

Abstract
Increasing burden of obesity world-wide and its effect on cardiovascular disease (CVD) risk is an opportunity for evaluation of preventive approaches. Both obesity and CVD have a genetic background and polymorphisms within genes that enhance expression of variant proteins that influence CVD in obesity, thus genome-based prediction may be a feasible strategy. However, identification of genetically driven risk factors for CVD manifesting as clinically recognized phenotypes is a major challenge. Clusters of such risk factors include hyperglycemia, hypertension, ectopic liver fat, and inflammation. All involve multiple genetic pathways having complex interactions with variable environmental influences. The factors that make significant contributions to CVD risk include altered carbohydrate homeostasis, ectopic deposition of fat in muscle and liver, and inflammation with contributions from the gut microbiome. Futuristic model depends on harnessing the predictive power of plausible genetic variants, phenotype reversibility, and effective therapeutic choices based on genotype–phenotype interactions. Inverting disease phenotypes into ideal cardiovascular health metrics could improve genetic and epigenetic assessment, and form the basis of a futuristic model for risk detection and early intervention.
Keywords: Obesity, Cardiometabolic disease, Genetics, Microbiome, Gene Environment Interactions
INTRODUCTION
The prevalence of obesity and its associated co-morbidities have increased world-wide over the past three decades. Further increases are predicted by 2030.1 Both, overweight and obesity continue to have serious implications for health2 including excess mortality and costs,3 in large part attributable to excess cardiovascular disease (CVD) in both men4 and women.5 Currently, over 78 million US adults (~one third of the US population) are obese, and 82 million are overweight. Obesity-associated morbidity is dependent on the rate at which midlife adults attain an increased body mass index (BMI) 6, suggesting that intervention to offset obesity-related disease early in life may help prevent its medical and economic consequences.7 Since an expanding role for preventative CVD medicine has been increasingly recognized,8,9an emphasis on earlier risk factor (biomarkers) identification is reasonable and could begin in children, adolescents or young adults especially before manifestation of the risk factors10,11.
This review highlights: molecular and genetic aspects of obesity and cardio-metabolic traits that are significantly associated with CVD, and mechanisms for the adverse metabolic effects of obesity on CVD. A futuristic screening model for effective and individualized interventions based on the degree of obesity, severity of the cardiovascular (CV) phenotype and response to available lifestyle and medical interventions has also been proposed.
GENETICS OF OBESITY
Obesity precedes the development of much of the CV risk4,5,12 and the early manifestation and progression of obesity due to genetic predisposition provides the environment for adverse CV outcomes. Genetically driven phenotypes of obesity have been sub-classified into three main types: syndromic, non-syndromic and polygenic obesity.13 All these have variable effects on body fat mass. Of the 79 forms of identified obesity syndromes, 19 have been fully elucidated, 11 have been partially elucidated, while 27 have been mapped to a chromosomal region but not characterized.14 Monogenic obesity presents as a rare condition with severe obesity due to recessive mutations coding for key metabolites in pathways for the hypothalamic control of appetite. Studies on animal models and on families carrying these genetic alterations have provided significant insights into mechanisms for the development of obesity in the general population15,16 such as the identification of the leptin-melanocortin pathway involved in monogenic obesity and satiation. 17
Large genome-wide association studies (GWAS) have helped in the identification of more than 250 genes/loci important in the biology of obesity18,19. These studies have led to the discovery of several new genes with previously unknown function. Detailed discoveries of GWAS identified obesity loci that have been reviewed previously. A summary of the association of common variants in major obesity loci and their pleiotropic effects on other cardiovascular risk factors (traits) is included in Table 1 and Figure 1. Discovery of FTO (fat mass and obesity-associated) gene by GWAS with robust replication of an intronic variant (rs9939609) in multiple independent studies, show that FTO variants have an important role in obesity and possibly in the pathogenesis of type 2 Diabetes (T2D). The association was abolished when adjusting for BMI suggesting that the FTO variant influences T2D via its effect on obesity 20. The associations of the same FTO variant (rs9939609) with obesity and T2D were explored in a large-scale meta-analyses study conducted on 96,551 individuals from East and South Asia, which suggested that the association of FTO with T2D was independent of obesity.21 Because of the strong association of FTO with T2D and its associated risk with CVD, the FTO gene has assumed priority for further studies beginning early in life. Indeed, the association of FTO with obesity has been replicated in longitudinal studies in childhood 22,23. Additionally, a Dutch study reported association of FTO variants with higher BMI, fat mass index, and leptin concentrations during puberty but declining at ages 13–14 years; a finding presumed to be consistent with hormonal effects at pubertal onset 24. Although considered a strong effect variant in polygenic obesity, the FTO gene has been predominantly associated with appetite regulation 25, as has been the case for most monogenic obesity genes. FTO mRNA transcripts have been observed in mouse hypothalamic nuclei encoding 2-oxoglutarate-dependent nucleic acid demethylase that supports a regulatory role in appetite and possibly energy balance and sympathetic outflow to the circulatory system 26. The mouse model studies further validated the role of FTO in controlling food intake, energy homeostasis, and energy expenditure 27 possibly via nearby genes such as RPGRIP1L and IRX3.25
Table 1.
Summary of common variants associations in major genes identified in GWAS of obesity showing pleiotropic association with two or more cardiovascular risk traits
| Obesity | Type 2 Diabetes | Fasting Blood Glucose | Dyslipidemia | Hypertension | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene/rs ID | Popul ation | Effect Allele | Beta or OR | P value | N | PMID | OR | P value | N | PMID | Beta | P value | N | PMID | Beta | P value | N | PMID | Beta | P value | N | PMID |
| ADAMTS9 rs6795735 | EUR | C | 0.025 | 9.8×10−14 | 161,642 | 20935629 | 1.07 | 4.4×10−4 | 69,033 | 24509480 | 2.03‡ | 0.042 | 77,167 | 20935629 | −2.5‡ | 0.013 | 77,167 | 20935629 | --- | --- | --- | --- |
| ADCY3 rs10182181 | EUR | G | 0.031 | 8.8×10−24 | 322,154 | 25673413 | 1.0 | 0.89 | 86,188 | 25673413 | 6×10−4 | 0.77 | 131,771 | 25673413 | 0.009 | 0.049 | 172,937 | 25673413 | 0.14 | 0.023 | 67,936 | 25673413 |
| ADCY3/ POMC rs713586 | EUR | C | 0.14 | 6.2×10−22 | 230,748 | 20935630 | − | 0.833 | 10,128 | 20935630 | 0.004 | 0.44 | 5,641 | 27530450 | − | 0.45 | 19,000 | 20935630 | --- | --- | --- | --- |
| rs713586 | EAS | C | 0.025 | 4.9×10−13 | 173,430 | 28892062 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| BDNF rs11030104 | EUR | A | 1.05† | 1.3×10−34 | 339,224 | 25673413 | 1.01 | 0.37 | 83,545 | 25673413 | 0.003 | 0.41 | 58,074 | 25673413 | 0.014 | 1.35×10−3 | 163,969 | 25673413 | 0.23 | 2.1×10−3 | 69,843 | 28135244 |
| FTO rs9939609 | EUR | A | 0.10‡ | 3×10−35 | 30,081 | 17434869 | 1.25 | 1×10−20 | 63,961 | 22693455 | 0.69 | 0.012 | 9,618 | 27790247 | --- | --- | --- | --- | 0.28 | 0.163 | 9,618 | 27790247 |
| rs9939609 | SAS | A | 1.25† | 9×10−19 | 39,774 | 22109280 | 1.15 | 5.5×10−8 | 18,112 | 22109280 | 0.24 | 0.03 | 1,666 | 21294771 | --- | --- | --- | --- | --- | --- | --- | --- |
| rs9939609 | EAS | A | 1.27† | 1.2×10−13 | 9,035 | 22109280 | 1.15 | 2.43×10−7 | 59,106 | 22109280 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| GIPR rs11671664 | EUR | A | 0.029 | 0.0012 | 194,562 | 22344221 | 1.08 | 2.4×10−5 | 20,298 | 28869590 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| rs11671664 | SAS | A | --- | --- | --- | --- | 1.08 | 1.29×10−2 | 42,071 | 28869590 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| rs11671664 | EAS | A | 0.046 | 7×10−14 | 62,245 | 22344221 | 1.08 | 6.3×10−7 | 50,981 | 28869590 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| GIPR rs10423928 | EUR | A | 1.16† | 4×10−13 | 95,632 | 23563607 | 1.07 | 1.8×10−4 | 57,599 | 20081857 | 0.08 | 2×10−15 | 41,099 | 20081857 | --- | --- | --- | --- | --- | --- | --- | --- |
| MC4R rs6567160 | EUR | C | 0.056 | 3.9×10−53 | 321,958 | 25673413 | 1.07 | 6×10−7 | 80,620 | 25673413 | 0.003 | 0.196 | 128,057 | 25673413 | -0.03 | 2.9×10−9 | 185,608 | 25673413 | 0 | 0.994 | 66,849 | 25673413 |
| MC4R rs12970134 | EUR | A | 1.12† | 9.9×10−16 | 51,151 | 23049848 | 1.26 | 0.006 | 4,561 | 18454146 | 0.004 | 0.083 | 889 | 21372613 | -1.34 | 0.01 | 4,561 | 18454146 | 0.11 | 0.64 | 4,561 | 18454146 |
| rs12970134 | SAS | A | 0.004 | 6.8×10−4 | 7,394 | 18454146 | 1.07 | 0.15 | 7,394 | 18454146 | --- | --- | --- | --- | -0.76 | 0.03 | 7,394 | 18454146 | -0.06 | 0.71 | 7,394 | 18454146 |
| rs12970134 | SAS- Sikhs | A | 0.67 | 0.0005 | 1,528 | 19680233 | --- | --- | --- | --- | --- | --- | --- | --- | 0.01 | 0.97 | 1,528 | 19680233 | --- | --- | --- | --- |
| PCSK1 rs6234/ rs6235 | EUR | G | 1.09† | 1.25×10−5 | 163,385 | 25784503 | --- | --- | --- | --- | −0.01 | 1.4×10−6 | 1,367 | 23903356 | --- | --- | --- | --- | --- | --- | --- | --- |
| rs6234/ rs6235 | EAS | G | 1.0† | 0.274 | 66,721 | 25784503 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| PEMT rs4646404 | EUR | G | 0.027 | 1.4×10−11 | 198,196 | 27195708 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| rs4646404 | SAS | G | -0.03 | 0.1 | 9,505 | 27195708 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| POMC rs1561288 | EUR | C | 0.055 | 5×10−8 | 28,998 | 23669352 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| SH2B1 rs7359397 | EUR | T | 0.15 | 1.9×10−20 | 204,309 | 20935630 | + | 0.306 | 10,128 | 20935630 | 0.018 | 9.8×10−4 | 5,641 | 27530450 | - | 0.099 | 19,000 | 20935630 | --- | --- | --- | --- |
| SH2B1 rs7498665 | EUR | G | 0.15 | 5.1×10−11 | 86,677 | 19079261 | 1.09 | 2.5×10−6 | 70,930 | 24528214 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| rs7498665 | EAS | G | --- | --- | --- | --- | 1.11 | 0.179 | 21,793 | 24528214 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| TBX15- WARS2 rs2645294 | EUR | T | 0.031 | 1.7×10−19 | 209,808 | 25673412 | --- | --- | --- | --- | -0.09 | 0.014 | 770 | 28257690 | 0.155 | 1.6×10−5 | 770 | 28257690 | -0.12 | 8.3×10−4 | 770 | 28257690 |
+/−: Effect Direction
OR Values
Z score
N: Sample Size; PMID: PubMed ID; EUR: European; EAS: East Asian; SAS: South Asian
Figure 1.
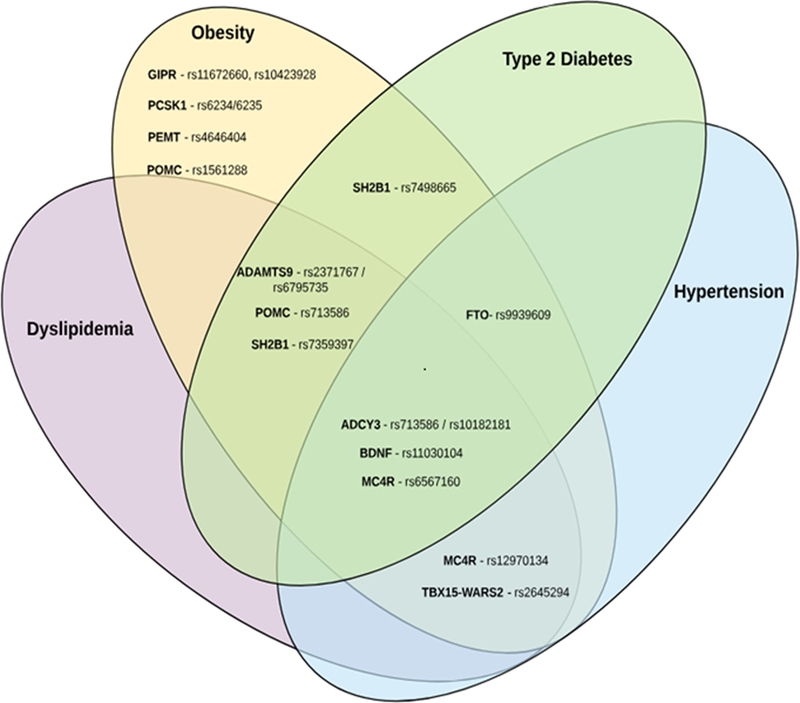
Venn diagram showing the pleiotropic associations of common variants among the major obesity loci identified by GWAS. Genetic variants identified in the GWAS for obesity are classified based on the results of the pleiotropic association reported of the same loci/variants with other cardiovascular traits. Each cardiovascular trait shown in different colors in the Venn diagram (yellow, purple, green, aqua) corresponds to each of the cardiovascular traits (obesity, dyslipidemia, type 2 diabetes, and hypertension), respectively. Effect sizes and pleiotropic association p-values are summarized in Table 1.
Another BMI locus detected by GWAS was near GIPR, the incretin receptor, which may indicate a causal contribution of variation in postprandial insulin secretion to the development of obesity.28 This is important because there is accumulating evidence that the gut microbiome is increasingly involved in obesity, and could interact with intestinal functions such as absorption, neural control and appetite.29 Most commonly encountered obese cases have a polygenic background. This accounts for about 60% of the BMI variance30 and suggests that polygenic obesity is also dependent on the genetic interactions with environmental, lifestyle and cultural factors. This argument is consistent with Mendelian randomization studies that have reported that BMI-associated increase in CVD risk is attributable to genetic variation. 31,32 A variant or combination of variants can be used as an instrumental variable for disease traits such as ‘BMI’ to evaluate a causal relationship of obesity to an outcome variable such as a ‘CV event’ or disease onset. Importantly, as gene variants have a lifetime effect, influence on the respective CV phenotypes would be of long duration, although this could be confounded by environment and lifestyles.33,34 Interestingly, these studies have also identified polymorphisms, within genes earlier known to be involved in monogenic obesity that also contribute to polygenic obesity.15,17 For instance, polymorphisms in PCSK1 contribute to extreme (monogenic) obesity (defined as having a BMI >40 kg/m2) in addition to common obesity (defined as a BMI ranging from 30 to 40), which was reported in a large size meta-analysis study including up to 331,175 individuals from diverse ethnic groups, suggesting that ethnicity, age and study design modulate the association of PCSK1 polymorphisms with obesity. Additionally, the study demonstrated the contribution of common variants in PCSK1 to contribute to BMI variation and obesity.35 One recent meta-analysis GWAS of BMI conducted on 123,865 individuals using 2.8 million single nucleotide polymorphisms (SNPs), and follow-up in significant numbers, identified 18 new obesity susceptibility loci and confirmed 14 known genes associated with obesity.28 Some variants identified by GWAS are near monogenetic loci, such as MC4R, POMC, SH2B1 and BDNF, and are hypothalamic regulators of energy balance.15,17
However, despite these successes, the common non-coding variants discovered using the GWAS approach explain only a small fraction of total genetic variance and these studies are often underpowered to locate those less common coding variants with a frequency of <5% [57]. Whole genome exome-wide sequencing or targeted sequencing studies have discovered low-frequency / rare variants with larger effect sizes within the earlier known obesity genes. A large meta-analysis study of exome and targeted sequence data using 718,734 individuals (80% Caucasians) discovered rare functional coding variants in 13 genes, including 5 known and 8 new obesity genes, and in silico gene set enrichment analysis predicted a strong role for neurobiology in body weight regulation. The effect sizes of rare variants were ~10 times larger than those observed in common variants. The carriers of a rare (MAF 0.01) MC4R mutation (Tyr35Ter) weighed 7 kg more than the non-carriers in this study36. Exome sequencing in a family-based design has detected novel functional variants for predicting childhood obesity in peroxisome biogenesis factor (PEX-1). PEX-1, an earlier identified gene by GWAS, is involved in childhood obesity through a novel mechanism of peroxisomal biogenesis and metabolism.37 Importantly, by far, the vast majority of whole genome, exome-wide, or targeted sequencing studies have been predominantly performed on Caucasian populations, more investigations on other major ethnic groups would be needed to identify causal variants with population-specific effects.
OBESITY AND CARDIOMETABOLIC RISK PHENOTYPES
Hyperglycemia
Hyperglycemia, a known cardiovascular risk factor and component of the metabolic syndrome joins other traits in preceding both diabetes and cardiovascular disease. Detectable changes in glucose and insulin metabolism precede T2D and have been studied as quantitative traits (QTs) in genetic studies and as targets for reversal or prevention of T2D onset. Therefore, there has been interest not only in searching for genetic association but also in finding the glucose levels which accurately reflect T2D and preceding risk. It is known that progression of IGT (Impaired glucose tolerance) to T2D is potentially reversible with lifestyle.36 The American Diabetes Association Expert Committee established the impaired fasting glucose and impaired glucose tolerance range.38 These cut-off points were selected to facilitate early diagnosis of risk and to initiate lifestyle interventions known to reduce risk.36 Approximately 60% of people who develop diabetes have either IGT or IFG (impaired fasting glucose) about 5 years before T2D onset, while 40% have normal glucose tolerance.39
Studies also suggest that IGT is strongly associated with hypertension, dyslipidemia and worse cardiovascular outcomes.38 The rs553668 of the ADRA2A gene predicts worsening of fasting glucose values in a prediabetic cohort.39 Variants associated with fasting glucose discovered through GWAS such as GCK, G6PC2, MTNR1B, and DGKB- TMEM195 40 in the normoglycemic population do not always influence risk for T2D (in contrast to TCF7L2 and SLC30A8), but their effect appears confined to fasting glucose homeostasis.40,41 The data support recognition of early hyperglycemic phenotypes derived from regulatory polymorphisms on the genes affecting interacting pathways leading to T2D. Meta-analysis of 21 GWA studies identified nine new loci influencing fasting blood glucose: ADCY5, MADD, ADRA2A, CRY2, FADS1, GLIS3, SLC2A2, PROX1 and C2CD4B. However, of these, only ADCY5 and PROX1 were associated with T2D. These data suggest that although there is overlap, the genetic background for fasting glucose is different from that for the T2D phenotype.42 Similarly, the 2-h glucose levels after a standard oral glucose load, can be defined as a separate trait to T2D with overlapping associated variants. Meta-analysis identified new loci, GIPR and UPS13C, uniquely influencing 2-h glucose 43 supporting the hypothesis that there are separate glucose-related QTs representing specific modes of carbohydrate metabolism.44
Based on population studies and animal models, it has been proposed that T2D has a progressive pathogenesis beginning with insulin resistance and advancing to β-cell failure 45, and that it may involve several genes, sometimes with significant epistatic interactions.46 For example, using knockout models for both IRS- 1 and the insulin receptor, it was shown that neither model alone had much effect on T2D onset, but the combined effect resulted in more than a 50% chance of developing diabetes at young ages.44 A recent study compared loci associated with multiple glycemic traits with those for T2D. Amongst 88 T2D risk loci and 72 glycemic trait loci, only 29 were shared and showed disproportionate magnitudes of phenotypic effects. These results lead to important insights regarding the role of T2D loci in disease predisposition through their contribution to glycemic trait variability.48
Hypertension
Obesity has been identified as an important risk factor for hypertension.49 The association of insulin resistance with high blood pressure 47 is one of several independent risk factors for CVD.48 However, evidence from genetic studies, specifically from GWAS, points to largely separate genetic backgrounds for hypertension and T2D. Because hypertension is attributed to enhanced sympathetic nervous system and renin-angiotensin-aldosterone system (RAAS) activity,47,49 cardiovascular effects are likely disparate.
Several studies have reported the association of genetic variation in RAAS with CVD risk factors, hypertension and coronary artery disease.50,51 The relationship of CVD with RAAS variants has been investigated and shows that multiple variants are associated with greater risk. Angiotensin II is also involved in triggering vascular inflammation and oxidative stress in the endothelium by stimulating NAPH/NADPH oxidase, protein kinase C, and mitogen-activated protein kinase (MAPK).52,53 It also has a direct effect on increasing insulin resistance independent of alterations in blood flow and interstitial insulin concentration,54 but angiotensin II is equally responsible for influencing the arterial wall through vascular inflammation.55,56 The insulin resistance is reversible by selective inhibitors of angiotensin II at AT1 receptors.58 Similar selective antagonism using irbesartan, an AT1 receptor blocker (ARB), has been shown to improve insulin action in the obese rat model associated with upregulation of GLUT4, the main glucose transporter in skeletal muscle.59 On investigating the association of common variants such as ACE (angiotensin-converting enzyme) and AGT (angiotensinogen) with hypertension, results have been inconclusive 57 and they have not been associated with T2D.61 However, variants in ACE and CYP11B2 genes have been associated with insulin resistance in hypertensive families in Taiwan.58
It appears likely that variation in genetic background for hypertension according to race is important since differences have provided insight on possible mechanisms and responses to treatments. Fine mapping of GWAS determined loci have revealed association of novel variants with blood pressure in Hispanics and African Americans, and were similar to variants such as KCNK3 and HOTTIP in populations of European descent.59 Data from the National Health and Nutrition Examination Survey (NHANES) showed the prevalence of hypertension to be 40% in African Americans compared to 27% in European Americans 60,61, leading to the hypothesis that part of the excess burden in African Americans suggests implication of genetic susceptibility that potentially manifests with the influence of environment (i.e. gene x environmental interactions).62 GWAS and candidate genes examined in the Candidate Gene Association Resource Consortium consisting of 8,591 African Americans identified novel associations for diastolic blood pressure on chromosome 5 near GPR98 and ARRDC3, and for systolic blood pressure on chromosome 21 in C21orf 91. However, none of these variants were associated with T2D.62
Certain blood pressure and hypertension loci under selective pressure or adaptation in some African American populations, and were likely advantageous in dry and/or salty environments. For instance, the CYP3A5 enzyme polymorphism CYP3A5*3 (which influences salt and water retention), showed extreme variation in allele frequency even within African populations, and correlated with the distance from the equator 63. Monogenic forms of hypertension have provided evidence for a regulatory role of key metabolic pathways and have been the basis for candidate gene population studies, but none have involved carbohydrate metabolism or insulin action. Using such an approach, 24-hour ambulatory blood pressure has been associated with five polymorphisms in the KCNJ1 gene, which has the potential to cause Bartter syndrome Type 2 when the abnormal allele is inherited.64 Ambulatory blood pressure is also associated with common variations in the WNK1 gene known to cause pseudohypoaldosteronism Type 2 or Gordon syndrome. Association of WNK1 with blood pressure in childhood underscores its possible association with evolving hypertension at young ages and emphasizes the role of WNK signaling pathways in blood pressure regulation.68 Additional association with variants in CASR, NR3C2, and SCNN1B, all of which are known to have had mutations causing rare Mendelian defects in blood pressure regulation, provide support for the hypothesis that relevant polymorphisms influence conventional pathways involved in blood pressure regulation.65 However, only a few variants have been discovered in GWAS in the earlier known genes, suggesting new pathways involved in hypertension.
A large meta-analysis performed by the International Consortium for Blood Pressure on 200,000 individuals of European descent, identified 16 loci of which only 6 contained genes that are known or suspected to regulate blood pressure, which include NPR3, GUCY1A3- GUCY1B3, ADM, GNAS- EDN3, NPPA- NPPB, and CYP17A1 66. CYP17A1 achieved the most robust GWAS significance and is the site for a known Mendelian-inherited mutation causing hypertension by increasing mineralocorticoids in the adrenal steroid pathway and causing a rare form of congenital adrenal hyperplasia attributed to 17-hydroxylase deficiency. Since diabetes and hypertension share common pathways there might be interaction with other genes and lifestyle factors. When the association of RAAS variants with CVD was scored by the Gensini system, multiple variants were associated with CVD and an AGTR1 variant encoding for the angiotensin II type 1 receptor was associated with CVD severity. There was gene-environment interaction for smoking status with an aldosterone synthase variant (CYP11B2) and CVD67.
Ectopic Liver Fat
The buildup of extra fat in liver cells that is not caused by alcohol or other drugs is termed non-alcoholic fatty liver disease (NAFLD) and has been defined as accumulation of fat in the hepatocyte exceeding 5%. The increase of the obesity epidemic parallels the rise in obesity associated insulin resistance68, which plays a role in the pathogenesis of NAFLD.69 Furthermore, it is associated with increased CVD risk over and above that attributed to obesity.70 This is also a risk factor for progression from a metabolically normal to abnormal state in obese and non-obese individuals71, supporting the need for early detection and prediction of severity.
Prevalence of NAFLD is dependent on the detection method and the study population. Serum aminotrasferases--ALT and AST levels are typically used to screen for the condition but are non-specific markers; liver scanning or biopsy, the gold-standard, is preferred. The mean prevalence in pediatric populations exceeds 7% but increases to above 30% in studies based on children attending obesity clinics.72 The prevalence varies dramatically among different ethnic groups despite similar rates of the metabolic syndrome and related risk factors, and there are possible environmental and genetic reasons.73 Male gender and Hispanic background increase risk 68, but African Americans are relatively spared.73
Accumulation of excessive diacylglycerol in the liver is associated with accumulation of liver fat leading to defective insulin action 74, particularly in genetically susceptible populations such as Asian Indian men. 75 Ectopic liver fat is highly associated with atherogenic dyslipidemia, even in adolescents. 76,77 Adiponectin, a fat cell hormone involved in lipid metabolism, is a possible mediator. Both +45T>G (rs2241766) and −11377C>G (rs577853790) have shown association with NAFLD in a meta-analysis,78 a finding that supports the hypothesis that overlapping genetic backgrounds contribute to both NAFLD and CVD, since the adiponectin +45T>G genotype has shown association with CVD in a separate meta-analysis.79 Increased visceral fat is associated with low adiponectin in adolescence80, supporting association with adiponectin action via adiponectin receptor 2 (ADIPOR2) in three independent Finnish cohorts.81 However, among Asians, a meta-analysis suggests that adiponectin variants might be risk factors for NAFLD while the +276G>T variant is protective.78 Simple steatosis progresses to inflammation with risk for cirrhosis and liver cancer 82, and is independently associated with increased risk of coronary artery disease (CAD).83 Large-sized VLDL has been observed in NAFLD in an adolescent population independent of adiposity and insulin resistance, and the NMR (nuclear magnetic resonance) lipid profile was characterized by small dense LDL and reduced number of large HDL particles. 84 An association of NAFLD with a lipid profile predisposing to atherosclerosis in adults 70, and with increased intima-media thickness (IMT) in adolescents 85, has been revealed. These data suggest pleiotropic effects, or alternatively, the effects arise from a biochemical cascade leading to excessive hepatic fat storage, inflammation, and lipoprotein abnormalities. Maturation of the VLDL particle in the Golgi, at the stage when triglyceride is transferred to apoB by microsomal triglyceride transfer protein encoded by MTTP, determines liver fat storage and if defective may lead to NAFLD.86 Carriers of the −493 G/T allele also have a more atherogenic lipid profile and the homozygous genotype of the less common T allele was associated with subclinical proinflammatory markers and CVD.87 The same genotype was also predicted to have a deleterious effect on β-cell function.88 Furthermore, the - I128T variant is associated with central obesity, elevated liver enzymes in fatty liver disease with and without association with alcoholism.89 In addition, a manganese superoxide dismutase (MnSOD) variant was associated, possibly working by reducing mitochondrial fatty acid oxidation. Genetic determinants of VLDL formation and disposal may result in both atherosclerosis and fatty liver disease. A study conducted on Asian Indian men revealed that the carriers of minor alleles of two APOCIII variants rs2854117 (C-482T), rs2854116 (T-455C), or both had a 30% increase in apoC-III levels and a 60% increase in serum triglyceride, as compared with the wild-type homozygotes. The prevalence of NAFLD was 38% among variant T allele of rs2854117 and C allele of rs2854116 carriers compared to 0% among wild-type carriers showing a significant correlation with insulin resistance.90 Furthermore, the apoC-III overexpression model is predisposed to diet-induced hepatic steatosis and hepatic insulin resistance.91
There also is evidence that inherited hepatic enzyme abnormalities are causative. PNPLA3 and TM6SF2 variants lower triglyceride92,93 by decreasing intrahepatic lipolysis thus promoting hepatic fat storage. These variants are associated with a distinct form of NAFLD 94 that may also co-exist with insulin resistance.95,96 Although, PNPLA3, TM6SF2 and GCK genes have shown the strongest associations with NAFLD in genome-wide association scans (GWAS) accounting for 10% of heritability, multiple genes with small additive effects are also causative 96. A GWAS of 2,111 participants in the Dallas Heart Study revealed a robust association of liver fat defined by magnetic spectroscopy with the I148M allele of the PNPLA3 gene 97, this association was independently replicated in children and adolescents 98. Another study investigated the effects of candidate gene SNPs and suggested joint effects between PNPLA3 and GCKR SNPs, explaining 32% of fatty liver disease in Caucasian children.99 A meta-analysis of 16 studies showed association of PNPLA3 with disease severity with strong effect on more aggressive disease susceptibility indicated by higher inflammation indices and progression to fibrosis. 100 The gene PNPLA3 codes for patatin-like phospholipase domain-containing protein 3, or adiponutrin, which plays a role in hepatic triglyceride hydrolysis catalyzing conversion of lysophosphatidic acid into phosphatidic acid, an important regulatory reaction in lipid synthesis. PNPLA3 is upregulated by sucrose feeding in the mouse model, and the l148M variant (rs738409 C/G) in PNPLA3 results in increased cellular lipid accumulation providing a plausible mechanism for its impressive association with NAFLD.101 In addition to PNPLA3, diet-induced obesity increases mRNA expression 102 which is associated with increased alanine amino transaminase (ALT), a marker of fatty liver disease, in Europeans, Hispanics, and Asian Indians.103,104 The homozygous carriers of the minor G allele of rs738409 C/G (I148M) variant showed increased fasting glucose levels89, and the minor T allele of rs6006460 G/T (S453I) variant was associated with lower hepatic fat content and was more frequent in African Americans who had the lowest hepatic fat content, suggesting protection from NAFLD.97
Inflammation and Gut Microbiota
There is strong evidence that inflammation plays a key role in increasing insulin resistance in obesity 105 and in the molecular mechanism underlying atherosclerosis.106 Presence of immune cells in adipose tissue suggests their major role in inflammation. Studies using genetic mouse models revealed key pathways in the immune process which ultimately led to the identification and targeting of key regulatory pathways.107 It is also becoming evident that the intestinal microbiome regulates immune cells108 and alters the permeability of the gut109 secondary to degradation of endothelial tight junctions, and thereby allows inflammatory products to circulate and cause systemic inflammation and activate insulin resistance. Composition of gut microbiota in obesity affect the epigenetic regulation of genes via the action of bacteria-derived short chain fatty acids on free fatty acid receptors,110 providing evidence for the hypothesis that dietary interventions to change microbial composition may ameliorate inflammation and insulin resistance. Experimental evidence has demonstrated that a high-fat diet increases expression of toll-like receptors (TLRs), mediators of chronic inflammation, and kruppel-like factors (KLFs) involved in adipocyte differentiation associated with an atherogenic lipid derangement111, suggesting that appropriate dietary modification can change obesity-associated inflammation.
It is also possible that changes in the microbiome and specific amino acids influence nutrient-specific appetite regulation via hypothalamic receptors based on evidence derived from non-mammalian as well as several recent mammalian and human studies.112 Recent metagenome-wide studies of gut microbiota were able to differentiate between useful and pathogenic microbes, and identified a set of bacteria associated with gut oxidative stress response linked with diabetes complications and inflammatory bowel disease113,114. Furthermore, evidence suggests that the severity of non-alcoholic fatty liver disease is influenced by a dysfunctional microbiome and that the dysbiosis is associated with hepatic inflammation leading to fibrosis115. These observations point to inflammation as being central to formation of metabolic traits that lead to atherosclerosis in obese individuals and are likely to have respective genetic interactions.
Endothelial Dysfunction
Endothelial dysfunction is characterized by impaired endothelium-dependent vasodilation and increased pro-coagulant and pro-inflammatory activity116. Vascular endothelial cells play a pivotal role in regulating blood flow in the entire circulatory system. Endothelial dysfunction has also been linked with obesity and elevated C-reactive protein (CRP). CRP is a pro-inflammatory marker whose concentrations are markedly increased in patients with T2D, hypertension and metabolic syndrome117. The development of atherosclerosis is considered to be a consequence of a chronic inflammatory process, perpetuated in part by LDL that is trapped and oxidized within the vessel wall118. Specifically, oxidative stress increases vascular endothelial permeability and promotes leukocyte adhesion (Figure 2), which is coupled with alterations in endothelial signal transduction and redox-regulated transcription factors 119. On the other hand, oxidized LDL may impair signal transduction activation of nitric oxide synthase, thus lowering the synthesis of nitric oxide. 120 Reduced nitric oxide could also stimulate the synthesis and release of endothelin, producing enhanced vasoconstrictor tone; promote the release and activity of growth factors, increasing smooth muscle cell migration into the intima and enhancing the synthesis and release of pro-inflammatory cytokines. Additionally, reduced nitric oxide could promote platelet attachment and release of growth factors in the vessel wall. These consequences of endothelial dysfunction such as lipid peroxidation along with reduced nitric oxide bioactivity, may be important in the initiation and progression of atherosclerosis and ultimately result in clinical manifestation of CVD. Angiotensin II is also involved in triggering vascular inflammation and oxidative stress in the endothelium by stimulating NAPH/NADPH oxidase, protein kinase C and mitogen-activated protein kinase (MAPK) 121,122. Peripheral endothelial function correlates well with coronary endothelial vasodilation and is reduced in patients with CVD risk factors such as obesity, hypercholesterolemia, hypertension and diabetes123.
Figure 2.
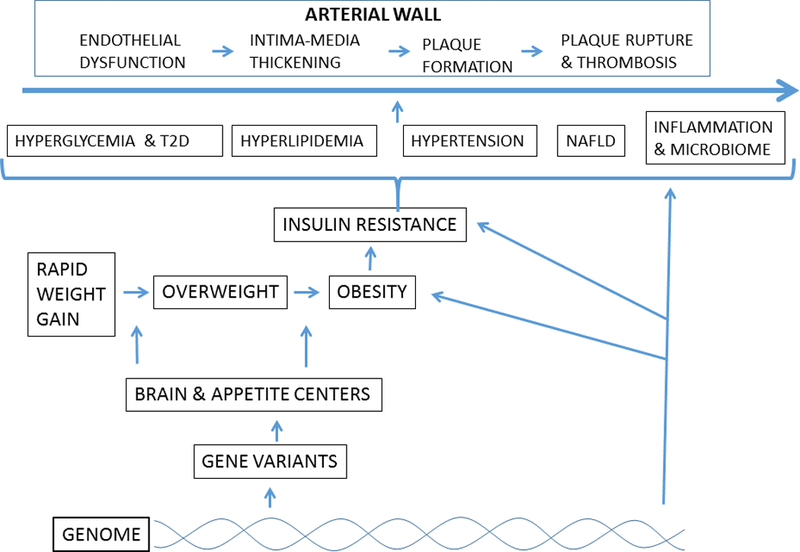
Genetic background for obesity resulting in CVD progression in three main stages. These include metabolic derangements due to genetic, non-genetic, dietary and lifestyle interactions; the development of cardiometabolic syndromic traits leading to fatty liver disease and inflammatory microbiome; and ultimately leading to vascular dysfunction and atherosclerotic plaque formation.
Diet, Physical Activity and Gene-Environment Interactions
Dietary and physical activity interventions for promoting metabolic health and reducing development of CVD are important. The cumulative effects of common variants and interaction of genetic markers with diet and exercise are approaches used to study predictors of weight change. Based on accumulating evidence that appetite-determining genes play a role in obesity these are targets of investigation.
A meta-analysis of 10 studies comprising 6,951 participants reported that individuals with heterozygous and homozygous FTO genotypes were predisposed to more weight loss during lifestyle intervention with a trend for a gene dose effect.124 Although a small effect, the association of FTO with appetite and components of dietary intake125 support the role of genotyping for predicting individual responses to dietary intervention. More recent studies show greater body weight and waist circumference reductions in individuals carrying a FTO risk variant in the Food4Me randomized control trial, adding support for the idea that gene-based advice can be helpful for weight loss.121 Leptin receptor (LEPR) variants contribute to BMI, LDL-C and HDL-C responses to a high carbohydrate diet in healthy Chinese adults. 126 Adiponectin, a key regulator of appetite and food intake, has been studied using a genetic risk score with divergent results on the effect of weight loss diets on adiponectin levels,127 suggesting that the levels could be a better predictor as shown in the POUNDS Lost Trial in which adiponectin increased in association with improved abdominal fat distribution and lipid metabolism independent of weight change.128,129 Fibroblast growth factor 21 (FGF21), although not directly involved in appetite, may interact with response to interventions for obesity. A low-calorie high carbohydrate diet had beneficial effects on body composition and abdominal obesity in obese individuals carrying an FGF21 ‘C’ allele in the 2-year POUNDS Lost Trial.130
A large body of data suggests strong interaction between obesity and 25 (OH)D deficiency. 131 Genetically determined vitamin D deficiency in certain populations has been reported to be linked with increased risk for obesity, diabetes, and cardiometabolic diseases. 132,133 Earlier it was speculated that the sequestration of vitamin D in adipose tissue reduces its bioavailability. 134 However, recent studies by Drincic et al. 135 could not find any evidence of sequestration of supplemental or endogenous 25(OH)D in fat cells. Their results suggested that the dosing for vitamin D in obese patients should be adjusted according to body size and not according to BMI to achieve desired serum 25(OH)D concentrations136. Additionally, hyperparathyroidism, secondary to hypovitaminosis D, augmented by obesity, could also be responsible for the observed association with obesity 137. These findings highlight the need to develop strategies to detect individuals at risk for the cardiometabolic effects of vitamin D deficiency. Essentially, ethnic variation in vitamin D deficiency, e.g., American Indians and their children beginning in infancy, whose dietary intake tends to be deficient in vitamin D138.
MC4R encoding the melanocortin-4 receptor has a common variant with a strong effect on obesity 139. Qi et al. reported association of MC4R variant (rs17782313) with higher intake of total energy and higher intake of dietary fat and protein. 140 The ‘C’ risk allele was associated with a 14% increased risk of T2D. The FGF21 polymorphism (rs838147) has been reported to interact with dietary carbohydrate/fat intake and changes with obesity.130 Since physical activity is a major modality used to enhance metabolic health and modulate obesity, it is a target for genetic association studies. A meta-analysis has shown that exercise attenuates the weight-gain effect of an FTO variant141 as shown in the Food4Me study.142 FTO variants interact with physical activity and dietary intake in the form of carbohydrate and fiber intake in an Asian Indian population, which are factors that are likely to be long-term beginning in youth.143 Since the MC4R receptor protein is a key step in the pathway for appetite control in the hypothalamus, it has been a target for studies on lifestyle interactions including physical activity. The effect of an MC4R variant on BMI was modified by physical activity in Chinese children135, and cardiometabolic risk in metabolically healthy obese Chinese children was predicted by a KCNQ1 variant, and it interacted with walking to school.144 The attenuating effect of physical activity on the effect of the FTO risk variant on BMI and waist circumference adds support for the variant as being helpful for weight loss in the Food4Me trial.145
Several genetic variants have been shown to interact with the effects of obesity, smoking and/or exercise on increasing triglyceride and lowering HDL-C 146, highlighting the importance of interactive lifestyle factors and their potential use in enhancing prediction. The LPL S447S (rs328) and Hind III (rs320) variants modified the effect of a high carbohydrate diet on triglyceride and HDL-C in a young Chinese population, especially in females suggesting that lifestyle intervention strategies for dietary intervention could be targeted according to genetic variants and gender.147
CLINICAL RELEVANCE OF GENETIC MODEL IN RISK PREDICTION
Genetic risk predictors represent loci or variants that contribute to polygenic obesity or may increase the risk of obesity in individuals who are not yet obese or are below obesity thresholds. Since over a hundred candidate genes have been linked to BMI, with most acting in the central nervous system and influencing food intake 148, identification of gene network pathways may facilitate prediction and can lead to therapies directed at modifying behaviors including appetite. However, existing literature suggests that predicting obesity using genetic models based on common variants (representing GWAS loci), still have poorer predictive value than traditional clinical predictors such as a family history or obesity in childhood.149 Incomplete understanding of the human genetic variation due to population heterogeneity, admixture, lack of population specific SNP arrays, incomplete mapping of regulatory domains etc., could likely contribute to the poor performance of genetic models. As data from more and more global populations are being added to the GWAS catalogue and the variant repositories, it may improve predictive power of genetic scores. This includes data obtained from targeted and whole genome sequencing in high-risk or susceptible populations.150 Furthermore, a potentially productive and accurate prediction strategy for complex traits like obesity or diabetes will only be accomplished when regulatory domains are identified.151 Therefore, in the current scenario, genetic information can only supplement the clinical history but cannot replace it. However, considering the usefulness of genetic models in better predicting the weight gain before middle age provides rationale for early screening which will be important for timely interventions.152 Also, the weight gain trajectory in early adult life is associated with greater risk for poor cardio-metabolic outcomes such as hypertension, diabetes and dyslipidemia, supporting the argument for using genetics for predicting weight gain 6, and degree of weight gain. However, identification of putative genetic predictors is still incomplete.
FUTURISTIC MODEL
Genetic variation collectively account for only 8–10 % of total heritability linked with cardiovascular traits in GWAS, hence efforts are needed to account for missing heritability using more biologically associated explanations. Accounting for the effects of environmental interactions in a time-sensitive manner may contribute to solving the problem. Ongoing research continues to improve design and tools for better evaluation of genetic and non-genetic factors. However, effective prediction early in the pathological sequence of atherosclerosis, is central to preventing or offsetting progression to recognized end-points. Obesity has a recognized benchmark, beginning with rapid weight gain clinically seen on childhood weight charts when percentiles are crossed with a trajectory towards overweight and obesity thresholds. This observation presents as an opportunity for detection of early obesity trends as signals for evaluation of genetic interaction. Similarly, insulin resistance has a progressive and graded presentation with corresponding effects on the risk factor cluster, including NAFLD and inflammation. Since genetic information on both monogenic and polygenic obesity has shown a predominance of appetite and brain-associated gene variants, intervention strategies have moved towards modifying the respective behaviors. Our futuristic model depends on knowing the predictive power of selected variants, phenotype reversibility and effective therapeutic choices based on genetic interactions. However, less is currently known about the role genes play in pathogenesis than how they predict treatment effects by interacting with nutritional, exercise and pharmacological interventions. Gene variants constitute the genetic background for obesity in part via the brain and appetite centers, subsequent insulin resistance, and the cluster of metabolic pathways influencing glucose intolerance, dyslipidemia, blood pressure, hepatic fat deposition and inflammation influence CVD progression in four main stages. These pathways can provide a road-map for intervention by using information from genetic variation that interact with nutrition, exercise and pharmacological treatments (Figure 2).
Interaction of gene variants influences effective primary, secondary and tertiary preventive strategies and CVD reversal. The ultimate goal is to improve end-points enabling treatment planning for timely interventions (Figure 3). Novel variant discovery by fine mapping, use of gene scores, whole exome sequencing and accounting for gene-gene and gene-environment interactions might contribute to improvements in prediction. However, the wide spectrum of clinical presentations for obesity and related complications presents as an organizational challenge.
Figure 3.
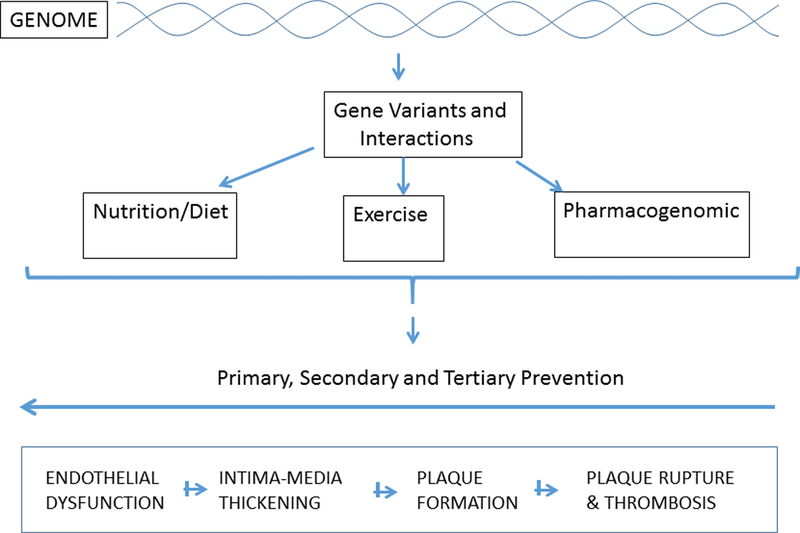
Influence of genetic interactions with various treatment modalities. Futuristic screening model suggesting the usefulness of clinically relevant genetic information and their interactions with dietary and lifestyle factors to design effective primary, secondary and tertiary preventive strategies for effective interventions for CVD reversal.
CONCLUSIONS
Adverse influence of obesity on CVD has the potential to be lowered by coordinated preventive approaches based on genetic prediction so that multiple risk factors can be simultaneously decreased. The genetic background of obesity and associated risk factors provides a basis for using genetic variants to offset their expression of CVD phenotypes and appears to be a feasible clinical strategy. The challenge is to select SNPs that can be identified as clinically recognized phenotypes. Altered carbohydrate homeostasis leading to insulin resistance and T2D appears to be central to this cluster that also includes hyperglycemia, hyperlipidemia and hypertension. Ectopic deposition of fat in muscle and in liver (manifesting as NAFLD), and inflammation with contributions from the gut microbiome are more recently recognized additions. Relatively little is known about the genetic background for inflammation and how it interacts with the microbiome, although they make significant contributions to CVD and present as possible therapeutic targets. Therefore, the plausible futuristic model would include the discovery of putative genetic and non-genetic factors that interact with phenotypes, and may help predict CVD risk and facilitate early detection, risk reversal, and effectual individualized intervention.
Acknowledgments
Funding
This work was supported by NIH grant -R01DK082766 funded by the National Institute of Health (NIDDK), and grants from Harold Hamm Diabetes Center and Presbyterian Health Foundation of Oklahoma.
Footnotes
Conflict of interest
We declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract January 2010;87(1):4–14. [DOI] [PubMed] [Google Scholar]
- 2.Stevens GA, Singh GM, Lu Y, et al. National, regional, and global trends in adult overweight and obesity prevalences. Popul Health Metr November 20 2012;10(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manson JE, Skerrett PJ, Greenland P, VanItallie TB. The escalating pandemics of obesity and sedentary lifestyle. A call to action for clinicians. Arch Intern Med February 09 2004;164(3):249–258. [DOI] [PubMed] [Google Scholar]
- 4.Rexrode KM, Buring JE, Manson JE. Abdominal and total adiposity and risk of coronary heart disease in men. Int J Obes Relat Metab Disord July 2001;25(7):1047–1056. [DOI] [PubMed] [Google Scholar]
- 5.Rexrode KM, Carey VJ, Hennekens CH, et al. Abdominal adiposity and coronary heart disease in women. JAMA December 02 1998;280(21):1843–1848. [DOI] [PubMed] [Google Scholar]
- 6.Xian H, Vasilopoulos T, Liu W, et al. Steeper change in body mass across four decades predicts poorer cardiometabolic outcomes at midlife. Obesity (Silver Spring) April 2017;25(4):773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bischoff SC, Boirie Y, Cederholm T, et al. Towards a multidisciplinary approach to understand and manage obesity and related diseases. Clin Nutr August 2017;36(4):917–938. [DOI] [PubMed] [Google Scholar]
- 8.Vine M, Hargreaves MB, Briefel RR, Orfield C. Expanding the role of primary care in the prevention and treatment of childhood obesity: a review of clinic- and community-based recommendations and interventions. J Obes 2013;2013:172035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith AW, Borowski LA, Liu B, et al. U.S. primary care physicians’ diet-, physical activity-, and weight-related care of adult patients. Am J Prev Med July 2011;41(1):33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.l’Allemand-Jander D Clinical diagnosis of metabolic and cardiovascular risks in overweight children: early development of chronic diseases in the obese child. Int J Obes (Lond) December 2010;34 Suppl 2:S32–36. [DOI] [PubMed] [Google Scholar]
- 11.Cook S, Weitzman M, Auinger P, Nguyen M, Dietz WH. Prevalence of a metabolic syndrome phenotype in adolescents: findings from the third National Health and Nutrition Examination Survey, 1988–1994. Arch Pediatr Adolesc Med August 2003;157(8):821–827. [DOI] [PubMed] [Google Scholar]
- 12.Haffner SM, Mykkanen L, Festa A, Burke JP, Stern MP. Insulin-resistant prediabetic subjects have more atherogenic risk factors than insulin-sensitive prediabetic subjects: implications for preventing coronary heart disease during the prediabetic state. Circulation March 07 2000;101(9):975–980. [DOI] [PubMed] [Google Scholar]
- 13.Cummings DE, Schwartz MW. Genetics and pathophysiology of human obesity. Annu Rev Med 2003;54:453–471. [DOI] [PubMed] [Google Scholar]
- 14.Kaur Y, de Souza RJ, Gibson WT, Meyre D. A systematic review of genetic syndromes with obesity. Obes Rev June 2017;18(6):603–634. [DOI] [PubMed] [Google Scholar]
- 15.Farooqi IS, O’Rahilly S. Genetic factors in human obesity. Obes Rev March 2007;8 Suppl 1:37–40. [DOI] [PubMed] [Google Scholar]
- 16.Adithan C, Gerard N, Vasu S, Rosemary J, Shashindran CH, Krishnamoorthy R. Allele and genotype frequency of CYP2C19 in a Tamilian population. British journal of clinical pharmacology September 2003;56(3):331–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farooqi IS, O’Rahilly S. New advances in the genetics of early onset obesity. Int J Obes (Lond) October 2005;29(10):1149–1152. [DOI] [PubMed] [Google Scholar]
- 18.Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature February 12 2015;518(7538):197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akiyama M, Okada Y, Kanai M, et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nature genetics October 2017;49(10):1458–1467. [DOI] [PubMed] [Google Scholar]
- 20.Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science May 11 2007;316(5826):889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Kilpelainen TO, Liu C, et al. Association of genetic variation in FTO with risk of obesity and type 2 diabetes with data from 96,551 East and South Asians. Diabetologia April 2012;55(4):981–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hallman DM, Friedel VC, Eissa MA, et al. The association of variants in the FTO gene with longitudinal body mass index profiles in non-Hispanic white children and adolescents. Int J Obes (Lond) January 2012;36(1):61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu G, Zhu H, Dong Y, Podolsky RH, Treiber FA, Snieder H. Influence of common variants in FTO and near INSIG2 and MC4R on growth curves for adiposity in African- and European-American youth. Eur J Epidemiol June 2011;26(6):463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rutters F, Nieuwenhuizen AG, Bouwman F, Mariman E, Westerterp-Plantenga MS. Associations between a single nucleotide polymorphism of the FTO Gene (rs9939609) and obesity-related characteristics over time during puberty in a Dutch children cohort. J Clin Endocrinol Metab June 2011;96(6):E939–942. [DOI] [PubMed] [Google Scholar]
- 25.Speakman JR. The ‘Fat Mass and Obesity Related’ (FTO) gene: Mechanisms of Impact on Obesity and Energy Balance. Curr Obes Rep March 2015;4(1):73–91. [DOI] [PubMed] [Google Scholar]
- 26.Gerken T, Girard CA, Tung YC, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science November 30 2007;318(5855):1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Church C, Lee S, Bagg EA, et al. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS genetics August 2009;5(8):e1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet November 2010;42(11):937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamilton MK, Raybould HE. Bugs, guts and brains, and the regulation of food intake and body weight. Int J Obes Suppl December 2016;6(Suppl 1):S8–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stunkard AJ, Harris JR, Pedersen NL, McClearn GE. The body-mass index of twins who have been reared apart. N Engl J Med May 24 1990;322(21):1483–1487. [DOI] [PubMed] [Google Scholar]
- 31.Nordestgaard BG, Palmer TM, Benn M, et al. The effect of elevated body mass index on ischemic heart disease risk: causal estimates from a Mendelian randomisation approach. PLoS Med 2012;9(5):e1001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagg S, Fall T, Ploner A, et al. Adiposity as a cause of cardiovascular disease: a Mendelian randomization study. Int J Epidemiol April 2015;44(2):578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Didelez V, Sheehan N. Mendelian randomization as an instrumental variable approach to causal inference. Stat Methods Med Res August 2007;16(4):309–330. [DOI] [PubMed] [Google Scholar]
- 34.Fall T, Ingelsson E. Genome-wide association studies of obesity and metabolic syndrome. Mol Cell Endocrinol January 25 2014;382(1):740–757. [DOI] [PubMed] [Google Scholar]
- 35.Nead KT, Li A, Wehner MR, et al. Contribution of common non-synonymous variants in PCSK1 to body mass index variation and risk of obesity: a systematic review and meta-analysis with evidence from up to 331 175 individuals. Hum Mol Genet June 15 2015;24(12):3582–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turcot V, Lu Y, Highland HM, et al. Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nature genetics January 2018;50(1):26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabo A, Mishra P, Dugan-Perez S, et al. Exome sequencing reveals novel genetic loci influencing obesity-related traits in Hispanic children. Obesity (Silver Spring) July 2017;25(7):1270–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Expert Committee on the D, Classification of Diabetes M. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care January 2000;23 Suppl 1:S4–19. [PubMed] [Google Scholar]
- 39.Unwin N, Shaw J, Zimmet P, Alberti KG. Impaired glucose tolerance and impaired fasting glycaemia: the current status on definition and intervention. Diabet Med September 2002;19(9):708–723. [DOI] [PubMed] [Google Scholar]
- 40.Reiling E, van ‘t Riet E, Groenewoud MJ, et al. Combined effects of single-nucleotide polymorphisms in GCK, GCKR, G6PC2 and MTNR1B on fasting plasma glucose and type 2 diabetes risk. Diabetologia September 2009;52(9):1866–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen WM, Erdos MR, Jackson AU, et al. Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. The Journal of clinical investigation July 2008;118(7):2620–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet February 2010;42(2):105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saxena R, Hivert MF, Langenberg C, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet February 2010;42(2):142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends in molecular medicine September 2010;16(9):407–416. [DOI] [PubMed] [Google Scholar]
- 45.Doria A, Patti ME, Kahn CR. The emerging genetic architecture of type 2 diabetes. Cell metabolism September 2008;8(3):186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell February 21 1997;88(4):561–572. [DOI] [PubMed] [Google Scholar]
- 47.Reaven GM. Insulin resistance: the link between obesity and cardiovascular disease. Med Clin North Am September 2011;95(5):875–892. [DOI] [PubMed] [Google Scholar]
- 48.Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet September 11–17 2004;364(9438):937–952. [DOI] [PubMed] [Google Scholar]
- 49.Cooper SA, Whaley-Connell A, Habibi J, et al. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am J Physiol Heart Circ Physiol October 2007;293(4):H2009–2023. [DOI] [PubMed] [Google Scholar]
- 50.Pilbrow AP, Palmer BR, Frampton CM, et al. Angiotensinogen M235T and T174M gene polymorphisms in combination doubles the risk of mortality in heart failure. Hypertension February 2007;49(2):322–327. [DOI] [PubMed] [Google Scholar]
- 51.Palmer BR, Pilbrow AP, Yandle TG, et al. Angiotensin-converting enzyme gene polymorphism interacts with left ventricular ejection fraction and brain natriuretic peptide levels to predict mortality after myocardial infarction. Journal of the American College of Cardiology March 5 2003;41(5):729–736. [DOI] [PubMed] [Google Scholar]
- 52.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res June 1994;74(6):1141–1148. [DOI] [PubMed] [Google Scholar]
- 53.Yamakawa T, Tanaka S, Numaguchi K, et al. Involvement of Rho-kinase in angiotensin II-induced hypertrophy of rat vascular smooth muscle cells. Hypertension January 2000;35(1 Pt 2):313–318. [DOI] [PubMed] [Google Scholar]
- 54.Richey JM, Ader M, Moore D, Bergman RN. Angiotensin II induces insulin resistance independent of changes in interstitial insulin. Am J Physiol November 1999;277(5 Pt 1):E920–926. [DOI] [PubMed] [Google Scholar]
- 55.Ogihara T, Asano T, Ando K, et al. Angiotensin II-induced insulin resistance is associated with enhanced insulin signaling. Hypertension December 2002;40(6):872–879. [DOI] [PubMed] [Google Scholar]
- 56.Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sci (Lond) June 2007;112(7):375–384. [DOI] [PubMed] [Google Scholar]
- 57.Norton GR, Brooksbank R, Woodiwiss AJ. Gene variants of the renin-angiotensin system and hypertension: from a trough of disillusionment to a welcome phase of enlightenment? Clin Sci (Lond) January 26 2010;118(8):487–506. [DOI] [PubMed] [Google Scholar]
- 58.Hsiao CF, Sheu WW, Hung YJ, et al. The effects of the renin-angiotensin-aldosterone system gene polymorphisms on insulin resistance in hypertensive families. J Renin Angiotensin Aldosterone Syst December 2012;13(4):446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franceschini N, Carty CL, Lu Y, et al. Variant Discovery and Fine Mapping of Genetic Loci Associated with Blood Pressure Traits in Hispanics and African Americans. PLoS One 2016;11(10):e0164132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hertz RP, Unger AN, Cornell JA, Saunders E. Racial disparities in hypertension prevalence, awareness, and management. Arch Intern Med October 10 2005;165(18):2098–2104. [DOI] [PubMed] [Google Scholar]
- 61.Cutler JA, Sorlie PD, Wolz M, Thom T, Fields LE, Roccella EJ. Trends in hypertension prevalence, awareness, treatment, and control rates in United States adults between 1988–1994 and 1999–2004. Hypertension November 2008;52(5):818–827. [DOI] [PubMed] [Google Scholar]
- 62.Fox ER, Young JH, Li Y, et al. Association of genetic variation with systolic and diastolic blood pressure among African Americans: the Candidate Gene Association Resource study. Hum Mol Genet June 01 2011;20(11):2273–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thompson EE, Kuttab-Boulos H, Witonsky D, Yang L, Roe BA, Di Rienzo A. CYP3A variation and the evolution of salt-sensitivity variants. American journal of human genetics December 2004;75(6):1059–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tobin MD, Tomaszewski M, Braund PS, et al. Common variants in genes underlying monogenic hypertension and hypotension and blood pressure in the general population. Hypertension June 2008;51(6):1658–1664. [DOI] [PubMed] [Google Scholar]
- 65.Tobin MD, Timpson NJ, Wain LV, et al. Common variation in the WNK1 gene and blood pressure in childhood: the Avon Longitudinal Study of Parents and Children. Hypertension November 2008;52(5):974–979. [DOI] [PubMed] [Google Scholar]
- 66.International Consortium for Blood Pressure Genome-Wide Association S, Ehret GB, Munroe PB, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature September 11 2011;478(7367):103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jia EZ, Xu ZX, Guo CY, et al. Renin-angiotensin-aldosterone system gene polymorphisms and coronary artery disease: detection of gene-gene and gene-environment interactions. Cell Physiol Biochem 2012;29(3–4):443–452. [DOI] [PubMed] [Google Scholar]
- 68.Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J Pediatr March 2013;162(3):496–500 e491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schwimmer JB, Pardee PE, Lavine JE, Blumkin AK, Cook S. Cardiovascular risk factors and the metabolic syndrome in pediatric nonalcoholic fatty liver disease. Circulation July 15 2008;118(3):277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Targher G, Arcaro G. Non-alcoholic fatty liver disease and increased risk of cardiovascular disease. Atherosclerosis April 2007;191(2):235–240. [DOI] [PubMed] [Google Scholar]
- 71.Hashimoto Y, Hamaguchi M, Fukuda T, Ohbora A, Kojima T, Fukui M. Fatty liver as a risk factor for progression from metabolically healthy to metabolically abnormal in non-overweight individuals. Endocrine July 2017;57(1):89–97. [DOI] [PubMed] [Google Scholar]
- 72.Anderson EL, Howe LD, Jones HE, Higgins JP, Lawlor DA, Fraser A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS One 2015;10(10):e0140908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kalia HS, Gaglio PJ. The Prevalence and Pathobiology of Nonalcoholic Fatty Liver Disease in Patients of Different Races or Ethnicities. Clin Liver Dis May 2016;20(2):215–224. [DOI] [PubMed] [Google Scholar]
- 74.Kumashiro N, Erion DM, Zhang D, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A September 27 2011;108(39):16381–16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petersen KF, Dufour S, Feng J, et al. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proceedings of the National Academy of Sciences of the United States of America November 28 2006;103(48):18273–18277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Targher G, Bertolini L, Padovani R, Zoppini G, Zenari L, Falezza G. Associations between liver histology and carotid intima-media thickness in patients with nonalcoholic fatty liver disease. Arterioscler Thromb Vasc Biol December 2005;25(12):2687–2688. [DOI] [PubMed] [Google Scholar]
- 77.Cali AM, Caprio S. Ectopic fat deposition and the metabolic syndrome in obese children and adolescents. Horm Res January 2009;71 Suppl 1:2–7. [DOI] [PubMed] [Google Scholar]
- 78.Wang J, Guo XF, Yu SJ, et al. Adiponectin polymorphisms and non-alcoholic fatty liver disease risk: a meta-analysis. J Gastroenterol Hepatol 2014;29(7):1396–1405. [DOI] [PubMed] [Google Scholar]
- 79.Zhou D, Jin Y, Yao F, Duan Z, Wang Q, Liu J. Association between the adiponectin +45T>G genotype and risk of cardiovascular disease: a meta-analysis. Heart Lung Circ February 2014;23(2):159–165. [DOI] [PubMed] [Google Scholar]
- 80.Burgert TS, Taksali SE, Dziura J, et al. Alanine aminotransferase levels and fatty liver in childhood obesity: associations with insulin resistance, adiponectin, and visceral fat. J Clin Endocrinol Metab November 2006;91(11):4287–4294. [DOI] [PubMed] [Google Scholar]
- 81.Kotronen A, Yki-Jarvinen H, Aminoff A, et al. Genetic variation in the ADIPOR2 gene is associated with liver fat content and its surrogate markers in three independent cohorts. Eur J Endocrinol April 2009;160(4):593–602. [DOI] [PubMed] [Google Scholar]
- 82.Kotronen A, Yki-Jarvinen H. Fatty liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol January 2008;28(1):27–38. [DOI] [PubMed] [Google Scholar]
- 83.Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med September 30 2010;363(14):1341–1350. [DOI] [PubMed] [Google Scholar]
- 84.Cali AM, Zern TL, Taksali SE, et al. Intrahepatic fat accumulation and alterations in lipoprotein composition in obese adolescents: a perfect proatherogenic state. Diabetes care December 2007;30(12):3093–3098. [DOI] [PubMed] [Google Scholar]
- 85.Pacifico L, Cantisani V, Ricci P, et al. Nonalcoholic fatty liver disease and carotid atherosclerosis in children. Pediatr Res April 2008;63(4):423–427. [DOI] [PubMed] [Google Scholar]
- 86.Sparks JD, Sparks CE. Overindulgence and metabolic syndrome: is FoxO1 a missing link? J Clin Invest June 2008;118(6):2012–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gambino R, Cassader M, Pagano G, Durazzo M, Musso G. Polymorphism in microsomal triglyceride transfer protein: a link between liver disease and atherogenic postprandial lipid profile in NASH? Hepatology May 2007;45(5):1097–1107. [DOI] [PubMed] [Google Scholar]
- 88.Musso G, Gambino R, Cassader M. Lipoprotein metabolism mediates the association of MTP polymorphism with beta-cell dysfunction in healthy subjects and in nondiabetic normolipidemic patients with nonalcoholic steatohepatitis. J Nutr Biochem September 2010;21(9):834–840. [DOI] [PubMed] [Google Scholar]
- 89.Jun DW, Han JH, Jang EC, et al. Polymorphisms of microsomal triglyceride transfer protein gene and phosphatidylethanolamine N-methyltransferase gene in alcoholic and nonalcoholic fatty liver disease in Koreans. Eur J Gastroenterol Hepatol June 2009;21(6):667–672. [DOI] [PubMed] [Google Scholar]
- 90.Petersen KF, Dufour S, Hariri A, et al. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N Engl J Med March 25 2010;362(12):1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee HY, Birkenfeld AL, Jornayvaz FR, et al. Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology November 2011;54(5):1650–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leung JC, Loong TC, Wei JL, et al. Histological severity and clinical outcomes of nonalcoholic fatty liver disease in nonobese patients. Hepatology January 2017;65(1):54–64. [DOI] [PubMed] [Google Scholar]
- 93.Wei JL, Leung JC, Loong TC, et al. Prevalence and Severity of Nonalcoholic Fatty Liver Disease in Non-Obese Patients: A Population Study Using Proton-Magnetic Resonance Spectroscopy. Am J Gastroenterol September 2015;110(9):1306–1314; quiz 1315. [DOI] [PubMed] [Google Scholar]
- 94.Hyysalo J, Gopalacharyulu P, Bian H, et al. Circulating triacylglycerol signatures in nonalcoholic fatty liver disease associated with the I148M variant in PNPLA3 and with obesity. Diabetes January 2014;63(1):312–322. [DOI] [PubMed] [Google Scholar]
- 95.Petaja EM, Yki-Jarvinen H. Definitions of Normal Liver Fat and the Association of Insulin Sensitivity with Acquired and Genetic NAFLD-A Systematic Review. Int J Mol Sci April 27 2016;17(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sookoian S, Pirola CJ. Genetic predisposition in nonalcoholic fatty liver disease. Clin Mol Hepatol March 2017;23(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nature genetics December 2008;40(12):1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Romeo S, Sentinelli F, Cambuli VM, et al. The 148M allele of the PNPLA3 gene is associated with indices of liver damage early in life. J Hepatol August 2010;53(2):335–338. [DOI] [PubMed] [Google Scholar]
- 99.Santoro N, Zhang CK, Zhao H, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology March 2012;55(3):781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology June 2011;53(6):1883–1894. [DOI] [PubMed] [Google Scholar]
- 101.Kumari M, Schoiswohl G, Chitraju C, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab May 02 2012;15(5):691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Oliver P, Caimari A, Diaz-Rua R, Palou A. Diet-induced obesity affects expression of adiponutrin/PNPLA3 and adipose triglyceride lipase, two members of the same family. Int J Obes (Lond) February 2012;36(2):225–232. [DOI] [PubMed] [Google Scholar]
- 103.Romeo S, Sentinelli F, Dash S, et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int J Obes (Lond) January 2010;34(1):190–194. [DOI] [PubMed] [Google Scholar]
- 104.Yuan X, Waterworth D, Perry JR, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet October 2008;83(4):520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee J Adipose tissue macrophages in the development of obesity-induced inflammation, insulin resistance and type 2 diabetes. Arch Pharm Res February 2013;36(2):208–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature May 19 2011;473(7347):317–325. [DOI] [PubMed] [Google Scholar]
- 107.Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta March 2014;1842(3):446–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol 2012;30:759–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Carvalho BM, Saad MJ. Influence of gut microbiota on subclinical inflammation and insulin resistance. Mediators Inflamm 2013;2013:986734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Remely M, Aumueller E, Merold C, et al. Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity. Gene March 01 2014;537(1):85–92. [DOI] [PubMed] [Google Scholar]
- 111.Wang C, Ha X, Li W, et al. Correlation of TLR4 and KLF7 in Inflammation Induced by Obesity. Inflammation February 2017;40(1):42–51. [DOI] [PubMed] [Google Scholar]
- 112.Leitao-Goncalves R, Carvalho-Santos Z, Francisco AP, et al. Commensal bacteria and essential amino acids control food choice behavior and reproduction. PLoS Biol April 2017;15(4):e2000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut October 2013;62(10):1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature October 4 2012;490(7418):55–60. [DOI] [PubMed] [Google Scholar]
- 115.Boursier J, Mueller O, Barret M, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology March 2016;63(3):764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Been LF, Hatfield JL, Shankar A, et al. A low frequency variant within the GWAS locus of MTNR1B affects fasting glucose concentrations: Genetic risk is modulated by obesity. Nutrition Metabolism and Cardiovascular Diseases November 2012;22(11):944–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Been LF, Ralhan S, Wander GS, et al. Variants in KCNQ1 increase type II diabetes susceptibility in South Asians: A study of 3,310 subjects from India and the US. Bmc Medical Genetics January 2011;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ross R The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature April 29 1993;362(6423):801–809. [DOI] [PubMed] [Google Scholar]
- 119.Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. American journal of physiology. Cell physiology April 2001;280(4):C719–741. [DOI] [PubMed] [Google Scholar]
- 120.Kugiyama K, Kerns SA, Morrisett JD, Roberts R, Henry PD. Impairment of endothelium-dependent arterial relaxation by lysolecithin in modified low-density lipoproteins. Nature March 8 1990;344(6262):160–162. [DOI] [PubMed] [Google Scholar]
- 121.Kooner JS, Saleheen D, Sim X, et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nature genetics October 2011;43(10):984–U994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rees SD, Islam M, Hydrie MZI, et al. An FTO variant is associated with Type 2 diabetes in South Asian populations after accounting for body mass index and waist circumference. Diabetic Medicine June 2011;28(6):673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Anderson TJ, Uehata A, Gerhard MD, et al. Close relation of endothelial function in the human coronary and peripheral circulations. Journal of the American College of Cardiology November 1 1995;26(5):1235–1241. [DOI] [PubMed] [Google Scholar]
- 124.Xiang L, Wu H, Pan A, et al. FTO genotype and weight loss in diet and lifestyle interventions: a systematic review and meta-analysis. Am J Clin Nutr April 2016;103(4):1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lappalainen T, Lindstrom J, Paananen J, et al. Association of the fat mass and obesity-associated (FTO) gene variant (rs9939609) with dietary intake in the Finnish Diabetes Prevention Study. Br J Nutr November 28 2012;108(10):1859–1865. [DOI] [PubMed] [Google Scholar]
- 126.Tang H, Zhang Z, Li Z, Lin J, Fang DZ. High-Carbohydrate/Low-Fat Diet-Induced Gender-Specific Serum Lipid Profile Changes Are Associated with LEPR Polymorphisms in Chinese Youth. Ann Nutr Metab 2017;70(1):1–8. [DOI] [PubMed] [Google Scholar]
- 127.Ma W, Huang T, Heianza Y, et al. Genetic Variations of Circulating Adiponectin Levels Modulate Changes in Appetite in Response to Weight-Loss Diets. J Clin Endocrinol Metab January 01 2017;102(1):316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ma W, Huang T, Wang M, et al. Two-year changes in circulating adiponectin, ectopic fat distribution and body composition in response to weight-loss diets: the POUNDS Lost Trial. Int J Obes (Lond) November 2016;40(11):1723–1729. [DOI] [PubMed] [Google Scholar]
- 129.Ma W, Huang T, Zheng Y, et al. Weight-Loss Diets, Adiponectin, and Changes in Cardiometabolic Risk in the 2-Year POUNDS Lost Trial. J Clin Endocrinol Metab June 2016;101(6):2415–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Heianza Y, Ma W, Huang T, et al. Macronutrient Intake-Associated FGF21 Genotype Modifies Effects of Weight-Loss Diets on 2-Year Changes of Central Adiposity and Body Composition: The POUNDS Lost Trial. Diabetes care November 2016;39(11):1909–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Braun TR, Been LF, Singhal A, et al. A Replication Study of GWAS-Derived Lipid Genes in Asian Indians: The Chromosomal Region 11q23.3 Harbors Loci Contributing to Triglycerides. PloS one May 2012;7(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang TJ, Zhang F, Richards JB, et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet July 17 2010;376(9736):180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sapkota BR, Hopkins R, Bjonnes A, et al. Genome-wide association study of 25(OH) Vitamin D concentrations in Punjabi Sikhs: Results of the Asian Indian diabetic heart study. The Journal of steroid biochemistry and molecular biology April 2016;158:149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wortsman J, Matsuoka LY, Chen TC, Lu Z, Holick MF. Decreased bioavailability of vitamin D in obesity. The American journal of clinical nutrition September 2000;72(3):690–693. [DOI] [PubMed] [Google Scholar]
- 135.Drincic A, Fuller E, Heaney RP, Armas LA. 25-Hydroxyvitamin D response to graded vitamin D(3) supplementation among obese adults. The Journal of clinical endocrinology and metabolism December 2013;98(12):4845–4851. [DOI] [PubMed] [Google Scholar]
- 136.Drincic AT, Armas LA, Van Diest EE, Heaney RP. Volumetric dilution, rather than sequestration best explains the low vitamin D status of obesity. Obesity (Silver Spring) July 2012;20(7):1444–1448. [DOI] [PubMed] [Google Scholar]
- 137.Parekh D, Sarathi V, Shivane VK, Bandgar TR, Menon PS, Shah NS. Pilot study to evaluate the effect of short-term improvement in vitamin D status on glucose tolerance in patients with type 2 diabetes mellitus. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists Jul-Aug 2010;16(4):600–608. [DOI] [PubMed] [Google Scholar]
- 138.Hoffhines H, Whaley KD, Blackett PR, et al. Early childhood nutrition in an American Indian community: educational strategy for obesity prevention. J Okla State Med Assoc February 2014;107(2):55–59. [PubMed] [Google Scholar]
- 139.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med March 20 2003;348(12):1085–1095. [DOI] [PubMed] [Google Scholar]
- 140.Qi L, Kraft P, Hunter DJ, Hu FB. The common obesity variant near MC4R gene is associated with higher intakes of total energy and dietary fat, weight change and diabetes risk in women. Human molecular genetics November 15 2008;17(22):3502–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kilpelainen TO, Qi L, Brage S, et al. Physical activity attenuates the influence of FTO variants on obesity risk: a meta-analysis of 218,166 adults and 19,268 children. PLoS Med November 2011;8(11):e1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Celis-Morales C, Marsaux CF, Livingstone KM, et al. Physical activity attenuates the effect of the FTO genotype on obesity traits in European adults: The Food4Me study. Obesity (Silver Spring) April 2016;24(4):962–969. [DOI] [PubMed] [Google Scholar]
- 143.Vimaleswaran KS, Bodhini D, Lakshmipriya N, et al. Interaction between FTO gene variants and lifestyle factors on metabolic traits in an Asian Indian population. Nutr Metab (Lond) 2016;13:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li L, Yin J, Cheng H, et al. Identification of Genetic and Environmental Factors Predicting Metabolically Healthy Obesity in Children: Data From the BCAMS Study. J Clin Endocrinol Metab April 2016;101(4):1816–1825. [DOI] [PubMed] [Google Scholar]
- 145.Celis-Morales C, Marsaux CF, Livingstone KM, et al. Can genetic-based advice help you lose weight? Findings from the Food4Me European randomized controlled trial. Am J Clin Nutr May 2017;105(5):1204–1213. [DOI] [PubMed] [Google Scholar]
- 146.Cole CB, Nikpay M, McPherson R. Gene-environment interaction in dyslipidemia. Curr Opin Lipidol April 2015;26(2):133–138. [DOI] [PubMed] [Google Scholar]
- 147.Huang X, Gong R, Lin J, et al. Effects of lipoprotein lipase gene variations, a high-carbohydrate low-fat diet, and gender on serum lipid profiles in healthy Chinese Han youth. Biosci Trends 2011;5(5):198–204. [DOI] [PubMed] [Google Scholar]
- 148.Yeo GS. Genetics of obesity: can an old dog teach us new tricks? Diabetologia May 2017;60(5):778–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Loos RJ, Janssens AC. Predicting Polygenic Obesity Using Genetic Information. Cell Metab March 07 2017;25(3):535–543. [DOI] [PubMed] [Google Scholar]
- 150.Wu Y, Wang W, Jiang W, Yao J, Zhang D. An investigation of obesity susceptibility genes in Northern Han Chinese by targeted resequencing. Medicine (Baltimore) February 2017;96(7):e6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Morrison AC, Huang Z, Yu B, et al. Practical Approaches for Whole-Genome Sequence Analysis of Heart- and Blood-Related Traits. Am J Hum Genet February 02 2017;100(2):205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rukh G, Ahmad S, Ericson U, et al. Inverse relationship between a genetic risk score of 31 BMI loci and weight change before and after reaching middle age. Int J Obes (Lond) February 2016;40(2):252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]