

Abstract
Introduction
While current standard of care (SOC) for idiopathic pulmonary fibrosis (IPF) slows disease progression, prognosis remains poor. Therefore, an unmet need exists for novel, well-tolerated agents that reduce lung function decline and improve quality of life. Here we report the design of two phase III studies of the novel IPF therapy, GLPG1690.
Methods and analysis
Two identically designed, phase III, international, randomised, double-blind, placebo-controlled, parallel-group, multicentre studies (ISABELA 1 and 2) were initiated in November 2018. It is planned that, in each study, 750 subjects with IPF will be randomised 1:1:1 to receive oral GLPG1690 600 mg, GLPG1690 200 mg or placebo, once daily, on top of local SOC, for at least 52 weeks. The primary endpoint is rate of decline of forced vital capacity (FVC) over 52 weeks. Key secondary endpoints are week 52 composite endpoint of disease progression or all-cause mortality (defined as composite endpoint of first occurrence of ≥10% absolute decline in per cent predicted FVC or all-cause mortality at week 52); time to first respiratory-related hospitalisation until end of study; and week 52 change from baseline in the St George’s Respiratory Questionnaire total score (a quality-of-life measure).
Ethics and dissemination
Studies will be conducted in accordance with Good Clinical Practice guidelines, Declaration of Helsinki principles, and local ethical and legal requirements. Results will be reported in a peer-reviewed publication.
Trial registration numbers
Keywords: interstitial fibrosis, rare lung diseases
Key messages.
Idiopathic pulmonary fibrosis (IPF) is associated with progressive loss of lung function and a median survival of 3–4 years.
Standard of care (SOC) with the antifibrotic agents pirfenidone or nintedanib may slow disease progression; however, more effective, well-tolerated IPF treatments are needed.
The autotaxin inhibitor, GLPG1690, is in development as a novel therapy for IPF and has demonstrated encouraging results in early clinical trials, warranting further studies.
GLPG1690 in addition to SOC will be assessed in patients with IPF in ISABELA 1 and 2, two identically designed, phase III, randomised, placebo-controlled, parallel-group trials.
The trials will provide valuable efficacy, safety and quality-of-life data, and positive findings may ultimately lead to a much-needed new treatment option for patients with IPF.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive condition characterised by fibrosis, dyspnoea, worsening lung function and impaired quality of life (QoL).1 2 IPF is one of the most common forms of interstitial lung disease,3 affecting approximately three million people worldwide.2 Patients are typically aged over 60 years at diagnosis4 and have a poor prognosis; the median survival is 3–4 years and the 5-year survival rate is 20%–40%.1 5 Respiratory failure is the most common cause of death.6
During the last decade, significant advances have been made in understanding the complex biological mechanisms responsible for IPF.1 7 It is currently accepted that chronic epithelial cell injury results in abnormal wound healing and widespread fibrosis, which replaces normal lung parenchyma.1 8 9 Consequently, treatment has moved from classical anti-inflammatory and immunosuppressant drugs to agents designed to inhibit the processes that drive fibrosis.1 7–9 Two antifibrotic agents, pirfenidone and nintedanib, were approved for the treatment of IPF and included under conditional recommendations in 2015 clinical practice guidelines10 11 after demonstrating a reduction in lung function decline, measured by forced vital capacity (FVC), in phase III trials.12–14 Subsequent pooled analyses of phase III trials have shown reductions in mortality.14 15 In addition, real-world registry data suggest improved survival in those treated with, versus without, antifibrotic treatments, regardless of baseline disease severity,16 and pooled registry and clinical trial data suggest that pirfenidone increases life expectancy compared with best supportive care.17
However, individuals may not tolerate adverse events (AEs) associated with pirfenidone (gastrointestinal and skin-related events)12 14 or nintedanib (diarrhoea)13; real-world data showed that AEs led to drug discontinuation in 20.9% and 26.3% of pirfenidone-treated and nintedanib-treated patients with IPF, respectively.18 QoL is often significantly impaired in IPF,1 9 19 and neither pirfenidone nor nintedanib has demonstrated benefits to QoL in clinical trials.20 21 While both agents may improve overall outcomes, treated patients continue to experience lung function loss and premature death. Therefore, new treatments are needed that reduce the decline in FVC to a greater degree than current therapies, hopefully halting the disease or, better yet, reversing its course. New treatments, ideally, would also improve or slow the rate of decline in patient-reported outcomes (PROs), including QoL, and must have an acceptable safety and tolerability profile, allowing for chronic use.
The aberrant wound healing responses that result in fibrosis in IPF are thought to be at least partially mediated by lysophosphatidic acid (LPA).22 23 Levels of LPA and autotaxin (ATX), an enzyme involved in its production,24 are upregulated in patients with IPF, confirming their role in disease pathogenesis and potential as targets for novel therapeutic agents.25 26 One such agent is GLPG1690 (Galapagos NV, Mechelen, Belgium), a first-in-class, small molecule ATX inhibitor.27 In a phase I, first-in-human trial, GLPG1690 was generally well tolerated and resulted in a maximum reduction in plasma LPA levels of approximately 90%.28 In the phase IIa FLORA study, conducted in 23 subjects with IPF, GLPG1690 exhibited a safety profile similar to placebo and demonstrated favourable effects on mean change from baseline in FVC at week 12 compared with placebo (25 mL vs –70 mL) (although the study was not powered to evaluate efficacy differences between groups).29
To further evaluate GLPG1690 for the treatment of IPF, two identically designed, phase III studies (ISABELA 1 and 2) will be conducted. The primary objective of the studies is to evaluate the efficacy of two doses of GLPG1690 compared with placebo, each given in addition to local standard of care (SOC), in subjects with IPF, as assessed by the rate of decline of FVC over 52 weeks. Secondary objectives relate to efficacy, mortality, QoL, safety, tolerability and pharmacokinetics. Here we report the design of the studies.
Methods and analysis
Study design and interventions
ISABELA 1 and 2 are identically designed, phase III, international, randomised, double-blind, placebo-controlled studies that will be conducted in parallel. Within each country, the study may be conducted at multiple centres. A significant proportion of subjects are expected to be recruited from the USA and Europe (see ClinicalTrials.gov for location details).
In each study, approximately 750 subjects will be randomised 1:1:1 to receive oral GLPG1690 600 mg, GLPG1690 200 mg or matching placebo, once daily, in addition to local SOC. SOC is defined as either pirfenidone or nintedanib, or neither pirfenidone nor nintedanib (for any reason). Treatment will continue for at least 52 weeks (subjects will continue to receive randomised treatment until the last patient reaches 52 weeks in the study). A follow-up visit will be conducted 4 weeks after the end-of-study visit (figure 1).
Figure 1.
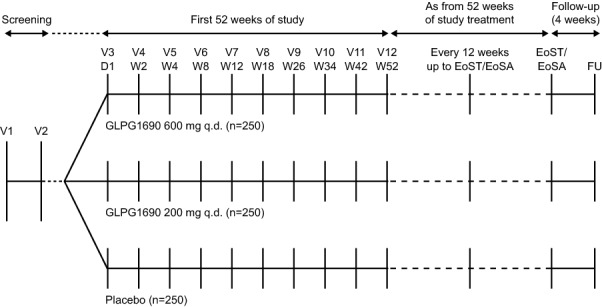
Design of the ISABELA 1 and 2 studies. D, day; EoSA, end-of-study assessment; EoST, end-of-study treatment; FU, follow-up; qd, once daily; V, visit; W, week.
Treatment will be allocated using a centralised electronic system (interactive web response system) and randomisation will be stratified by local SOC for IPF. Both subjects and study personnel will be blinded to treatment assignment. The blind can be broken only if deemed necessary for safety reasons by the investigator. GLPG1690 doses were selected based on efficacy, safety, tolerability, and pharmacokinetic and pharmacodynamic data from previous clinical and preclinical studies,28 29 and are expected to allow plasma concentrations >80% inhibitory concentration of LPA to be attained for ≥60% of the dosing interval. Investigators may reduce the dose, interrupt or permanently discontinue GLPG1690 for safety concerns at any point. Downtitration will be performed as a single step to reduce the dose to the lowest level. Re-escalation from the lowest dose to the original dose will be conducted in two steps using an intermediate dose. Subjects who discontinue early (unless lost to follow-up/withdrawal of consent) will be encouraged to complete the remaining visits and evaluations.
ISABELA 1 and 2 are registered on ClinicalTrials.gov (NCT03711162 and NCT03733444, respectively). Enrolment began in November 2018; both studies are currently open to recruitment.
Patient population
Eligible subjects will be men or women aged ≥40 years diagnosed with IPF within the previous 5 years (based on applicable international guidelines11 30). IPF diagnosis will be confirmed by central reading of a chest high-resolution computed tomography performed within the previous 12 months, plus central reading of lung biopsy (if available), based on the Fleischner Society white paper.31 Subjects should be receiving local SOC, defined as a stable dose of either pirfenidone or nintedanib, for at least 2 months prior to screening, or neither pirfenidone nor nintedanib; and have an FVC ≥45% predicted of normal, forced expiratory volume in 1 s (FEV1):FVC ≥0.7, and diffusing capacity of the lung for carbon monoxide (DLCO) corrected for haemoglobin ≥30% predicted of normal. Subjects will be excluded if they have had an acute IPF exacerbation (determined by the investigator) within the previous 6 months; lower respiratory tract infection requiring antibiotics within the previous 4 weeks; interstitial lung disease associated with known primary diseases, environmental exposures or drugs; a history of lung volume reduction surgery or lung transplant; a diagnosis of severe pulmonary hypertension; or unstable cardiovascular, pulmonary or other disease within 6 months prior to screening or during the screening period. Full inclusion and exclusion criteria are included in box 1.
Box 1. Full inclusion and exclusion criteria.
Inclusion criteria.
Male or female aged ≥40 years.
Diagnosis of IPF within 5 years prior to screening based on the ATS/ERS/JRS/ALAT guidelines.
Chest HRCT performed within 12 months prior to screening and according to minimum requirements for IPF diagnosis by central review based on HRCT only (if no lung biopsy is available) or HRCT and lung biopsy. If no HRCT is available prior to screening, an HRCT can be performed at screening.
Subjects receiving local SOC for IPF, defined as either pirfenidone or nintedanib, at a stable dose for at least 2 months prior to screening, or neither pirfenidone nor nintedanib.
The total extent of fibrotic changes is greater than the extent of emphysema on the most recent HRCT scan (investigator-determined).
-
Meeting all of the following criteria during the screening period:
FVC ≥45% predicted of normal.
FEV1:FVC ≥0.7.
DLCO corrected for Hb ≥30% predicted of normal.
In a stable condition and suitable for study participation based on the results of a medical history, physical examination, vital signs, 12-lead ECG and laboratory evaluation.
Estimated minimum life expectancy of at least 30 months for non–IPF-related disease in the opinion of the investigator.
Male subjects and female subjects of childbearing potential agree to use highly effective contraception/preventive exposure measures.
Able to walk at least 150 m during the 6MWT at screening visit 1; at visit 2, for the oxygen titration test, resting SpO2 should be ≥88% with a maximum of 6 L O2/min; during the walk, SpO2 should be ≥83% with 6 L O2/min or ≥88% with 0, 2, or 4 L O2/min.
Able to read and complete the EQ-5D, SGRQ, LCQ, K-BILD questionnaire and VAS by themselves.
Able to understand the importance of adherence, and willing to comply to study treatment, study procedures and requirements as per study protocol, including the concomitant medication restrictions.
Exclusion criteria.
Investigator or other study staff or relative thereof who is directly involved in the conduct of the study.
Any condition or circumstance that, in the opinion of the investigator, may make a subject unsuitable for inclusion or unlikely or unable to complete the study or comply with study procedures and requirements.
Previous participation in a clinical study with GLPG1690.
Known hypersensitivity to any of the investigational medicinal product’s ingredients or a history of a significant allergic reaction to any drug as determined by the investigator.
Current immunosuppressive condition.
Positive serology for hepatitis B (antigen) or C (antibody).
History of malignancy within the past 5 years (except for carcinoma in situ of the uterine cervix, basal cell carcinoma of the skin that has been treated with no evidence of recurrence, prostate cancer that has been medically managed through active surveillance or watchful waiting, squamous cell carcinoma of the skin if fully resected, and ductal carcinoma in situ).
Clinically significant abnormalities detected on ECG of either rhythm or conduction, a QTcF >450 ms or a known long QT syndrome.
Currently taking medication known to be a substrate mainly metabolised by CYP2C8.
Currently taking medication known to be strong inducers of CYP3A4, and also including St John’s wort.
Currently taking medication known to be strong inhibitors of CYP3A4.
Currently taking medication known to be potent inducers of P-gp.
Currently taking medication known to be potent inhibitors of P-gp.
Acute IPF exacerbation within 6 months prior to screening and/or during the screening period (investigator-determined).
Lower respiratory tract infection requiring antibiotics within 4 weeks prior to screening and/or during the screening period.
Interstitial lung disease associated with known primary diseases (eg, sarcoidosis and amyloidosis), exposures (eg, radiation, silica, asbestos and coal dust) or drugs (eg, amiodarone).
History of lung volume reduction surgery or lung transplant. Note: being on a transplant list is allowed.
Diagnosis of severe pulmonary hypertension (investigator-determined).
Unstable cardiovascular, pulmonary (other than IPF) or other disease within 6 months prior to screening or during the screening period (eg, acute coronary disease, heart failure and stroke).
Had gastric perforation within 3 months prior to screening and/or underwent major surgery within 3 months prior to screening or have major surgery planned during the study period.
A history of being admitted to an institution under an administrative court order, if applicable by local legislation.
Moderate to severe hepatic impairment (Child-Pugh B or C) and/or abnormal LFT at screening, defined as AST and/or ALT and/or total bilirubin ≥1.5× ULN, and/or GGT ≥3× ULN. Retesting is allowed once.
Abnormal renal function defined as estimated creatinine clearance, calculated according to Cockcroft-Gault calculation <30 mL/min. Retesting is allowed once.
Hb level <10 g/dL. Retesting is allowed once.
Participation in a drug, device or biological investigational research study, concurrently with the current study, or within five half-lives of the agent (or within 8 weeks when half-life is unknown) prior to screening, or prior participation in an investigational drug antibody study within 6 months prior to screening.
Use of any of the following therapies within 4 weeks prior to screening and during the screening period, or planned during the study: warfarin, imatinib, ambrisentan, azathioprine, cyclophosphamide, ciclosporin A, bosentan, methotrexate, sildenafil (except for occasional use), and prednisone at steady dose >10 mg/day or equivalent.
Current alcohol or substance abuse in the opinion of the investigator.
Pregnant, breastfeeding or planning to become pregnant or breastfeed during the study treatment or within 30 days after the last dose of investigational medicinal product.
6MWT, 6-Minute Walk Test; ALAT, Latin America Thoracic Association; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ATS, American Thoracic Society; CYP, cytochrome P450; DLCO, diffusing capacity of the lung for carbon monoxide; ERS, European Respiratory Society; EQ-5D, EuroQoL 5-Dimensions Questionnaire; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; GGT, gamma glutamyl transferase; Hb, haemoglobin; HRCT, high-resolution computed tomography; IPF, idiopathic pulmonary fibrosis; JRS, Japanese Respiratory Society; K-BILD, King’s Brief Interstitial Lung Disease; LCQ, Leicester Cough Questionnaire; LFT, liver function test; P-gp, P-glycoprotein; QTcF, QT interval corrected for heart rate using Fridericia’s formula; SGRQ, St George’s Respiratory Questionnaire; SOC, standard of care; SpO2, arterial oxygen saturation; ULN, upper limit of normal; VAS, Visual Analogue Scale.
Endpoints
The primary endpoint is rate of FVC decline over 52 weeks. Key secondary endpoints are disease progression, defined as the composite endpoint of first occurrence of ≥10% absolute decline in per cent predicted FVC (%FVC) or all-cause mortality at week 52; time to first respiratory-related hospitalisation until the end of the studies; and change from baseline in the St George’s Respiratory Questionnaire (SGRQ)32 total score at week 52. Other endpoints will capture additional efficacy, mortality, QoL, safety, pharmacokinetic, pharmacodynamic and biomarker data (box 2).
Box 2. Endpoints to be assessed.
Primary endpoint.
Rate of decline of FVC (in mL) over 52 weeks.
Key secondary endpoints.
Disease progression defined as the composite endpoint of first occurrence of ≥10% absolute decline in per cent predicted FVC or all-cause mortality at 52 weeks.
Time to first respiratory-related hospitalisation until EoS.
Change from baseline in the SGRQ total score at 52 weeks.
Other secondary endpoints.
Rate of decline of FVC (in mL) until EoS.
Disease progression defined as the composite endpoint of first occurrence of ≥10% absolute decline in per cent predicted FVC or all-cause mortality until EoS.
Change from baseline in the SGRQ total score at EoS.
Time to first all-cause non-elective hospitalisation until EoS.
Time to respiratory-related mortality until EoS.
Time to lung transplant until EoS.
Time to first acute IPF exacerbation (determined by the clinical endpoint adjudication committee) until EoS.
Time to all-cause mortality or lung transplant until EoS.
Time to all-cause mortality, or lung transplant or qualifying for lung transplant until EoS.
Time to all-cause mortality, ≥10% absolute decline in per cent predicted FVC or respiratory-related hospitalisations until EoS.
Time to all-cause mortality or respiratory-related hospitalisations until EoS.
-
FVC analyses at 52 weeks and until EoS:
Absolute and relative change from baseline of FVC and per cent predicted FVC.
Absolute categorical change of per cent predicted FVC until EoS: decrease by >5, increase by >5 and change within ≤5.
Absolute categorical change of per cent predicted FVC until EoS: decrease by >10, increase by >10 and change within ≤10.
Safety and tolerability over time until EoS.
Changes from baseline in cough-related quality of life, assessed by the LCQ total score and domains over time, and the VAS Cough and Urge to Cough at 52 weeks and until EoS.
Changes from baseline in quality of life, assessed by the EQ-5D, K-BILD total score and domains over time at 52 weeks and until EoS.
Plasma concentration of GLPG1690, pirfenidone and nintedanib (as appropriate) at 52 weeks and until EoS.
Change from baseline in functional exercise capacity, assessed by the 6MWT distance, at 52 weeks and until EoS.
Change from baseline in DLCO (corrected for Hb) at 52 weeks and until EoS.
Other endpoints.
Changes in target biomarkers/pharmacodynamics in blood over time compared with baseline until EoS.
Changes in disease-specific biomarkers in blood over time compared with baseline until EoS.
Efficacy and biomarker endpoints by genotype subgroups.
Change from baseline in Borg Scale before and after 6MWT at 52 weeks and until EoS.
Change from baseline in SpO2 until EoS.
6MWT, 6-Minute Walk Test; DLCO, diffusing capacity of the lung for carbon monoxide; EoS, end of study; EQ-5D, EuroQoL 5-Dimensions Questionnaire; FVC, forced vital capacity; Hb, haemoglobin; IPF, idiopathic pulmonary fibrosis; K-BILD, King’s Brief Interstitial Lung Disease; LCQ, Leicester Cough Questionnaire; SGRQ, St George’s Respiratory Questionnaire; SpO2, arterial oxygen saturation; VAS, Visual Analogue Scale.
Key assessments
The assessment schedule is shown in online supplementary file table 1. Prebronchodilator spirometry will be performed at screening and throughout the treatment period to assess pulmonary function. Spirometers will be provided to study centres and used according to standards outlined in international guidelines.33 The parameters measured or calculated as part of the spirometry assessments will be FVC (mL) and %FVC, FEV1 (mL) and per cent predicted FEV1, FEV1:FVC ratio, and forced expiratory flow between 25% and 75% of exhaled volume. Predicted values will be calculated using the 2012 Global Lung Function Initiative equations.34 At specified time points during screening and treatment, the DLCO test, corrected for haemoglobin, will be performed according to local practice, with appropriate prediction formulas, and in line with international guidelines (study centres may use different prediction formulas, as appropriate based on the local method used to measure DLCO), and at-rest arterial oxygen saturation will be measured by pulse oximetry. PROs will be assessed during screening and at specified weeks during treatment using the EuroQoL 5-Dimensions Questionnaire (EQ-5D),35 SGRQ, King’s Brief Interstitial Lung Disease (K-BILD) questionnaire,36 Leicester Cough Questionnaire (LCQ),37 and Visual Analogue Scale (VAS) Cough and Urge to Cough. PRO questionnaires will be completed electronically by subjects using a tablet device at clinical study centres. The 6-Minute Walk Test, performed using a standardised manual designed based on clinical guidelines and published literature,38–40 will be used to assess pulmonary function at specified time points during screening and treatment periods. Safety will be assessed throughout via reporting of AEs, clinical laboratory evaluations, physical examination, vital signs and electrocardiogram. Other assessments will include pharmacokinetic, pharmacodynamic, biomarker and genotype evaluations.
bmjresp-2019-000422supp001.docx (48.9KB, docx)
Statistical analyses
Primary and safety analyses will be performed on the full analysis set (all randomised subjects who received at least one dose of study drug). The primary time point is week 52; analyses conducted at the end of the studies are secondary or supportive. An interim analysis to assess futility will be conducted when a reasonable number of subjects (eg, 25% from the two studies combined) have completed 52 weeks of treatment. An independent data monitoring committee (IDMC) will review the interim analysis results and make a recommendation to the sponsor, who will remain blinded. The study will not be terminated early for positive interim efficacy results.
The rate of decline of FVC will be analysed using a random coefficient regression model including sex, age, height and stratification factor as covariates and a random slope and intercept. The proportion of subjects who have an absolute decline ≥10% in %FVC at least once during the study, or who die, will be analysed using logistic regression analysis using data up to week 52. A time-to-event analysis may be used to analyse the composite endpoint of %FVC decline and mortality up to the end of the studies. FVC and %FVC will be analysed by subgroups (eg, baseline characteristics, background treatment and stratum).
Time-to-event data (including hospitalisations and mortality) will be presented as Kaplan-Meier estimates. In addition, a Cox proportional hazards model with terms for age, sex, height and stratum will be used to estimate and test hazard ratios for each dose compared with placebo. A mixed-effects model with treatment, time (as a categorical factor), time–treatment interaction and baseline total score as factors will be applied to the SGRQ total score, and descriptive statistics will be used for other PRO data.
Sensitivity analyses including multiple imputation methods and a per-protocol analysis will assess the impact of missing data, non-compliance and protocol violations on primary efficacy analyses. To account for multiple testing with respect to the primary endpoint within each study, due to two dose comparisons being compared with placebo, a Bonferroni approach will be applied to the alpha level with a focus on the higher dose. Using this approach, the GLPG1690 600 mg and 200 mg doses will be tested versus placebo at a 0.04 and 0.01 level, respectively. A sample size of 250 per group will have 80% power to show a significant effect, assuming GLPG1690 600 mg has an effect ≥80 mL. Further details regarding the statistical power of the studies are included in table 1.
Table 1.
Probability of statistical significance of zero, one or two treatment comparisons with placebo
| mL | 0 successful, % | 1 successful, % | 2 successful, % | Power, % |
| ∆600=80 ∆200=80 | 6.8 | 23.4 | 69.9 | 93.2 |
| ∆600=80 ∆200=60 | 10.1 | 46.9 | 43.0 | 89.9 |
| ∆600=80 ∆200=20 | 11.5 | 84.4 | 4.1 | 88.5 |
| ∆600=80 ∆200=0 | 11.2 | 88.0 | 0.7 | 88.8 |
| ∆600=90 ∆200=60 | 5.1 | 51.1 | 43.8 | 94.9 |
| ∆600=0 ∆200=0 | 95.2 | 4.5 | 0.3 | 4.8 |
Denoting ∆600 and ∆200, the true treatment differences of the two GLPG1690 treatment groups with placebo, and assuming a common standard deviation on the week 52 decline in forced vital capacity of 275 mL, the probability that zero, one or two of the treatment comparisons with placebo will be statistically significant and the power for each scenario are provided in the table.
A sample size of 250 patients in each treatment group will have at least 80% power to show a significant effect, assuming the GLPG1690 600 mg group has a treatment effect of at least 80 mL in the overall population of treatment-naïve patients and patients on standard of care.
Patient and public involvement
Patients or the public were not involved in the design of the studies, but may be involved in the reporting of the research.
Ethics and dissemination
All subjects will provide written informed consent (obtained by investigator/designated personnel) and a subject identification code list will ensure patient confidentiality. The IDMC will regularly assess potential safety risks and will also assess the effect of GLPG1690 on lung function, determined by rate of FVC decline, as part of a benefit–risk evaluation. Based on interim futility analysis results, the IDMC will make recommendations to the sponsor regarding study progress. A clinical endpoint adjudication committee comprising an independent group of experts will review and adjudicate clinical endpoints in a blinded manner. Details regarding data dissemination are provided in online supplementary file 1.
Discussion
ISABELA 1 and 2, two identically designed, phase III studies, will provide efficacy, safety and QoL data from subjects with IPF treated with GLPG1690 in addition to local SOC. Although antifibrotic agents are recommended for the treatment of IPF, approximately 40% of patients do not receive them.41 42 Furthermore, while both pirfenidone and nintedanib slow physiological disease progression, substantial declines in FVC still occur (–145 mL with pirfenidone in ASCEND43; –114.7 mL and –113.6 mL with nintedanib in INPULSIS-1 and INPULSIS-2,13 respectively), neither benefits QoL, and while survival is likely prolonged by treatment with these agents, IPF-related mortality remains high and the majority of patients still die due to respiratory failure.44 Therefore, there is a need for novel IPF therapies that further prevent worsening in lung function, benefit QoL and improve life expectancy. In addition, therapies must have an acceptable safety and tolerability profile. Data from early clinical studies of GLPG1690 are encouraging.28 29 In the phase IIa FLORA study, GLPG1690 was generally well tolerated and preliminary efficacy signals were observed,29 warranting further evaluation of the drug in a larger population, over a longer period.
ISABELA 1 and 2 will allow a wider range of subjects to be enrolled compared with previous phase III IPF clinical studies due to less restrictive eligibility criteria.12–14 For example, subjects in ISABELA 1 and 2 must have an FVC ≥45% predicted of normal, whereas predicted FVC was required to be ≥50% in the CAPACITY and INPULSIS studies, and 50%–90% in the ASCEND study.12–14 In addition, unlike in CAPACITY and ASCEND, which excluded subjects aged >80 years,12 14 there is no upper age limit in ISABELA 1 and 2. The broad eligibility criteria mean that only subjects with very severe IPF will be excluded, more closely reflecting how the agent may be used in the real world. Furthermore, in contrast to previous phase III IPF trials, ISABELA 1 and 2 will evaluate GLPG1690 when added to local SOC. Such data are important as experience of late-stage clinical studies to evaluate IPF treatments as an add-on to background therapy is limited. With multiple pathways thought to be involved in the pathogenesis of IPF, combination therapy is a logical treatment approach. In addition, given the benefits of pirfenidone and nintedanib over placebo, future clinical trials will likely assess new IPF treatments as add-on therapies to these agents, and consequently combination regimens may become SOC in clinical practice.45 Nintedanib plus sildenafil was recently assessed in patients with IPF with DLCO ≤35% of predicted in the INSTAGE trial. However, no significant difference in change from baseline in SGRQ total score versus nintedanib alone (the primary endpoint) was observed in this advanced patient population.46
ISABELA 1 and 2 will have a minimum treatment period of 52 weeks, allowing for long-term collection of blinded efficacy and safety data. Depending on recruitment duration, a proportion of subjects will remain on study for substantially longer than 52 weeks, providing an opportunity to assess less frequently occurring clinical and safety events that may not be captured during studies with a conventional 1-year duration. A potential limitation of the ISABELA studies is that outcomes beyond 52 weeks will not be captured for all subjects. Furthermore, FVC data at week 52, a key element of the primary outcome, will likely be missing for a proportion of subjects; this is a limitation of trials in subjects with progressive, fatal diseases, such as IPF. The impact of missing data in the ISABELA trials will be assessed using sensitivity analyses. As IPF is a rare disease, recruiting the planned number of subjects may be difficult. In addition, GLPG1690 is not being evaluated against a specific comparator agent, and treatment in the comparator arms could change during the studies, making comparisons with existing SOC more challenging.
The studies include a wide range of endpoints allowing for the collection of clinically relevant data related to safety, tolerability, efficacy, mortality, QoL, pharmacokinetics, pharmacodynamics and biomarkers. Given the potential challenge of enrolling subjects with a rare disease, it is advantageous to collect such an array of data from one study. Change in FVC over time, the studies’ primary endpoint, is an accepted surrogate for mortality, and a 10% decline in FVC is an indicator of disease progression, as reflected in certain key secondary endpoints.4 47 48 The inclusion of the EQ-5D, SGRQ, K-BILD questionnaire, LCQ, and VAS Cough and Urge to Cough endpoints allows for the assessment of treatment effects on PROs. While the key aim of treatment is to prevent disease progression, palliating symptoms and improving QoL are key aspects of IPF management.49 Conducting two identically designed studies in parallel enables outcomes to be confirmed between studies and allows for secondary data to be pooled, increasing the probability of detecting treatment effects.
Conclusions
GLPG1690, the first ATX inhibitor in clinical development for the treatment of IPF, will be evaluated in two parallel, identically designed, phase III studies (ISABELA 1 and 2) with a 52-week minimum treatment period. For the first time, GLPG1690 will be assessed as an addition to SOC, reflecting potential real-world use. The studies will provide a wide range of clinically relevant data, building on positive clinical findings to date.28 29 Positive results may ultimately mean that GLPG1690, alone or in combination, will become a much-needed new treatment option for patients with IPF.—
Acknowledgments
Medical writing support (including development of a draft outline and subsequent drafts in consultation with the authors, assembling tables and figures, collating author comments, copy editing, fact checking and referencing) was provided by Debbie Sherwood BSc, Aspire Scientific (Bollington, UK), and funded by Galapagos NV (Mechelen, Belgium).
Footnotes
Contributors: All authors were involved in the study design, reviewed and revised drafts of the manuscript, and approved the final draft.
Funding: This work is supported by Galapagos NV (Mechelen, Belgium). TMM is supported by an NIHR Clinician Scientist Fellowship (NIHR Ref: CS-2013-13-017) and a British Lung Foundation Chair in Respiratory Research (C17-3).
Competing interests: TMM has received personal fees from AstraZeneca, Bayer, Biogen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Galapagos, GlaxoSmithKline, Indalo Therapeutics, Pliant Therapeutics, ProMetic, Roche, Samumed and UCB, and stock options from Apellis outside the submitted work. His institution received grants or research fees from AstraZeneca, GlaxoSmithKline and UCB outside the submitted work. MK reports grants and personal fees from Galapagos during the conduct of the study, and grants and personal fees from Boehringer Ingelheim and Roche outside the submitted work. DJL reports personal fees from Galapagos during the conduct of the study, and personal fees from Philips Respironics, Roche, Sanofi Genzyme and Veracyte, grants and personal fees from Boehringer Ingelheim, FibroGen and Global Blood Therapeutics, unpaid Steering Committee membership for Galecto, and institutional fees for consultancy work from the Pulmonary Fibrosis Foundation outside the submitted work. KKB reports personal fees from Galapagos during the conduct of the study, and conversation under CDA with Genoa, grants from National Heart, Lung, and Blood Institute, and personal fees from Aeolus, AstraZeneca, aTyr, Biogen, Boehringer Ingelheim, Galapagos, Galecto Biotech, MedImmune, Novartis, Roche/Genentech, ProMetic, Patara and Third Pole outside the submitted work. WW reports no personal fees; the University Hospitals Leuven received consultancy fees from Galapagos during the conduct of the study and grants from Boehringer Ingelheim and Roche outside the submitted work. NV, SS, PF and WA-S are employees of, and have received warrants from, Galapagos. AF is an employee of Galapagos. MW reports no personal fees; the Erasmus MC received an advisory board fee from Galapagos, and speaker and advisory board fees from Boehringer Ingelheim and Hoffmann-La Roche outside the submitted work.
Patient consent for publication: Not required.
Ethics approval: The studies will be conducted in accordance with Good Clinical Practice guidelines, Declaration of Helsinki principles, and local ethical and legal requirements. Study protocols will be approved by the independent ethics committee/institutional review board for each site or country as applicable.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Sharif R. Overview of idiopathic pulmonary fibrosis (IPF) and evidence-based guidelines. Am J Manag Care 2017;23(11 Suppl):S176–82. [PubMed] [Google Scholar]
- 2. Martinez FJ, Collard HR, Pardo A, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers 2017;3:17074 10.1038/nrdp.2017.74 [DOI] [PubMed] [Google Scholar]
- 3. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol Mech Dis 2014;9:157–79. 10.1146/annurev-pathol-012513-104706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824. 10.1164/rccm.2009-040GL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li S, Zhao J, Shang D, et al. Ubiquitination and deubiquitination emerge as players in idiopathic pulmonary fibrosis pathogenesis and treatment. JCI Insight 2018;3:e120362 10.1172/jci.insight.120362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183:431–40. 10.1164/rccm.201006-0894CI [DOI] [PubMed] [Google Scholar]
- 7. Spagnolo P, Tzouvelekis A, Bonella F. The management of patients with idiopathic pulmonary fibrosis. Front Med 2018;5:148 10.3389/fmed.2018.00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chambers RC. Abnormal wound healing responses in pulmonary fibrosis: focus on coagulation signalling. Eur Respir Rev 2008;17:130–7. 10.1183/09059180.00010905 [DOI] [Google Scholar]
- 9. Scelfo C, Caminati A, Harari S. Recent advances in managing idiopathic pulmonary fibrosis. F1000Res 2017;6:2052 10.12688/f1000research.10720.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Raghu G. Pharmacotherapy for idiopathic pulmonary fibrosis: Current landscape and future potential. Eur Respir Rev 2017;26:pii: 170071 10.1183/16000617.0071-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med 2015;192:e3–19. 10.1164/rccm.201506-1063ST [DOI] [PubMed] [Google Scholar]
- 12. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011;377:1760–9. 10.1016/S0140-6736(11)60405-4 [DOI] [PubMed] [Google Scholar]
- 13. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2071–82. 10.1056/NEJMoa1402584 [DOI] [PubMed] [Google Scholar]
- 14. King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083–92. 10.1056/NEJMoa1402582 [DOI] [PubMed] [Google Scholar]
- 15. Richeldi L, Cottin V, du Bois RM, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS® trials. Respir Med 2016;113:74–9. 10.1016/j.rmed.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 16. Jo HE, Glaspole I, Grainge C, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian idiopathic pulmonary fibrosis registry. Eur Respir J 2017;49:1601592 10.1183/13993003.01592-2016 [DOI] [PubMed] [Google Scholar]
- 17. Fisher M, Nathan SD, Hill C, et al. Predicting life expectancy for pirfenidone in idiopathic pulmonary fibrosis. J Manag Care Spec Pharm 2017;23(3-b Suppl):S17–S24. 10.18553/jmcp.2017.23.3-b.s17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galli JA, Pandya A, Vega-Olivo M, et al. Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: tolerability and adverse drug reactions. Respirology 2017;22:1171–8. 10.1111/resp.13024 [DOI] [PubMed] [Google Scholar]
- 19. Kreuter M, Swigris J, Pittrow D, et al. Health related quality of life in patients with idiopathic pulmonary fibrosis in clinical practice: insights-IPF registry. Respir Res 2017;18:139 10.1186/s12931-017-0621-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Loveman E, Copley VR, Scott DA, et al. Comparing new treatments for idiopathic pulmonary fibrosis – a network meta-analysis. BMC Pulm Med 2015;15:37 10.1186/s12890-015-0034-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Manen MJG, Geelhoed JJM, Tak NC, et al. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Ther Adv Respir Dis 2017;11:157–69. 10.1177/1753465816686743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tager AM. Autotaxin emerges as a therapeutic target for idiopathic pulmonary fibrosis: limiting fibrosis by limiting lysophosphatidic acid synthesis. Am J Respir Cell Mol Biol 2012;47:563–5. 10.1165/rcmb.2012-0235ED [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Funke M, Zhao Z, Xu Y, et al. The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am J Respir Cell Mol Biol 2012;46:355–64. 10.1165/rcmb.2010-0155OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ninou I, Magkrioti C, Aidinis V. Autotaxin in pathophysiology and pulmonary fibrosis. Front Med 2018;5:180 10.3389/fmed.2018.00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oikonomou N, Mouratis M-A, Tzouvelekis A, et al. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol 2012;47:566–74. 10.1165/rcmb.2012-0004OC [DOI] [PubMed] [Google Scholar]
- 26. Tager AM, LaCamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med 2008;14:45–54. 10.1038/nm1685 [DOI] [PubMed] [Google Scholar]
- 27. Desroy N, Housseman C, Bock X, et al. Discovery of 2-[[2-Ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methylimidazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)thiazole-5-carbonitrile (GLPG1690), a first-in-class autotaxin inhibitor undergoing clinical evaluation for the treatment of idiopathic pulmonary fibrosis. J Med Chem 2017;60:3580–90. 10.1021/acs.jmedchem.7b00032 [DOI] [PubMed] [Google Scholar]
- 28. van der Aah EM, Fagard L, Desrivot J, et al. Favorable human safety, pharmacokinetics and pharmacodynamics of the autotaxin inhibitor GLPG1690, a potential new treatment in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016;193:A2701. [Google Scholar]
- 29. Maher TM, van der Aar EM, Van de Steen O, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2A randomised placebo-controlled trial. Lancet Respir Med 2018;6:627–35. 10.1016/S2213-2600(18)30181-4 [DOI] [PubMed] [Google Scholar]
- 30. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018;198:e44–68. 10.1164/rccm.201807-1255ST [DOI] [PubMed] [Google Scholar]
- 31. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society white paper. Lancet Respir Med 2018;6:138–53. 10.1016/S2213-2600(17)30433-2 [DOI] [PubMed] [Google Scholar]
- 32. Jones PW, Quirk FH, Baveystock CM, et al. A self-complete measure of health status for chronic airflow limitation. The St. George's Respiratory Questionnaire. Am Rev Respir Dis 1992;145:1321–7. 10.1164/ajrccm/145.6.1321 [DOI] [PubMed] [Google Scholar]
- 33. Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J 2005;26:319–38. 10.1183/09031936.05.00034805 [DOI] [PubMed] [Google Scholar]
- 34. Quanjer PH, Stanojevic S, Cole TJ, et al. Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J 2012;40:1324–43. 10.1183/09031936.00080312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rabin R, de Charro F. EQ-5D: a measure of health status from the EuroQol group. Ann Med 2001;33:337–43. 10.3109/07853890109002087 [DOI] [PubMed] [Google Scholar]
- 36. Patel AS, Siegert RJ, Brignall K, et al. The development and validation of the King's Brief Interstitial Lung Disease (K-BILD) health status questionnaire. Thorax 2012;67:804–10. 10.1136/thoraxjnl-2012-201581 [DOI] [PubMed] [Google Scholar]
- 37. Birring SS, Prudon B, Carr AJ, et al. Development of a symptom specific health status measure for patients with chronic cough: Leicester Cough Questionnaire (LCQ). Thorax 2003;58:339–43. 10.1136/thorax.58.4.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brown AW, Nathan SD. The value and application of the 6-minute-walk test in idiopathic pulmonary fibrosis. Ann Am Thorac Soc 2018;15:3–10. 10.1513/AnnalsATS.201703-244FR [DOI] [PubMed] [Google Scholar]
- 39. American Association for Respiratory Care AARC clinical practice guideline. Exercise testing for evaluation of hypoxemia and/or desaturation. American Association for respiratory care. Respir Care 1992;37:907–12. [PubMed] [Google Scholar]
- 40. ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002;166:111–7. 10.1164/ajrccm.166.1.at1102 [DOI] [PubMed] [Google Scholar]
- 41. Maher TM, Molina-Molina M, Russell A-M, et al. Unmet needs in the treatment of idiopathic pulmonary fibrosis—insights from patient chart review in five European countries. BMC Pulm Med 2017;17:124 10.1186/s12890-017-0468-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maher TM, Swigris JJ, Kreuter M, et al. Identifying barriers to idiopathic pulmonary fibrosis treatment: a survey of patient and physician views. Respiration 2018;96:514–24. 10.1159/000490667 [DOI] [PubMed] [Google Scholar]
- 43. Lederer DJ, Bradford WZ, Fagan EA, et al. Sensitivity analyses of the change in FVC in a phase 3 trial of pirfenidone for idiopathic pulmonary fibrosis. Chest 2015;148:196–201. 10.1378/chest.14-2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maher TM. Combination therapy and the start of a new epoch for idiopathic pulmonary fibrosis? Am J Respir Crit Care Med 2018;197:283–4. 10.1164/rccm.201709-1939ED [DOI] [PubMed] [Google Scholar]
- 45. Wuyts WA, Antoniou KM, Borensztajn K, et al. Combination therapy: the future of management for idiopathic pulmonary fibrosis? Lancet Respir Med 2014;2:933–42. 10.1016/S2213-2600(14)70232-2 [DOI] [PubMed] [Google Scholar]
- 46. Kolb M, Raghu G, Wells AU, et al. Nintedanib plus sildenafil in patients with idiopathic pulmonary fibrosis. N Engl J Med 2018;379:1722–31. 10.1056/NEJMoa1811737 [DOI] [PubMed] [Google Scholar]
- 47. Collard HR, King TE, Bartelson BB, et al. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003;168:538–42. 10.1164/rccm.200211-1311OC [DOI] [PubMed] [Google Scholar]
- 48. Flaherty KR, Mumford JA, Murray S, et al. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2003;168:543–8. 10.1164/rccm.200209-1112OC [DOI] [PubMed] [Google Scholar]
- 49. Spagnolo P, Tonelli R, Cocconcelli E, et al. Idiopathic pulmonary fibrosis: diagnostic pitfalls and therapeutic challenges. Multidiscip Respir Med 2012;7:42 10.1186/2049-6958-7-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjresp-2019-000422supp001.docx (48.9KB, docx)