

 The Cu(i)-binding protein NosL functions specifically as an assembly factor for the unique CuZ centre of nitrous oxide reductase (N2OR).
The Cu(i)-binding protein NosL functions specifically as an assembly factor for the unique CuZ centre of nitrous oxide reductase (N2OR).
Abstract
Nitrous oxide reductase (N2OR) is the terminal enzyme of the denitrification pathway of soil bacteria that reduces the greenhouse gas nitrous oxide (N2O) to dinitrogen. In addition to a binuclear CuA site that functions in electron transfer, the active site of N2OR features a unique tetranuclear copper cluster bridged by inorganic sulfide, termed CuZ. In copper-limited environments, N2OR fails to function, resulting in truncation of denitrification and rising levels of N2O released by cells to the atmosphere, presenting a major environmental challenge. Here we report studies of nosL from Paracoccus denitrificans, which is part of the nos gene cluster, and encodes a putative copper binding protein. A Paracoccus denitrificans ΔnosL mutant strain had no denitrification phenotype under copper-sufficient conditions but failed to reduce N2O under copper-limited conditions. N2OR isolated from ΔnosL cells was found to be deficient in copper and to exhibit attenuated activity. UV-visible absorbance spectroscopy revealed that bands due to the CuA center were unaffected, while those corresponding to the CuZ center were significantly reduced in intensity. In vitro studies of a soluble form of NosL without its predicted membrane anchor showed that it binds one Cu(i) ion per protein with attomolar affinity, but does not bind Cu(ii). Together, the data demonstrate that NosL is a copper-binding protein specifically required for assembly of the CuZ center of N2OR, and thus represents the first characterised assembly factor for the CuZ active site of this key environmental enzyme, which is globally responsible for the destruction of a potent greenhouse gas.
Introduction
Nitrous oxide (N2O) is a significant greenhouse gas with a ∼300 fold greater global warming potential than CO2 and an ability to deplete stratospheric ozone.1 Agriculture produces 65% (6.8 Tg N–N2O per year) of the total N2O emitted each year. The main contributor within this sector is the soil microbial community, which produces 40% of these emissions.2 The surge in atmospheric N2O from 270 ppb to 324 ppb over the last 100 years correlates strongly with the use of anthropogenic nitrogen-based fertilisers in farming to improve crop yield.3 The doubling of available nitrogen in the environment has enriched a class of soil dwelling microorganisms called denitrifiers, which respire anaerobically by reducing nitrate to dinitrogen (N2) gas via the free intermediates nitrite, nitric oxide and N2O using different metallo-enzymes. Environmental factors such as soil pH, moisture, carbon to nitrogen ratio, temperature and a lack of copper4–6 have all been identified as factors leading to increased N2O emissions from these microbes.
Encoded by the nosZ gene, the cupro-enzyme nitrous oxide reductase (N2OR or NosZ) catalyses the 2-electron reduction of N2O to N2. Two distinct and approximately equally abundant clades of N2OR-containing bacteria and archaea have been identified.7 Importantly, clade II members act as an N2O sink, while members of clade I, such as α-, β- and γ-proteobacteria, are able to produce and remove N2O under optimum conditions. In order to assist with future strategies for the control of emissions from soil ecosystems, a key task is to explore how the enzyme is produced and matured in N2O emitting bacteria.3
N2OR in the denitrifying α-proteobacterium Paracoccus denitrificans (PdN2OR) is exported to the periplasm via the twin-arginine translocation (TAT) pathway,8,9 where its two copper centers, CuA and CuZ, are assembled. Several crystal structures are now available for N2OR,10–13 revealing that the CuA site is housed in a C-terminal cupredoxin domain, while the CuZ site lies within the N-terminal seven bladed β-propeller domain. The CuA center contains two copper ions that are bridged by two conserved Cys residues and further coordinated by Met, His and Trp ligands to form a site that closely resembles the electron transfer CuA site present in subunit II of cytochrome c oxidase.14 In N2OR the CuA site also acts as an electron shuttle, accepting electrons from small electron donors such as cytochrome c550 (ref. 15) or pseudoazurin, in P. denitrificans,16 for the reduction of N2O at the CuZ site. This center comprises four copper atoms coordinated by seven conserved His residues and bridged by one ([4Cu:S]) or two ([4Cu:2S]) sulfides, depending on the presence or absence of O2, respectively, during purification.10,12
A major challenge that is particularly important for addressing N2O emissions from soil is to understand how the cofactor sites of N2OR are assembled and, in doing so, identify assembly/chaperone systems that are involved. For clade I organisms, including P. denitrificans, any such systems must be periplasmic, as mutations in the TAT leader sequence of N2OR results in the protein remaining in the cytoplasm, in a folded but copper-free state.17 Studies of the biogenesis of copper cofactor sites of eukaryotic cytochrome c oxidase have identified the proteins Cox17 and ScoB, which are involved in the assembly of the CuA site,18 and prokaryotic homologues PCuAC and SenC have been implicated in the maturation of the CuA site of the aa3-type cytochrome c oxidase from Rhodobacter sphaeroides.19
In comparison, little is known about the assembly of the CuZ site. The nos gene cluster of clade I N2O reducing bacteria vary between denitrifying phyla but contain, in addition to nosZ, five other core genes (nosRZDFYL), while predominantly α- and β-proteobacteria members also contain a further two genes (nosC and nosX). nosDFY are predicted to encode an ABC-type transporter homologous to ATM1 from eukaryotes, which transports a sulfur-containing species out of the mitochondrion for Fe–S cluster assembly in the cytoplasm,20 suggesting that these proteins are likely involved in supplying sulfide for assembly of the CuZ center. In P. denitrificans, downstream of nosDFY are two further accessory genes that are part of the same operon, nosL and nosX. The only mutation analysis of nosL has been in P. stutzeri9 and of nosX in P. denitrificans,21 both of which led to the conclusion they alone are not important for whole-cell N2O reduction. However, nosL is part of the denitrification core gene cluster, its expression is responsive to cellular copper status,22 and the NosL protein has been shown to bind copper,23 suggesting that it plays a role in maturation or activation of N2OR.
Here, we report genetic and biochemical studies of P. denitrificans nosL/NosL. The data demonstrate that NosL is a Cu(i)-binding protein that is required for efficient assembly of the N2OR CuZ center, and thus represents the first characterised assembly factor for this unique metal center in biology.
Results
NosL is required for N2OR activity under copper-limited conditions
Wild type PD1222 (WT), ΔnosZ (lacking the gene encoding N2OR) and ΔnosL strains (Table S1†) were cultured under both Cu-sufficient and Cu-limited anaerobic conditions and their growth characteristics investigated. For the WT strain, growth yield was affected under Cu-limited conditions (Fig. 1A). For the ΔnosZ strain, growth rate and yield were affected under both conditions, consistent with the importance of N2OR for optimal growth under anaerobic nitrate-sufficient conditions (Fig. 1B). Growth of the ΔnosL strain was similar to WT under Cu-sufficient conditions, but was attenuated under Cu-limited conditions, exhibiting a maximum OD600 nm similar to the ΔnosZ strain (Fig. 1C).
Fig. 1. Growth and N2O emission profiles for P. denitrificans WT and mutant strain cultures. (A)–(C) OD600 nm as a function of time for WT PD1222 (A), ΔnosZ deletion mutant PD2303 (B), and ΔnosL deletion mutant PD2501 (C). (D)–(F) N2O emissions as N·N2O (millimolar N in the form of N2O) for PD1222 (D), PD2303 (E), and PD2501 (F). Strains were grown in anaerobic batch culture conditions in Cu-sufficient media (red diamonds) and Cu-limited media (blue squares). Bars represent SE.

The impact of nosL deletion on N2OR activity in vivo was assessed by measuring N2O levels in the headspace of cultures. Consistent with previous studies, no N2O was accumulated by the WT strain under Cu-sufficient conditions and only a transient low level of N2O (<1 mM at ∼16 h) was observed under Cu-limited conditions (Fig. 1D), where the latter is likely due to lower transcription of nosZ under these conditions.22 In contrast, the ΔnosZ mutant exhibited a marked Nos-negative phenotype (Nos–) regardless of copper levels, with N2O emitted from cultures to >4 mM after 24 h (Fig. 1E). Like the WT, essentially no N2O was emitted from ΔnosL cultures under Cu-sufficient conditions. Importantly, however, after 24 h of growth and once the cells had reached stationary phase, up to 4 mM N2O was detected in the headspace of ΔnosL cultures under Cu-limited conditions, similar to the ΔnosZ strain (Fig. 1F).
The Nos– phenotype of the ΔnosL strain under Cu-limited conditions was almost fully complemented by expression of a plasmid-borne functional nosL gene copy from a taurine inducible promoter (Fig. S1†). In particular, the extent of N2O release from the complemented ΔnosL mutant closely resembled the WT strain in the transient accumulation of N2O at ∼16–20 h.
Absence of NosL results in a copper- and catalytically-deficient N2OR
To investigate the functional properties of NosL in relation to N2OR biogenesis, N2OR (NosZ) with a C-terminal Strep-II tag was overproduced in P. denitrificans strains grown under Cu-sufficient conditions and purified. N2OR from ΔnosL cells contained on average 4 Cu atoms per monomer, compared to ∼6 for the enzyme from ΔnosZ cells (Table 1). N2OR from a ΔnosZL double mutant contained ∼3.5 copper atoms per monomer, demonstrating that the presence of the chromosomal nosZ gene in the ΔnosL cells had little effect on the copper content of the tagged N2OR (NosZ) (Table 1).
Table 1. Copper content and activities of N2OR enzymes from different P. denitrificans strains.
| Strain | Cu-sufficient |
Cu-limited | |
| Cu/monomer a | Maximum specific activity b (μmol N2O min–1 mg–1) | Cu/monomer | |
| ΔnosL/pMSL002 (StrepII tagged-N2OR) | 4.1 ± 0.43 | 95.7 ± 3.9 | N.D. |
| ΔnosZ/pMSL002 | 5.9 ± 0.56 | 196.2 ± 9 | 4.8 ± 0.42 |
| ΔnosZL/pMSL002 | 3.4 ± 0.26 | 93.6 ± 0.8 | N.D. |
aTotal copper contents per monomer were determined using a BCS copper assay.
bN2O reductase activity was determined using a reduced methyl viologen assay (μmol N2O min–1 mg–1 enzyme). Proteins were pre-incubated with a 500-fold excess reduced methyl viologen for 150 min prior to activity assay. All reactions were carried out in triplicate and SD is shown. N.D., not detectable.
As purification of N2OR was carried out aerobically, the enzyme from ΔnosZ cells contained the 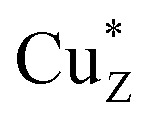 (pink, form II) form of the active site, which is catalytically inactive.24 To activate the isolated N2OR to the fully reduced form, the enzyme was incubated with excess reduced methyl viologen at room temperature25,26 for 150 min, at which point N2O reductase activity had reached a maximum. N2OR from ΔnosL and ΔnosZL cells exhibited significantly lower maximum activities than N2OR from ΔnosZ, see Table 1.
(pink, form II) form of the active site, which is catalytically inactive.24 To activate the isolated N2OR to the fully reduced form, the enzyme was incubated with excess reduced methyl viologen at room temperature25,26 for 150 min, at which point N2O reductase activity had reached a maximum. N2OR from ΔnosL and ΔnosZL cells exhibited significantly lower maximum activities than N2OR from ΔnosZ, see Table 1.
Absence of NosL results in N2OR deficient in the CuZ center
The UV-visible absorbance spectrum of as isolated air-oxidized (pink, form II) strep-tagged N2OR purified from ΔnosZ P. denitrificans displayed bands at 480, 535 and 645 nm (Fig. 2A and S2†), in agreement with literature for N2OR enzymes from a range of bacteria.11,26–29 These features arise from S2– to Cu(ii) charge-transfer transitions and transitions due to interactions between Cu(i) and Cu(ii) ions.27 The bands at 480 and 550 nm correspond to the CuA centre, while that at 645 nm is characteristic of the CuZ centre (in its 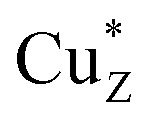 form).29
form).29
Fig. 2. UV-Visible absorbance spectra of N2OR purified from Cu-sufficient P. denitrificans mutant strains. (A) N2OR isolated from ΔnosZ PD2303 (blue line), ΔnosL PD2501 (black line) and ΔnosZL PD2505 (red line) following oxidation using ferricyanide. (B) As in (A) but following reduction using sodium dithionite. (C) Oxidized minus reduced difference spectrum. Oxidized spectra were normalised to ε580 nm 5000 M–1 cm–1 per monomer as described by Rasmussen et al.27 Sample buffer contained 20 mM HEPES, 150 mM NaCl, pH 7.2.

The spectra of N2OR enzymes purified from ΔnosL and ΔnosZL strains were very similar in the 450–550 nm region, but exhibited significantly reduced intensity beyond 550 nm (Fig. 2A). The apparent absorbance maximum was shifted to ∼635 nm, consistent with the enhanced relative influence of the underlying absorbance due to CuA (maximum at 535 nm). Reduction with sodium dithionite resulted in the spectra shown in Fig. 2B. Bands at 480 and 540 nm were lost, consistent with the reduction of the CuA center to its colorless diamagnetic Cu(i)/Cu(i) state. The remaining band is characteristic of the CuZ center following addition of dithionite.27 N2OR from both ΔnosL and ΔnosZL strains exhibited a much less intense CuZ absorbance than that from ΔnosZ, indicating diverse occupancies of the center. Ferricyanide-oxidized minus dithionite-reduced difference spectra (Fig. 2C) closely overlay, particularly in the 450–550 nm region, demonstrating that the N2OR CuA centers of the different enzymes are essentially identical, and close to fully populated, as estimated by the measured extinction coefficients.27 Together, these data demonstrate that N2OR isolated from a ΔnosL background is specifically deficient in its CuZ center.
Absence of NosL under copper-limited growth conditions results in complete absence of CuA and CuZ centers in N2OR
The copper determinations, activity assays and spectroscopic characterizations of N2OR described above were performed with samples isolated from cultures grown in copper-sufficient media, in which only a minor growth phenotype for the ΔnosL strain was observed (Fig. 1). Thus, N2OR characteristics under low copper, where N2O is generated from cultures, were investigated. N2OR from ΔnosZ cells contained ∼5 Cu per monomer (Table 1) with an absorbance spectrum indicative of complete, or near complete, CuA center population, but a less than stoichiometric population of CuZ (Fig. 3). In contrast, N2OR from ΔnosL and ΔnosZL cells contained no detectable copper (Table 1), and gave UV-visible absorbance spectra with no absorbance in the visible region (Fig. 3), indicating complete failure to assemble either of the copper centers of N2OR in the absence of NosL.
Fig. 3. UV-Visible absorbance spectra of N2OR purified from Cu-limited P. denitrificans mutant strains. (A) N2OR isolated from ΔnosZ PD2303 (blue line), ΔnosL PD2501 (black line) and ΔnosZL PD2505 (red line) following oxidation using ferricyanide. (B) As in (A) but following reduction with sodium dithionite. (C). Oxidised minus reduced difference spectra. Spectra were normalised to the protein concentration. Sample buffer contained 20 mM HEPES, 150 mM NaCl, pH 7.2.

NosL binds Cu(i) with attomolar affinity
NosL contains a Type-II signal peptidase recognition sequence that, when cleaved, produces a protein with an N-terminal Cys residue that is predicted to bind lipid and anchor NosL into the outer membrane.23,30 The NMR solution structure of NosL, lacking its membrane anchor sequence, from the β-proteobacterium Achromobacter cycloclastes revealed two independent homologous domains with an unusual ββαβ topology.31 The same authors showed that the protein binds Cu(i) specifically and XAFS data were consistent with a Cu(i) coordination consisting of S and N/O ligands.23 To determine the biochemical/biophysical properties of P. denitrificans NosL, the protein lacking its periplasmic export signal sequence and its predicted N-terminal Cys residue23 was purified resulting in a metal-free form of the protein, which gave a mass of 18 890 Da (predicted mass 18 891 Da) by LC-ESI-MS (Fig. S3†). The final, gel filtration step of purification resulted in a broad elution band that suggested a mixture of monomer/dimer association states for NosL, a result confirmed by native PAGE (Fig. S4†), which showed two species of NosL. Similar observations were made for apo-NosL from A. cycloclastes.23
Titration of apo-NosL with Cu(i) resulted in the series of spectra shown in Fig. 4A, in which broad absorbance in the near-UV region of the spectrum was observed to gradually increase and saturate at a level of 1 Cu(i) per NosL (Fig. 4B). The absorbance is characteristic of charge transfer transitions involving Cu(i) coordinated to a cysteine thiolate.32 Cu(i)-binding was also investigated by CD spectroscopy, which confirmed the tight association of one Cu(i) per protein but also suggested that further Cu(i) can associate with NosL, albeit weakly (Fig. S5†). Gel filtration and native PAGE analysis of Cu(i)-NosL (Fig. S4†) also demonstrated that Cu(i) binding does not significantly affect the association state equilibrium.
Fig. 4. Copper-binding to NosL. (A) UV-visible absorbance titration of NosL (21 μM) with Cu(i). (B) Plots of Δε260 nm (black circles) and Δε265 nm (red circles) as a function of Cu(i) ions per protein molecule. NosL was in 100 mM MOPS, 100 mM NaCl, pH 7.5.

Equivalent titrations with Cu(ii) followed by absorbance spectroscopy resulted in spectra very similar to those for Cu(i) (Fig. S6†), suggesting that Cu(ii) may undergo auto-reduction upon binding to NosL. This possibility was investigated using EPR (Fig. S7 and Table S2†). Addition of Cu(ii) to NosL resulted in a characteristic S = 1/2 Cu(ii) signal that, by comparison with a Cu(ii) standard, corresponded to only ∼8% of the Cu(ii) initially added. The same sample in the presence of EDTA (a Cu(ii) chelator), which prevents Cu(ii) ions in solution dimerizing to form EPR-inactive species, resulted in a Cu(ii) concentration of ∼11%. Similar experiments but with the addition of Cu(i) in place of Cu(ii) resulted in 4% and 8% in the absence and presence of EDTA, respectively (Table S2†). As isolated NosL contained <1% Cu(ii). Together, these data indicate that Cu(ii) undergoes auto-reduction to EPR silent Cu(i) upon binding to NosL.
To further characterise Cu(i)-binding to NosL, ESI-MS under non-denaturing conditions, where non-covalent interactions are preserved, was also employed. Fig. 5 shows the deconvoluted spectrum of apo-NosL with the major peak at 18 890 Da (as observed by LC-ESI-MS, Fig. S3†), along with a number of lower intensity peaks to the higher mass side, due to non-covalent sodium and ammonium adducts. Attempts to remove these adducts from the non-denaturing MS, via buffer exchange, changes in pH and ionic strength, were unsuccessful. NosL was loaded with a single Cu(i) ion and the peak envelope of the resulting deconvoluted mass spectrum was at +63 Da relative to that of the apo-NosL spectrum (Fig. 5), consistent with the binding of a single Cu(i) ion.
Fig. 5. Deconvoluted ESI-MS of NosL (20 μM) in the apo-form and containing 1 Cu(i) per protein, as indicated, in 50 mM ammonium acetate, pH 7.8.

Competition binding experiments using the high-affinity chelator BCS were used to determine the dissociation constant for Cu(i)-binding to NosL. Cu(i)-NosL was titrated with BCS, and the partition of Cu(i) between NosL and BCS determined from measured A483 nm, due to [Cu(BCS)2]3–, together with the well-characterised formation constant for Cu(BCS2)3–, β2 = 1019.8 (Fig. S8†)33. From these, an average Kd value of ∼4 × 10–18 M was determined34 (Table S3†), demonstrating very tight binding of Cu(i) to NosL.
Discussion
Despite nosL being a core component of the nos gene clusters in a range of microorganisms, mutational studies9 have so far failed to reveal a function in N2O reduction. A nosL mutant of Pseudomonas stutzeri (containing an insertion towards the 3′ end of the gene) exhibited a slightly lower growth rate but produced active, holo-N2OR.9 The presence of a CXXC motif in the encoded protein prompted the suggestion that NosL might be a protein disulfide isomerase, but analysis of the sequence of NosL from Sinorhizobium meliloti35 showed that one of the Cys residues of the P. stutzeri protein is not conserved.
Here, a nosL deletion mutant of P. denitrificans was generated and compared to the WT and nosZ deletion strains, as growth and N2O benchmarks, under both Cu-limited and Cu-sufficient conditions. The nosL mutant exhibited a Cu-limited growth phenotype relative to WT, associated with a deficiency in the activity of N2OR, leading to accumulation of N2O in the culture headspace. The phenotype was complemented in trans by nosL and also under copper replete conditions, strongly suggesting that NosL functions in an aspect of copper metabolism. The reason why the previously reported P. stutzeri nosL mutant did not present a phenotype is unclear; one possibility is that there was sufficient copper in the growth medium to mask the phenotype, but growth conditions were not reported.9
Strep-tagged N2OR enzymes purified from ΔnosZ, ΔnosL and ΔnosZL mutants exhibited clear differences. Under Cu-sufficient conditions, N2OR from ΔnosL cells contained only ∼4 Cu per N2OR monomer and was substantially less active (though it was apparently sufficiently active in vivo to mask any obvious in vivo phenotype). Importantly, spectroscopic characterisation revealed that the N2OR protein from ΔnosL was specifically deficient in its CuZ center, demonstrating that NosL functions in the assembly of this unique biological metal center required for N2O destruction.
Under Cu-limited conditions, tagged N2OR purified from the ΔnosZ mutant contained nearly 5 Cu/monomer, while those from ΔnosL or ΔnosZL mutant strains lacked copper entirely, indicating complete failure to assemble either N2OR copper center in the absence of NosL. Thus, although the CuA can be reconstituted using copper alone,36 when copper is limited, NosL may also supply Cu for incorporation into CuA.
The N-terminal sequence of NosL contains a periplasmic export signal and a lipobox that is predicted to be processed by Lgt, Lsp and Lnt enzymes, resulting in a mature protein that is membrane-anchored via a triacylated N-terminal Cys residue (indicated by a yellow arrowhead in Fig. S9†).37 This is likely a substrate of the Lol system,37 such that it is located in the outer membrane, with its soluble domains facing the periplasm. Studies of a soluble form of NosL, lacking its periplasmic targeting sequence and the N-terminal Cys residue that is acylated, revealed that it binds Cu(i) with attomolar affinity, a characteristic of many copper chaperones.
Our data are consistent with Cys coordination, in agreement with previous studies of A. cycloclastes NosL using thiol-specific reagents and EXAFS, with data from the latter consistent with a three or four coordinate Cu(i) center. The best fit was obtained for three coordinate Cu(i) with (O/N)S2 ligands.23 Alignment of NosL sequences from the two clades of N2O reducing bacteria identifies two conserved Cys residues: one through which the protein is believed to be anchored to the membrane (see, Fig. S9,† yellow arrowhead); and, one within a CXM motif that is likely to participate in Cu(i)-binding.23 The Met residue of this motif is also strictly conserved and was proposed to provide the second sulfur ligand identified by EXAFS.23 There is one other absolutely conserved Met residue but, as acknowledged by McGuirl et al., there is no absolutely conserved His residue (Fig. S9†).
Separation of NosL proteins according to the clade to which the organism belongs may shed some further light on this, as it reveals residues that are conserved within, but not between, clades. Clade I NosL proteins, which include P. denitrificans and A. cycloclastes, contain a well-conserved His residue close to the CXM motif, resulting in a CXMX3H motif. Clade II NosL proteins do not contain this His but instead contain a conserved Cys residue at the N-terminal side of the CXM motif (resulting in a CXXCXM motif). P. stutzeri NosL is the exception to this, as it comes from a Clade I organism but is more similar to Clade II NosL proteins.
In summary, we identify here the first Cu chaperone with a specific role in CuZ center assembly. Our data indicate that NosL, which binds Cu(i) with attomolar affinity, is a key part of a high-affinity Cu trafficking pathway that enables assembly of the CuZ center of N2OR. The pathway functions under Cu-sufficient conditions, but becomes essential under Cu-limited conditions. During copper limitation, the NosL pathway may also play a key role in supplying Cu for the CuA center. How NosL delivers Cu to N2OR is unknown; it is likely that NosL functions directly in the transfer of Cu(i) to N2OR, and is thus a copper chaperone, but further studies are now needed to investigate this. The identification of NosL as a key component of a Cu-trafficking pathway for the assembly of the active holoreductase N2OR, the sole pivotal enzyme for N2O destruction, is a significant advance towards the long-term aim of mitigating microbial emissions of N2O into the atmosphere.
Materials and methods
Construction of mutant nosL and nosZL deficient strains of P. denitrificans
Unmarked deletions of nosL in P. denitrificans wild type or ΔnosZ backgrounds (Table S1†) were produced by the method of allelic replacement.22 Briefly, regions flanking nosL (Table S4†) were cloned into pK18mobsacB using EcoRI and PstI sites to generate a suicide plasmid (pSPBN1, Table S1†) that was subsequently conjugated into P. denitrificans PD1222 or ΔnosZ PD2303 using the helper E. coli pRK2013 strain. Single cross-over recombination events were screened using spectinomycin (25 μg ml–1) and kanamycin (50 μg ml–1). Primary SpecR/KmR transconjugants were grown to stationary phase in Luria Bertani broth (LB) with no antibiotic. Double cross over events were selected for using a high-salt modified LB agar supplemented with 6% (w/v) sucrose. Sucrose resistant colonies with SpecR were screened using colony PCR and gene deletion confirmed by sequencing. Deletion strains were named PD2501 (ΔnosL) and PD2505 (ΔnosZL). For complementation of ΔnosL cells the coding sequence of Pden_4215 was synthesised (Genscript) with flanking 5′ NdeI and 3′ EcoRI restriction sites and sub-cloned into a taurine-inducible modified pLMB509 plasmid with gentamicin resistance (20 μg ml–1) to generate pSPBN2. The complementation plasmid was conjugated into the mutant strain using the helper E. coli pRK2013 strain and successful conjugants were SpecR/GmR. Expression of nosL from the plasmid was induced by adding 1 mM taurine to the medium at the start of the growth experiment.
Growth and phenotype analysis of cultures
Anaerobic minimal media batch cultures (400 ml) were grown in sealed Duran flasks (500 ml total volume), fitted with a septum to allow for gas-tight sample extraction. Minimal media consisted of: 30 mM succinate, 20 mM nitrate, 11 mM dihydrogen orthophosphate, 29 mM di-sodium orthophosphate, 0.4 mM magnesium sulfate, 1 mM ammonium chloride, pH 7.5. The minimal media was supplemented with a 2 ml l–1 Vishniac and Santer trace element solution38 where copper sulfate was present (Cu-sufficient, 12.8 μM) or excluded from the original recipe (Cu-limited, <0.5 μM), as previously described.22 Media were inoculated using a 1% inoculum from a starter culture to give a starting OD600 nm of 0.02 and incubated at 30 °C. Samples of the liquid culture were taken in 1 ml aliquots and OD600 nm measured. 3 ml gas samples were removed from the headspace of the cultures and stored in pre-evacuated 3 ml Exetainer® vials. A 50 μl gas sample was injected into a Clarus 500 gas chromatograph (PerkinElmer) with an Elite-PLOT Q (30 m × 0.53 mm internal diameter) and an electron capture detector. Carrier gas was N2, make-up gas was 95% (v/v) argon, 5% (v/v) methane. Standards containing N2O at 0.4, 5, 100, 1000, 5000, and 10 000 ppm (Scientific and Technical Gases) were measured and total N2O was determined as previously described.22
Purification and characterisation of affinity-tagged N2OR from P. denitrificans strains
N2OR (NosZ) was expressed in trans in P. denitrificans using pLMB511, a derivative plasmid of the taurine-inducible expression vector pLMB509 for α-proteobacteria (Table S1†).39 The EcoRI site at position 1107 bps in pLMB509 was removed by PCR-based site-directed mutagenesis to generate pMSL001 (see Table S4† for primers). Subsequently, pMSL001 was modified by cloning of an NdeI–EcoRI fragment (Table S1† for sequence) to yield pLMB511, which has a unique NdeI–BamHI–XmaI–EcoRI multiple cloning site that also contains the Strep-II tag sequence. As high-GC content precluded PCR gene amplification, the coding sequence of Pden_4219 (nosZ) was synthesised (GenScript) and cloned into pLMB511 as a NdeI–XmaI fragment, yielding pMSL002 from which N2OR (NosZ) with a C-terminal Strep-tag II was overproduced. The pMSL002 plasmid with GenR (20 μl ml–1) was conjugated into PdΔnosZ (PD2303), PdΔnosL (PD2501) and PdΔnosZL (PD2505) using the E. coli pRK2013 helper strain. Conjugants were screened for both GenR/SpecR and first cultured in LB and subsequently in 4 L minimal media supplemented with 2 ml l–1 trace element solution, at 30 °C. Overproduction of strep-tagged N2OR was initiated by the addition of 10 mM taurine when the culture reached OD600 nm ∼ 0.6 and cultures were incubated at 30 °C for 24 h. Cells were harvested by centrifugation at 5000 × g and resuspended in binding buffer (20 mM HEPES, 150 mM NaCl, pH 7.2) with a protease inhibitor (cOmplete™ from Roche, 1 tablet per 50 ml resuspended cells) and lysed using a French pressure cell at 1000 psi. The cell lysate was centrifuged at 205 000 × g for 1 h at 4 °C and the supernatant applied to a Hi-Trap HP Strep II affinity column (5 ml, GE Healthcare). N2OR-Strep-tag II was eluted using elution buffer (20 mM HEPES, 150 mM NaCl and 2.5 mM desthiobiotin, pH 7.2) and exchanged back into binding buffer using a 30 kDa MWCO Centricon filter unit. Purity of the sample was confirmed using SDS-PAGE analysis and LC-MS. Protein concentrations were determined using the Bradford assay (BioRad)40 and bovine serum albumin as a protein standard.
UV-visible absorbance spectra of N2OR-Strep-tag II were recorded on a Jasco V-550 spectrophotometer. Circular dichroism spectra were recorded using Jasco J-810 Spectropolarimeter. Samples were made anaerobic by sparging with nitrogen gas for 5 min and oxidised or reduced with 5 mg ml–1 stocks of potassium ferricyanide and sodium dithionite, respectively, in 20 mM HEPES, 150 mM NaCl, pH 7.2, by titrating concentration equivalents. Total copper content of the protein was determined using a colorimetric bathocuproinedisulfonic acid (BCS) assay. A 100 μl protein sample was heated to 95 °C for 1 h with an equal volume of 20% (v/v) HNO3. The reaction was cooled and neutralised using 0.6 ml saturated ammonium sulfate solution. Copper was reduced using 100 μl hydroxylamine (100 mM) and 100 μL BCS (10 mM) added. The absorbance at 483 nm was recorded after 30 min. A standard curve was produced using a standard copper sulfate solutions (Sigma).
Activities of N2OR-Strep-tag II enzymes was determined using an adapted methyl viologen assay25,41 in which samples were incubated with a 500-fold excess of reduced methyl viologen. Reaction was initiated by adding N2O saturated buffer and the oxidation of blue (reduced) methyl viologen to its oxidised colourless form was followed at 600 nm as a function of time and data converted to specific activity using ε600 nm = 13 600 M–1 cm–1 for the reduced methyl viologen cation radical.41
Purification and characterisation of NosL
A codon-optimised gene encoding an N-terminally truncated version of PdNosL (Pden_4215) was synthesised (Genscript) and sub-cloned into pET-21a(+) using 5′ NdeI and 3′ EcoRI restriction sites to generate pSPBN3. The truncation, which resulted in the replacement of the first 16 residues with a Met, was designed to simplify the expression and maturation of the protein in E. coli as it yielded a soluble protein located in the cytoplasmic fraction. The pSPBN3 plasmid was used to transform E. coli BL21 (DE3) to ampicillin (100 μg ml–1) resistance. Typically, 2 L flasks containing 500 ml LB supplemented with 100 μg ml–1 ampicillin were inoculated with 1% (v/v) of an overnight culture and grown for 2 h, 180 rpm, 37 °C until OD600 nm reached ∼0.6. Expression of the NosL-encoding gene was induced by addition of 500 μM IPTG and cultures were subsequently incubated at 37 °C, 180 rpm for 5 h. Cells were harvested by centrifugation at 4000 × g for 15 min at 4 °C, resuspended with buffer A (50 mM MES, pH 6.5) and lysed by three rounds of sonication, each for 8 min 20 s (0.2 s intervals, 50% power), on ice. The cell lysate was centrifuged at 40 000 × g for 45 min at 4 °C and the supernatant applied to a DEAE column (HiPrep DEAE FF 16/10; GE Healthcare) equilibrated in buffer A. NosL was eluted using a 0–50% gradient of buffer B (50 mM MES, 1 M NaCl, pH 6.5). Fractions containing NosL were buffer exchanged using a 10 kDa MWCO Centricon into buffer A and applied to a Q-sepharose column (HiPrep Q FF 16/10; GE Healthcare) and eluted using a 20–50% gradient of buffer B. NosL-containing fractions (as determined by SDS-PAGE) were pooled and subsequently applied to an S-100 gel filtration column (120 ml, GE Healthcare) equilibrated in 100 mM MOPS, 100 mM NaCl, pH 7.5 (buffer C) and eluted in the same buffer. Fractions containing pure NosL were combined and dialysed overnight against buffer C containing 1 mM EDTA at 4 °C, and subsequently back into buffer C.
An extinction coefficient at 280 nm of 11 923 ± 5.2 M–1 cm–1, determined using a guanidine hydrochloride assay,42 was used to quantify the NosL protein concentration. Copper analysis revealed that the protein was purified in a copper-free form. Copper titrations were carried out using a 1 M stock solution of Cu(i)Cl (in 1 M NaCl and 0.1 M HCl) or CuCl2 dissolved in water. The protein was titrated with copper to give increases in the ratio of Cu : NosL of 0.1 per addition and spectra were recorded between 240 and 600 nm after each addition. Competition assays between Cu(i)-NosL and BCS were carried out to measure the dissociation constant, Kd, for Cu(i)-binding to NosL, using the extinction coefficient ε483nm = 13 300 M–1 cm–1 to determine the concentration of [CuBCS2]3–, as previously described,33 with 10 min incubation after each addition. Absorbance spectra were recorded using a Jasco V550 spectrophotometer.
LC-MS was conducted using a Bruker microQTof-QIII electrospray ionisation time of flight (TOF) mass spectrometer calibrated in the m/z range 300–2000 using ESI-L Low Concentration Tuning Mix (Agilent Technologies). Samples were prepared by ten-fold dilution of 50 μM protein solution with 2% (v/v) acetonitrile and 0.1% (v/v) formic acid to 0.5 ml. Samples were loaded into the LC-MS via an autosampler using an UltiMate 3000 HPLC system (Dionex). A 20 μl injection volume of the protein was applied to a ProSwift reversed phase RP-1S column (4.6 × 50 mm; Dionex) at 25 °C. A gradient elution was performed at a flow rate of 200 ml min–1 using solvents A (0.1% formic acid) and B (acetonitrile, 0.1% formic acid). Once loaded the following chromatographic method was used: isocratic wash (2% B, 0–2 min), linear gradient from 2–100% B (2–12 min), followed by an isocratic wash (100% B, 12–14 min) and column re-equilibration (2% B, 14–15 min). Mass spectra were acquired throughout using the following parameters: dry gas flow 8 l min–1, nebuliser gas pressure 0.8 bar, dry gas 240 °C, capillary voltage 4500 V, offset 500 V, collision RF 650 Vpp. Mass spectra from manually chosen elution volumes were averaged and deconvoluted using a maximum entropy deconvolution algorithm in Compass DataAnalysis version 4.1 (Bruker Daltonik).
Samples for non-denaturing ESI-MS were prepared in volatile buffer, ammonium acetate, 50 mM, pH 7.8. Lines were washed with anaerobic buffer prior to sample loading to ensure all O2 was removed and protein samples were loaded into a Hamilton syringe and directly infused into the ESI source at a rate of 300 μl h–1. Data was acquired in 5 min increments with ion scans between 500 and 3000 m/z. NosL mass spectra (m/z 1000–3000) were recorded with acquisition controlled by Bruker qTOF Control software, with parameters as follows: dry gas flow 4 l min–1, nebulise gas pressure 0.8 Bar, dry gas 180 °C, capillary voltage 4000 V, offset 500 V, quadrupole voltage 5 V, collision RF 1000 Vpp, collision cell voltage 20 V. Spectra were deconvoluted as above. Exact masses are reported from peak centroids representing the isotope average neutral mass. Predicted masses are given as the isotope average of the neutral protein or protein complex, in which Cu(i)-binding is expected to be charge compensated.43
Continuous wave X-band electronic paramagnetic resonance (EPR) measurements were recorded using a Bruker EMX EPR Spectrometer equipped with an ESR-900 liquid helium flow cryostat (Oxford Instruments). Spectra were recorded at 10 K with the following instrumental settings: microwave frequency, 9.4652 GHz; microwave power, 3.18 mW; modulation frequency, 100 kHz; modulation amplitude, 5 G; time constant, 82 ms; scan rate, 22.6 G s–1; single scan per spectrum. A 98 μM Cu(ii)–EDTA standard was used to estimate Cu(ii) concentrations for protein samples by spin integration of signal area for spectra versus that of the standard, where all spectra were recorded under non-saturating power conditions.
Author contributions
S. P. B., D. J. R., A. J. G. and N. L. B. conceived the study. S. P. B., M. J. S. L., J. M. B. and D. A. S. performed the experiments. S. P. B., D. A. S., A. J. G. and N. L. B. analysed the data. S. P. B., D. J. R., A. J. G. and N. L. B. wrote the manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Supplementary Material
Acknowledgments
This work was supported by Biotechnology and Biological Sciences Research Council through the award of a DTP PhD studentship to SPB, grants BB/L022796/1 and BB/M00256X/1, and by UEA through the purchase of the ESI-MS instrument. MJS-L was funded by the Marie Sklodowska Curie Initial Training Network, Nitrous Oxide Research Alliance (NORA), grant 316472, under EU's 7th framework programme.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01053j
References
- Ravishankara A. R., Daniel J. S., Portmann R. W. Science. 2009;326:123–125. doi: 10.1126/science.1176985. [DOI] [PubMed] [Google Scholar]
- Hu H. W., Chen D., He J. Z. FEMS Microbiol. Rev. 2015;39:729–749. doi: 10.1093/femsre/fuv021. [DOI] [PubMed] [Google Scholar]
- Thomson A. J., Giannopoulos G., Pretty J., Baggs E. M., Richardson D. J. Philos. Trans. R. Soc., B. 2012;367:1157–1168. doi: 10.1098/rstb.2011.0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeignoz-Horta L. A., Philippot L., Peyrard C., Bru D., Breuil M.-C., Bizouard F., Justes E., Mary B., Léonard J., Spor A. Global Change Biology. 2018;24:360–370. doi: 10.1111/gcb.13853. [DOI] [PubMed] [Google Scholar]
- Liu B., Frostegard A., Bakken L. R. mBio. 2014;5:e01383. doi: 10.1128/mBio.01383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffis T. J., Chen Z., Baker J. M., Wood J. D., Millet D. B., Lee X., Venterea R. T., Turner P. A. Proc. Natl. Acad. Sci. U. S. A. 2017;114:12081–12085. doi: 10.1073/pnas.1704552114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. M., Graf D. R., Bru D., Philippot L., Hallin S. ISME J. 2013;7:417–426. doi: 10.1038/ismej.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkila M. P., Honisch U., Wunsch P., Zumft W. G. J. Bacteriol. 2001;183:1663–1671. doi: 10.1128/JB.183.5.1663-1671.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreusch A., Riester J., Kroneck P. M., Zumft W. G. Eur. J. Biochem. 1996;237:447–453. doi: 10.1111/j.1432-1033.1996.0447k.x. [DOI] [PubMed] [Google Scholar]
- Haltia T., Brown K., Tegoni M., Cambillau C., Saraste M., Mattila K., Djinovic-Carugo K. Biochem. J. 2003;369:77–88. doi: 10.1042/BJ20020782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraskevopoulos K., Antonyuk S. V., Sawers R. G., Eady R. R., Hasnain S. S. J. Mol. Biol. 2006;362:55–65. doi: 10.1016/j.jmb.2006.06.064. [DOI] [PubMed] [Google Scholar]
- Pomowski A., Zumft W. G., Kroneck P. M., Einsle O. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2010;66:1541–1543. doi: 10.1107/S1744309110038832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L. K., Einsle O. Biochemistry. 2016;55:1433–1440. doi: 10.1021/acs.biochem.5b01278. [DOI] [PubMed] [Google Scholar]
- Farrar J. A., Neese F., Lappalainen P., Kroneck P. M. H., Saraste M., Zumft W. G., Thomson A. J. J. Am. Chem. Soc. 1996;118:11501–11514. [Google Scholar]
- Richardson D. J., Bell L. C., McEwan A. G., Jackson J. B., Ferguson S. J. Eur. J. Biochem. 1991;199:677–683. doi: 10.1111/j.1432-1033.1991.tb16170.x. [DOI] [PubMed] [Google Scholar]
- van Spanning R. J. M., Wansell C., Harms N., Oltmann L. F., Stouthamer A. H. J. Bacteriol. 1990;172:986–996. doi: 10.1128/jb.172.2.986-996.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreusch A., Burgisser D. M., Heizmann C. W., Zumft W. G. Biochim. Biophys. Acta. 1997;1319:311–318. doi: 10.1016/s0005-2728(96)00174-0. [DOI] [PubMed] [Google Scholar]
- Horng Y. C., Cobine P. A., Maxfield A. B., Carr H. S., Winge D. R. J. Biol. Chem. 2004;279:35334–35340. doi: 10.1074/jbc.M404747200. [DOI] [PubMed] [Google Scholar]
- Thompson A. K., Gray J., Liu A., Hosler J. P. Biochim. Biophys. Acta. 2012;1817:955–964. doi: 10.1016/j.bbabio.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan V., Pierik A. J., Lill R. Science. 2014;343:1137–1140. doi: 10.1126/science.1246729. [DOI] [PubMed] [Google Scholar]
- Saunders N. F., Hornberg J. J., Reijnders W. N., Westerhoff H. V., de Vries S., van Spanning R. J. J. Bacteriol. 2000;182:5211–5217. doi: 10.1128/jb.182.18.5211-5217.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan M. J., Gates A. J., Appia-Ayme C., Rowley G., Richardson D. J. Proc. Natl. Acad. Sci. U. S. A. 2013;110:19926–19931. doi: 10.1073/pnas.1314529110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuirl M. A., Bollinger J. A., Cosper N., Scott R. A., Dooley D. M. J. Biol. Inorg Chem. 2001;6:189–195. doi: 10.1007/s007750000190. [DOI] [PubMed] [Google Scholar]
- Carreira C., Pauleta S. R., Moura I. J. Inorg. Biochem. 2017;177:423–434. doi: 10.1016/j.jinorgbio.2017.09.007. [DOI] [PubMed] [Google Scholar]
- Ghosh S., Gorelsky S. I., Chen P., Cabrito I., Moura J. J., Moura I., Solomon E. I. J. Am. Chem. Soc. 2003;125:15708–15709. doi: 10.1021/ja038344n. [DOI] [PubMed] [Google Scholar]
- Prudencio M., Pereira A. S., Tavares P., Besson S., Cabrito I., Brown K., Samyn B., Devreese B., Van Beeumen J., Rusnak F., Fauque G., Moura J. J., Tegoni M., Cambillau C., Moura I. Biochemistry. 2000;39:3899–3907. doi: 10.1021/bi9926328. [DOI] [PubMed] [Google Scholar]
- Rasmussen T., Berks B. C., Butt J. N., Thomson A. J. Biochem. J. 2002;364:807–815. doi: 10.1042/BJ20020055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle C. L., Zumft W. G., Kroneck P. M., Korner H., Jakob W. Eur. J. Biochem. 1985;153:459–467. doi: 10.1111/j.1432-1033.1985.tb09324.x. [DOI] [PubMed] [Google Scholar]
- Dell'Acqua S., Pauleta S. R., Moura J. J., Moura I. Philos. Trans. R. Soc., B. 2012;367:1204–1212. doi: 10.1098/rstb.2011.0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S., Tokuda H. Annu. Rev. Microbiol. 2011;65:239–259. doi: 10.1146/annurev-micro-090110-102859. [DOI] [PubMed] [Google Scholar]
- Taubner L. M., McGuirl M. A., Dooley D. M., Copie V. Biochemistry. 2006;45:12240–12252. doi: 10.1021/bi061089+. [DOI] [PubMed] [Google Scholar]
- Pountney D. L., Schauwecker I., Zarn J., Vasak M. Biochemistry. 1994;33:9699–9705. doi: 10.1021/bi00198a040. [DOI] [PubMed] [Google Scholar]
- Xiao Z., Loughlin F., George G. N., Howlett G. J., Wedd A. G. J. Am. Chem. Soc. 2004;126:3081–3090. doi: 10.1021/ja0390350. [DOI] [PubMed] [Google Scholar]
- Zhou L., Singleton C., Le Brun N. E. Biochem. J. 2008;413:459–465. doi: 10.1042/BJ20080467. [DOI] [PubMed] [Google Scholar]
- Chan Y. K., McCormick W. A., Watson R. J. Microbiology. 1997;143:2817–2824. doi: 10.1099/00221287-143-8-2817. [DOI] [PubMed] [Google Scholar]
- Savelieff M. G., Wilson T. D., Elias Y., Nilges M. J., Garner D. K., Lu Y. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7919–7924. doi: 10.1073/pnas.0711316105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckert W. R. Biochim. Biophys. Acta. 2014;1843:1509–1516. doi: 10.1016/j.bbamcr.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishniac W., Santer M. Bacteriol. Rev. 1957;21:195–213. doi: 10.1128/br.21.3.195-213.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tett A. J., Rudder S. J., Bourdès A., Karunakaran R., Poole P. S. Appl. Environ. Microbiol. 2012;78:7137–7140. doi: 10.1128/AEM.01188-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M. M. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Kristjansson J. K., Hollocher T. C. J. Biol. Chem. 1980;255:704–707. [PubMed] [Google Scholar]
- Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. Prot. Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay K. L., Hamilton C. J., Le Brun N. E. Metallomics. 2016;8:709–719. doi: 10.1039/c6mt00036c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.