

Abstract
We have developed a high resolution correlative method involving cryo-soft X-ray tomography (cryo-SXT) and stochastic optical reconstruction microscopy (STORM), which provides information in 3 dimensions on large cellular volumes at 70 nm resolution. Cryo-SXT morphologically identified and localized aggregations of carbon-rich materials, and STORM identified specific markers on the desired epitopes, enabling co-localization between the identified objects, in this case cholesterol crystals, and the cellular environment. The samples were studied under ambient and cryo-conditions without dehydration or heavy metal staining. The early events of cholesterol crystal development were investigated in relation to atherosclerosis, using as model macrophage cell cultures enriched with LDL particles. Atherosclerotic plaques build up in arteries in a slow process involving cholesterol crystal accumulation. Cholesterol crystal deposition is a crucial stage in the pathological cascade. Our results show that cholesterol crystals can be identified and imaged at a very early stage on the cell plasma membrane and in intracellular locations. This technique can in principle be applied to other biological samples where specific molecular identification is required in conjunction with high resolution 3D-imaging.
Keywords: Cholesterol, Pathological crystallization, Atherosclerosis, Correlative microscopy
Graphical Abstract
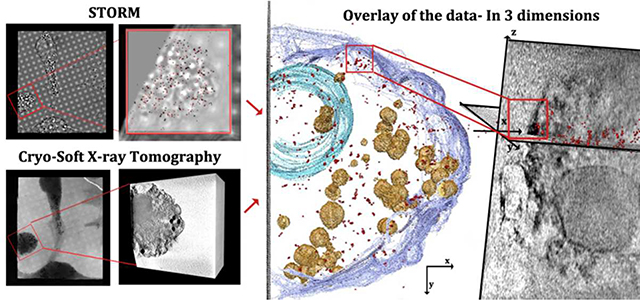
INTRODUCTION
Atherosclerosis is characterized by lipid and cholesterol accumulation in the innermost layer of the artery wall 1–4. The slowly progressing disease starts with the penetration of low density lipoprotein (LDL) particles into the artery wall. The LDL particles are then taken up by macrophage cells and are processed in lysosomes where LDL protein is degraded and LDL’s cholesteryl ester is hydrolyzed. Much of the cholesterol is then transferred and accumulated in the plasma membrane 5–7. Excess plasma membrane cholesterol can be shuttled back into the cell and stored in lipid droplets 8,9. With time, this accumulation process leads to the maturation and subsequent death of foam cells, resulting in growing pools of extracellular lipid and cholesterol monohydrate crystal deposits, the atherosclerotic plaque 10. The plaque can be stable for years until it ruptures 9. It is well accepted that precipitation of cholesterol crystals can increase the inflammatory response, cause expansion of the lesion core and induce plaque disruption 11–13. Macrophage cells are actively involved in the process of cholesterol crystallization 14–16. However, what initiates cholesterol crystal formation is still not well-understood9,16.
Ong et al 17 showed that accumulation of acetylated LDL (acLDL) particles by macrophages in cell culture leads to the generation of cholesterol structured domains in cell membranes and of particles in the intracellular and/or extracellular space. It is not clear whether these particles comprise 2-dimensional (2D) structured cholesterol domains, or 3 dimensional (3D) crystals. In parallel with these studies, structured cholesterol 2D domains segregating inside hydrated lipid bilayers in vitro were shown to serve as nucleation sites for the formation of 3D cholesterol crystals 18. Cholesterol segregation in bilayers into secluded domains is a well-known phenomenon. Micrometer size cholesterol domains were observed by fluorescence microscopy and atomic-force microscopy (AFM) to form in vitro in cholesterol rich bilayers 19,20. The size of the cholesterol crystalline domains nucleating cholesterol 3D crystals was measured on supported lipid bilayers by Grazing Incidence X-ray Diffraction (GIXD) to be tens of nanometers 21–23. The combined evidence from these studies led to the hypothesis that cholesterol crystals in vivo can nucleate from segregated 2D cholesterol domains in cell membranes, in a process analogous to directed templated nucleation 24,25
High resolution imaging is required in order to directly visualize cholesterol domains and identify the initial stages of cholesterol accumulation and crystal formation in cell membranes. Fluorescence light microscopy is conventionally used to identify and localize specific molecular structures and their accumulation in cell cultures and tissues at a resolution up to the diffraction limit of light, ~300nm. The recent development of super-resolution fluorescence imaging techniques exceeds the diffraction limit of resolution allowing biological structures to be imaged at nanometer scale resolution 26. Super-resolution microscopy however, can only detect labeled targets, whereas it cannot detect the unlabeled cellular environment.
Cryo-soft X-ray Tomography (cryo-SXT) is an attractive complementary technique to super-resolution microscopy, whereby dense molecular aggregates can be detected in intact cells and tissues, in 3 dimensions also at nanometer scale resolution 27–34. By making measurements with cryo-SXT at the so-called water window energy range, structures such as proteins, lipid membranes, or in the present case cholesterol crystals, can be directly visualized without staining or processing 34. Cryo-SXT was recently used in correlation with cryo-fluorescence microscopy, to localize and identify cells or sub-cellular organelles at a resolution of 350 nm 29,30,33,35,36. These studies inspired us to develop a correlative technique between cryo-SXT and super-resolution microscopy, to allow the detection of nascent cholesterol crystals that may be only tens of nanometers wide and not thicker than a lipid bilayer.
Herein we show a proof of concept of the feasibility of the newly developed correlative stochastic optical reconstruction microscopy (STORM) and cryo-SXT technique, and use it to provide detailed insights on the first stages of cholesterol crystal formation in cells. The combined resolution of the correlative imaging is ~70nm.
RESULTS
For identification and labeling of cholesterol segregated ordered domains and crystals we used specific antibodies, which were detected using STORM. For crystal localization and imaging inside the cell we used 3D cryo-soft X-ray tomography. Neither of the techniques alone can provide a complete image of the crystals, including their unambiguous identification side-by-side with the determination of their morphology, their size and their exact location in the cellular environment. For this purpose a correlative approach had to be developed, combining the information provided by STORM and cryo-SXT. We shall first describe the development of each of the applications separately, and then together.
STORM
Macrophage-like RAW 264.7 cells were incubated with or without acLDL for 48 h 17 on glass slides. Cells were fixed and incubated with a unique structural antibody (MAb 58B1) that recognizes cholesterol 2D and 3D crystals 37,38. The recognition is based on the interaction between the antibody binding site and an ordered surface composed of 15–20 cholesterol molecules. The antibody specifically interacts also with cholesterol crystalline domains which form inside cell membranes 8,17. When cells are fixed with paraformaldehyde (PFA) without permeabilization, the structural antibodies penetrate into the intra-cellular cavity, where they specifically label the respective antigens, albeit at lower concentration than in the extra-cellular space 39.
After incubation with the primary structural antibody, the cells were incubated with secondary fluorescently labeled antibody and the super-resolution imaging was performed using STORM.
Cholesterol-crystalline domains were generated in large amounts in cells that were incubated with acLDL (Figure 1 A–C), but were not detected in comparable amount on cultures incubated without acLDL (Figure 1 D, E). Similarly, cultures incubated with acLDL, followed by incubation only with secondary antibody, did not produce observable labeling (Figure 1 F, G). It is therefore assured that the antibody specifically labels cholesterol ordered domains that form during cholesterol enrichment.
Figure 1:

(A, D, F): Bright field images of cells after 48 h incubation with (A, F) or without (D) 50 μg/ml acLDL, overlaid with the conventional fluorescence data. Cells were fixed and incubated with (A-E) or without (F, G) 3 μg/ml 58B1 antibody that specifically recognizes 2D and 3D cholesterol crystals (Green) (B, E, G,): A magnified area of the fluorescence data from (A), (D) and (F), respectively. (C): Localization map of the resolved super-resolution image of (B). (H): Magnified area from (C) (red rectangle). Small (~100 nm) densely labelled aggregates (red arrows) and features labeled mainly on the edges (white arrows,) are detected. This labeling may be an indication for the presence of 3D crystals in the cell area. Inset H1 and H2 show magnifications of the putative crystals indicated by 1 and 2 in H. The point size is presented in localization precision, i.e. a label with smaller diameter corresponds to a higher precision in the localization of the signal.
The labeled domains can be seen in the conventional fluorescence microscope (Figure 1B) as punctate micrometer-wide structures. Clearly, the features are below the limit of resolution for light microscopy. The super-resolution map reveals that each large micro-domain is composed of sub-domain aggregation. Each subdomain is a few tens of nanometers in diameter, possibly again limited by the effective resolution for these measurements, i.e. 20–40 nm (Figure 1H). The nano-domains are spread over the cell area in a non-uniform manner. There are several labeling centers where the sub-domains aggregate into larger quadrilateral clusters with a typical size of 100 nm (Figure 1H red arrows, inset H1). Their regular quadrilateral shape suggests that the clusters may be labelled 3D cholesterol crystals that nucleated and grew to a certain extent, enough to assume the morphology of mature cholesterol crystal plates. Other clusters are seen within a wider size range (100 – 300 nm). The latter are often labeled only at the boundaries of the structure (Figure 1H white arrows, inset H2). Taking into account that MAb 58B1 characteristically interacts with 3D cholesterol crystals on the {h0l} and {0kl} side faces 37, the latter observation is thus in agreement with the presence of 3D cholesterol crystals associated with the cell. So far, no information is provided on the location of the putative crystals, whether inside the cell or at the surface of the cell. The super resolution map, however, does show higher labelling at the outer rim compared with the rest of the cell (Figure 1C). Since more labeling is expected on the cell surface due to lower accessibility into the cell cavity, the specific plane in Figure 1C is most probably located inside the cell.
Cryo-soft X-ray tomography
Macrophage-like RAW 264.7 cells were grown on gold grids. The cells were then incubated with or without acLDL and vitrified on the grids. The frozen samples were imaged using soft X-ray tomography (MISTRAL beam line in Alba, 40,41) under cryogenic conditions and were aligned to yield a reconstructed volume of 18×18×18 μm3 with an effective resolution of 71.1 nm half pitch. Representative reconstructions of cell tomograms are presented in Figure 2.
Figure 2:

(A): Slice through a 3D reconstruction of soft X-ray tomogram of a macrophage cell after incubation with acLDL. (A): High contrast sharp feature (arrow) attached to lipid-rich objects inside the cell. (B, C): Volume rendering (orange color-map) of organelles found in the volume of the cell presented in (A). The volume rendering in (B) is overlaid on a slice of the reconstructed data. The sharp feature attached (arrow) has slightly higher contrast than the lipid objects, seen from the orange color gradient: Bright white= high contrast (higher X-ray absorbance) and dark orange = lower contrast (less X-ray absorbance).
The cell cytoplasm contains many spherical objects with sizes of 1–2μm having high contrast (Figure 2A). The spherical high contrast objects were not detected in comparable amount in cells incubated without acLDL (Figure S1 in the Supporting Material). These are most likely organelles filled with high levels of lipids. Lipids have X-ray absorption coefficient of ~ 1 μm−1, high compared to the surrounding media and almost ten times higher than that of vitreous ice, ~0.11 μm−1 28,30,31,33,42,43. Furthermore, lipid accumulation is expected for acLDL-loaded cells, which are known to store high levels of cholesterol and fatty materials in lysosomes and lipid droplets 9,44. Cryo-scanning electron micrographs of cells (Figure 3A, B) and TEM of embedded sections from the same culture (Figure S2) confirm the presence of objects of the same size, often membrane-limited, with the typical smooth texture of lipid-rich particles, i.e. lysosomes and/or lipid droplets. Sharp and thin features with high X-ray absorbance were detected closely associated with the lipid-rich spherical objects (Figure 2A, B, arrow) and with the plasma membrane of the cells (Figure S3, arrows). Three-dimensional rendering revealed that these features have morphologies similar to those of cholesterol crystals with thicknesses as low as 80nm (resolution limited) and lateral dimension of 0.5–2μm (Figure 2B, C). In the volume rendering color-map obtained after 3D reconstruction, the orange gradient represents the contrast, showing that the sharp features have higher contrast than the lipid-rich objects (contrast increases from orange to white). In a side view, the cell plasma membrane also appears decorated by square features closely adhering to it (Figure S3). Unambiguous identification of these features as cholesterol crystals is, however, impossible with the tools available in soft X-ray tomography alone, therefore the correlation with an additional technique that provides molecular specificity at high resolution is essential.
Figure 3:

(A, B): Cryo SEM micrographs of high pressure frozen and freeze fractured macrophage cells after 48 h incubation with 50 μg/ml acLDL. The samples were etched for 5–20 min at −105°C in order to expose the outer surface features and enhance the contrast between the cytoplasm and the unetched lipid objects. The cells are filled with objects (indicated by arrows) that have the typical texture of lipid-rich particles, most probably lysosomes and/or lipid droplets.
Correlative cryo-soft X-ray tomography/super-resolution fluorescence microscopy
Super-resolution fluorescence microscopy identifies cholesterol organized structures but does not provide information on their location in the cell relative to other unlabeled organelles and sub-structures, neither does it unambiguously attribute them to crystals. Cryo-SXT shows thin features closely associated with cellular membranes, but does not unambiguously identify them as composed of cholesterol. Coupling between the two techniques should, therefore, provide a complete information on the putative crystals and their locations. To this end, a correlative approach between the two techniques was developed (Figure 4). In order to image the same sample positions with the two techniques, the cells were grown on gold finder-grids. The cells were cultured, enriched with cholesterol, fixed and labelled on the grids (Figure 4, steps 1–2). The position of individual cells was recorded before imaging with STORM. STORM imaging was performed in the upper part of each cell over a volume of 18×18×1 μm3 in x, y and z, respectively. (Figure 4, steps 3). Following analysis with STORM, the grids were vitrified and imaged using the soft X-ray beam (Figure 4, step 4 and 5). Tomograms were taken from the same cells that were analyzed by STORM and were aligned to yield a reconstructed volume of 18×18×18 μm3 (Figure 4, step 5).
Figure 4:

Correlative microscopy flow-chart. Step1: Cells were grown on gold finder grids with fiducial markers that allow navigation to desired locations on the grid. The cells were incubated for 48 h with acLDL. Step2: Cells were fixed (4 % PFA, 0.1% GTA, see methods) and incubated with primary and secondary antibodies. Step 3: super-resolution fluorescence signal was resolved by STORM. Step4: Grids were high pressure frozen. Step5: X-ray tomograms were taken of the same cells that were analyzed by STORM. Data were reconstructed (field of view 15 μm3). Step6: Overlay of the data was done manually using the grid mesh as fiducial markers for the xy plane. For the z plane, each of the X-ray and fluorescence volume stacks was divided into layers of 80 nm and overlaid (see experimental section).
The super-resolution fluorescence images which were acquired at xy resolution of 36nm were then overlaid on to the reconstructed X-ray data (Figure 4, step 6), that were taken at xy resolution of 69 nm half pitch. The xy location and the cell orientation were tracked first using the fiducial markers provided by the finder grid, including the labels, the metal grating, the holes in the holy coating, and the cell morphology imaged in both techniques (Figure 4, step 5). The z-resolution of STORM is twice the resolution in xy, i.e. 72nm, and we estimate the z resolution of soft X-ray tomography to be the same 34,35. To reliably overlay the data in z, a script was consequently developed that shifts each of the data sets in steps of 80 nm (see complete description in experimental section). After each z- shift the software calculates the percentage of the super resolution signal coming from the area in each plane delimited by the cell contour seen at high contrast in cryo-SXT. In addition, the coverage area of the super resolution data is calculated relative to the X-ray cell area. The optimal z position is located where the maximum of the super-resolution signals are located within the cell area, and are distributed over most of the area (excluding in the cell nucleus). Fine adjustments in xy are then performed, taking into account that the xy sections at different z are not independent of each other. We note that both data sets suffer from deformations in the z axis due to the missing wedge in the SXT, and from the point spread function in STORM. Both types of deformation result in elongation in the z axis, but the amount by which they are deformed may be different. This may in principle cause a mismatch in the overlay in the z direction. However, the superimposition was done by overlay and evaluation of individual slices in the xy plane. We therefore estimate that the deformations in the z direction should have only minor effects on the correlation of the data over the 1μm depth that was evaluated.
Segmentation of the reconstructed X-ray data (Figure 5A) allows observation of the cell volume (Figure 5C–E) together with the overlaid super resolution 3D localization map. The super resolution map is located at the upper part of the cell above the nucleus (Figure 5C and 5E, red spots).
Figure 5:

(A, B): Slices through the 3D reconstruction of a soft X-ray tomogram of a cell after incubation with acLDL. The localization map of the resolved super-resolution image (red spots, 180 nm in depth) is superimposed on the corresponding X-ray data slice in (B). (C): 3D reconstruction of the cell in A, B, showing the location of the slices in (A) and (B): The slice in (A) comes from the middle part of the cell and the slice in (B) comes from the upper part of the cell, where STORM was performed. (D, E): 3D reconstruction of the same cell from a top view (D) and a side view (E). The localization map of the corresponding resolved super-resolution image (red spots) is 1μm in thickness and is located on the upper part of the cell, above the nucleus. Arrow indicates cluster of STORM signals that is magnified in (F). (F): High magnification of a well-defined cluster of the STORM signal (arrow). The cluster was detected on the plasma membrane envelope, and is presented also in (B) and (D) from different orientations (arrows). Segmentation based on contrast was used for the 3D reconstruction. The different features are described using arbitrary colors: plasma membrane (purple) lipid objects (yellow-orange) and cell nucleus (blue).
Observation of the overlay results on the individual xy sections (Figure 5B) shows high contrast SXT areas corresponding to lipid droplets and/or lysosomes and other cell membrane delimited organelles distributed over the whole area. There are a few clusters of STORM signals conceivably corresponding to 3D cholesterol crystals. These are interspersed among individual signals spread over the whole section, which may correspond to isolated 2D cholesterol domains (Figure S4). Because the antibodies have little access to the intracellular space, this limits the intracellular labeling.
As a consequence of the heterogeneity of the SXT signals and the limitations to the antibody access, the overlay, even though it appears to be not completely random, does not unequivocally identify regions of interest. Evaluation of a random spot distribution of the STORM signal by a cross correlation function (for more details of the cross correlation function see experimental section) yielded overlay values differing by only 6% from the observed data (i.e. 34% random distribution compared to 40% in the observed data).
In contrast to the many sources of intracellular high contrast SXT features, the plasma membrane is uniform, free from organelles, and easily accessible to the antibodies. Few well defined clusters of STORM signals are detectable on the plasma membrane envelope (Figure 5E arrow and 5F, arrow), which appear to correspond to high contrast SXT features. Evaluation of a random spot distribution of the STORM signal by the same cross correlation function used above yields on the plasma membrane envelope values of 26% overlay for a statistical distribution, and values of 94% for the observed data. It was thus decided to concentrate first on the cluster signals associated with the plasma membrane.
Observation was performed first from a top view (xy plane) on the superimposed data (Figure 6A–C, arrows). The top views were then combined with views from the relevant perpendicular planes (xz or yz planes, Figure 6D). The SXT In the locations corresponding to the STORM-labelled features (Figure 6D) show objects with a rectangular shape (Figure 6E and 6F) with lengths and widths in the range of 200–400 nm. The combination of the information contained in the two sets of data converges to identify these objects as cholesterol crystals. It is not completely clear why the crystals have high contrast on the edges, and low contrast in the center. We estimate that the crystal plate dimensions are in the order of 200×200nm with a thickness of ≤70nm, and density of 1.05 g/cm3, which complicates their absorption detection in the surrounding cellular structure. The detection would then probably depend on their orientation relative to the impinging X-ray beam, with the photon path through the longer dimensions being more favorable for detection. This could explain the fact that for many of them the edges are enhanced and become clearly visible. In addition we note that the crystal lateral sides are coated by antibodies, which certainly enhance their corresponding absorption contrast.
Figure 6:

(A1, A2, A3): Parts of slices through the 3D reconstruction of a cell tomogram, taken after incubation with AcLDL; top views (xy plane). High contrast sharp features (arrows) appear on the cell plasma membrane. (B1, B2, B3): Localization map of the corresponding resolved super-resolution image (red spots). (C1, C2, C3): Superimposition of the X-ray data in A1 A2 A3 with the super-resolution fluorescence data in B1, B2, B3. The inset in each reconstructed picture shows the high contrast feature marked in yellow for clearer visualization. (D1, D2, D3): side orientation of the superimposed data presented in (C1, C2, C3) combined with a perpendicular slice (zy plane, profiled in green); in D2 a slice in the zx plane was also introduced. The arrows point to the same labeled features presented in the corresponding top view. (E1, E2, E3): Magnified areas of the labelled features presented in the corresponding views in (D1, D2, D3). (F1, F2, F3): Corresponding super resolution fluorescence data. (G1, G2, G3): Superimposition of the X-ray data in E1, E2, E3 with the super-resolution fluorescence data in F1, F2, F3. (H1, H2, H3): the profiles of the high contrast features in (G1, G2, G3) are marked in yellow for easier visualization. This was done manually, based on the contrast.
After alignment is performed on the plasma membrane, better interpretation of the signals from the intracellular compartments is possible. The intra-cellular sections are much noisier, but where STORM signals cluster, they do often overlap with sharp-edged, high contrast features inside the cell (Figure S4, white arrows). The antibody accessibility to the intracellular compartments is variable, depending on the individual cell fixation. In cases such as in Figure 7, where the antibody labelling is higher inside the cell, it is easy to differentiate between background signals and intracellular cholesterol crystals that are clearly detected by large clusters of STORM signals. The crystals are mostly, but not exclusively associated with the surface of lipid bodies.
Figure 7:

(A) Slices through the 3D reconstruction of a soft X-ray tomogram of a cell after incubation with acLDL. The cell slice is a mosaic composed of area 1, area 2 and area 3 that were taken at different z inside the cell, to match the locations of the labelled features (arrows). The localization map of the resolved super-resolution image (red spots 360 nm in depth) is superimposed on the corresponding X-ray data slice. (A1, A2): Magnified areas of two features labelled 1, 2, respectively, inside the cell from a top view (xy plane). (B1, B2): Corresponding super resolution fluorescence data. (C1, C2): Superimposition of the X-ray data in A1, A2 with the super-resolution fluorescence data in B1, B2. (D1,D2): Side orientations of the superimposed data presented in C1, C2, combined with perpendicular slices in the zy plane. The inset in each reconstructed picture show the high contrast feature marked in yellow for clearer visualization. This was done manually, based on the contrast the feature. The inset in D2 is enlarged by 30%.
DISCUSSION
We have developed a correlative technique using STORM and cryo-SXT, which provides visualization of cholesterol crystals as small as 200 × 200nm on the plasma membrane, as well as in intracellular locations, of macrophage cells incubated with LDL particles.
Cryo-SXT provides a picture in 3 dimensions of large cellular volumes, identifying any aggregation of carbonrich materials on the background of the X-ray transparent cytoplasm rich in water (within the energy range of the water window). The introduction of a specific marker that identifies desired epitopes by STORM enables colocalization between the identified objects and the local environment morphologically characterized by the cryo-SXT, at 70 nanometer resolution.
With a voxel size of 18 × 18 × 18 nm3 in SXT and of 36 × 36 × 72 nm3 in the STORM measurements, we estimate that the minimum crystal size that can be unambiguously detected and identified in the correlative measurements is 100–150 nm on the plate crystal face.
Theoretically, cryo-SXT might have been sufficient also for identification of the cholesterol crystals, without STORM, if it were possible to accurately measure the X-ray absorption coefficient of the unidentified objects. This would be, however, a challenging task because the absorption coefficient of the cholesterol crystals is expected to be less than 12% different from that of the lipid membrane. Under these conditions, the existence of background noise and of geometric restrictions to the maximal tilt of the sample grid (the missing wedge problem) makes the task virtually impossible.
The volume of each data acquisition in the STORM measurements reported here was limited to 18 × 18 × 1 μm3, whereas SXT data were collected over 18 × 18 × 18 μm3. The two volumes were first roughly aligned in the z direction using the outline of the cell nucleus and then refined using the cell contour of the plasma membrane over the stack of sections within the 1-μm thickness. The addition of external fiducial markers could conceivably minimize the difficulties involved in the overlay procedure.
Data correlation and overlay in intracellular locations proved to be challenging, especially in the direction of the incoming beam (z direction in the figures). Cryo SXT can detect all membranes, lipid bodies and organelles inside the cell to which cholesterol crystals can be associated with. Intracellular labelling by the antibody, however, is limited by the antibody’s penetration without permeabilization. The antibodies that do enter, label both 3D crystals and 2D membrane-domains, which are spread over the sections. Therefore, inside the cell, correlating and identifying every labelled feature to every cryo-SXT signal proved to be less straightforward. Clearly defined labelled clusters of size 200–400 nm (Figure 7) can be easily identified and overlaid to the XST images. Identification of smaller crystals would be most probably unreliable. The method presented here, even though limited by the probe accessibility, can still allow detection of small crystals in their hydrated biological environment and without dissolution.
In the procedure developed here, STORM imaging was performed in solution and was followed by vitrification and SXT imaging with reference to an internal coordinate system. Cell vitrification (cryofixation) was performed either by high pressure freezing (Figures 2 and 7) or by plunge freezing (Figures 5 and 6) in liquid ethane. Both techniques provide adequate specimens, but both techniques can introduce minor sample deformations that may create additional difficulties in the image overlay. The correlation between the two techniques would have been much more straightforward if STORM had been also performed on frozen samples. Cryo-STORM is indeed under development. Its feasibility was demonstrated in a home-made set-up, albeit at lower resolution than STORM in solution 45.
The specific volume to be examined in each observation was chosen out of a specimen grid with a volume of 20 × 3000 × 3000 μm3. The correlative approach developed here is thus not limited only to detect cholesterol crystals in cell cultures. The method can in principle be applied to tissue sections or to whole organisms within sizes of several tens of micrometers in thickness and millimeters in width. The cholesterol structural antibody was shown to detect cholesterol crystals in atherosclerotic tissue sections 17. Generalization of the technique to other specimens using STORM and SXT will depend on the existence of specific probes, and on the development of appropriate sample preparation and freezing techniques, which still ensure a reasonable signal to noise ratio in SXT. The detection of structures in cryo-SXT depends on several factors such as the density of the desired materials, on the atomic content, on the surrounding material (are they embedded in vitreous ice or other organic content), on the dose applied and on other factors that can reduce the sensitivity such as the missing wedge and misalignment.
To the best of our knowledge, this is the first example of a technique providing 3D-imaging and target identification within large volumes of hydrated cellular systems at resolution of tens of nanometers. Cryo-electron imaging in the TEM combined with immuno-labeling allows achieving much higher lateral resolution, but observation is limited to slices ≤ 700nm inside the cells 46. Cryo-SEM can provide topographic images of fracture surfaces at high resolution that can be correlated with fluorescence imaging of appropriately labeled specimens, but images are limited to an arbitrary fracture surface 47. Cryo-focused ion beam milling (FIB) and SEM block face imaging can provide 3D reconstructions over large volumes, and could conceivably be combined with STORM in a correlative manner. The Cryo-FIB SEM serial imaging is currently still under development, and the evaluation of its potential in this direction will need to be checked 48.
Using the newly developed correlative technique, we have imaged and unambiguously identified cholesterol crystals at the early stage of development in their cellular environment, in 3D over relatively thick samples, without dehydration and dissolution of the crystals, and exploiting the density contrast of SXT. The crystals are as small as 200 nm wide and ≤70nm thick, and appear in close association with the cell plasma membrane, as well as with intracellular membranes and/or lipid droplets.
Cholesterol is known to accumulate inside lysosomes of macrophage cells after cholesterol enrichment by lipoproteins 44. TEM studies have shown micrometer size crystals associated with areas of acid phosphatase activity and therefore the crystals were suggested to grow within secondary lysosomes 15. However, the crystals observed were at a mature stage, leaving the door open for further investigations on the initial crystal growth stages and the chemical events that may trigger the practically irreversible process of crystal precipitation. Our results show that it is possible to image crystals in a less mature state at their exact locations inside the cell and on the cell plasma membrane. The crystals observed had dimensions of 200–400nm, and we estimated that the minimum size detectable with the present correlative technique is 100–150nm. These are crystals well beyond the nucleation stage, but still at an initial stage of growth. It is thus conceivable that the crystals nucleated at the location where they were observed, or very close to it. This would support the hypothesis that the crystals may have nucleated from cholesterol domains segregated from lipid mixtures in cell membranes 17,18,23.
Formation of cholesterol crystals with association to the cell plasma membrane might be directly related to cholesterol crystal invasion of the extracellular space in atherosclerotic lesions. Cholesterol crystals have been shown to activate complement and to stimulate inflammation by activating the inflammasome pathway 49–51. Thus, learning how cholesterol crystals form and gaining insight into preventing cholesterol crystal formation could be highly significant for limiting the cholesterol crystal-induced inflammatory component of the atherosclerosis disease process.
EXPERIMENTAL SECTION
Materials
DMEM culture media was obtained from Gibco (Grand Island, NY, USA); 35 mm Dish, No. 1.5 Coverslip, 14 mm Glass Diameter, uncoated (Cat# P35G-1.5–14-C) were obtained from MatTek Corporation Homer Ave Ashland, MA, USA); G200F1 200 mesh Pitch 125 μm, Bar Width 35–25 μm, Hole Width 90–100 μm gold TEM grids (Cat# BNG200F1-Au-50) were obtained from EMS (Hatfield, PA, USA). QUANTIFOIL® R 2/2 on Au G200F1 200 mesh finder grids were obtained from Quantifoil Micro Tools (Jena, Germany); acetylated LDL (acLDL) (Cat# BT-906) was obtained from Alfa Aesar (Heysham, UK); Alexa Fluor® 115–605-020 647-AffiniPure Goat Anti-Mouse IgM, μ-Chain Specific (Cat# 115–605-020), was obtained from Jackson ImmunoResearch (West Grove, PA, USA). Dulbecco’s phosphate-buffered saline (DPBS) with Ca 2+ and Mg 2+ (Cat# 02–020-1A) was obtained from Biological Industries (Kibbutz Beit-Haemek,, Israel); Bovine Serum Albumin and Fetal Bovine Serum were from Sigma- Aldrich (St Louis, Missouri, MO).
Antibody Purification
The monoclonal 58B1 antibodies were purified from the supernatant hybridoma fluid by affinity chromatography using an ImmunoPure IgM purification kit according to the manufacturer’s instructions. The purified fractions were checked by UV absorbance at 280 nm, and those with optical density higher than 0.2 were collected and dialyzed (Spectra/Por membrane with cutoff 12000−14000 Da) in 2 L of phosphate-buffered saline (PBS) at 4 °C (the PBS solution was changed three times every 4 h). The antibodies were used for a maximum of 5 days after purification. The antibody concentration was checked by UV absorbance at 280 nm each day before use.
Cell culture
Adherent murine macrophage-like RAW 264 cells were used as cells for uptake of acLDL particles following the reporting protocol 52. RAW 264.7 cells were used as obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA), and were cultured in DMEM (Dulbecco’s modified Eagle’s medium, Gibco), supplemented with10% fetal calf serum (FCS), 100 units/ml penicillin, 100 g/ml streptomycin and 1% L-glutamine. Maintaining of the culture was done in a water saturated atmosphere of 5% CO2.
Immunolabeling
Cells were cultured on glass bottom culture dishes at a seeding density of 105 cells/cm2. Then, cells were incubated with or without 50 μg/ml acetylated LDL (acLDL) for 48 h (17). This concentration is known to saturate the scavenger receptor that mediates its uptake by macrophage cells 53. During this process of cholesterol enrichment, serum was not added into the culture media so no cholesterol acceptors will be introduced into the model system 17. Cells were rinsed three times in PBS and fixed by incubating with 4% paraformaldehyde (PFA), 0.1% glutaraldehyde (GA) in PBS at room temperature for 1 hour. An additional three washes in PBS containing 0.1% bovine serum albumin (BSA) were done to remove any excess of fixative. Cells were then incubated 60 min with 3 μg/ml purified MAb 58B1 IgM diluted in PBS containing 0.1% BSA. After three rinses in PBS (5 min each), cultures were incubated 30 min with 0.4 μg/ml Alexa Fluor 647 Goat Anti-Mouse IgM secondary antibody diluted in PBS containing 0.1% BSA.
Cultures were then rinsed three times with PBS containing 0.1% BSA and observed on the same day. Control cultures were incubated without acLDL or incubated with acLDL but stained only with secondary antibody and measured in parallel to the sample under the same experimental conditions.
STORM imaging
Super-resolution images were recorded with Vutara SR 200 (Bruker) commercial microscope based on the single-molecule localization biplane technology. Cholesterol ordered areas labeled with AlexaFluor 647 were imaged using 647 nm excitation laser and 405 nm activation laser in an imaging buffer composed of 5 mM cysteamine, oxygen scavengers (7 μM glucose oxidase and 56 nM catalase) in 50 mM Tris with 10 mM NaCl and 10% glucose at pH 8.0. Images were recorded using a 60x, NA 1.2 water immersion objective (Olympus) and Evolve 512 EMCCD camera (Photometrics) with gain set at 50, frame rate at 50 Hz, and maximal power of 647 and 405 nm lasers set at 6 and 0.05 kW/cm2, respectively. The total number of frames acquired was 10000. Data were analyzed by the Vutara SRX software. The image resolution capable of experimentally being achieved is 20 nm laterally (x and y) and 40–50 nm axially (in z).
Cryo-soft X-ray tomography
For the 3D imaging of the cell and associated cholesterol crystals we used cryo-SXT with the 40 nm zone plate lens at 520 eV which has a measured depth of field (DOF) of 3.3 μm. Gold 3.05 mm TEM -grids were coated with formvar film and carbon or quantifoil holey carbon. Then the grids were glow-discharged and sterilized under UV light. Cells were cultured on the grids and incubated with or without AcLDL for 48 hours as described above. For high pressure freezing (Figures 2 and 7) the grids were subsequently lifted and placed between two 15-μm thick stainless steel shims, followed by cryo-immobilization in HPM 010 high pressure freezer (Bal-Tec, Balzers, Liechtenstein), more on high pressure freezing in Ref 54. For plunging (Figures 5 and 6), the grids were lifted and held by tweezers inside the plunge freezer chamber at 70% relative humidity and 37 °C (Leica EM-GP plunger, Leica Microsystems). The grids were blotted for 4 s and were plunge frozen in liquid ethane. Frozen samples were mounted on a cryogenically cooled sample holder at MISTRAL beamline, ALBA synchrotron, Barcelona, Spain 40,41.
The soft X-ray projection tilt series were collected from a number of locations on several grids at the X-ray energy of 520 eV over the sample tilt range between 106 and 141 degrees with steps of one degree. The collection time per complete tomogram ranged from 5 minutes to 30 minutes, depending on the tilt range and the exposure time, which was higher for thicker vitrified ice samples. For figure 2: exposure times were varied from 4s to 2s, for figures 5 and 6: exposure times were varied from 7s to 2s and for figure 7: exposure times were varied from 10s to 4s, depending on the tilt angle.
The alignment of the tomographic projections was performed in bsoft 55 using lipid-rich cellular organelles as fiducial markers. The aligned projection tilt series were reconstructed using tomo3d 56,57. The presented 3D reconstructed volumes have a resolution of 76 nm half pitch (Figures 2 and 7) and 71.1 nm half pitch (Figures 5 and 6) (FSC calculation with 30 iterations of SIRT, at 0.25). This value is higher than the 50nm expected experimental one on other cell types32 because the sample thickness is 3 times larger than the DOF of the lens, and because the alignment accuracy of the tilt series degrades the final resolution of the reconstructed volume.
Correlative cryo-soft X-ray tomography/super-resolution fluorescence microscopy
Cells were cultured on gold TEM finder-grids and incubated with or without AcLDL for 48 hours as described above. Fixation and labeling were performed as above and then observed on the same day in the VUTARA SR200 system. Cells were chosen for observation based on their location on the grid. Since the same cells should be suitable for tomogram imaging, only cells that sit at the center of the grid and approximately far from the gold mesh were imaged. Fixed cells were then kept in PBS at 4°C overnight and vitrified the next day as described above. Frozen samples were mounted on a cryogenically cooled sample holder at MISTRAL, ALBA. Mosaic projection scans were collected over the large areas centered at the recorded positions based on the lettering-fiducials on the grid. The cells from which the STORM data were collected were located and the tilt images were acquired by the soft X-ray beam on the desired cell.
Overlay of the data was performed as follows: light microscopy and STORM images of Alexa-647 signal were overlaid onto images from the X-ray mosaic projection scans (Figure S5, Step 1). Several markers were used for this alignment: the goldmesh, the gold fiducial lettering and the regular array of the holes in the grid quantifoil coating. Using this alignment the angle between the super resolution map and the reconstructed tomogram stack was obtained (Figure S5, Step 1, Orange lines). For the alignment in z we were aided by the outline of the nucleus and plasma membrane. Then, each of the data sets were sliced into layers of 80 nm (based on the z resolution, (Figure S5, Step 2). The sliced tomogram layers were loaded into the tailor-made Correlator script (available for download) that finds the cell’s contour (Figure S5, Step 2, blue outline). The super resolution sliced data is then loaded and the software overlays the data for every possible z-shift. The output is a graph with two parameters: the percentage of the super resolution signal comes from the cell area for every given z shift and the coverage area of the super resolution data relative to the cell area for every shift (Figure S5, Step 2, graph). The optimal z position is chosen where most of the super-resolution signals are located within the cell and the coverage area of the signal is 100%. The signal is enveloped by the cell contour, with some signal superimposed to the plasma membrane area (Figure S5, Step 2H, red arrow). The resulting overlay can then be evaluated with a cross correlation function which works as follows: both of the data sets are superimposed such that the outline of the cell plasma membrane signal from the cryo-SXT data is overlaid on the outline of the signal coming from the super resolution data. The amount of super resolution spots coming from the outlined cell area is calculated. The cross correlation is evaluated by counting the relative amount of super resolution spots that co-localized (with a radius of 1 pixel) with a signal from the cryo-SXT membrane and compares it with a random spot distribution. The random case is evaluated by spreading the same amount of total spots over the same area and then evaluates the relative amount that randomly overlaid with cryo-SXT signal. For the data presented here: the membrane was segmented based on contrast and were overlaid over the SR signal. 1514 out of 1604 spots (94.389%) were colocalized with the cryo-SXT segmented membrane, relative to random spot distribution cases of about 419 spots out of 1604 which randomly overlaid on the membrane (26.122%). The STORM and SXT data sets were finally overlaid in Aviso for 3D observation. Both data sets are overlaid using AVIZO 3D software. The nucleus, the plasma membrane and the lipid bodies were then segmented based on their contrast and rendered in 3D.
Cryo-SEM microscopy
Macrophage-like RAW 264 cells were grown on tissue culture plates and were gently removed by scraping.
The pellet was centrifuged (290g, 4 min) and washed with PBS. High pressure freezing was performed as follows: A 4μl drop of a suspension of cells was placed on a grid and sandwiched between two aluminum plates, followed by cryo-immobilization in the same high pressure freezing device mentioned above. Frozen samples were mounted on a holder under liquid N2 and transferred to a Freeze Fracture BAF 60 device (Bal-Tec) using a Vacuum Cryo Transfer unit VCT 100 (Bal-Tec). Samples were fractured at a temperature of −120°C, etched for 5–20 min at −105°C at a vacuum of <5 ·10−7 mbar. Samples were observed using an Ultra 55 SEM (Zeiss) with a secondary electron in-lens detector in the frozen-hydrated state by use of a cryostage operating at a temperature of at −120°C.
TEM microscopy
Macrophage-like RAW 264 cells were grown on tissue culture plates and were fixed with Karnovsky fixative (4% paraformaldehyde, 2% glutaraldehyde 5 mM CaCl2 in 0.1 M Cacodylate buffer, pH 7.4) 58.
After extensive washing with 0.1 M Cacodylate buffer, cells were gently removed by scraping, centrifuged (290g, 4 min) and embedded in 3.4% agar. The embedded pellet was post-fixed with 1% osmium tetraoxide, 0.5% potassium dichromate,and 0.5% potassium hexacyanoferrate in cacodylate buffer and then with 2% aqueous uranyl acetate (1 h each). Dehydrated through ethanol series was done followed by embedding in EMBED 812 (EMS), Sections of 70 nm were cut from the block, placed on a TEM grid and stained with 2% uranyl acetate and Reynolds’ lead.
Samples were viewed on T12-Tecnai TEM microscope operating at 120 kV. Images were recorded on an Eagle 2 K × 2 K FEI camera (Eindhoven, Netherlands).
Supplementary Material
ACKNOWLEDGMENTS
We thank prof. Benjamin Geiger and Michal Shemesh for their support in the cell culture growth and maintenance. We thank Dr. Sharon G. Wolf for her help with freezing and handling cryo-samples and for many fruitful discussions. We thank Helena Sabanay for her help and guidance with the TEM sample preparation. We thank Moshe Varsano for his great help with programming. Super resolution microscopy was performed at the Irving and Cherna Moskowitz Center for Nano and Bio-Nano Imaging at the Weizmann Institute of Science. Cryo-SXT experiments were performed at MISTRAL beamline at ALBA Synchrotron with the collaboration of ALBA staff. The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007–2013) under grant agreement nº 283570. This research was supported by the Binational Science Foundation (Grant 2013045 to LA and HSK). L.A. is the incumbent of the Dorothy and Patrick Gorman Professorial Chair of Biological Ultrastructure at the Weizmann Institute of Science.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Material
Figure S1: Representative reconstruction of cell tomograms after incubation without acLDL. STROM signal from the same untreated cell. Figure S2: TEM micrographs of embedded cell culture after 48h incubation with acLDL Figure S3: Soft X-ray tomogram with high contrast feature attached to its plasma membrane Figure S4: High magnification of intracellular labeling. Figure S5: Step by step overlay streamline The correlator script is available for download using the following link: https://drive.google.com/file/d/0B6247JiJFhITV0l3X0FMODhnZUU/view?usp&ts=57eb9fc8 This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Insull W Jr; Bartsch GJ Clin. Invest 1966, 45, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kruth HS Am J Pathol 1984, 114, 201. [PMC free article] [PubMed] [Google Scholar]
- (3).Katz S; Shipley GG; Small DJ Clin. Invest 1976, 58, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kruth HS Curr. Mol. Med 2001, 1, 633. [DOI] [PubMed] [Google Scholar]
- (5).Tabas I; Rosoff W; Boykow GJ Biol. Chem 1988, 263, 1266. [PubMed] [Google Scholar]
- (6).Brasaemle DL; Attie AD J. Lipid Res 1990, 31, 103. [PubMed] [Google Scholar]
- (7).Liscum L; Munn NJ Biochim. Biophys. Acta 1999, 1438, 19. [DOI] [PubMed] [Google Scholar]
- (8).Kruth HS; Ifrim I; Chang J; Addadi L; Perl-Treves D; Zhang W-YJ Lipid Res. 2001, 42, 1492. [PubMed] [Google Scholar]
- (9).Tabas I Trends Cardiovasc. Med 1997, 7, 256. [DOI] [PubMed] [Google Scholar]
- (10).Kruth HS Front. Biosci. 2001, 6, D429. [DOI] [PubMed] [Google Scholar]
- (11).Small D Arterioscler. Thromb. Vasc. Biol 1988, 8, 103. [Google Scholar]
- (12).Rajamäki K; Lappalainen J; Öörni K; Välimäki E; Matikainen S; Kovanen PT; Eklund KK PloS one 2010, 5, e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shah PK; Falk E; Badimon JJ; Fernandez-Ortiz A; Mailhac A; Villareal-Levy G; Fallon JT; Regnstrom J; Fuster V Circulation 1995, 92, 1565. [PubMed] [Google Scholar]
- (14).Klinkner AM; Waites CR; Kerns WD; Bugelski PJ J. Histochem. Cytochem 1995, 43, 1071. [DOI] [PubMed] [Google Scholar]
- (15).Tangirala RK; Jerome WG; Jones N; Small DM; Johnson W; Glick J; Mahlberg F; Rothblat GJ Lipid Res. 1994, 35, 93. [PubMed] [Google Scholar]
- (16).Kellner-Weibel G; Yancey P; Jerome W; Walser T; Mason R; Phillips M; Rothblat G Arterioscler. Thromb. Vasc. Biol 1999, 19, 1891. [DOI] [PubMed] [Google Scholar]
- (17).Ong DS; Anzinger JJ; Leyva FJ; Rubin N; Addadi L; Kruth HS J. Lipid Res 2010, 51, 2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Varsano N; Fargion I; Wolf SG; Leiserowitz L; Addadi LJ Am. Chem. Soc 2015, 137, 1601. [DOI] [PubMed] [Google Scholar]
- (19).Tabaei SR; Jackman JA; Kim S-O; Liedberg B; Knoll W; Parikh AN; Cho N-J Langmuir 2014, 30, 13345. [DOI] [PubMed] [Google Scholar]
- (20).Tabaei SR; Jackman JA; Liedberg B; Parikh AN; Cho N-JJ Am. Chem. Soc 2014, 136, 16962. [DOI] [PubMed] [Google Scholar]
- (21).Ziblat R; Kjaer K; Leiserowitz L; Addadi L Angew. Chem. Int. Ed 2009, 48, 8958. [DOI] [PubMed] [Google Scholar]
- (22).Ziblat R; Leiserowitz L; Addadi LJ Am. Chem. Soc 2010, 132, 9920. [DOI] [PubMed] [Google Scholar]
- (23).Ziblat R; Fargion I; Leiserowitz L; Addadi L Biophys. J 2012, 103, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Frincu MC; Fleming SD; Rohl AL; Swift JA J. Am. Chem. Soc 2004, 126, 7915. [DOI] [PubMed] [Google Scholar]
- (25).Frincu MC; Sharpe RE; Swift JA Cryst. Growth Des 2004, 4, 223. [Google Scholar]
- (26).Huang B; Bates M; Zhuang X Annu. Rev. Biochem 2009, 78, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kapishnikov S; Weiner A; Shimoni E; Schneider G; Elbaum M; Leiserowitz L Langmuir 2013, 29, 14595. [DOI] [PubMed] [Google Scholar]
- (28).Kapishnikov S; Weiner A; Shimoni E; Guttmann P; Schneider G; Dahan-Pasternak N; Dzikowski R; Leiserowitz L; Elbaum M Proc. Natl. Acad. Sci. USA 2012, 109, 11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cinquin BP; Do M; McDermott G; Walters AD; Myllys M; Smith EA; Cohen-Fix O; Le Gros MA; Larabell CA J. Cell. Biochem 2014, 115, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Smith EA; McDermott G; Do M; Leung K; Panning B; Le Gros MA; Larabell CA Biophys. J. 2014, 107, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Le Gros MA; McDermott G; Larabell CA Curr. Opin. Struct. Biol 2005, 15, 593. [DOI] [PubMed] [Google Scholar]
- (32).Pérez-Berná AJ; Rodríguez MJ; Chichón FJ; Friesland MF; Sorrentino A; Carrascosa JL; Pereiro E; Gastaminza P ACS nano 2016, 10, 6597. [DOI] [PubMed] [Google Scholar]
- (33).McDermott G; Le Gros MA; Knoechel CG; Uchida M; Larabell CA Trends Cell Biol. 2009, 19, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Schneider G; Guttmann P; Heim S; Rehbein S; Mueller F; Nagashima K; Heymann JB; Müller WG; McNally JG Nat. Methods 2010, 7, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hagen C; Guttmann P; Klupp B; Werner S; Rehbein S; Mettenleiter TC; Schneider G; Grünewald KJ Struct. Biol 2012, 177, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Duke EM; Razi M; Weston A; Guttmann P; Werner S; Henzler K; Schneider G; Tooze SA; Collinson LM Ultramicroscopy 2014, 143, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Perl-Treves D; Kessler N; Izhaky D; Addadi L Chem. Biol 1996, 3, 567. [DOI] [PubMed] [Google Scholar]
- (38).Izhaky D; Addadi L Adv. Mater 1998, 10, 1009. [Google Scholar]
- (39).Goldschmidt-Arzi M; Shimoni E; Sabanay H; Futerman AH; Addadi LJ Struct. Biol 2011, 175, 21. [DOI] [PubMed] [Google Scholar]
- (40).Pereiro E; Nicolas J; Ferrer S; Howells MJ Synchrotron Rad 2009, 16, 505. [DOI] [PubMed] [Google Scholar]
- (41).Sorrentino A; Nicolás J; Valcárcel R; Chichón FJ; Rosanes M; Avila J; Tkachuk A; Irwin J; Ferrer S; Pereiro EJ Synchrotron Rad 2015, 22, 1112. [DOI] [PubMed] [Google Scholar]
- (42).Uchida M; Sun Y; McDermott G; Knoechel C; Le Gros MA; Parkinson D; Drubin DG; Larabell CA Yeast 2011, 28, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Weiss D; Schneider G; Niemann B; Guttmann P; Rudolph D; Schmahl G Ultramicroscopy 2000, 84, 185. [DOI] [PubMed] [Google Scholar]
- (44).Tangirala R; Mahlberg F; Glick J; Jerome W; Rothblat GJ Biol. Chem 1993, 268, 9653. [PubMed] [Google Scholar]
- (45).Kaufmann R; Schellenberger P; Seiradake E; Dobbie IM; Jones EY; Davis I; Hagen C; Grünewald K Nano Lett. 2014, 14, 4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Medalia O; Weber I; Frangakis AS; Nicastro D; Gerisch G; Baumeister W Science 2002, 298, 1209. [DOI] [PubMed] [Google Scholar]
- (47).Khalifa GM; Kirchenbuechler D; Koifman N; Kleinerman O; Talmon Y; Elbaum M; Addadi L; Weiner S; Erez JJ Struct. Biol 2016, DOI: 10.1016/j.jsb.2016.01.015. [DOI] [PubMed] [Google Scholar]
- (48).Vidavsky N; Masic A; Schertel A; Weiner S; Addadi LJ Struct. Biol 2015, 192, 358. [DOI] [PubMed] [Google Scholar]
- (49).Vogt W; Von Zabern I; Damerau B; Hesse D; Lühmann B; Nolte R Mol. Immunol 1985, 22, 101. [DOI] [PubMed] [Google Scholar]
- (50).Adams C; Bayliss O Atherosclerosis 1975, 22, 629. [DOI] [PubMed] [Google Scholar]
- (51).Lundberg B Atherosclerosis 1985, 56, 93. [DOI] [PubMed] [Google Scholar]
- (52).Ylitalo R; Jaakkola O; Lehtolainen P; Ylä-Herttuala S Life Sci. 1999, 64, 1955. [DOI] [PubMed] [Google Scholar]
- (53).Basu SK; Goldstein JL; Anderson G; Brown MS Proc. Natl. Acad. Sci. USA 1976, 73, 3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Weiner A; Kapishnikov S; Shimoni E; Cordes S; Guttmann P; Schneider G; Elbaum MJ Struct. Biol 2013, 181, 77. [DOI] [PubMed] [Google Scholar]
- (55).Heymann JB; Cardone G; Winkler DC; Steven AC J. Struct. Biol 2008, 161, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Agulleiro J; Fernandez J-J Bioinformatics 2011, 27, 582. [DOI] [PubMed] [Google Scholar]
- (57).Agulleiro J-I; Fernandez J-JJ Struct. Biol 2015, 189, 147. [DOI] [PubMed] [Google Scholar]
- (58).Reef S; Zalckvar E; Shifman O; Bialik S; Sabanay H; Oren M; Kimchi A Mol. Cell 2006, 22, 463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.