

Abstract
Advances in understanding and treatment of cardiac disorders have been thwarted by inability to study beating human cardiac cells in vitro. Induced pluripotent stem cells (iPSCs) bypass this hurdle by enabling the creation of patient-specific iPSC-derived cardiomyocytes (iPSC-CMs). These cells provide a unique platform to study cardiac diseases in vitro, especially hereditary cardiac conditions. To date, iPSC-CMs have successfully been used to model arrhythmic disorders showing excellent recapitulation of cardiac channel function and electrophysiologic features of Long QT syndrome types 1, 2, 3, and 8 and catecholaminergic polymorphic ventricular tachycardia (CPVT). Similarly, iPSC-CM models of dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM) have shown robust correlation of predicted morphologic, contractile, and electrical phenotypes. In addition, iPSC-CMs have shown some features of the respective phenotypes for arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C), LEOPARD syndrome, Pompe’s disease, and Friedriech’s ataxia. In this review, we examine the progress of utilizing iPSC-CMs as a model for cardiac conditions and analyze the potential for the platform in furthering the biology and treatment cardiac disorders.
Keywords: human induced pluripotent stem cells, dilated cardiomyopathy, hypertrophic cardiomyopathy, channelopathy, drug discovery
Introduction
Cardiac disease remains the most common cause of death in developed countries.1 Over the past four decades, there have been remarkable advances in the understanding and treatment of ischemic heart disease, while our understanding of cardiomyopathies and arrhythmic disorders has lagged behind. Indeed, the realization that many cardiomyopathies and sudden cardiac deaths are in fact related to genetic disorders has heralded a remarkable era for hereditary cardiac disorders.2 With advances in genetic sequencing methods, more genetic targets are being identified as pathogenic variants of these disorders. Unfortunately, understanding the biology of how these putative genetic variants result in observed phenotypic changes has proven challenging.3
A big hurdle to understanding cardiac molecular and cellular physiology is the inability to maintain and study functional (beating) human cardiomyocytes in culture. This leaves scientists to rely on animal models or transfected cell models. Furthermore, the invasive nature of obtaining primary human cardiac tissue has hindered the field compared to other fields such as hematology or oncology. Induced pluripotent stem cells (iPSCs) have emerged as a reliable method to produce patient-specific somatic tissue lines via directed differentiation. By enabling the analysis and manipulation of human cardiac cells in culture, iPSC-derived cardiomyocytes (iPSC-CMs) have provided a robust platform to study genetic cardiac disorders (Table 1). The model has already proven complementary or superior in compared to transfected cell or animal models of cardiac genetic disorders. In this review, we aim to examine the growing body of work that has utilized iPSC-CMs to study cardiac disorders, offer some insights as to the strength of iPSC-CMs as a modeling tool, and how this could affect cardiovascular clinical practice and biomedical research (Figure 1).
Table 1.
Summary of major efforts utilzing iPSC-CM to model hereditary cardiac disorders.
| Disorder | Study | Gene (Variant/s) | Histological or Cellular Abnormalities |
Electrophysiologic Abnormalities |
Response to Standard Pharmacologic Agents |
Force Generation Abnormalities |
Novel drug testing |
|---|---|---|---|---|---|---|---|
| LQT1 | Moretti et al. (2010) | KCNQ1 (p.R190Q) | - | + | + | ||
| Egashira et al. (2012) | KCNQ1 (p.C1893del) | - | + | + | |||
| LQT2 | Itzhaki et al. (2011) | KCNH2 (p.A614V) | - | + | + | ||
| Matsa et al. (2011) | KCNH2 (p.G1681A) | - | + | + | |||
| Lahi et al. (2012) | KCNH2 (p.R176W) | - | + | + | |||
| LQT3 | Terrenoire et al. (2013) | SCN5A (p.F1473C) | - | + | + | ||
| Ma et al. (2013) | SCN5A (p.V1763M) | - | + | + | |||
| LQT8 | Yazawa et al. (2011) | CACNA1C (p.G406R) | - | + | + | ||
| CPVT | Itzhaki et al. (2012) | RYR2 (p.M4109R) | - | + | + | ||
| Fatima et al. (2011) | RYR2 (p.T7447A) | - | + | + | |||
| Novak et al. (2012) | CASQ2 (p.D307H) | +* | + | + | |||
| Jung et al. (2012) | RYR2 (p.S406L) | - | + | + | + | ||
| DCM | Sun et al. (2012) | TNNT2 (p.R173W) | + | - | + | + | |
| Siu et al. (2012) | LMNA (p.R225x, p.S18fs) | + | + | ||||
| Tse et al. (2013) | DES (p.A285V) | + | |||||
| HCM | Lan et al. (2013) | MYH7 (p.R663H) | + | + | + | + | |
| ARVD/C | Kim et al. (2013) | PKP2 (c.2484C>T, p.G828G, r.[2483_2489del]) | - | + | |||
| Ma et al. (2013) | PKP2 (p.L614P) | + * | + | ||||
| LEOPARD | Carvajal-Vergara (2010) | PTPN11 (p.T468M) | + | ||||
| Pompe’s Disease | Huang et al. (2011) | p.D645E/p.D645E, c.1935C.A/c.2040+1G>T | + | + | |||
| Friedreich’s Ataxia | Hick et al. (2011) | Expanded GAA repeats in 1st intron of FXN | + | - | |||
| Other | Liang et al. (2013) | KCNQ1 (p.G269S), MYH7 (p.R663H), TNNT2 (p.R173W) | + |
(Using Electron Microscopy)
Figure 1:
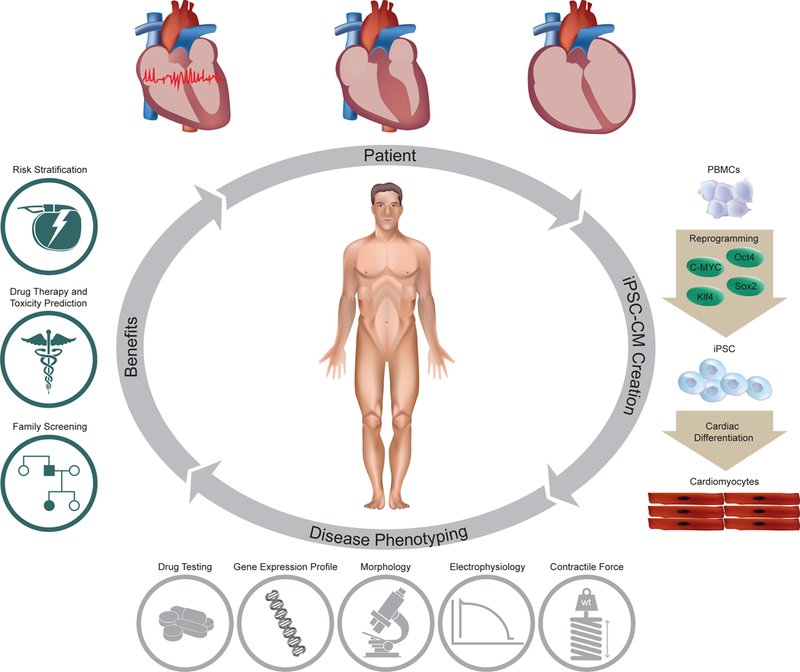
Future role iPSC-CMs can play in clinical care of patients with hereditary cardiac disorders. iPSC-CMs can be profiled using molecular techniques, electrical and mechanical measures, and response to different pharmacological agents. This data can help provide input on the predicted severity of disease, associated risks and response to therapy; which can be used as adjuvant in clinical decision making. In addition, functional characterization of the tissue might help elucidate causative candidate variants and assist in screening family members for the disease. iPSC-CMs, induced pluripotent stem cell-derived cardiomyocytes; PMBCs, peripheral blood mononuclear cells; iPSCs, induced pluripotent stem cells.
History of iPSCs
iPSCs are made by reprogramming somatic cells into a more pluripotent phenotype that is capable of self renewal and differentiating into all three germ layers. This was first described by Takahashi et al. and later by Yu et al.4,5 Shortly thereafter, several groups showed that directed differentiation can produce somatic cell derivatives such as neurons, hematopoietic cells, and cardiomyocytes (Figure 1).6–8 The technology offered several advantages from the prior state of biomedical research. First, it bypasses the ethical concerns about the use of other stem cell sources. Second, because they are self-renewing, iPSCs can provide an unlimited supply of differentiated somatic tissues. Lastly, iPSCs as well as their derivatives recapitulate the genotype of their original somatic donor, which offers the unprecedented potential to obtain patient-specific tissue types non-invasively.
Since the first reports, there have been major advances in the methods of making iPSCs and lineage-specific differentiation protocols, which are outside the scope of this review. However, it is fair to say that obtaining patient-specific iPSCs and iPSC-CMs has become more reliable and more efficient.9–11 Given the difficulty in studying cardiac disorders in vitro, interest in utilizing iPSC-CMs as a model for cardiac disorders has grown very rapidly. Here we will highlight some of the studies that illustrate the advantages and limitations of the model.
Inherited Channelopathy Models
The most extensively studied iPSC-CM models are those of arrhythmic disorders. This is in part because the mechanism by which gene variants lead to perturbations in ion flux is better defined compared to that of sarcomeric myopathies. Furthermore, patch clamp and multielectrode array (MEA) platforms reliably measure cellular action potential (AP) properties, ion channel currents, and arrhythmic potential.12 This detailed electrophysiologic assessment enables direct comparison and correlation with in vivo phenotypic changes such as ECG abnormalities or arrhythmia formation.
Long QT syndrome (LQT) is a disorder that is characterized clinically by prolongation of the QT interval on surface ECG and increased burden of ventricular arrhythmias and sudden cardiac death. Advances in genetics and functional classification were able to further classify the syndrome into multiple subtypes based on common genetic loci and functional disturbances in ion channel flux.3 While 13 loci associated with LQT have been identified, variants in 3 loci account for 60–70% of the cases seen clinically.13,14 LQT is probably the most extensively studied arrhythmic disorder given its prevalence and the fact the disorder has been described for over 50 years, compared to more recently described arrhythmic disorders such as Short QT syndrome or catecholaminergic polymorphic ventricular tachycardia (CPVT). Because of the relatively advanced understanding of LQT compared to other arrhythmic disorder, LQT has been an attractive first option to investigate the utility of iPSC-CMs as a functional model.
Long QT 1
Long QT 1 (LQT1) is most commonly caused by variants in KCNQ1 gene resulting in disruption in the slow delayed-rectifier potassium current (IKs). Classically, patients are described to develop exercise induced arrhythmia, which presumably results from inability of the cell to shorten QT interval with exercise.15 In a seminal paper by Moretti et al., the authors reprogrammed fibroblasts from a patient with a variant in KCNQ1 gene (p.R190Q) into iPSCs, which were subsequently differentiated into iPSC-CMs.16 The authors determined that the levels of KCNQ1 were similar between control and affected iPSC-CMs, but affected iPSC-CMs showed localization of KCNQ1 protein in the endoplasmic reticulum, suggesting that the variant resulted in a trafficking defect rather than an ion channel function defect. Furthermore, the authors observed that LQT1 iPSC-CMs exhibited a longer AP duration with a reduced repolarization velocity compared to control cells. This was due to reduced IKs current density in affected iPSC-CMs, whereas IKr and Ito were unaffected, supporting the hypothesis that the identified KCNQ1 variant led to reduced IKs and was responsible for the prolonged repolarization and action potential duration (APD) observed. Lastly, iPSC-CMs recapitulated clinical response of the phenotype to β-agonists and β-blockers by demonstrating that stimulation with isoproterenol resulted in blunting of the expected reduction in APD duration and development of early after depolarizations (EAD), which was abated by the use of propranolol.
Long QT 2
Another defect in potassium channel flow results in Long QT 2 (LQT2). LQT2 is most commonly caused by variants in KCNH2 gene, also called human Ether-à-go-go-Related Gene (hERG), which result in abnormal rapid cardiac delayed rectifier potassium current (IKr). Patients who develop ventricular arrhythmia or sudden death in response to auditory stimuli most commonly have LQT2.15 Another group published an iPSC-CM model of LQT2 due to a known missense mutation in KCNH2 gene (p.A614V).17 Similar to the LQT1 model, the iPSC-CMs exhibited prolonged APD and FPD at the single cell and tissue levels, respectively. In addition, the cells exhibited reduced IKr current that also supports the proposed mechanism for LQT2, namely that the prolonged repolarization is caused by the loss of function of rapid acting inward rectifying potassium channels. The authors found that at the multicellular level, 38% of LQT2 iPSC-CMs exhibited ectopic single and multiple triggered beats. This is the same mechanism hypothesized to trigger premature beats that occur during the repolarization phase and to result in the ventricular arrhythmia of torsades de pointes (TdP) seen in LQT2 patients.
Timothy Syndrome (LQT8)
Timothy syndrome is a genetic disorder caused by mutation in L-type calcium channel (Cav1.2) first described by Splawski and colleagues in 2004.18 The disorder is characterized by syndactyly, immune deficiency, autism, and LQT (classified as LQT 8). Yazawa et al. created iPSC lines from 2 Timothy syndrome patients and 2 control patients, all of which were differentiated into beating embryoid bodies (EBs).19 There were no gross differences on immunohistochemistry staining of major cardiac proteins among lines. As predicted by the genetic variant, patch clamp analysis revealed reduced L-type calcium channel voltage-dependent inactivation in Timothy syndrome iPSC-CMs. This led to prolongation of the APD and increased incidence of delayed after depolarizations (DAD) in ventricular iPSC-CMs from Timothy syndrome patients compared to controls. Roscovitine, which increased the voltage-dependent inactivation of the L-type calcium channels, reduced the timing and amplitude of calcium transients and APD in Timothy syndrome iPSC-CMs, bringing them closer to control cell levels. This model demonstrated that calcium handling properties of iPSC-CMs recapitulated the in vivo phenotype and not just potassium currents.
Catecholaminergic Polymorphic Ventricular Tachycardia
CPVT is a hereditary arrhythmic disorder thought to be caused by abnormal calcium handling in cardiomyocytes.20 Clinically, the disorder is associated with states of catecholamine surge that precipitate bidirectional ventricular tachycardia and sudden death. The majority of patients harbor variants in ryanodine receptor gene (RYR2) and cardiac calsequestrin (CASQ2) that result in calcium leak potentiating DAD on a cellular level. The exact mechanism by which the variants lead to calcium handling defects is not completely understood. This makes CPVT an especially interesting disorder to study with an iPSC-CM platform.
Itzhaki et al. reprogrammed iPSCs from dermal fibroblasts of a CPVT patient with a heterozygous mutation in RYR2 (p.M4109R).21 The authors noted no difference in baseline AP properties between CPVT and control iPSC-CMs. However, pacing at faster intervals resulted in development of DAD in 69% of CPVT iPSC-CMs compared to 11% of healthy control iPSC-CMs. Furthermore, the authors chemically simulate adrenergic surges by adding forskolin (adenylate cyclase activator) and isoproterenol, both of which increased the frequency of DAD and were associated with the development of triggered activity. Flecainide, which is thought to be clinically useful in CPVT patients, completely eliminated DAD. To confirm the proposed mechanism of CPVT as being due to increased intracellular levels of calcium, the authors applied a sarcoplasmic reticulum (SR) calcium ATPase pump inhibitor (thapsigargin) that depletes intracellular calcium stores. As predicted, depleting the cells of intracellular calcium decreased the incidence of DAD. Furthermore, the authors performed calcium imaging demonstrating that abnormal calcium transients were exacerbated by faster pacing, adrenergic stimulation, and higher calcium concentration. Similar results were published by Fatima et al. and Novak et al., who made iPSC-CMs from another RYR2 variant (p.F24831) and a CASQ2 variant (p.D307H) respectively.22,23 The results were congruent, demonstrating the increased propensity to arrhythmia and diastolic calcium rise in response to isoproterenol.
Inherited Cardiomyopathy Models
All of the above models relied on studying arrhythmic disorders by taking advantage of the reliable tools available to measure cellular electrophysiology (EP). However, there was considerable doubt whether myopathies could be functionally modeled by iPSC-CM and if abnormalities in contractile function could be analyzed in vitro to validate the cellular phenotype of cardiomyopathy.
Familial Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is a disorder of progressive ventricular dilation and decreased systolic function that usually leads to the heart failure syndrome and possibly the development of lethal arrhythmias. Familial DCM refers to genetic cases of the disorder, but the term DCM is sometimes used interchangeably to refer to those cases. The pathophysiology of progression of DCM is thought to be related to upregulation of catecholamines, renin-angiotensin axis, and loading conditions. However, the pathophysiology by which individual causative variants lead to the observed phenotype is not well defined. The complexity in simulating systemic in vivo conditions, the limitations of measuring cellular phenotype, and incomplete understanding of cellular pathophysiology were all barriers to creating successful myopathy models.
The first successful model of a cardiomyopathy that tested the functional phenotype of iPSC-CMs was by Sun et al., who recruited a three-generation family with a point mutation TNNT2 mutation (p.R173W) to make patient specific iPSC-CMs.24 The authors saw clear morphological differences between DCM iPSC-CMs and controls (Figure 2). Functionally, this translated into decreased contractility compared to control iPSC-CMs as measured by atomic force microscopy. In addition, the authors mirrored the clinical impact of various drugs in DCM by demonstrating that treatment with norepinephrine resulted in exacerbation of the DCM phenotype, whereas treatment with metoprolol was protective.
Figure 2: iPSC–CMs from familial dilated cardiomyopathy (DCM) patients show an abnormal sarcomeric a-actinin distribution.
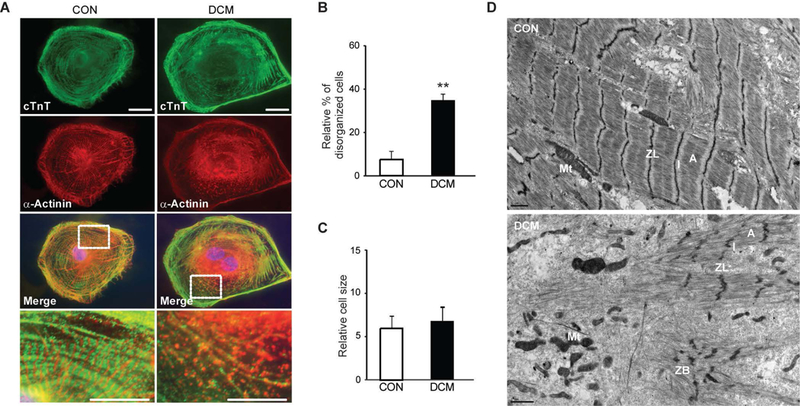
(A) Immunostaining of sarcomeric a-actinin and cardiac troponin T (cTnT) at day 30 after differentiation. (B) Compared to control iPSC-CMs, a higher percentage of DCM iPSC–CMs showed a punctate sarcomeric a-actinin staining pattern in greater than one-fourth of the total cellular area (**P = 0.008). (C) No significant difference was observed in cell size between control and DCM iPSC–CMs. (D) Transmission electron microscopy demonstrate that compared to controls, DCM iPSC-CMs exhibited an increased variability in the degree of organization. ZL, Z line; ZB, Z bodies; A, A band; I, I band; Mt, mitochondria.
Another effort to model DCM involved a lamin A/C (LMNA) variant, which causes a characteristic pattern of progressive conduction system disease and DCM.25 The authors created iPSCs from a patient with LMNA mutation (p.R225X). Affected iPSC-CMs demonstrated nuclear senescence compared to control iPSC-CM. Electrical stimulation increased the prevalence of nuclear senescence and induced apoptosis in LMNA iPSC-CMs. The finding of tachycardia resulting in cell death is consistent with the prevailing hypothesis of the disorder that increased stimulation induces damage to myocytes with fragile nuclear properties.26
Familial Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is a disorder of abnormal thickening of primarily the left ventricular wall. The condition is predominantly genetic in nature, and although it usually has a more benign course than DCM, it can lead to the heart failure syndrome and lethal arrhythmias. There are no approved life prolonging drug therapies for HCM, and predicting any given patient’s course in terms of development of symptoms or lethal arrhythmias remains a challenge.27 Lan et al. sought to evaluate the mechanism by which a well-reported genetic variant in MYH7 gene (p.R663H) results in cardiac hypertrophy.28 HCM iPSC-CMs showed a clear hypertrophic phenotype on the single cell level (Figure 3). In addition, electrophysiologic assessment showed frequent DAD in HCM iPSC-CMs compared to control iPSC-CMs. The authors showed that impaired calcium handling and increased diastolic intracellular calcium concentration were implicated in cellular hypertrophy and arrhythmic burden. Interestingly, the administration of L-type calcium channel blocker verapamil improved calcium handling and ameliorated the hypertrophic and arrhythmic profile. This data suggest that impaired calcium regulation is actually causative of the arrhythmia and hypertrophy rather than a secondary effect, contrary to the notion that the pro-arrhythmic effect was solely a function of tissue-level structural hypertrophy or intracavitary obstruction.
Figure 3. iPSC-CM from familial hypertrophic cardiomyopathy (HCM) patients show hypertrophy on a single cell level.
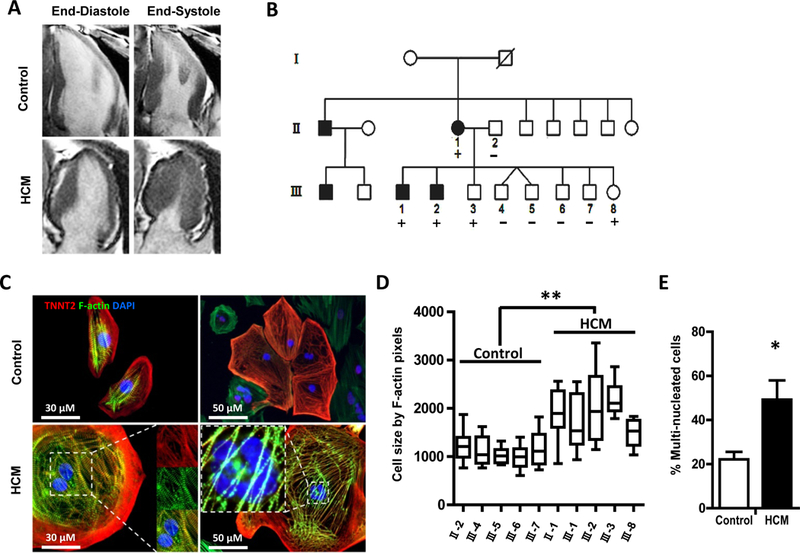
(A) Representative long-axis MRI images of the proband and a control matched family member at end systole and end diastole demonstrating asymmetric hypertrophy of the inferior wall. (B) Schematic pedigree of the proband carrying the Arg663His mutation in MYH7 recruited for this study (II-1) as well as her husband (II-2) and eight children (III-1 through III-8). (C) Representative immunostaining for cardiac troponin T and F-actin demonstrating increased cellular size and multinucleation in HCM iPSC-CMs as compared to control iPSC-CMs. (D) HCM iPSC-CM lines show increase in cell size compared to controls at day 40 after induction of cardiac differentiation. (E) Quantification of multinucleation in control and HCM iPSC-CMs.
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C)
ARVD/C is an inherited primary cardiomyopathy that results in predominantly right ventricular aneurysms and fibro fatty infiltration. The disorder is associated with ventricular arrhythmias and sudden cardiac death. Approximately half of cases have been linked to the desmosomal proteins: desmoplakin, junction plakoglobin (PKG), plakophilin-2 (PKP2), desmoglein 2, and desmocollin.2 The mechanism by which the abnormal cell-to-cell desmosal abnormalities lead to tissue level myopathy, fatty infiltration, and increased arrhythmic burden is still poorly understood.
Kim et al. reprogrammed iPSC lines from fibroblasts from an ARVD patient homozygous for a PKP2 variant (p.G828G).29 The iPSC lines were differentiated into iPSC-CMs and the authors observed abnormal nuclear translocation of PKG and very low β-catenin activity and expression in affected iPSC-CMs. At baseline, this did not result in any changes in lipid content or apoptosis levels of ARVD iPSC-CMs. In an attempt to induce iPSC-CMs to switch to fatty acid oxidation (i.e., metabolic state of adult cardiomyocytes rather than fetal), the authors utilized a lipogenic medium in addition to rosiglitazone and indomethacin. The use of these agents was due to the compounds’ ability to upregulate PPAR-γ, which is overexpressed in explanted ARVD heart tissue. Indeed, adding rosiglitazone and indomethacin to an inducing medium increased lipogenesis and apoptosis of ARVD iPSC-CMs compared to controls. The authors also used 6-bromoindirubin-39-oxime (BIO) or Dickkopf-1 (Dkk1) to modify the number of Isl1+ cells, which are right ventricular cardiomyocyte progenitor cells. When simulated by lipogenic factors, Isl1+ cells developed more lipogenesis and apoptosis compared to EBs with low level Isl1+ cells. This supports the hypothesis that the Isl1+ derived right ventricular-like cardiomyocytes confer the dominant pathology seen in ARVD/C.
Another model by Ma et al. created iPSC-CMs from a patient heterozygous for a different mutation in the PKP gene (p.L614P).30 At baseline, iPSC-CMs expressed lower levels of PKP2 RNA and proteins but similar levels of other desmosomal proteins. But much like the Kim et al. paper, no significant differences in lipid content were found at baseline conditions. Also consistent with the findings of Kim et al., growing the cells in a lipogenic medium resulted in increased lipid content as detected by Oil Red O staining.
Although these studies were commendable first steps in attempting to model ARVD/C, they also revealed some of the significant challenges and limitations to iPSC-CM modeling. First, ARVD/C is a primarily adult onset disease, usually manifesting by the 3rd−4th decade, and observing any changes at a fetal stage of cardiomyocytes is more challenging. Second, ARVD/C is a tissue-level disorder characterized by multi-cellular abnormalities of right ventricular morphologic changes, fatty infiltration, and arrhythmias that localize to the RVOT. Furthermore, understanding the systemic and cellular conditions that contribute to the disorder is important to creating successful models. While the manipulations involving activation of lipid pathways are conceptually useful and may help answer specific questions, they do not represent well-described mechanisms of disease pathogenesis and do not necessarily mimic the physiologic process in vivo. This is not to say that there are not useful insights that could be gained from studying multi-cellular diseases at the single cell level, only that given our current tools, the limitations described above have yet to be overcome.
Other Cardiac Models
LEOPARD Syndrome
The first study to explore cardiac modeling of iPSC derivatives tackled LEOPARD syndrome.31 LEOPARD syndrome is a genetic disorder marked by multi-organ abnormalities resulting from mutations in PTPN11 gene. Up to 80% of patients with the disorder have HCM. LEOPARD iPSC-CMs were found to have a significantly higher median surface area compared to wild type counterparts. In addition, the authors noticed an increased nuclear expression of the hypertrophy-associated transcription factor NFATC4. The authors acknowledged their iPSC-CM population was not pure enough to carry out more detailed molecular analysis for hypertrophy. Despite these limitations, this landmark paper was a proof of concept that iPSC-CMs recapitulate the genetic profile of their somatic derivatives and possibly some morphological features.
Pompe’s Disease
Pompe’s disease is an autosomal recessive metabolic disorder resulting from a mutation in gene encoding α-glucosidase (GAA). The disorder has an incidence of approximately 1 in 40,000 individuals and can present in an infantile or late-onset form. The infantile form is characterized by cardiomyopathy and severe hypotonia, whereas the adult onset form presents more as a skeletal myopathy that eventually leads to respiratory failure.32 Huang et al. demonstrated that both the iPSC lines and the iPSC-CMs derived from two different patients had lower GAA activity, lower markers of metabolism (oxygen consumption rate and extracellular acidification), and higher glycogen content.33 The iPSC-CMs contained large glycogen vacuoles, deteriorating mitochondria, and autophagosome-like structures, all of which are histological characteristics of tissue from patients with Pompe’s disease. The authors also showed that supplementation of recombinant human GAA, an existing therapy, rescued the phenotype.
Friedreich’s Ataxia (FA)
FA is a genetic disorder caused by mutations in the frataxin gene, which encodes the frataxin mitochondrial protein, resulting in cardiac and neurologic abnormalities. Clinically the disorder is most notable for severe ataxia and dysarthria caused by neurodegeneration of cerebellar pathways. However, over 50% of patients with the disorder die from congestive heart failure or arrhythmias, which are products of cardiac manifestation of the mitochondrial disorder.34 Hick et al. created iPSC-CM models from patients with FA and showed the cells contained higher frequency of structurally abnormal nuclei.35 Interestingly, the prevalence of abnormal nuclei increased over time, though this did not result in worsening electrical properties of the cell. However, the study was limited to image recordings and did not perform patch clamp analysis as they did for neurons. Thus, iPSC-CMs are not only useful for studying mechanical and electrical disorders of the heart, but also metabolic and mitochondrial disorders.
Potential and future directions of utilizing iPSC-CMs
Functional classification of genotypes
One crucial requirement in treating heritable conditions, including cardiac disorders, is the identification of causative genetic variants. This is necessary to enable screening of family members and may also play a role in the diagnosis. However, the results of genetic testing are not binary positive or negative, but are subject to interpretation of the clinician evaluating the candidate variant and the existing evidence for the disease-causing mechanism. Clinicians who care for these patients often struggle with variants of uncertain significance..2 iPSC-CMs can play a key role in improving future medical evaluation by functionally characterizing these genetic variants.
Tse et al. investigated a novel variant in the desmin gene identified in a patient with DCM (p.A285V).36 The authors created iPSC-CM models that demonstrated failure of co-localization of desmin with other sarcomeric proteins. The authors then used a viral transfection method to transfect iPSC-CMs with A285-DES, which confirmed the above phenotype. Although the confirmation of the DCM phenotype was molecular and not functional, it demonstrated an early potential for studying novel variants or variants of unclear significance identified in individuals.
Subsequently, Egashira et al. created iPSC-CMs from a young patient who presented with sudden cardiac arrest and exhibited features of LQT1.37 The patient was found to have a novel variant in KCNQ1 (p.C1893del). Similar to the study by Moretti et al., the patient cells exhibited prolonged FPD and exhibited a trafficking defect of KCNQ1. One critique of the paper is the authors used a transfection model in HEK cells that demonstrated reduced IKs current to confirm variant pathogenicity. However, iPSC-CMs may offer an improved standard of testing pathogenicity especially by applying genetic editing techniques, such as TALEN or CRISPR, and observing phenotypic changes. This has already been done in an iPSC model of Parkinson’s disease, but has yet to be shown in cardiac disorders.38
Personalized Medicine
Over the past two decades, our understanding of LQT syndrome has changed, the key realization being that different subtypes are caused by variants in different loci. The subtypes have different natural histories and treatment approaches. We now recognize that even within subtypes, different variants can predict different disease courses.39 Furthermore, it is recognized that arrhythmic disorders sometimes do not have well demarcated borders, but there is a spectrum of overlap syndromes.40 The ability to define and use the functional effect of de novo mutations for making therapeutic decisions is one of the promising features of iPSC-CMs.
This was elegantly demonstrated by Terrenoire et al., who created iPSC-CMs from a proband with LQT syndrome and severe arrhythmia phenotype (QTc of 825ms) that was refractory to multiple treatments.41 Genetic testing identified a de novo mutation in SCN5A (p.F1473C), which normally leads to LQT Type 3 (LQT3). In addition, the patient was heterozygous for a common polymorphism in KCNH2 (p.K897T). LQT3 is caused by a gain of function variation in SCN5A, resulting in decreased voltage dependent inactivation of sodium channels. Prior work by the authors showed that transfection of this variant in HEK293 cells resulted in defects in inactivation (producing a gain of function) consistent with the LQT3 phenotype.42 However, the patient was particularly refractory to therapy, and the role of the KCNH2 polymorphism on the phenotype remained unclear. The effect of this polymorphism is controversial, whether it exerts a protective versus harmful effect.43,44 Using patch clamp and current clamp techniques, the authors clearly showed a defect in sodium channel inactivation kinetics, but did not find any abnormalities in IKr current or activation properties of the channel. Furthermore, high dose mexiletine, a sodium channel blocker, as well as faster pacing were able to significantly reduce the late sodium current. This is precisely how the patient was treated clinically, pacing at a higher rate and administering high-dose mexiletine therapy. This model of personalized medicine will likely emerge as a powerful clinical application for iPSC-CMs in cases of complex genetic disorders with poorly defined therapeutic pathways. A more classic model of a de novo SCN5A variant (p.V1763M) causing LQT3 was later published, showing similar findings.45
Risk stratification
One of the challenges in treating many hereditary cardiac disorders is the variable expression of phenotype and severity of phenotype even in carriers from the same family.3 Thus, even in cases of well defined variants within the same family, one family member will exhibit severe disease, while others will be completely asymptomatic. This makes risk stratification based on genotype alone challenging, and the creation of a platform that can better risk stratify carriers can prove very helpful clinically. This then raises the question whether patient-specific iPSC-CMs can be that platform by recapitulating disease severity and not just gross features of the disease.
This issue was examined in study by Matsa et al. using iPSC lines from an affected LQT2 patient and an asymptomatic gene carrier.46 The symptomatic patient had a more prolonged surface ECG and markedly symptomatic arrhythmias, whereas the asymptomatic gene carrier had a less prolonged surface ECG but otherwise no evidence of arrhythmias. Interestingly, the APD of iPSC-CMs from the asymptomatic carrier patient was more prolonged than control cells, but still significantly shorter compared to the symptomatic patient’s iPSC-CMs. Furthermore, the asymptomatic carrier’s iPSC-CMs did not exhibit the large EAD burden with isoproterenol stimulation that the symptomatic LQT patient’s iPSC-CMs did. Similarly, Lahti et al. derived iPSC-CMs from an asymptomatic carrier of a known LQT2 variant (p.R176W).47 The iPSC-CMs showed a prolonged APD, but interestingly did not exhibit more spontaneous EAD compared to control cells correlating with the patient’s clinical phenotype of prolonged surface ECG QT interval but lack of arrhythmia. This is in contrast to other LQT2 iPSC-CM models derived from more symptomatic LQT2 patients that showed higher incidence of spontaneous EADs.17,46
Taken together, these results suggest that there may be a correlation between the observed clinical phenotype and iPSC-CM phenotype. These observations are very preliminary and need to be further tested on a larger scale and across multiple disease models. However, if indeed iPSC-CMs could recapitulate severity of phenotype, this would redefine the clinical approach to risk stratifying patients with LQT and possibly other hereditary cardiac disorders.
Studying molecular mechanisms of disease
Beyond genotype-phenotype investigations, other groups have used iPSC-CMs to study long standing questions that were impaired by the inability to culture and grow functional human cardiomyocytes in vitro. For example, clinicians often recognized that some patients will experience drastic prolongation of QT with certain hERG blocking drugs, while others have minimal symptoms. This phenomenon led to the hypothesis of repolarization reserve, namely the ability of the cell to compensate for the reduced current via other channels and a degree of built-in redundancy between repolarization currents.48 Given that iPSC-CMs express virtually all channels expressed by human myocytes, they offered a platform with some advantages over animal models and HEK293 transfection models to study the concept of repolarization reserve.
Braam et al. investigated the effects of 9 anti-arrhythmic drugs on the field potential duration (FPD) of iPSC-CMs as measured by MEA.49 Most compounds showed responses in terms of prolonging or shortening field potential that are congruent with those of other models. The exceptions were HMR1556 and JNJ303 both of which were expected to prolong the FP by blocking the IKs current, but actually had a minor effect in wild type cells. When the repolarization reserve was challenged by administering sotalol (IKr blocker) or administering the compounds in LQT2 iPSC-CMs (reduced IKr), a clear prolongation of the FPD was observed. This iPSC-CM platform supported the hypothesis that there is a certain redundancy in the repolarization currents (IKs and IKr), and that there is a compound effect to genetic and exogenous modifying factors that affect the FPD.
Toxicity testing
Cardiac toxicity remains one of the leading causes of withdrawal of candidate drugs either during development or after market release.50 The current approach to cardiovascular safety assessment focuses on animal testing to test systemic cardiovascular effects and evaluation of hERG channel blockade, which most often involve drug testing in cell transfection models expressing the hERG channel. The transfected cellular models have several limitations. First, the cardiac action potential involves complex and sequential activation of multiple ion channels, and the evaluation of a single channel (even an influential and commonly affected one) fails to predict the effects on the entire AP. Of equal importance, there are non-electrophysiological mechanisms of cardiac drug toxicity that have become increasingly clinically significant. To date, iPSC-CMs are the closest surrogates for human in vivo cardiac cells that can be manipulated and studied in culture, and thus would appear to be a much more predictive model of human cardiac toxicity.
This direct comparison was addressed in a study in which the hERG channel transfected HEK293 cells (hERG-HEK293) were directly matched to iPSC-CMs in terms of predicting drug toxicity.51 Liang et al. was able to demonstrate the superiority of iPSC-CMs compared to hERG-HEK293 cells in predicting toxicity of multiple drugs including verapamil and alfuzosin. The authors also screened diseased iPSC-CMs derived from individuals with LQT, HCM, and DCM with a panel of drugs known to interact with different cardiac ion channels, including cisapride which is a potent hERG blocking drug removed from the market in 2000 (Figure 4). Another study showed that iPSC-CMs were equally effective to hERG-HEK293 cells in predicting zolpidem toxicity.52 Thus, the data suggest iPSC-CMs represent an advanced model for predicting cardiac toxicity compared to hERG transfection models.
Figure 4. Differences in susceptibilities of disease-specific iPSC-CMs to cisapride-induced cardiotoxicity.
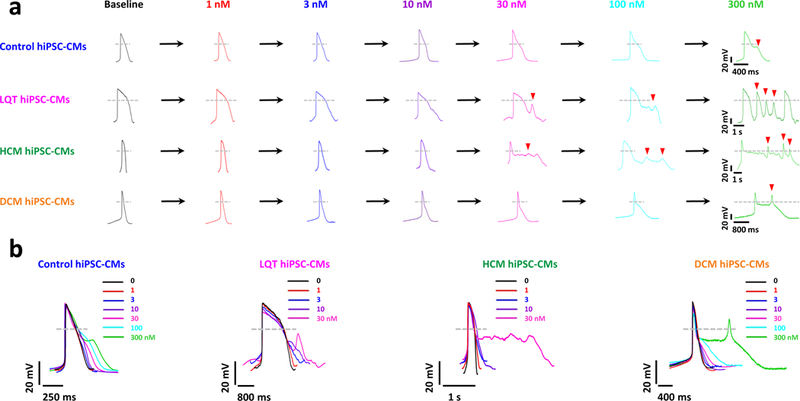
(A) Representative tracings showing dose-dependent effect of cisapride on the AP of control iPSC-CMs, LQT iPSC-CMs, HCM iPSC-CMs, and DCM iPSCCMs (Baseline: black, 1 nmol/L: red, 3 nmol/L: blue, 10 nmol/L: purple, 30 nmol/L: magenta, 100 nmol/L: cyan, 300 nmol/L: green). Arrowheads indicate cisapride-induced EADs. LQT iPSC-CMs and HCM iPSC-CMs were observed to exhibit cisapride-induced EADs at lower concentrations (30 nmol/L) as compared with family-matched control iPSC-CMs and DCM iPSC-CMs (300 nmol/L; n=10, 3 patient or control subject lines per cohort). (B) Aligned APs (A) showing the effects of cisapride on control, LQT, HCM, and DCM iPSC-CMs (n=10, 3 patient or control subject lines per cohort).
In addition to toxicity and efficacy testing, iPSC-CMs provide a platform to study the mechanism of efficacy and toxicity in human cells, which can be difficult to ascertain. Sunitinib is a chemotherapeutic agent used to treat renal cell carcinoma and gastrointestinal stromal tumors, and is known to be associated with left ventricular dysfunction. Cohen et al. administered sunitinib to iPSC-CMs and demonstrated that it induced cell death by ATP depletion, increasing oxidative stress, and apoptosis.53 Animal models have suggested aminoimidazole carboxamide ribonucleotide (AMPK) or ribosomal S6 kinase (RSK) as possible mediators of sunitinib-induced cardiotoxicity. The authors used iPSC-CMs as a platform to test these hypotheses and showed that neither AMPK nor RSK were mediators in sunitinib cytotoxicity.
Identifying new drug targets
In most iPSC-CM disease modeling studies to date, researchers have utilized drugs with well-defined clinical responses to validate the respective iPSC-CM models. In other words, they were retrospective tests to validate predicted human in vivo clinical response. Jung et al. studied whether iPSC-CMs can be used to prospectively test for drug efficacy.54 The investigators created iPSC lines from a patient with CPVT and documented the abnormal calcium handling and arrhythmic potential seen by other groups.21–23 The cells were then treated with dantrolene, a drug used to treat a skeletal muscle ryanodine receptor condition, which restored normal calcium handling and abated the DAD seen in CPVT iPSC-CMs. Similar work done by Siu et al. showed that blockers of the MEK1 pathway, U0126 and selumetinib, could ameliorate cell death in LMNA iPSC-CMs.25 Care should be taken to avoid the inference that such data necessarily signify clinical efficacy, systemic safety, or an alternative to clinical testing. However, these studies do provide examples of the potential of iPSC-CMs for novel drug discovery based on improved understanding of mechanism of action. When combined with a high-throughput platform, iPSC-CM can be especially useful in screening large chemical libraries for novel drug discovery.
Limitations of iPSC-CM Model
Maturity and modeling late-onset disorders
One of the main criticisms of iPSC-CMs is that they are an immature phenotype and as such represent an inadequate model of pediatric and adult disorders. This statement is more deduction than empirical science, as some data suggest that LQT syndrome phenotype is already expressed at the fetal stage, reaffirming some of the findings of the above model.55 It is certainly true that iPSC-CMs are mostly still in fetal stage of development, and ongoing efforts to induce maturation via prolonged culturing, 3-dimensional growth, and pacing have shown promise in advancing maturation.56–60 Nonetheless, the immature phenotype issue may make it difficult to draw correlations with adult cells and to successfully model some late-onset disorders such as ARVD. However, it should be noted that iPSC-CMs are a model or surrogate for functional human cardiomyocytes, and not an equivalent or full replacement. Animal models have limitations in modeling human cardiac disorders as well, but they still play an important role in understanding the disorders. Ultimately, the success of a model should be based on how it performs against the actual disorder. Finally, so far the relevant data have shown that iPSC-CMs perform fairly well in modeling most genetic cardiac disorders. There could be a publication bias affecting some these results, but it is worth noting that the iPSC-CM phenotype for LQT1, LQT2, LQT3, and CPVT has been reproduced by independent investigators.16,17,21–23,37,41,45–47,54
Recapitulating cardiac-systemic interactions
Another limitation of iPSC-CMs is that they are not analogous to a whole heart. The progression of cardiac disorders is a product of different loading conditions, complex hormonal milieu, and environmental insults faced by human cardiomyocytes. Recapitulating all of these conditions and complex multi-organ interactions is impossible to do in vitro. This is not to say that simple interventions can unmask the cardiomyopathy or arrhythmia phenotype, but such simple interventions do not reflect the in vivo complexity of the whole body. Thus, iPSC-CMs are not a replacement for in vivo studies that aim to study the complex interactions of different systemic conditions
Modeling multi-cellular disorders
The use of iPSC-CM modeling has largely been limited to hereditary disorders, especially those in which the pathology is reproducible at the single cell level. This limitation makes it difficult to model disorders that would appear to be more multi-cellular, particularly if the genetic component is less pronounced as in the case of atrial fibrillation. Furthermore, iPSC-CMs do not replicate complex cardiac embryogenesis processes, making it difficult to model congenital heart disorders. Lastly, the behavior of monolayer or dissociated single cells of myocytes do not replicate the behavior of human cardiac tissue.
This was challenged by Kadota et al., who differentiated iPSC-CMs and plated them in cell sheet formations that were capable of electric coupling.61 The authors confirmed that the propagation of depolarization was not only lateral but to abutting ends of the cells. With careful titration of cell density and stimulation frequency, the authors were able to induce a spiral wave that resembled the development of reentrant arrhythmias in human myocardium. Furthermore, the authors were able to demonstrate that nifekalant, E-4031, sotalol, and quinidine terminated the spiral wave. Utilizing iPSC-derived tissues in 3-dimensional models has already yielded powerful models in other tissue types.57,62 The above study is a pioneer on multiple levels and reveals more potential for iPSC-CMs by 1) modeling a non-hereditary disorder of cardiomyopathy; 2) incorporating novel engineering methods to allow for modeling of multi-cellular disorder, including those that are dependent on 2- or 3-dimensional spatial organization; and 3) proving that drug testing on a multi-cellular cardiac model is feasible.
Conclusions
In less than 4 years, iPSC-CMs have been used to model over 10 cardiac hereditary conditions. In most cases, the model has shown robust recapitulation of the phenotype. While the cells are not an in vivo model and are of a fetal phenotype, most models have offered novel insights or confirmed known pathogenesis of the disorder. Some early work also shows an emerging role for iPSC-CMs in functional evaluation of genes, risk stratification of patients, drug testing, and personalized medicine. This promising work likely represents the scratching of the surface of the full potential of iPSC-CM. While large studies will be needed to confirm these applications, the platform of iPSC-CM represents a remarkable advance for biomedical research in cardiovascular medicine with great promise for tremendous clinical impact.
Acknowledgement
We gratefully acknowledge Blake Wu for critical reading, Ms. Amy Thomas for her assistance with illustration, and funding support from National Institute of Health (NIH) T32 (KS), he Uehara Memorial Foundation Research Fellowship (KK), Leducq Foundation, American Heart Association 13EIA14420025, NIH R01 HL113006, NIH U01 HL099776, California Institute for Regenerative Medicine (CIRM) TR3-05556, and CIRM DR2-05394 (JCW).
Footnotes
Conflict of Interest
JCW: scientific advisory board and co-founder of Stem Cell Theranostics
References
- 1.World Health Organization. The top 10 causes of death. (July 2013). [Google Scholar]
- 2.Ackerman MJ, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart rhythm 8, 1308–1339 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Giudicessi JR & Ackerman MJ Genetic testing in heritable cardiac arrhythmia syndromes: differentiating pathogenic mutations from background genetic noise. Current Opinion in Cardiology 28, 63–71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi K & Yamanaka S Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Dimos JT, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218–1221 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Kambal A, et al. Generation of HIV-1 resistant and functional macrophages from hematopoietic stem cell-derived induced pluripotent stem cells. Molecular Therapy 19, 584–593 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mordwinkin NM, Burridge PW & Wu JC A review of human pluripotent stem cell-derived cardiomyocytes for high-throughput drug discovery, cardiotoxicity screening, and publication standards. Journal of Cardiovascular Translational Research 6, 22–30 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burridge PW, Keller G, Gold JD & Wu JC Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell 10, 16–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tohyama S, et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12, 127–137 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Fujiwara M, et al. Induction and enhancement of cardiac cell differentiation from mouse and human induced pluripotent stem cells with cyclosporin-A. PloS One 6, e16734 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navarrete EG, et al. Screening drug-induced arrhythmia events using human induced pluripotent stem cell-derived cardiomyocytes and low-impedance microelectrode arrays. Circulation 128, S3–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tester DJ, Will ML, Haglund CM & Ackerman MJ Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2, 507–517 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Splawski I, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102, 1178–1185 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Schwartz PJ, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103, 89–95 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. The New England Journal of Medicine 363, 1397–1409 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Itzhaki I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471, 225–229 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Splawski I, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Yazawa M, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471, 230–234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaufman ES Mechanisms and clinical management of inherited channelopathies: long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, and short QT syndrome. Heart Rhythm 6, S51–55 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Itzhaki I, et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. Journal of the American College of Cardiology 60, 990–1000 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Fatima A, et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cellular physiology and biochemistry 28, 579–592 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novak A, et al. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to beta-adrenergic stimulation. Journal of Cellular and Molecular Medicine 16, 468–482 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun N, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Science Translational Medicine 4, 130ra147 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siu CW, et al. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging 4, 803–822 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sylvius N & Tesson F Lamin A/C and cardiac diseases. Current Opinion in Cardiology 21, 159–165 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Jacoby DL, DePasquale EC & McKenna WJ Hypertrophic cardiomyopathy: diagnosis, risk stratification and treatment. CMAJ : Canadian Medical Association Journal 185, 127–134 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lan F, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 12, 101–113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim C, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494, 105–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma D, et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. European Heart Journal 34, 1122–1133 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Carvajal-Vergara X, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 465, 808–812 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Ploeg AT & Reuser AJ Pompe’s disease. Lancet 372, 1342–1353 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Huang HP, et al. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Human Molecular Genetics 20, 4851–4864 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Tsou AY, et al. Mortality in Friedreich ataxia. Journal of the Neurological Sciences 307, 46–49 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Hick A, et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Disease Models & Mechanisms 6, 608–621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tse HF, et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Human Molecular Genetics 22, 1395–1403 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Egashira T, et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovascular Research 95, 419–429 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Soldner F, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146, 318–331 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruan Y, Liu N, Napolitano C & Priori SG Therapeutic strategies for long-QT syndrome: does the molecular substrate matter? Circulation. Arrhythmia and Electrophysiology 1, 290–297 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Remme CA & Wilde AA SCN5A overlap syndromes: no end to disease complexity? Europace 10, 1253–1255 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Terrenoire C, et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. The Journal of General Physiology 141, 61–72 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bankston JR, et al. A novel and lethal de novo LQT-3 mutation in a newborn with distinct molecular pharmacology and therapeutic response. PloS One 2, e1258 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X, et al. Protective effect of KCNH2 single nucleotide polymorphism K897T in LQTS families and identification of novel KCNQ1 and KCNH2 mutations. BMC Medical Genetics 9, 87 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Linna EH, et al. Functional significance of KCNH2 (HERG) K897T polymorphism for cardiac repolarization assessed by analysis of T-wave morphology. Annals of Noninvasive Electrocardiology 11, 57–62 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma D, et al. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. International Journal of Cardiology 168, 5277–5286 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Matsa E, et al. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. European Heart Journal 32, 952–962 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lahti AL, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Disease Models & Mechanisms 5, 220–230 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roden DM Repolarization reserve: a moving target. Circulation 118, 981–982 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Braam SR, et al. Repolarization reserve determines drug responses in human pluripotent stem cell derived cardiomyocytes. Stem Cell Research 10, 48–56 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Mordwinkin NM, Lee AS & Wu JC Patient-specific stem cells and cardiovascular drug discovery. JAMA 310, 2039–2040 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang P, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 127, 1677–1691 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jehle J, et al. Mechanisms of zolpidem-induced long QT syndrome: acute inhibition of recombinant hERG K(+) channels and action potential prolongation in human cardiomyocytes derived from induced pluripotent stem cells. British Journal of Pharmacology 168, 1215–1229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen JD, et al. Use of human stem cell derived cardiomyocytes to examine sunitinib mediated cardiotoxicity and electrophysiological alterations. Toxicology and Applied Pharmacology 257, 74–83 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jung CB, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Molecular Medicine 4, 180–191 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuneo BF, et al. Arrhythmia phenotype during fetal life suggests long-QT syndrome genotype: risk stratification of perinatal long-QT syndrome. Circulation. Arrhythmia and Electrophysiology 6, 946–951 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ivashchenko CY, et al. Human-induced pluripotent stem cell-derived cardiomyocytes exhibit temporal changes in phenotype. American Journal of Physiology. Heart and Circulatory Physiology 305, H913–922 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Mihic A, et al. The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes. Biomaterials (2014). [DOI] [PubMed] [Google Scholar]
- 58.Chan YC, et al. Electrical stimulation promotes maturation of cardiomyocytes derived from human embryonic stem cells. Journal of Cardiovascular Translational Research 6, 989–999 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Yu T, et al. In vivo differentiation of induced pluripotent stem cell-derived cardiomyocytes. Circulation Journal 77, 1297–1306 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Kamakura T, et al. Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circulation Journal 77, 1307–1314 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Kadota S, et al. Development of a reentrant arrhythmia model in human pluripotent stem cell-derived cardiac cell sheets. European Eeart Journal 34, 1147–1156 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Lau TT, Ho LW & Wang DA Hepatogenesis of murine induced pluripotent stem cells in 3D micro-cavitary hydrogel system for liver regeneration. Biomaterials 34, 6659–6669 (2013). [DOI] [PubMed] [Google Scholar]