21. Hayashi T, Noshita N, Sugawara T, Chan PH. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab 2003;23:166–180. [DOI] [PubMed] [Google Scholar]
- 22. American Psychiatric Association . Group to Revise DSM‐III. Diagnostic and statistical manual of mental disorders, DSM‐III‐R. 3 ed Washington, DC: American Psychiatric Association, 1987. [Google Scholar]
- 23. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 24. Roman GC, Tatemichi TK, Erkinjuntti T, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS‐AIREN International Workshop. Neurology 1993;43:250–260. [DOI] [PubMed] [Google Scholar]
- 25. Geser F, Wenning GK, Poewe W, McKeith I. How to diagnose dementia with Lewy bodies: state of the art. Mov Disord 2005;20(Suppl 12):S11–S20. [DOI] [PubMed] [Google Scholar]
- 26. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134(Pt 9):2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 28. Santillo AF, Martensson J, Lindberg O, et al. Diffusion tensor tractography versus volumetric imaging in the diagnosis of behavioral variant frontotemporal dementia. PLoS ONE 2013;8:e66932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palmqvist S, Zetterberg H, Blennow K, et al. Accuracy of brain amyloid detection in clinical practice using cerebrospinal fluid beta‐amyloid 42: a cross‐validation study against amyloid positron emission tomography. JAMA Neurol 2014;71:1282–1289. [DOI] [PubMed] [Google Scholar]
- 30. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004;256:183–194. [DOI] [PubMed] [Google Scholar]
- 31. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dopper EG, Rombouts SA, Jiskoot LC, et al. Structural and functional brain connectivity in presymptomatic familial frontotemporal dementia. Neurology 2014;83:e19–e26. [DOI] [PubMed] [Google Scholar]
- 33. Hansson O, Janelidze S, Hall S, et al. Blood‐based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology 2017;88:930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Core R, Team R. A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; Available at: http://www.R-project.org/. [Google Scholar]
- 35. Baldeiras I, Santana I, Leitao MJ, et al. Cerebrospinal fluid Abeta40 is similarly reduced in patients with Frontotemporal Lobar Degeneration and Alzheimer's Disease. J Neurol Sci 2015;358:308–316. [DOI] [PubMed] [Google Scholar]
- 36. Bian H, Van Swieten JC, Leight S, et al. CSF biomarkers in frontotemporal lobar degeneration with known pathology. Neurology 2008;70(19 Pt 2):1827–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Struyfs H, Niemantsverdriet E, Goossens J, et al. Cerebrospinal fluid P‐Tau181P: biomarker for improved differential dementia diagnosis. Front Neurol 2015;6:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Landqvist Waldo M, Frizell Santillo A, Passant U, et al. Cerebrospinal fluid neurofilament light chain protein levels in subtypes of frontotemporal dementia. BMC Neurol 2013;13:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 2016;3:623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meeter LHH, Vijverberg EG, Del Campo M, et al. Clinical value of neurofilament and phospho‐tau/tau ratio in the frontotemporal dementia spectrum. Neurology 2018;90:e1231–e1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Janelidze S, Lindqvist D, Francardo V, et al. Increased CSF biomarkers of angiogenesis in Parkinson disease. Neurology 2015;85:1834–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kelly BD, Hackett SF, Hirota K, et al. Cell type‐specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia‐inducible factor 1. Circ Res 2003;93:1074–1081. [DOI] [PubMed] [Google Scholar]
- 43. Kim YW, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood 2014;123:625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003;9:677–684. [DOI] [PubMed] [Google Scholar]
- 45. Martinez A, Carmona M, Portero‐Otin M, et al. Type‐dependent oxidative damage in frontotemporal lobar degeneration: cortical astrocytes are targets of oxidative damage. J Neuropathol Exp Neurol 2008;67:1122–1136. [DOI] [PubMed] [Google Scholar]
- 46. Rohrer JD, Woollacott IO, Dick KM, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology 2016;87:1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol 2014;75:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hall S, Ohrfelt A, Constantinescu R, et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol 2012;69:1445–1452. [DOI] [PubMed] [Google Scholar]
- 49. Zerr I, Schmitz M, Karch A, et al. Cerebrospinal fluid neurofilament light levels in neurodegenerative dementia: evaluation of diagnostic accuracy in the differential diagnosis of prion diseases. Alzheimers Dement 2018;14:751–763. [DOI] [PubMed] [Google Scholar]
- 50. Alladi S, Xuereb J, Bak T, et al. Focal cortical presentations of Alzheimer's disease. Brain 2007;130(Pt 10):2636–2645. [DOI] [PubMed] [Google Scholar]
- 51. Forman MS, Farmer J, Johnson JK, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol 2006;59:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Knopman DS, Boeve BF, Parisi JE, et al. Antemortem diagnosis of frontotemporal lobar degeneration. Ann Neurol 2005;57:480–488. [DOI] [PubMed] [Google Scholar]
- 53. Rosen HJ, Hartikainen KM, Jagust W, et al. Utility of clinical criteria in differentiating frontotemporal lobar degeneration (FTLD) from AD. Neurology 2002;58:1608–1615. [DOI] [PubMed] [Google Scholar]
- 54. Skillback T, Farahmand BY, Rosen C, et al. Cerebrospinal fluid tau and amyloid‐beta1‐42 in patients with dementia. Brain 2015;138(Pt 9):2716–2731. [DOI] [PubMed] [Google Scholar]
- 55. Tang W, Huang Q, Wang Y, et al. Assessment of CSF Abeta42 as an aid to discriminating Alzheimer's disease from other dementias and mild cognitive impairment: a meta‐analysis of 50 studies. J Neurol Sci 2014;345:26–36. [DOI] [PubMed] [Google Scholar]
- 56. van Harten AC, Kester MI, Visser PJ, et al. Tau and p‐tau as CSF biomarkers in dementia: a meta‐analysis. Clin Chem Lab Med 2011;49:353–366. [DOI] [PubMed] [Google Scholar]
- 57. Claassen DO, Parisi JE, Giannini C, et al. Frontotemporal dementia mimicking dementia with Lewy bodies. Cogn Behav Neurol 2008;21:157–163. [DOI] [PubMed] [Google Scholar]
- 58. Jung NY, Kim HJ, Kim YJ, et al. Neuropsychiatric characteristics of PiB‐negative subcortical vascular dementia versus behavioral variant frontotemporal dementia. Arch Gerontol Geriatr 2016;67:86–91. [DOI] [PubMed] [Google Scholar]
- 59. Lleo A, Irwin DJ, Illan‐Gala I, et al. A 2‐step cerebrospinal algorithm for the selection of frontotemporal lobar degeneration subtypes. JAMA Neurol 2018;75:738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Demographic data, clinical characteristics, and CSF levels of PlGF in another group of 14 cognitively healthy controls and 8 patients with FTD where CSF levels of PlGF were measured on a separate occasion and using different PlGF assay.
Table S2. Sensitivities, specificities, and maximized Youden index for PlGF as FTD biomarker in the discovery cohort.
Table S3. Receiver Operating Characteristic (ROC) analysis of PlGF and NfL as biomarkers of FTD in the discovery cohort.
Figure S1. CSF levels of PlGF in cognitively healthy controls and FTD patients in another group of 14 cognitively healthy controls and eight patients with FTD where CSF levels of PlGF were measured on a separate occasion and using different PlGF assay.
Figure S2. CSF levels of PlGF in FTD subtypes.