

Abstract
Background
MicroRNA (miRNA) expression in the serum of multiple sclerosis (MS) patients has been correlated with white matter (WM) magnetic resonance imaging (MRI) abnormalities. The expression levels and cellular specificity of the target genes of these miRNAs are unknown in MS brain.
Objective
The aim of this study was to analyze and validate the expression of miRNAs, previously reported as dysregulated in sera of MS patients, in white‐matter lesions (WMLs) of progressive MS brains.
Methods
We performed global miRNA expression profiling analysis in demyelinated WMLs of progressive MS brains (n = 5) and compared the significantly altered miRNAs to previously identified miRNAs from sera of MS patients. Top dysregulated miRNAs common between the two datasets were validated in an independent cohort of MS brains by quantitative PCR (qPCR) and in situ hybridization.
Results
Among the miRNAs that were significantly changed in WML tissues, 11 were similar to pathogenic and 12 were common to protective miRNAs previously identified in sera and correlating with WM MRI abnormalities. Importantly, the expression levels of 58% of the protective miRNAs (7 of 12) were decreased in MS lesions compared to surrounding normal‐appearing tissue. Target genes of these miRNAs were also altered in MS lesions and queries of cell‐specific databases identified astrocytes and microglia as the key cellular expressers of these genes in MS brains.
Conclusions
We identified miRNAs that correlate with MRI abnormalities in lesioned tissue from MS brains.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) characterized by multiple focal demyelination, axonal loss, remyelination failure, and gliosis. Several lines of evidence suggest that the immune system plays a major role in MS disease initiation and progression.1 The precise etiology and exact disease mechanisms underlying MS pathogenesis are largely unknown. Genome‐wide expression profiling in MS brains has identified regulatory elements like transcription factors, chromatin regulators, and small RNA molecules within MS lesions.2, 3, 4, 5, 6
MicroRNAs (miRNAs) are small regulatory molecules (19–23 nucleotides) that are key regulators of gene expression. They bind to the 3′UTR region of target genes and cause translational inhibition or mRNA degradation at the posttranscriptional level.7 Comparative miRNA profiling has identified several miRNAs in different body fluids as well as brain tissues.8, 9 In a recent study, Regev et al.10 correlated serum miRNA levels and magnetic resonance imaging (MRI) measures of disease severity in three MS disease categories (95 relapsing‐remitting MS, 17 secondary‐progressive MS, and 8 primary‐progressive MS) across two different cohorts. Between these two cohorts of MS patients, 93 miRNAs were predicted to have a protective correlation and 48 miRNAs were predicted to be pathogenic based on brain T1 hypo and T2 hyper‐intense lesion volumes (T2LV and T1/T2 ratio). Interestingly, none of the identified miRNAs were associated with MRI measures of atrophy, suggesting differing mechanisms of tissue loss reflected by the miRNA profile and detected by these two imaging modalities. While this study was comprehensive, presence of these miRNAs in MS brain tissues are unknown.
In the present study, we therefore sought to identify the expression of miRNAs in MS white‐matter lesions (WMLs) that were previously reported in sera and correlated with imaging abnormalities. The expression levels of significantly changed miRNAs identified in MS brains were further validated in a separate cohort of MS brains using RT‐qPCR and in situ hybridization (ISH). mRNA levels of the target genes of the identified miRNAs were also determined in MS lesions. Queries via cell‐specific databases identified astrocytes and microglia as the major cell types expressing these mRNAs in MS lesions. Collectively, these findings identify a group of miRNAs that can be detected in both sera and MS brain tissues in MS patients.
Material and Methods
Human subjects and regulatory compliance
Patient demographics and MS disease stages are listed in Table 1. All brains were collected as part of the tissue procurement program approved by the Cleveland Clinic Institutional Review Board. Brains were removed according to a rapid autopsy protocol and sliced (1 cm thick) using a guided box. Slices were either rapidly frozen for biochemical analysis or long fixed in 4% paraformaldehyde for morphological studies.
Table 1.
Demographics and disease types of study cohorts
| Case # | MS type | Myelin status | Disease duration (years) | Age (years) | Sex | EDSS | PMI (h) | Study method |
|---|---|---|---|---|---|---|---|---|
| MS01 | SPMS | Myelinated/demyelinated | 36 | 45 | M | 7 | 3 | Microarray |
| MS02 | SPMS | Demyelinated | 25 | 52 | M | 9.5 | 5 | |
| MS03 | PPMS | Myelinated/demyelinated | 9 | 63 | F | 7.5 | 5 | |
| MS04 | SPMS | Myelinated | 19 | 53 | F | 9 | 6 | |
| MS05 | SPMS | Myelinated/demyelinated | 21 | 53 | F | 8 | 7 | |
| MS06 | SPMS | Myelinated/demyelinated | 20 | 60 | F | 8 | 6 | |
| MS07 | PPMS | Myelinated/demyelinated | 23 | 75 | M | 9 | 5 | qPCR |
| MS08 | SPMS | Myelinated | 26 | 69 | F | 8 | 6 | |
| MS09 | SPMS | Demyelinated | 30 | 60 | F | 9 | 8 | |
| MS010 | SPMS | Myelinated | 27 | 71 | F | 9.5 | 5 | |
| MS011 | SPMS | Myelinated/demyelinated | 35 | 74 | M | 8 | 9 | |
| MS012 | SPMS | Demyelinated | 37 | 59 | F | 9 | 5 |
SPMS, secondary‐progressive multiple sclerosis; PPMS, primary‐progressive multiple sclerosis; EDSS, expanded disability status scale; PMI, postmortem interval.
Global miRNA expression profiling and pathway analysis
Five myelinated and five demyelinated MS WML samples were selected from the frozen MS brain slices (Table 1, MS1‐6). WMLs were identified based on immunohistochemical analysis using proteolipid protein (PLP) and were separated from surrounding normal‐appearing white matter (NAWM) tissue using previously described methods.4, 11 Fourteen micron sections from frozen blocks were used to characterize demyelination using myelin (PLP) immunohistochemistry (Fig. 1A). Serial sections (60 μm thick) from characterized tissues were collected and used for small RNA isolation for microarray‐based miRNA analysis by LC Sciences, Houston, TX, using Qiagen miRNA isolation kits (miRNeasy Mini Kit, #217004) as per manufacturer's instructions4 (Details of the array, miRNA probe sequences, and quality control measurements are listed at www.lcsciences.com). Resultant scan intensity data were analyzed by subtracting the background, normalizing the signals using a LOWESS filter, and using locally weighted regression analysis to remove system‐related variations. These include variations in sample amounts, dye labeling biases and signal gain differences between scanners, so that biologically relevant variations can be detected. Finally, intensity values were subjected to a false discovery rate (FDR) correction in order to eliminate false positives and significance was established at P < 0.05.4 For fold change (FC), normalized intensity values of individual miRNAs were selected from microarray expression output files and compared between WMLs and NAWM. Significant differences were calculated using a nonparametric (Mann–Whitney) test and a P‐value <0.05 was considered to be significant. To identify miRNA‐associated molecular pathways, we used online DIANA‐miRPath v3.0, which utilizes predicted or validated miRNA target interactions derived from DianaTarBase (http://www.microrna.gr/miRPathv3).12 For this analysis, we applied default parameters (P‐value threshold‐0.05 and MicroT threshold‐0.08) and the human gene database to find associated KEGG pathways.
Figure 1.
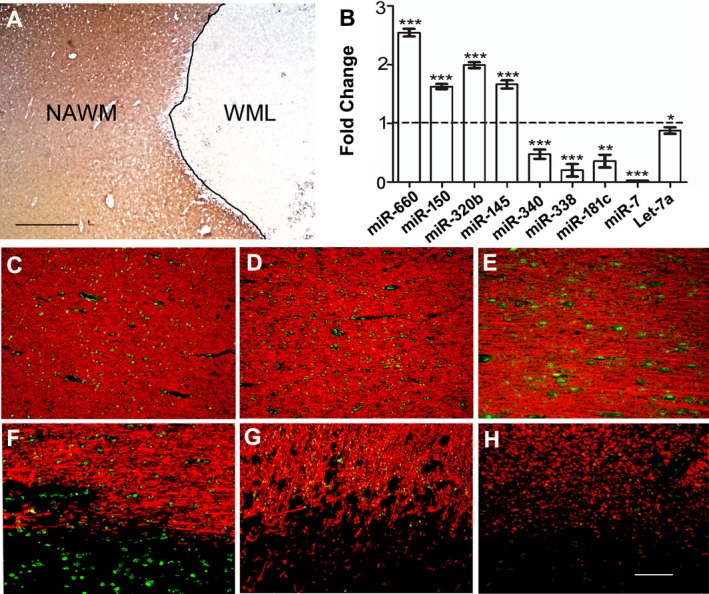
(A) Representative immunohistochemical proteolipid protein (PLP) image showing a demyelinated white‐matter lesion (WML) and surrounding normal‐appearing white matter (NAWM) from a progressive MS brain. Scale bar‐1 mm. (B) Validation of reported serum‐specific miRNAs expression in demyelinated WMLs of progressive MS brains. Relative expression of selected circulatory miRNAs was determined using specific TaqMan miRNAs assays for upregulated and downregulated miRNAs. Relative expression was calculated using ΔΔCt methods and normalized against RNU43 and U6 snRNA. The horizontal broken line represents the expression level of NAWM tissue (FC‐1), which was used as a control to compare respective miRNA expression levels. Error bars represent mean ± standard error of the mean (±SEM). *P < 0.05, **P < 0.01, ***P < 0.001. (C–H) Immuno‐fluorescent in situ hybridization (immune‐in situ) of miRNAs (green) and myelin basic protein (MBP, red) showing miR‐145 (C and F), miR‐181c (D and G), and miR‐340 (E and H) in myelinated (C–E, NAWM) and demyelinated (F–H, WML) areas of MS brain. miR‐145 showed increased expression in lesioned areas, whereas miR‐181c and miR‐340 were decreased in WMLs. WML‐white‐matter lesion, NAWM‐normal‐appearing white matter, FC‐Fold change, Scale bar‐100 μm.
Comparison of miRNAs in MS WM lesions with data from previously published datasets
We used the classification of miRNAs from Regev and colleagues10 as “protective” (serum miRNA expression correlating with low T1/T2 ratio and T2LV in MRI) and “pathogenic” (serum miRNA expression correlating with high T1/T2 ratio and T2LV) in serum samples. Fifty miRNAs correlated with MRI measures in cohort 1 and 91 miRNAs in cohort 2, with only nine miRNAs common between the two cohorts. We used these datasets with our comparison of significantly changed miRNAs in WMLs compared to NAWM from MS brains. The resultant comparisons identified 12 miRNAs with “protective” and 11 miRNAs with “pathogenic” associations in MS lesions (Table 2).
Table 2.
Expression of miRNAs having protective and pathogenic association10 in progressive MS brain
| miRs # | miRNAs | WML/NAWM | P‐value |
|---|---|---|---|
| Protective | |||
| 1 | hsa‐miR‐199a‐3p | 2.71 | 0.0088 |
| 2 | hsa‐miR‐143 | 2.26 | 0.0080 |
| 3 | hsa‐miR‐199a‐5p | 1.81 | 0.0084 |
| 4 | hsa‐miR‐30d | 1.53 | 0.0240 |
| 5 | hsa‐miR‐362‐5p | 1.24 | 0.0227 |
| 6 | hsa‐miR‐574‐3p | −0.49 | 0.0080 |
| 7 | hsa‐miR‐23b | −0.54 | 0.0280 |
| 8 | hsa‐miR‐18a | −1.25 | 0.0070 |
| 9 | hsa‐miR‐27b | −1.51 | 0.0007 |
| 10 | hsa‐miR‐181c | −1.53 | 0.0193 |
| 11 | hsa‐miR‐338‐3p | −2.97 | 0.0080 |
| 12 | hsa‐miR‐219‐5p | −14.67 | 0.0142 |
| Pathogenic | |||
| 1 | hsa‐miR‐320b | 2.80 | 0.0040 |
| 2 | hsa‐miR‐320a | 2.58 | 0.0162 |
| 3 | hsa‐miR‐22 | 2.49 | 0.0120 |
| 4 | hsa‐miR‐188‐5p | 2.05 | 0.0319 |
| 5 | hsa‐miR‐145 | 1.63 | 0.0047 |
| 6 | hsa‐miR‐30d | 1.53 | 0.0240 |
| 7 | hsa‐miR‐222 | 1.41 | 0.0336 |
| 8 | hsa‐miR‐628‐3p | −0.75 | 0.0080 |
| 9 | hsa‐miR‐27b* | −0.87 | 0.0026 |
| 10 | hsa‐miR‐15a | −0.96 | 0.0193 |
| 11 | hsa‐miR‐7 | −4.10 | 0.0004 |
miRNAs in bold letters were also validated in independent cohorts by qPCR. miR‐145 and miR‐181c was additionally validated using in situ hybridization.
Quantitative RT‐PCR
Expression levels of miRNAs were conducted in a separate validation cohort consisting of six progressive MS brain samples (Table 1, MS7‐12). To evaluate and validate miRNA expression, small‐sized RNA was isolated from demyelinated lesions (WML) and surrounding NAWM with a Qiagen miRNA isolation kit as per manufacturer's instructions. Isolated miRNAs were reverse‐transcribed to cDNA with a TaqMan miRNA RT Kit (Applied Biosystems, #4366596) as recommended by the supplier. Each sample was run in triplicate. The expression of selected miRNAs (Table S1) was validated using TaqMan miRNA assays (cat# 4427975) with PCR efficiency of 100% (±10%). RNU43 (assay ID #001095) and U6 snRNA (assay ID #001973) were used as endogenous controls in the reaction. To rule out inter‐assay variation, endogenous controls were also profiled in each plate separately. Delta Ct values were used to determine relative expression changes (fold change, 2−ΔΔCT). Groups in RT‐PCR analysis were compared using Student's t‐tests and a P‐value <0.05 was considered as significant. All quantitative data are expressed as mean ± standard error of the mean (±sem).
In situ hybridization
In situ hybridization was performed using a modified in situ protocol with locked nucleic acid‐modified oligonucleotide probes (Exiqon, Denmark) as previously described.13 Briefly, well‐characterized formalin‐fixed, paraffin‐embedded (FFPE) 7 μmol/L sections were de‐paraffinized and rehydrated. Section was washed in phosphate‐buffered saline (PBS) followed by treatment with proteinase K (60 ng) at 37°C/30 min and then treated in 4% paraformaldehyde (PFA). Next, washed sections were incubated in imidazole buffer, followed by incubation in EDC‐Imidazole solution for 90 min at room temperature. After washing the sections, a DIG‐labeled probe was hybridized to each section overnight (56–60°C). The next morning, sections were washed in 0.1 mol/L SSC, followed by endogenous peroxidase activity and blocking by 3% H2O2. Sections were then placed in blocking solution (Roche) for 1 h and incubated in α‐DIG antibody (Roche) and 1° antibody (MBP, Dako, cat#A0623) overnight at room temperature. The next morning, sections were washed (PBS/Tris‐HCl/Triton buffer) and incubated with fluorescent‐tagged TSA (Perkin Elmer, cat# NEL741E001KT) to label the probe. After washing, sections were incubated with alexa‐594‐tagged secondary antibody (ThermoFisher Scientific, cat#A11037) for 1 h at room temperature. Slides were then washed in 1 × PBS, fixed in filtered auto‐fluorescent eliminator regent (Millipore, cat#2160), and subjected to a series of 70% ethanol washes (6×), with a final wash in PBS. Sections were then mounted in prolong gold antifade reagent (Invitrogen, cat#P36930) and micrographed under a fluorescent microscope (Leica DM5500 B).
miRNA target genes and cell type identification
miRNA regulates gene expression by binding to the 3′UTR of target genes. To identify potential target genes that may be involved in MS pathogenesis, we searched for predicted target genes in three different miRNAs databases (miRDB, TargetScan, and DIANA‐TarBase v7.0)14, 15, 16 and selected top hits that were present in at least two of the databases. To further determine cellular identity, selected genes were queried against human and mouse cell‐specific RNA sequencing databases.17, 18 Using a previously published dataset from MS WMLs,6 we retrieved the mRNA levels of the target genes of the identified miRNAs and selected genes that were significantly altered in demyelinated lesions and having an opposite expression pattern as the target miRNA. For assessment of cell types, we used our previously published strategy to select cellular expression above the 50th percentile across all cell types.3
Results
miRNA expression profile of progressive MS brain tissue
To identify miRNA expression profiles of demyelinated WMLs in progressive MS brains, we used global miRNA expression profiling to quantify a total of 362 human miRNAs (listed in Table S2) between normal and demyelinated MS white‐matter tissue (Fig. 1A). Comparative analysis showed that 101 (28%) miRNAs were differentially expressed (P < 0.05) between normal myelinated and demyelinated regions in MS brains. Of these 101 miRNAs, 48 were upregulated and 53 were downregulated in WMLs (Table S2). A previous study reported that 56 miRNAs were altered in chronic MS lesions compared to NAWM tissues,9 with 22 miRNAs in common with the current data. Searching through miRNA target gene‐enriched molecular pathways (DIANA‐miRPath v3.0),12 the 101 miRNAs were found to be targeting several diverse pathways such as extracellular matrix‐receptor interactions, adherence junctions, axonal guidance, etc. (for the complete list, see Table S3).
To further validate these data, we selected four significantly upregulated miRNAs and five significantly downregulated miRNAs (Table S2) and measured their expression levels in a separate cohort of MS brains (Table 1, MS7‐12). Using a RT‐qPCR technique, we found that all nine miRNAs (miR‐660, miR‐150, miR‐320b, miR‐145, miR‐340, miR‐338, miR‐181c, miR‐7, and let‐7a) showed similar expression patterns as those observed in global microarray expression analysis (Fig. 1B). The cellular specificity of miRNAs dictates their biological functions. Using an immune‐in situ hybridization technique, we validated the cellular localization of miR‐145, miR‐181c, and miR‐340 in MS brains. In line with the upregulation of miR‐145 in MS lesions, the results also showed an increase in miR‐145‐expressing cells in MS lesions (Fig. 1F) compared to NAWM (Fig. 1C). On the other hand, we detected a significant decrease in the levels of miR‐181c and miR‐340 in demyelinated WM regions (Fig. 1G and H) compared to NAWM (Fig. 1D and E). Thus, miRNA expression detected by microarray and RT‐qPCR was also supported by cellular expression data in MS brains.
Identification and validation of reported circulatory miRNA expression in progressive MS brains
Regev and colleagues10 had identified clinically associated miRNAs (protective/pathogenic) in the sera of MS patients using significant associations between serum miRNA levels and WM‐MRI measurements (T2LV and T1/T2 ratio). As there can be significant difference between the miRNAs detected in peripheral circulation versus brain, we compared our current data from MS lesions to the previously published study.10 Comparing the two datasets, we found that 81% (292 of 362) of the miRNAs were common between the two different miRNA detection platforms used in this and the previous study. Of the 101 miRNAs found to be significant in MS WMLs, 88% (89 miRNAs) were also detected as circulatory miRNAs.10 Based on the MRI association (T1/T2 and T2 only), circulatory miRNA expression was classified as either “protective” or “pathogenic.” Using this classification, of the 89 miRNAs common between the datasets, we identified 12 protective and 11 pathogenic miRNAs to be significantly dysregulated in WMLs of progressive MS brains (Table 2). Among the 23 that were significantly changed, five miRNAs (miR‐181c, miR‐338, miR‐320b, miR‐145, and miR‐7) were also validated in independent samples using RT‐qPCR (Fig. 1B). In addition, results from the immuno‐in situ (miR‐145 (Fig. 1C,F); miR‐181c (Fig. 1D,G) and miR‐340 (Fig. 1E,H)) experiments provide evidence for the presence of circulating miRNAs in MS brains.
Among the 12 protective miRNAs that were significantly changed in MS lesions (Table 2), five were upregulated while seven were downregulated. Of the upregulated miRNAs that have protective associations, miR‐199a was increased by 2.7‐fold (P = 0.0088) in MS lesions compared to NAWM tissue. Interestingly, this miRNA, identified to be correlated with MS susceptibility genes,19 raises the possibility of miRNAs involvement in MS pathogenesis. On the other hand, miR‐219 is one of the critical miRNA that is necessary for the maturation of oligodendrocytes20 and it was found to be decreased in the cerebrospinal fluid of MS patients. This miRNA was found to be decreased by greater than 14‐fold (P = 0.0142) in MS lesions (Table 2) supporting a critical role of this miRNA in glial cell survival in MS lesions.9
Conversely, among the pathogenic miRNAs identified in the sera of MS patients (Table 2), seven were found to be increased in MS lesions, whereas four were deceased. Majority of the pathogenic miRNAs detected in MS lesions have also been reported to be associated with MS.9, 21, 22 Among the miRNAs increased in MS lesions with pathogenic association, miR‐320 was upregulated by 2.8‐fold (P = 0.004). Increase in the expression of this miRNA could be associated with exacerbations in multiple sclerosis through action of matrix metalloproteinases as shown previously.21 On the other hand, there was a decrease (−4.1 folds; P = 0.0004) in the levels of miR‐7 in WML tissue. The significant decrease in levels of miR‐7, together with pathogenic association in the previous study,10 raises the possibility of a protective effect rendered by the targets of this miRNA in MS lesions.
Common pathways as targets of pathogenic and protective miRNAs that are altered in MS brains
A single miRNA can regulate a large number of gene products belonging to a particular pathway network, and likewise a cluster of miRNAs may cooperatively target various genes of the same molecular pathway.23, 24 To identify protective and pathogenic miRNA target gene‐enriched molecular pathways in progressive MS lesions, we used DIANA‐miRPath v3.0.12 We found increased similarity between the pathways targeted by the downregulated protective miRNAs and upregulated pathogenic miRNAs. These included MAPK signaling, Ras signaling, Rap1 signaling, focal adhesion, cAMP signaling, FoxO signaling, etc. (Fig. 2A). On the other hand, pathways like ErbB signaling, Wnt signaling, long‐term depression, were identified by pathway analysis of upregulated protective and downregulated pathogenic miRNAs (Fig. 2B, Table S4). Collectively, the results indicate possible involvement of these pathways in either having a “protective” or “pathogenic” role underlying the pathogenesis of MS lesions.
Figure 2.
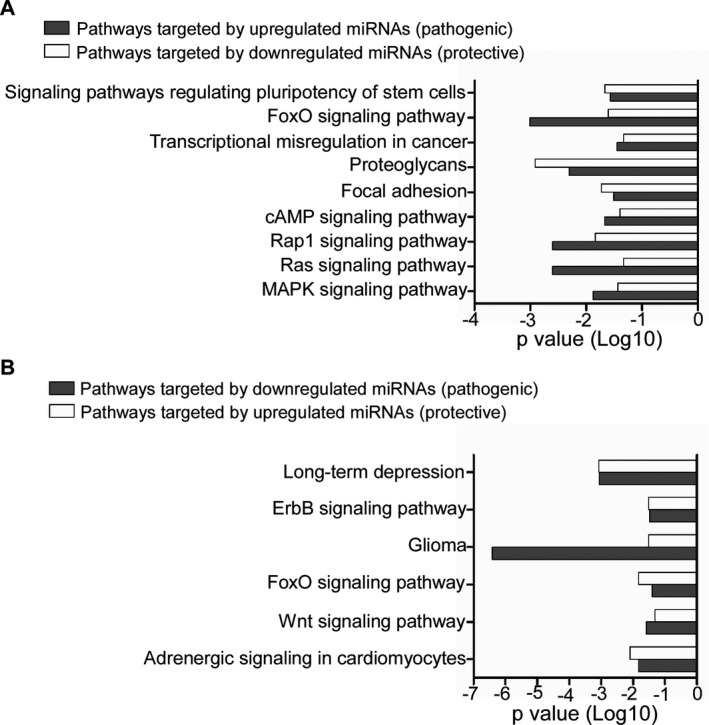
Representative bar graph showing common KEGG pathways of genes targeted by pathogenic and protective miRNAs in WMLs. Common pathways of genes targeted by (A) upregulated pathogenic and downregulated protective miRNAs, and (B) downregulated pathogenic and upregulated protective miRNAs.
Identification of the cell types expressing transcripts regulated by clinically associated miRNAs
As miRNAs regulate gene expression by binding to the 3′UTR of target genes,7 we asked whether the mRNA levels of the target genes of these miRNAs (Table S2) were also altered in MS WMLs. Target genes were identified based on their presence across three separate miRNA databases (miRDB, TargetScan, and DIANA‐TarBase v7.0)14, 15, 16 as well as identified as top hits (miRDB target score >90). mRNA levels of these target genes were searched using our previously reported WML mRNA dataset.6 Genes with significant changes in mRNA levels and inverse correlations in expression to the miRNAs are presented in Table S5. The role of these candidate genes and their biological validation could therefore lead to novel insights into their protective and pathogenic roles in MS. In addition to the expression levels, we also searched for the cellular source of these target genes in human and mouse RNAseq databases using previously published criteria.3 Results showed that the identified genes were expressed by all major CNS cell populations (astrocytes, neurons, oligodendrocytes, and microglia) in both human and mouse (Table S5). While the target genes of upregulated miRNAs were expressed by all four of these cell types, the target genes of the downregulated miRNAs were enriched mostly in astrocytes and microglia.
Discussion
miRNAs have been under intense scrutiny in several diseases, as they are among the most effective regulators controlling gene expression. The present study compared miRNAs in WMLs from progressive MS brains and validated the data by using separate groups of MS patients. Many of these miRNAs have been detected in other biological fluids of human MS patients.9, 21, 25, 26 A group of these miRNAs also correlated with miRNAs previously identified in sera from MS patients that have been hypothesized to have a “pathogenic” or “protective” function based on T2LV and T1/T2 MRI measures.10 In addition to determining the expression levels of the miRNAs in MS lesions, our results identified several common pathways that are targeted by these pathogenic and protective miRNAs. Taken together, this study identifies a unique group of miRNAs that can be detected in both sera as well as MS lesions. Expression levels of the target genes of these miRNAs, as well as their cellular specificity, provide unique insights into the pathogenesis of MS.
The study conducted by Regev et al.10 provides an important step in determining the role of miRNAs as biomarkers in MS. Their study compared MRI measures of tissue injury with the changes in miRNA levels in serum to classify the miRNA functions as pathogenic or protective. While MRI has been the only method to detect lesions in MS patients, the results do not provide biological evidence as to whether these miRNAs are also altered in actual sites of tissue injury, that is, in MS WMLs. Using comparative analysis, we found 23 miRNAs that were significantly changed in MS lesions (Table S2). These were classified as either pathogenic or protective based upon the previous report.10 Among these miRNAs, we found that miR‐320a (“identified as pathogenic”) was significantly upregulated in WMLs (Table S2) compared to NAWM and has been previously reported to be highly expressed in B cells of MS patients contributing toward increased blood–brain barrier permeability through regulation of matrix metallopeptidase.21 Similarly, miR‐219 and miR‐338, (designated as protective) were significantly downregulated in WMLs and shown to play a major role during CNS myelination and myelin repair.20
Interestingly, the pathways targeted by pathogenic miRNAs (increased in WMLs) were very similar to those targeted by the downregulated protective miRNAs (in WMLs). Among these common pathways (Fig. 2A), MAPK signaling pathway is involved in regulating key immunopathogenic mechanisms in multiple sclerosis and experimental allergic encephalomyelitis (EAE).27 On the other hand, ErbB signaling pathway (Fig. 2B) identified as a common pathway between downregulated pathogenic and upregulated protective miRNAs has been shown to be critical for the process of myelination in both peripheral and central nervous system. Most importantly, insufficient ErbB signaling has also been found to be correlated with development of MS.28 As the brain is a heterogeneous mixture of cells, we extended our analysis to ascertain the cellular specificity of the target genes in MS lesions. These results lay the foundation for future studies related to targeting these genes and pathways to investigate mechanisms underlying lesion formation in MS.
It is now accepted that neuronal and axonal loss are main factors underlying the progressive neurological disability observed in MS patients. Our miRNA data have been generated in progressive MS lesions and provide important clues to the correlation of these miRNAs (common between two studies) with MRI measures of WM tissue injury. The previous study10 did not find any significant correlation between the miRNAs involved in predominantly WM injury (T2LV and T1/T2 measures) versus those related to predominantly gray matter (GM) injury (atrophy and brain GM fraction measures). In a recent study, cortical demyelination and neuronal loss were found to be independent of WMLs.29 Analysis of miRNAs and other regulatory factors in well‐characterized cortical lesions would therefore be the next steps toward determining the role of miRNAs in neuronal and axonal degeneration.
Author Contributions
R. D. conceived the idea and designed the study. A. T., C. V., and U. D. planned and performed the experiments. K. R. provided the circulating miRNA data. A. T., C. V., K. R., and R. D. participated in writing of the manuscript. All authors discussed the results and commented on the manuscript.
Conflict of Interest
None reported.
Supporting information
Table S1. TaqMan assay IDs and expression level of selected miRNAs in WMLs validated by qPCR.
Table S2. Expression level of 362 miRNAs in WMLs compared to NAWM.
Table S3. Pathways of genes targeted by significantly changed miRNAs in WMLs.
Table S4. KEGG pathway analysis of genes targeted by pathogenic and protective miRNAs.
Table S5. Inverse correlation between miRNAs and target gene mRNA levels. Clinically associated circulatory miRNA expression was significantly dysregulated in WM lesions as analyzed by microarray expression analysis. miRNAs highlighted in bold were validated by qPCR in independent MS samples. Predicted target genes of respective miRNAs were selected from miRDB (target score > 90) and also present in the TargetScan and/or the DIANA‐TarBase v7.0 database.14, 15, 16 Expression levels of target genes were extracted using previously published microarray‐based analysis6 and are presented as upregulated (+)/downregulated (−) in white‐matter lesions (WMLs) compared to normal‐appearing white matter (NAWM) microarray gene expression values. *P‐value represents level of significance. Identified genes were mapped to astrocytes (A), neurons (N), oligodendrocytes (O), or microglia/macrophage lineage (M) cells in human and mouse CNS cell‐specific databases. To increase confidence in cellular identity, matching genes showing expression levels above the 50th percentile were selected.3
Acknowledgments
The authors thank C. Nelson for editorial assistance. This work was supported by grants from NINDS (NS096148) and the National Multiple Sclerosis Society, USA (RG 5298) to RD. The MS brain collection program is supported by NINDS grant R35NS097303 to Dr. Bruce D. Trapp.
Funding Information
This work was supported by grants from NINDS (NS096148) and the National Multiple Sclerosis Society, USA (RG 5298) to RD. The MS brain collection program is supported by NINDS grant R35NS097303 to Dr. Bruce Trapp.
Funding Statement
This work was funded by NINDS grants NS096148 and NS097303; National Multiple Sclerosis Society USA grant RG 5298.
References
- 1. Baecher‐Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron 2018;97:742–768. [DOI] [PubMed] [Google Scholar]
- 2. Arthur AT, Armati PJ, Bye C, et al. Genes implicated in multiple sclerosis pathogenesis from consilience of genotyping and expression profiles in relapse and remission. BMC Med Genet 2008;9:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chomyk AM, Volsko C, Tripathi A, et al. DNA methylation in demyelinated multiple sclerosis hippocampus. Sci Rep 2017;7:8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dutta R, Chomyk AM, Chang A, et al. Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR‐124 and reduced AMPA receptors. Ann Neurol 2013;73:637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Freiesleben S, Hecker M, Zettl UK, et al. Analysis of microRNA and gene expression profiles in multiple sclerosis: integrating interaction data to uncover regulatory mechanisms. Sci Rep 2016;6:34512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huynh JL, Garg P, Thin TH, et al. Epigenome‐wide differences in pathology‐free regions of multiple sclerosis‐affected brains. Nat Neurosci 2014;17:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 2011;12:99–110. [DOI] [PubMed] [Google Scholar]
- 8. Gandhi R. miRNA in multiple sclerosis: search for novel biomarkers. Mult Scler 2015;21:1095–1103. [DOI] [PubMed] [Google Scholar]
- 9. Junker A, Krumbholz M, Eisele S, et al. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain 2009;132(Pt 12):3342–3352. [DOI] [PubMed] [Google Scholar]
- 10. Regev K, Healy BC, Khalid F, et al. Association between serum MicroRNAs and magnetic resonance imaging measures of multiple sclerosis severity. JAMA Neurol 2017;74:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang A, Staugaitis SM, Dutta R, et al. Cortical remyelination: a new target for repair therapies in multiple sclerosis. Ann Neurol 2012;72:918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vlachos IS, Zagganas K, Paraskevopoulou MD, et al. DIANA‐miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res 2015;43(W1):W460–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dutta R, McDonough J, Chang A, et al. Activation of the ciliary neurotrophic factor (CNTF) signalling pathway in cortical neurons of multiple sclerosis patients. Brain 2007;130(Pt 10):2566–2576. [DOI] [PubMed] [Google Scholar]
- 14. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015;4:e05005: 1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vlachos IS, Paraskevopoulou MD, Karagkouni D, et al. DIANA‐TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015;43(Database issue):D153–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Database issue):D146–D152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y, Chen K, Sloan SA, et al. An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014;34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y, Sloan SA, Clarke LE, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 2016;89:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luo D, Fu J. Identifying characteristic miRNAs‐genes and risk pathways of multiple sclerosis based on bioinformatics analysis. Oncotarget 2018;9:5287–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang H, Moyano AL, Ma Z, et al. miR‐219 cooperates with miR‐338 in myelination and promotes myelin repair in the CNS. Dev Cell 2017;40:566–582 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aung LL, Mouradian MM, Dhib‐Jalbut S, Balashov KE. MMP‐9 expression is increased in B lymphocytes during multiple sclerosis exacerbation and is regulated by microRNA‐320a. J Neuroimmunol 2015;278:185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu Z, Cui Y, Qiao X, et al. Silencing miR‐150 ameliorates experimental autoimmune encephalomyelitis. Front Neurosci 2018;12:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Y, Luo J, Zhang H, Lu J. microRNAs in the same clusters evolve to coordinately regulate functionally related genes. Mol Biol Evol 2016;33:2232–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mestdagh P, Bostrom AK, Impens F, et al. The miR‐17‐92 microRNA cluster regulates multiple components of the TGF‐beta pathway in neuroblastoma. Mol Cell 2010;40:762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Regev K, Healy BC, Paul A, et al. Identification of MS‐specific serum miRNAs in an international multicenter study. Neurol Neuroimmunol Neuroinflamm 2018;5:e491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Regev K, Paul A, Healy B, et al. Comprehensive evaluation of serum microRNAs as biomarkers in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm 2016;3:e267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krementsov DN, Thornton TM, Teuscher C, Rincon M. The emerging role of p38 mitogen‐activated protein kinase in multiple sclerosis and its models. Mol Cell Biol 2013;33:3728–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang J, Kramer EG, Asp L, et al. Promoting myelin repair and return of function in multiple sclerosis. FEBS Lett 2011;585:3813–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trapp BD, Vignos M, Dudman J, et al. Cortical neuronal densities and cerebral white matter demyelination in multiple sclerosis: a retrospective study. Lancet Neurol 2018;17:870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. TaqMan assay IDs and expression level of selected miRNAs in WMLs validated by qPCR.
Table S2. Expression level of 362 miRNAs in WMLs compared to NAWM.
Table S3. Pathways of genes targeted by significantly changed miRNAs in WMLs.
Table S4. KEGG pathway analysis of genes targeted by pathogenic and protective miRNAs.
Table S5. Inverse correlation between miRNAs and target gene mRNA levels. Clinically associated circulatory miRNA expression was significantly dysregulated in WM lesions as analyzed by microarray expression analysis. miRNAs highlighted in bold were validated by qPCR in independent MS samples. Predicted target genes of respective miRNAs were selected from miRDB (target score > 90) and also present in the TargetScan and/or the DIANA‐TarBase v7.0 database.14, 15, 16 Expression levels of target genes were extracted using previously published microarray‐based analysis6 and are presented as upregulated (+)/downregulated (−) in white‐matter lesions (WMLs) compared to normal‐appearing white matter (NAWM) microarray gene expression values. *P‐value represents level of significance. Identified genes were mapped to astrocytes (A), neurons (N), oligodendrocytes (O), or microglia/macrophage lineage (M) cells in human and mouse CNS cell‐specific databases. To increase confidence in cellular identity, matching genes showing expression levels above the 50th percentile were selected.3