

Abstract
Gene therapy holds great promise for the treatment of many diseases, but clinical translation of gene therapies has been slowed by the lack of safe and efficient gene delivery systems. Here, we report two versatile redox-responsive polyplexes (i.e., crosslinked and non-crosslinked) capable of efficiently delivering a variety of negatively charged payloads including plasmid DNA (DNA), messenger RNA (mRNA), Cas9/gRNA ribonucleoprotein (RNP), and RNP-donor DNA complexes (S1mplex) without any detectable cytotoxicity. The key component of both types of polyplexes is a cationic poly(N,N’-bis(acryloyl)cystamine-co-triethylenetetramine) (p(BAC-TET)) polymer (a type of poly(N,N’-bis(acryloyl)cystamine-poly(aminoalkyl)) (PBAP) polymer) containing disulfide bonds in the backbone and bearing imidazole groups. This composition enables efficient encapsulation, cellular uptake, controlled endo/lysosomal escape, and cytosolic unpacking of negatively-charged payloads. To further enhance the stability of non-crosslinked PBAP polyplexes, adamantane (AD) and β-cyclodextrin (β-CD) were conjugated to the PBAP-based polymers. The crosslinked PBAP (CLPBAP) polyplexes formed by host–guest interaction between β-CD and AD were more stable than non-crosslinked PBAP polyplexes in the presence of polyanionic polymers such as serum albumin, suggesting enhanced stability in physiological conditions. Both crosslinked and non-crosslinked polyplexes demonstrated either similar or better transfection and genome editing efficiencies, and significantly better biocompatibility than Lipofectamine 2000, a commercially available state-of-the-art transfection agent that exhibits cytotoxicity.
Keywords: Redox-responsive polymer, crosslinked polyplex, gene editing, gene therapy, nonviral delivery
GRAPHICAL ABSTRACT
INTRODUCTION
Gene therapy has tremendous therapeutic potential to prevent and treat a wide range of pathological conditions over the last two decades 1. However, clinical translation has limited due to various technical barriers, particularly, the lack of safe and efficient gene delivery systems 1–2. Both plasmid DNA (DNA) and messenger RNA (mRNA) have been widely investigated for gene therapy 1, 3, and can be used to express functional proteins 2, 4–5. To function, mRNA needs to reach the cytosol of the target cell, while DNA usually translocates to the cell nucleus for transgene expression. Both DNA and mRNA can result in relatively safe and rapid protein production for disease treatment 2, 4, 6. However, due to their relatively large sizes and high negative charge densities, naked DNA and mRNA exhibit low cellular uptake efficiency 7–9. Furthermore, naked plasmid DNA and mRNA are also susceptible to chemical and/or enzymatic degradation 1, 10–12. Similar issues are encountered when payloads encode or contain genome editors. The clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 system constitute a powerful class of genome editors 13–14. The Cas9/sgRNA ribonucleoprotein (RNP) can edit a target gene with high efficiency and specificity 13–15. Moreover, the combination of a RNP with a single-stranded oligonucleotide DNA (ssODN) donor template (i.e., RNP-ssODN complex, for example, the recently reported S1mplex 16) can achieve precise genome editing to incorporate exogenous sequences from the ssODN template. However, safe and efficient delivery of RNP and S1mplex remains a significant challenge for their potential application owing to their relatively large and complex structures 17–18. Similar to plasmid DNA and mRNA, unpackaged RNP and S1mplex are also susceptible to chemical degradation. Furthermore, in comparison to DNA and mRNA delivery, the delivery of protein/nucleic acid complexes such as RNP and S1mplex is more challenging due to their mixed charges (e.g., positively charged Cas9 protein and negatively charged sgRNA and ssODN) and more sophisticated structures 19–20. Therefore, judiciously designed delivery systems for these genetic materials (e.g., DNA, mRNA, RNP, and S1mplex) are essential to achieve high transfection or genome editing efficiencies.
Viral vectors have been employed to deliver both nucleic acids and the CRISPR gene editing system, however, the application of viral vectors is limited due to their potential immunogenicity, carcinogenesis, broad tropism, poor reproducibility and high cost 1, 21–22. Non-viral nanovectors, including lipid-based 19, 23–25 nanoparticles (NPs), cationic polymer-based NPs 5, 26–28 and functionalized inorganic NPs 29–31 have also been actively investigated for the delivery of nucleic acids and gene editing agents. However, current state-of-the-art non-viral nanovectors often have lower transfection efficiencies than viral vectors, and non-viral nanovectors also face other challenges including insufficient in vivo stability 32–33.
Due to their chemical versatility, high reproducibility, low immunogenicity, and low cost, cationic polymer-based nanovectors offer a promising approach for gene therapy. Cationic polymers can interact with negatively charged genetic materials via electrostatic charge to form polyplexes. Desirable gene delivery nanovectors should not only protect payloads from chemical degradation, but also chaperone the payloads to the desired subcellular space (e.g., nucleus for DNA, Cas9 RNP, and RNP-repair template complex; cytosol for mRNA) of the target cells 16. This requires the nanovectors to overcome critical extracellular and intracellular barriers while maintaining biocompatibility. For instance, an ideal nanovector for systemic delivery must be stable during circulation in the bloodstream. In fact, insufficient in vivo stability of nanovectors, including polyplexes and lipoplexes, in the presence of polyanions (e.g., serum albumin and heparin) is one of the major stumbling blocks for their in vivo application 32–33. The nanovectors also need to be efficiently taken up by the target cells, followed by efficient endo/lysosomal escape and rapid release of the payloads in the desired subcellular location. Besides insufficient stability under physiological conditions, common polyplexes also exhibit relatively high positive zeta potential and cytotoxicity due to the use of cationic polymers, which hinders their clinical application. Only a few reports have used polyplexes for the nonviral delivery of Cas9 RNP and other RNP complexes 1, 34.
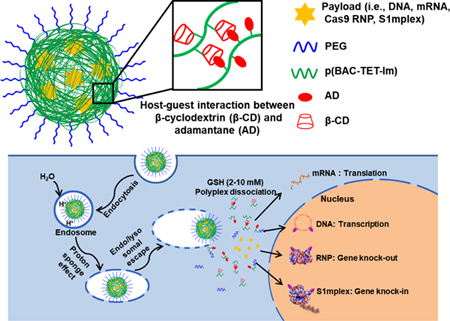
Here we report two (i.e., crosslinked vs. non-crosslinked) bioreducible nanovectors made of a family of cationic poly(N,N’-bis(acryloyl)cystamine-co-triethylenetetramine) (p(BAC-TET)) based polymers (a type of poly(N,N’-bis(acryloyl)cystamine-poly(aminoalkyl)) (PBAP) polymer) that are capable of delivering DNA, mRNA, Cas9 RNP, and S1mplex with high transfection efficiency and low cytotoxicity (Figure 1). These PBAP-based polymers contain redox-responsive disulfide bonds in their main chain for the rapid release of payloads in the cytosol. They also bear imidazole groups that enable rapid endo/lysosomal escape. The non-crosslinked PBAP polyplexes were formed by mixing the payloads with PBAP first and then PEG (polyethylene glycol)–PBAP–PEG (Figure 1(C)). PEGylation can shield the positive surface charge, reduce cytotoxicity, allow for versatile ligand conjugations when desirable, and potentially enhance the stability and circulation time of the polyplexes in vivo 35. The crosslinked PBAP (i.e., CLPBAP) polyplexes were formed by first mixing the payloads with a mixture of PBAP bearing adamantane (AD) and PBAP bearing β-cyclodextrin (β-CD), followed by mixing with PEG–PBAP–PEG bearing AD (Figure 1 (A) and (B)). The host-guest interaction between β-CD and AD greatly enhanced the stability of the polyplexes against polyanions, which could be advantageous for both in vitro and in vivo applications.
Figure 1.

(A) Illustration of a CLPBAP polyplex for the delivery of various negatively charged payloads. (B) Chemical structures of the PBAP-based polymers used to form crosslinked polyplexes including CLPBAP polymers (i.e., p(BAC-TET-Im/AD) and p(BAC-TET-Im/β-CD)), and PEG–p(BAC-TET-Im/AD)–PEG. (C) Chemical structures of the PBAP-based polymers used to prepare non-crosslinked PBAP polyplexes including the PBAP polymer (i.e., p(BAC-TET-Im)) and PEG–p(BAC-TET-Im)–PEG. (D) Formation of the crosslinked CLPBAP polyplexes and their proposed intracellular trafficking pathways. CLPBAP: crosslinked poly(N,N’-bis(acryloyl)cystamine-poly(aminoalkyl)); p(BAC-TET): poly(N,N’-bis(acryloyl)cystamine-co-triethylenetetramine; Im:imidazole; AD: adamantane; β-CD: β-cyclodextrin; PEG: poly(ethylene glycol, Mw = 5000 Da); GSH: glutathione
MATERIALS AND METHODS
Materials
Triethylenetetramine (TET), triethylamine (TEA), carboxymethyl-β-cyclodextrin sodium salt (β-CD, degree of carboxyl substitution = 3), 4-imidazolecarboxylic acid, N,N’-dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), L-glutathione (reduced, GSH) and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (USA). Ethyl trifluoroacetate, N,N’-bis(acryloyl)cystamine (BAC), and 1-adamantanecarboxylic acid (AD) were purchased from TCI America (USA). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was purchased from Acros (USA). A poly(ethylene glycol) (PEG) derivative, H2N-PEG-OCH3 (Mw = 5000 Da), was purchased from JenKem Technology (USA). The nuclear localization signal (NLS) peptide (sequence: PKKKKRKV) was purchased from Abi Scientific (USA). Bovine serum albumin (BSA) was purchased from Fisher (USA). NLS-tagged Cas9 protein (sNLS–Cas9–sNLS) was purchased from Aldevron (USA).
Synthesis of N,N’-[1,2-ethanediylbis(imino-2,1-ethanediyl)]bis[2,2,2-trifluoroacetamide] (Compound 1)
Ethyl trifluoroacetate was used to protect the primary amines in TET. TET (219 mg, 1.5 mmol, 1 equiv.) and TEA (455 mg, 4.5 mmol, 3 equiv.) were dissolved in 30 ml of methanol. Ethyl trifluoroacetate (532 mg, 3.75 mmol, 2.5 equiv.) dissolved in 20 ml of methanol was added dropwise to the mixture and stirred at 20 °C. After 24 h, the solvent was evaporated, and the mixture was purified by silica gel flash chromatography using an eluent of 1:1 v/v ethyl acetate and methanol to yield the product as a white solid (406 mg, 80 % yield). 1H NMR (400 MHz, DMSO-d6): δ 2.56 (s, 4H, CF3CONHCH2CH2NHCH2CH2NHCH2CH2NHCOCF3); δ 2.63 (t, 4H, CF3CONHCH2CH2NHCH2CH2NHCH2CH2NHCOCF3); δ 3.24 (t, 4H, CF3CONHCH2CH2NHCH2CH2NHCH2CH2NHCOCF3).
Synthesis of poly(N,N’-bis(acryloyl)cystamine-co-triethylenetetramine) (p(BAC-TET))
p(BAC-TET) was synthesized via a Michael addition reaction between Compound 1 and N,N’-bis(acryloyl)cystamine (BAC). BAC (28.6 mg, 0.11 mmol) and Compound 1 (33.8 mg, 0.1 mmol) were dissolved in DMF and reacted at a 1.1:1 molar ratio at 90 °C for 96 h. Polymers were then precipitated in cold ether three times to remove any unreacted species and dried under vacuum. The precipitate was then dissolved in 1 M NaOH and stirred for 2 h to deprotect the trifluoroacetate groups. Then the polymer solution was neutralized by 1 M HCl and purified by dialysis against deionized (DI) water (molecular weight cut-off (MWCO): 2 kDa). The final p(BAC-TET) product was obtained after lyophilization. 1H NMR (400 MHz, DMSO-d6): δ 2.15–2.40 (m, 68 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 2.52–2.96 (m, 100 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)-CH2CH2CONHCH2CH2S); δ 3.20–3.46 (m, 70 H, SCH2CH2NHCOCH2CH2-(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 5.55 (dd, 2 H, CH1H2=CH-CONH-, terminal acryloyl group); δ 6.03 (dd, 2 H, CH1H2=CH-CONH-, terminal acryloyl group); δ 6.11 (dd, 2 H, CH1H2=CH-CONH-,terminal acryloyl group).
Synthesis of PEG–p(BAC-TET) –PEG
PEG was conjugated to the terminals of p(BAC-TET) by Michael addition. p(BAC-TET) (15 mg, 0.0018 mmol) with protected primary amines and mPEG-NH2 (22 mg, 0.0044 mmol) were dissolved in anhydrous DMF and stirred at 50 °C for 24 h. Then the polymers were precipitated in cold ether three times to remove any unreacted species and vacuum dried. The precipitate was dissolved in 1 M NaOH and stirred for 2 h to deprotect the trifluoroacetate groups 36. The final PEG–p(BAC-TET)–PEG polymer solution was purified by dialysis against deionized(DI) water (molecular weight cut-off: 8 kDa) followed by lyophilization. 1H NMR (400 MHz, D2O): δ 2.67–2.79 (m, 36 H, SCH2CH2NHCOCH2CH2-(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 3.01–3,10 (m, 34 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 3.15–3.28 (m, 100 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2) CH2CH2CONHCH2CH2S); δ 3.35–3.55 (m, 70 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 3.60 (s, 900 H, CH2CH2O-, PEG).
Synthesis of p(BAC-TET-Im/AD) and PEG–p(BAC-TET-Im/AD) –PEG
Imidazole and adamantane (AD) groups were conjugated to either p(BAC-TET) or PEG–p(BAC-TET) –PEG by DCC/NHS catalyzed amidation. The feed molar ratio of p(BAC-TET) (or PEG–p(BAC-TET) –PEG : Im was controlled as 1:4, while the feed ratio of p(BAC-TET) (or PEG–p(BAC-TET) –PEG) : AD was 1:5. Typically, p(BAC-TET) or PEG-p(BAC-TET)-PEG (0.0011 mmol), 4-imidazolecarboxylic acid (0.5 mg, 0.0044 mmol), 1-adamantanecarboxylic acid (1 mg, 0.0055 mmol), DCC (2.1 mg, 0.01 mmol), and NHS (1.2mg, 0.01 mmol) were dissolved in 4 ml of anhydrous DMSO and stirred at room temperature. After 24 h, the mixture was filtered through a Büchner funnel to remove the byproduct dicyclohexylurea and dialyzed against DI water (MWCO 8 kDa). PEG– p(BAC-TET-Im) –PEG for non-crosslinked polyplex was synthesized with the same PEG– p(BAC-TET) –PEG: Im feed ratio without 1-adamantanecarboxylic acid addition. 1H NMR: p(BAC-TET-Im/AD) (400 MHz, DMSO-d6): δ 1.61–1.81 (d, 64 CHCH2, adamantane); δ 2.14–2.43 (m, 68 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 2.55–2.92 (m, 108 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 3.21–3.46 (m, 70 H, SCH2CH2NHCOCH2CH2-(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)-CH2CH2CONHCH2CH2S); δ 5.55 (dd, 2 H, CH1H2=CH-CONH-, terminal acryloyl group); δ 6.03 (dd, 2 H, CH1H2=CH-CONH-, terminal acryloyl group); δ 6.11 (dd, 2 H, CH1H2=CH-CONH-,terminal acryloyl group). δ 7.82–8.32 (m, 7 H, imidazole). PEG-p(BAC-TET-Im/AD)-PEG (400 MHz, D2O): δ 1.60–1.81 (d, 63 CHCH2, adamantane); δ 2.65–2.80 (m, 36 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 3.01–3.10 (m, 40 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 3.15–3.28 (m, 100 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2) CH2CH2CONHCH2CH2S); δ 3.35–3.55 (m, 70 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 3.60 (s, 900 H, CH2CH2O-, PEG); δ 7.82–8.22 (m, 7 H, imidazole).
Synthesis of p(BAC-TET-Im/β-CD)
Imidazole and β-CD groups were conjugated to p(BAC-TET) by EDC/NHS catalyzed amidation in water. The feed molar ratio of p(BAC-TET): Im and p(BAC-TET): β-CD were both controlled at 1:4. p(BAC-TET) (9.4 mg, 0.0011 mmol), 4-imidazolecarboxylic acid (0.5mg, 0.0044 mmol), β-CD (4.5 mg, 0.004 mmol), EDC (1.6 mg, 0.01 mmol), and NHS (1.2 mg, 0.01 mmol) were dissolved in 4 ml of DI water and stirred at room temperature for 24 h. The final product was obtained by dialysis against DI water (MWCO 8 kDa). p(BAC-TET-Im) for the non-crosslinked polyplex was synthesized with the same p(BAC-TET)):Im feed ratio without β-CD addition. 1H NMR: (400 MHz, DMSO-d6): δ 2.14–2.43 (m, 68 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 2.58–2.92 (m, 103 H, SCH2CH2NHCOCH2CH2(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)CH2CH2CONHCH2CH2S); δ 3.21–3.70 (m, 200 H, SCH2CH2NHCOCH2CH2-(NH2CH2CH2)NCH2CH2N(CH2CH2NH2)-CH2CH2CONHCH2CH2 S and β-CD); δ 5.55 (dd, 2 H, CH1H2=CH-CONH-, terminal acryloyl group); δ 6.03 (dd, 2 H, CH1H2=CH-CONH-, terminal acryloyl group); δ 6.11 (dd, 2 H, CH1H2=CH-CONH-,terminal acryloyl group). δ 7.62–8.22 (m, 7 H, imidazole).
Characterization
The chemical structures of the polymers were analyzed by 1H-NMR spectroscopy. The molecular weights of the PBAP polymers were studied by gel permeation chromatography (GPC) system equipped with a refractive index detector, a viscometer detector, and a light scattering detector (Viscotek, USA). The hydrodynamic diameter and zeta potential of the polyplexes were characterized by a dynamic light scattering (DLS) spectrometer (Malvern Zetasizer Nano ZS) at a 90° detection angle with a polyplex concentration of 0.1 mg/ml. Polyplex morphologies were characterized by transmission electron microscopy (TEM, Philips CM200 Ultra Twin).
Preparation of the Non-Crosslinked and Crosslinked Polyplexes with Various Nucleic Acids and CRIPR-Cas9 Genome Editing Machinery
Polymer/payload polyplexes at various weight ratios were prepared using a two-step method. To form the non-crosslinked PBAP polyplexes, a sodium acetate buffer (NaOAc, 25 mM, pH 5.5) solution of p(BAC-TET-Im) polymer (5 mg/ml) was added to a payload solution (0.5 mg/ml) with varying p(BAC-TET-Im)-to-payload weight ratios. Sodium acetate buffer (NaOAc, 25 mM, pH 5.5) was then added to adjust the payload concentration to 50 μg/ml. The mixture was vortexed for 15 s and incubated for 30 min at room temperature to form the primary polyplexes. Then a sodium acetate buffer (NaOAc, 25 mM, pH 5.5) solution of PEG–p(BAC-TET-Im) –PEG polymer (5 mg/ml) and sodium acetate buffer (NaOAc, 25 mM, pH 5.5) were added to the primary polyplex solution to keep the payload concentration at 25 μg/ml. The mixture was vortexed (15 s) and incubated (30 min) at room temperature to obtain the final non-crosslinked PBAP polyplexes. To form the crosslinked PBAP (i.e., CLPBAP) polyplexes, sodium acetate buffer (NaOAc, 25 mM, pH 5.5) solutions of p(BAC-TET-Im/β-CD) and p(BAC-TET-Im/AD) polymers (5 mg/ml) were added to a payload solution (0.5 mg/ml) with a pre-determined molar ratio of β-CD and AD as well as sodium acetate buffer (NaOAc, 25 mM, pH 5.5) to adjust the payload concentration to 50 μg/ml, followed by vortexing (15 s) and incubation (30 min) at room temperature to obtain the primary crosslinked polyplexes. Then PEG–p(BAC-TET-Im/AD) –PEG polymer solution (5 mg/ml) and sodium acetate buffer (NaOAc, 25 mM, pH 5.5) were added to the primary crosslinked polyplex solution to adjust the final payload concentration to 25 μg/ml, followed by vortexing (15 s) and incubation (30 min) at room temperature to obtain the final CLPBAP polyplexes. To complex DNA with NLS, DNA (0.5 mg/ml) was incubated with an NLS solution (1 mg/ml in NaOAc buffer) at an N/P ratio of 0.25 (the ratio of moles of the amine groups of NLS to those of the phosphate groups of DNA) for 20 min, then formed polyplexes with PBAP polymers.
To study the stability of DNA polyplexes in the presence of different polyanions including albumin, non-crosslinked and crosslinked DNA polyplexes were prepared and incubated under different conditions (i.e., cell culture medium containing 10 % FBS, and 40 mg/ml BSA solution in PBS. The size change of non-crosslinked and crosslinked DNA polyplexes over time was studied by DLS.
Cell Culture
Cells were cultured in a humidified cell culture incubator (Thermo Fisher, USA) at 37°C with 5% carbon dioxide. HEK 293 cells (a human embryonic kidney cell line) and RAW 264.7 cells (a mouse monocyte cell line) were purchased from ATCC (USA) and cultured with DMEM medium (Gibco, USA) with 10% (v/v) Fetal Bovine Serum (FBS, Gibco, USA) and 1% (v/v) Penicillin-Streptomycin (Gibco, USA). HCT 116 cells (a human colon cancer cell line) were cultured with 89% McCoy’s 5A medium, 10% FBS, and 1% penicillin−streptomycin. NHDF (normal human dermal fibroblast) cells were cultured with Fibroblast Growth Medium (Sigma-Aldrich, USA).
DNA Transfection Efficiency Study
Twenty-four hour prior to treatment, HEK 293 and HCT116 cells were seeded onto a 96 well plate at the amount of 20,000 per well, NHDF cells were seeded onto a 96 well plate at the amount of 10,000 per well. The cell culture medium volume was 100 μl/well. Cells were transfected with green fluorescence protein (GFP) plasmid (Addgene #40259, USA, 200 ng/well) using Lipofectamine™ 2000 (Lipo 2000, Thermo Fisher, USA) loaded with DNA, non-crosslinked PBAP polyplexes with different polymer/DNA weight ratios, and crosslinked CLPBAP polyplexes with different AD:β-CD molar ratios (4:2, 4:3, 4:4, 4:5 and 4:6). An untreated group was used as the control group. DNA was also transfected using Lipo 2000 following the manufacturer’s instructions, with a Lipo 2000 dosage of 0.5 μl/well. Cells were harvested with 0.25% trypsin (Thermo Fisher, USA) 24 h and 48 h post-treatment, spun down, and resuspended with 500 μl PBS (Thermo Fisher, USA). GFP expression efficiencies were obtained with an Attune NxT flow cytometer system (Thermo Fisher, USA) and analyzed with FlowJo 7.6.
To study the stability of the non-crosslinked and crosslinked DNA polyplexes in the presence of GSH, transfection experiments were carried out under similar conditions, using GSH containing media. The GSH concentration in media varied from 0.001 to 20 mM.
To study the effects of the PEG and imidazole groups, two special non-crosslinked DNA polyplexes were prepared: (1) polyplexes formed by PEG-lacking p(BAC-TET-Im) polymers with a p(BAC-TET-Im):DNA weight ratio of 60:1 (i.e., the same p(BAC-TET-Im) content as the polyplex formulation with a p(BAC-TET-Im): PEG–p(BAC-TET-Im)–PEG:DNA weight ratio of 48:28:1); (2) polyplexes formed by imidazole-lacking polymers p(BAC-TET) and PEG–p(BAC-TET) –PEG. The transfection experiments were carried out under similar conditions.
mRNA Transfection Efficiency Study
HEK 293, RAW 264.7, HCT116 and NHDF cells were used as mRNA transfection model cells. Twenty-four hours before treatment, HEK 293, RAW 264.7 and HCT116 cells were seeded onto a 96–well plate at the amount of 20,000 per well, NHDF cells were seeded onto a 96 well plate at the amount of 10,000 per well. Cells were transfected with GFP mRNA (OZ Biosciences, USA, 200 ng/well) using Lipo 2000 (0.5 μl/well), as well as non-crosslinked and crosslinked mRNA polyplexes prepared at varying polymer/mRNA weight ratios. HEK 293 cells were harvested at 4, 6, 10, 24 and 48 h with 0.25% EDTA-trypsin, while RAW 264.7 cells were harvested at the same time points by repeatedly pipetting. The cells were spun down and resuspended with 500 μl of PBS. GFP expression percentages were obtained with flow cytometry and analyzed with FlowJo 7.6. In a GSH stability study, gradient concentrations of GSH (0 to 20 mM) were added into mRNA treatment solution. HEK 293 cells were harvested 24 h later and GFP transfection efficiencies were analyzed by flow cytometry.
RNP Genome Editing Efficiency Study
mCherry-expressing HEK 293 cells (HEK-H2B-mCherry) were generated as described previously 16, 37 and used as a RNP transfection cell model. Twenty-four hours prior to treatment, mCherry HEK 293 cells were seeded onto a 96 well plate at 15,000 cells per well. RNP was prepared as previously reported 16, by mixing NLS-tagged Cas9 protein and in vitro transcribed sgRNA (mCherry targeting guide sequence: GGAGCCGTACATGAACTGAG) at a 1:1 molar ratio. Cells were treated with complete medium, Lipo 2000 (0.5 μl/well) loaded with RNP, and non-crosslinked PBAP and crosslinked CLPBAP polyplexes at varying polymer/RNP weight ratios. The amount of RNP for each treatment was maintained at 156 ng Cas9 RNP (i.e., 125 ng Cas9 protein) per well. A quantity of 100 μl of fresh culture medium was added into each well two days after treatment and thereafter, half of the culture medium was refreshed every day. Six days after treatment, cells were treated with trypsin, neutralized with serum, spun down and re-suspended with 500 μl of PBS. The RNP gene editing efficiencies were quantified via gating for mCherry negative cells with flow cytometry and data were analyzed with FlowJo 7.6.
Genome Editing with Polyplexes
S1mplex are complexes made of Cas9 protein, sgRNA with a S1m aptamer, streptavidin, and a ssODN donor template. Both S1m aptamer and ssODN can bind to streptavidin to form a complex 16. S1mplex was prepared as reported previously by mixing the four components at 4°C for 5 min 16. To study the genome editing efficiency of S1mplex complexed with the non-crosslinked and crosslinked polyplexes, blue fluorescence protein (BFP) HEK 293 cells generated through lentiviral transduction of a BFP dest clone (Addgene, Cambridge, MA) were employed 16. When cells are transfected with S1mPlex containing sgRNA targeting BFP (target sequence: GCTGAAGCACTGCACGCCAT) and ssODN, if precise editing occurs, three nucleotides within BFP are edited to generate a green fluorescent protein (GFP) as described previously 16. BFP HEK 293 cells were seeded onto a 96 well plate (15,000 per well) 24 h prior to treatment. Cells were treated with Lipo 2000 (0.5 μl/well) loaded with S1mplex, as well as non-crosslinked and crosslinked polyplexes complexed with S1mplex at varying polymer to S1mplex ratios. For each treatment, the S1mplex dosage was kept at 235 ng/well (i.e., an equivalent Cas9 protein dosage of 125 ng/well). The gene editing knock-in efficiencies were quantified six days after treatment using flow cytometry for green fluorescent cells.
Intracellular Trafficking of DNA Polyplexes
Intracellular trafficking of DNA polyplexes were studied by confocal laser scanning microscopy (CLSM). Cy3.5-conjugated DNA was used for subcellular tracking. Crosslinked CLPBAP polyplexes were prepared with or without NLS being complexed with DNA. Twenty-four hours before treatment, HEK 293 cells were seeded onto a Nunc™ Lab-Tek™ II CC2™ Chamber Slide (Thermo Fisher, USA, 50,000 per well). At each time point (i.e., 0.5, 2, and 6 h) after crosslinked DNA polyplexes treatment, the cells were stained with endosome/lysosome marker LysoTracker Green DND-26 (100 nM) and nucleus marker Hoechst 33342 (5 μg/mL) for 30 min at 37 oC. DNA subcellular localization images were obtained with a confocal laser scanning microscope (CLSM, Nikon, Japan).
Cell Viability of the Non-Crosslinked and Crosslinked Polyplexes with Different Types of Payloads (i.e., mRNA, DNA, RNP and S1mplex)
HEK 293, mCherry HEK 293, and BFP HEK 293 cells were seeded onto a 96 well plate (20,000 per well). Cells were treated with complete medium, Lipo 2000 loaded with a payload, and non-crosslinked or crosslinked polyplexes containing a specific payload (e.g., DNA, mRNA, RNP, or S1mplex) fabricated as described in the transfection assays. For the non-crosslinked and crosslinked polyplexes, optimized formulations that provided the highest transfection or genome editing efficiencies were used for the cell viability studies. Cell viability was measured using a standard MTT assay 48 h after treatment (Thermo Fisher,USA). Briefly, cells were treated with media containing 500 μg/ml MTT and incubated for 4h. Then the MTT-containing media was aspirated. Next, the purple precipitates were dissolved in 150 μl DMSO. The absorbance at 560 nm was obtained with a micro-plate reader (GloMax®-Multi Detection System, Promega, USA).
Statistical Analysis
Results are presented as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) with Tukey’s multiple comparisons was used to determine the difference between independent groups. Statistical analyses were conducted using GraphPad Prism software version 6.
RESULTS AND DISCUSSION
Synthesis and Characterization of a Family of PBAP-Based Polymers
The non-crosslinked PBAP polyplexes were formed by p(BAC-TET-Im) (i.e., PBAP) and PEG–PBAP–PEG (Figure 1(C)), while the crosslinked CLPBAP polyplexes were formed by p(BAC-TET-Im/AD) and p(BAC-TET-Im/β-CD) (i.e., CLPBAP), as well as PEG–p(BAC-TET-Im/AD)–PEG (i.e., PEG–CLPBAP–PEG) (Figure 1(A), (C) and (D)). Both crosslinked and non-crosslinked polyplexes were made of PBAP-based polymers containing redox-responsive disulfide bonds in the backbone to facilitate the release of payloads in the cytosol as well as imidazole groups for efficient endo/lysosomal escape. PBAP-based polymers conjugated with β-CD and AD that are capable of forming crosslinks via the host-guest interaction between β-CD and AD, were used to form stable crosslinked polyplexes. As demonstrated in Figure 1 (D), once the polyplexes are taken up by cells through endocytosis, they can escape quickly from endo/lysosomal compartments due to the proton sponge effect enhanced by the imidazole groups. The polyplexes fall apart once they enter the cytosol, where the disulfide bond-containing polymer backbone is degraded by high-concentration GSH, thereby releasing the payloads. mRNA functions in cytosol, while DNA, RNP and S1mplex, facilitated by NLS, will be transported into the nucleus.
Figure 2 shows the synthesis scheme for the three PBAP-based polymers used to form the CLPBAP polyplexes. First, trifluoroacetate (TFA)-protected TET (Compound 1) was polymerized with disulfide bond-containing monomer BAC through Michael addition. To ensure that both terminal groups are acrylamides, the feed molar ratio of BAC:Compound 1 was set at 1.1:1. After the removal of the TFA protecting groups, imidazole groups and crosslinking groups (i.e., AD and β-CD) were conjugated onto the p(BAC-TET) polymer via DCC/NHS catalyzed amidation to yield p(BAC-TET-Im/AD) and p(BAC-TET-Im/β-CD). To synthesize the PEG–p(BAC-TET) –PEG polymers, methoxy(m)PEG-NH2 was conjugated to TFA-protected PBAP polymer through Michael addition, followed by TFA deprotection. To ensure that PEG–p(BAC-TET)–PEG polymer molecules can also be integrated into the crosslinked polyplexes via β-CD and AD host–guest interactions, PEG–p(BAC-TET) –PEG was conjugated with both AD and imidazole groups to form PEG–p(BAC-TET-Im/AD)–PEG. PBAP-based polymers used to form non-crosslinked PBAP polyplexes, namely, p(BAC-TET-Im) and PEG-p(BAC-TET-Im)-PEG, were synthesized similarly without the conjugation of AD and β-CD (Figure S1) The chemical structures of all intermediate and final polymer products were confirmed by 1H NMR (Figure S2). The number-average molecular weight (Mn) of p(BAC-TET), characterized by GPC, was 8.5 kDa with a polydispersity index (PDI) of 1.6. The numbers of AD and β-CD groups conjugated onto p(BAC-TET-Im) were controlled to 4 and 3, respectively, per polymer chain, as confirmed by NMR (Figure S2 (A)) and GPC (Table S1). The host–guest interactions between β-CD and AD in CLPBAP polymers were confirmed by 1H NMR (Figure S3). A downfield shift of the AD peaks was observed from 1H NMR spectrum when the AD: β-CD molar ratio decreased from 4:2 to 4:5, indicating hydrophobic interaction within the host–guest complex 38.
Figure 2.

Synthesis scheme for the three PBAP-based polymers (i.e., CLPBAP polymers) used to form the crosslinked CLPBAP polyplexes.
Preparation and Characterization of the Non-Crosslinked and Crosslinked Polyplexes
Non-crosslinked PBAP polyplexes were fabricated by mixing p(BAC-TET-Im) (i.e., PBAP) with payloads to form the primary polyplexes first, followed by the addition of PEG– p(BAC-TET-Im)–PEG (i.e., PEG–PBAP–PEG) to yield the final PEGylated polyplexes. To fabricate the crosslinked CLPBAP polyplexes, CLPBAP polymers (i.e., p(BAC-TET-Im/AD) and p(BAC-TET-Im/β-CD)) were mixed with payloads and then incubated for 30 min to allow for complete complexation between AD and β-CD. Thereafter, the PEG–p(BAC-TET-Im/AD)–PEG (i.e., PEG–CLPBAP–PEG) polymer was added to the primary polyplexes to yield the PEGylated crosslinked polyplexes. The sizes and morphologies of the non-crosslinked and crosslinked polyplexes with different payloads were studied by transmission electron microscopy (TEM) as shown in Figure 3 (A) and (B), respectively. The size distribution and zeta potential of the various polyplexes were studied by DLS (Table S2). The average hydrodynamic diameters of the non-crosslinked polyplexes ranged from 136 to 151 nm, depending on the type of payload, while the average hydrodynamic diameters of the crosslinked polyplexes ranged from 168 to 191 nm. Both non-crosslinked and crosslinked polyplexes had nearly neutral surface charges with zeta potentials ranging from −1.5 to 8.6 mV.
Figure 3.

(A-B) TEM images of (A) non-crosslinked and (B) crosslinked polyplexes with different payloads. From left to right: DNA, mRNA, RNP, and S1mplex. The scale bar is 200 nm. (C) Stability of the non-crosslinked and crosslinked polyplexes in FBS-containing cell culture medium and a polyanion solution (BSA, 40 mg/ml in PBS). (D) Effects of GSH concentration in cell culture media on the mRNA transfection efficiency of both crosslinked and non-crosslinked polyplexes. Non-crosslinked polyplexes were formed with a PBAP:PEG–PBAP–PEG:mRNA weight ratio of 48:28:1; while the crosslinked polyplexes were formed with a CLPBAP:PEG–CLPBAP–PEG:mRNA weight ratio of 48:28:1 and an AD:β-CD molar ratio of 4:3. ****: p < 0.0001; n = 3.
Stability Study of the Non-Crosslinked PBAP and Crosslinked CLPBAP Polyplexes
Nanoparticles (NPs) formed by cationic polymers and cationic lipids are commonly used for the delivery of negatively charged nucleic acids and proteins. However, NPs formed solely by electrostatic interactions possess insufficient stability in vitro and in vivo. During circulation, the stability of such NPs can be affected by several factors including dilution, flow stress, and interaction with serum proteins. For instance, polyanions such as serum albumin and heparin can destabilize NPs and cause premature release of payloads 32–33, 39–40. To overcome the potentially poor stability of non-crosslinked PBAP polyplexes, β-CD and AD were conjugated to the p(BAC-TET-Im) polymer backbone. The host–guest interactions between β-CD and AD, in addition to the electrostatic interactions, may enhance the stability of the resulting crosslinked CLPBAP polyplexes.
To investigate the stability of the non-crosslinked and crosslinked polyplexes against polyanions, non-crosslinked and crosslinked DNA polyplexes formed by PBAP and CLPBAP polymers, respectively, were incubated in a cell culture medium (DMEM containing 10% FBS) and a polyanion solution (BSA solution, 40 mg/ml in PBS, mimicking the albumin concentration in vivo) 41. The sizes of the polyplexes were monitored over time. As shown in Figure 3(C), the sizes of both non-crosslinked and crosslinked polyplexes remained unchanged in FBS-containing cell culture media, indicating that both types of polyplexes exhibited good stability in cell culture media. However, when the non-crosslinked PBAP polyplexes were exposed to a BSA solution with a BSA concentration similar to the blood albumin concentration, their sizes increased significantly after 2 h, indicating the interruption of the polyplex structure by polyanions. In contrast, the size of the crosslinked CLPBAP polyplexes did not change notably throughout the study period, thus indicating enhanced stability, which is desirable for in vivo applications.
Disulfide bonds were integrated into the PBAP-based polymer backbone to facilitate payload release into the cytosol where the GSH concentration (2–10 mM) is much higher than in the extracellular spaces (0.001–0.02 mM). While it is highly desirable to rapidly release the payload into the cytosol of the target cell, where the payload can function (e.g., mRNA) or be transported to the nucleus (e.g., DNA, RNP, and S1mplex), it is also essential to keep the polyplexes intact in the blood stream and other extracellular spaces. To study the stability of the polyplexes at different GSH levels, GFP mRNA polyplexes were prepared and incubated with HEK 293 cells for 24 h in culture media with GSH concentrations ranging from 0–20 mM. GFP transfection efficiency was measured to determine the functionality of the polyplexes. As shown in Figure 3(D), mRNA transfection was not affected at GSH concentrations lower than 0.1 mM for both non-crosslinked and crosslinked polyplexes, indicating that both types of polyplexes exhibited good stability at extracellular GSH levels. A significant decrease in transfection efficiencies was observed at a GSH concentration of 1 mM or higher, suggesting that GSH induced polyplex degradation occurred in the cell culture media, which may cause mRNA release before cellular uptake. These findings have demonstrated that these GSH-responsive polyplexes are stable at extracellular spaces, but can fall apart and release payloads effectively in the cytosol of the target cells.
Effects of Polyplex Formulations on DNA Delivery
To study the effects of various polyplex formulations on DNA transfection efficiencies, the weight ratio of PBAP: PEG–PBAP–PEG:DNA was optimized initially using the non-crosslinked polyplexes (Figure 4 (A)) in HEK 293 cells. Three weight ratios, namely, PBAP:PEG–PBAP–PEG:DNA = 24:14:1, 48:28:1, and 72:42:1, were studied. GFP-expressing plasmid DNA was used to measure the transfection efficiency of these polyplex formulations. Among the three formulations studied, the two formulations with higher PBAP polymer ratios showed comparable transfection efficiencies to Lipofectamine 2000, a commercially available liposome-based delivery system. The mean fluorescence intensity (MFI) data indicated a similar trend (Figure S4). Among the three different weight ratios, PBAP:PEG–PBAP–PEG:DNA weight ratios of 48:28:1 and 72:42:1 induced similar, but higher transfection efficiency as compared to the PBAP:PEG-PBAP-PEG:DNA weight ratio of 24:14:1. However, the polyplexes with a PBAP:PEG–PBAP–PEG:DNA weight ratio of 48:28:1 exhibited significantly lower cytotoxicity than the one with a weight ratio of 72:42:1(Figure 4 (B)). Therefore, the PBAP:PEG–PBAP–PEG:DNA weight ratio of 48:28:1 was used for further optimization of DNA delivery. At a DNA dosage of 200 ng/well, the corresponding PBAP and PEG-PBAP-PEG concentrations were 96 μg/ml and 56 μg/ml, respectively.
Figure 4. Efficient delivery of plasmid DNA by the polyplexes.

(A) The effects of polyplex formulation on the DNA transfection efficiency of polyplexes. Cells were treated with Lipo 2000, non-crosslinked polyplexes with three different PBAP:PEG–PBAP–PEG: DNA weight ratios, as well as the polyplex with a PBAP:PEG–PBAP–PEG: DNA weight ratio of 48:28:1 together with NLS (N/P ratio = 0.25). (B) Effects of polyplex formulation on the cytotoxicity of non-crosslinked DNA polyplexes with different weight ratios. (C) Optimization of the AD:β-CD molar ratio in crosslinked CLPBAP polyplexes. HEK 293 cells were treated with Lipo 2000, non-crosslinked PBAP polyplexes, and crosslinked CLPBAP polyplexes with different crosslinker molar ratios. The transfection efficiency was measured 48 h post treatment. (D) Intracellular trafficking of the crosslinked DNA polyplex with and without NLS. Scale bar: 10 μm. (E-F) Effects of PEG and imidazole groups on the transfection efficiency of the crosslinked polyplexes. (E) Transfection efficiency of PEG- or imidazole-lacking crosslinked polyplexes. (F) Cell viability of PEG-lacking crosslinked polyplexes. NS: not significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001; n = 3.
The intracellular trafficking pathway is important for proper function of the payload. The nuclear membrane acts as a barrier to prevent free passage of macromolecules, making nuclear entry challenging but crucial for DNA and CRISPR-Cas9 genome editing machinery. To induce efficient nuclear translocation and ultimately, efficient transfection, NLS (positively charged) was complexed with negatively charged DNA through electrostatic interactions before polyplex formation. As shown in Figure 4(A), complexation of DNA with a small amount of NLS (N/P ratio = 0.25) before polyplex formation can significantly enhance DNA transfection efficiency, owing to NLS’s nuclear translocation capability. To study the intracellular trafficking of the crosslinked DNA polyplexes, imaging using CLSM was performed at different time points. As shown in Figure 4(D), the co-localization of DNA and endo/lysosomes was observed as early as 0.5 h post treatment, indicating the uptake of polyplexes through endocytosis. The co-localization of DNA and endo/lysosomes considerably decreased 2 h post treatment, indicating the efficient endo/lysosomal escape capability of the polyplexes facilitated by the imidazole groups. The crosslinked DNA polyplexes with NLS showed greater overlapping of DNA signals and nucleus 6 h post treatment, indicating that NLS effectively facilitated the nuclear entry of DNA. Non-crosslinked DNA polyplexes exhibited a similar intracellular trafficking behavior as crosslinked polyplexes (Figure S8(A)). Furthermore, mRNA polyplexes also exhibited endo/lysosomal escape after a 2 h treatment (Figure S8(B)).
The effects of the AD: β-CD molar ratio for the crosslinked CLPBAP polyplexes were subsequently studied. By fixing the weight ratio of CLPBAP:PEG–CLPBAP–PEG:DNA to 48:24:1, the molar ratio of the crosslinkers (AD:β-CD) was adjusted from 4:2 to 4:6. As shown in Figure 4(C), the highest transfection efficiency was achieved with an AD:β-CD molar ratio = 4:3, and the transfection efficiency of the crosslinked CLPBAP polyplexes was comparable to the non-crosslinked PBAP and Lipo 2000. The MFI data also indicated a similar trend (Figure S5). This AD:β-CD molar ratio was used for further studies.
To study the function of PEG and imidazole groups in the polyplex delivery system, crosslinked CLPBAP polyplexes were prepared using PBAP-based polymers without PEG and imidazole groups, respectively. The DNA transfection efficiencies of the polyplexes were studied by flow cytometry (Figure 4(E)). The crosslinked polyplexes without imidazole groups showed significantly lower transfection efficiency than those with imidazole. The enhanced transfection efficiency associated with imidazole conjugation can be attributed to the enhanced endo/lysosomal escape capability afforded by the imidazole groups whose pKa value is around 6.0. Imidazole groups can be quickly protonated in acidic endocytic compartments, leading to the so-called proton sponge effect. We also found that crosslinked CLPBAP polyplexes without PEG (i.e., polyplexes made of CLPBAP and DNA only, without the PEG-CLPBAP-PEG component) exhibited a higher transfection efficiency, but this was accompanied with a significantly higher cytotoxicity (Figure 4(F)). Without PEG on the surface of the polyplexes, the crosslinked polyplexes had a higher positive surface charge (17.2 mV), which may lead to elevated binding and endocytosis of the polyplexes by cells; however, the dense positive surface charge may also cause cell membrane disruption, leading to cell death 42–43.
mRNA Delivery Using Polyplexes
The weight ratio between the PBAP-based polymers and mRNA was first optimized (Figure S6) using GFP mRNA, and the formulations with the highest transfection efficiency for non-crosslinked (PBAP: PEG–PBAP–PEG:mRNA = 48:28:1) and crosslinked (CLPBAP: PEG–CLPBAP–PEG:mRNA = 48:28:1) were used for further studies.
The transfection efficiencies of both non-crosslinked and crosslinked mRNA polyplexes were studied in two cell lines (HEK 293 and RAW 264.7), as shown in Figure 5(A) and (B). In HEK 293 cells, GFP expression was detected as early as 4 h post treatment and peaked at 24 h; in RAW 264.7 cells, GFP expression reached a plateau much earlier at 10 h. The non-crosslinked PBAP polyplexes exhibited comparable transfection efficiencies to Lipo 2000 in HEK 293 cells, and higher transfection efficiencies in RAW 264.7 cells 10 h post treatment. The crosslinked CLPBAP polyplexes exhibited higher transfection efficiencies than Lipo 2000 in both cell lines, particularly in RAW 264.7 cells, where they showed an approximately 2-fold higher transfection efficiency. In HEK 293 cells, the transfection efficiencies of non-crosslinked and crosslinked polyplexes were similar; however, in RAW 264.7 cells, the crosslinked polyplexes exhibited an approximately 1.5-fold higher transfection efficiency. The MFI data also exhibited a similar trend (Figure S7). The high mRNA delivery efficacy observed with the crosslinked CLPBAP polyplexes in RAW 264.7 cells may be attributed to its crosslinked nature, which may keep the polyplexes and mRNA—which is chemically less stable than some of the other genetic materials such as DNA—from disassembling and degrading before it reaches the cytosol. The enhanced stability exhibited by the crosslinked CLPBAP polyplexes makes it potentially advantageous for mRNA delivery.
Figure 5. Efficient delivery of mRNA by the polyplexes.

Transfection efficiency of the non-crosslinked and crosslinked mRNA polyplexes in (A) HEK 293 cells and (B) RAW 264.7 cells at various time points. The weight ratios of PBAP:PEG–PBAP–PEG:mRNA or CLPBAP:PEG–CLPBAP–PEG:mRNA in the PBAP and CLPBAP polyplexes, respectively, were fixed at 48:24:1. *: p < 0.05; **: p < 0.01; n = 3.
We further investigated the delivery efficiency of the polyplexes in two other cell lines (i.e., HCT 116 and NHDF cells). As shown in Figure 6(A) and (B), in HCT116 cells, both non-crosslinked and crosslinked polyplexes showed similar DNA transfection efficiencies to Lipo 2000; however, a significantly higher mRNA transfection efficiency was observed with the crosslinked CLPBAP polyplexes. In NHDF cells (Figure 6(C) and (D)), crosslinked PBAP polyplexes induced higher DNA and mRNA transfection efficiencies than non-crosslinked PBAP polyplexes, while non-crosslinked polyplexes showed similar DNA and mRNA transfection efficiencies to Lipo 2000. These results suggest that both non-crosslinked and crosslinked polyplexes are suitable for DNA and mRNA delivery in a variety of cells.
Figure 6. Efficient delivery of DNA and mRNA by the polyplexes in multiple cell lines.

(A) and (B): Transfection efficiency of the (A) DNA and (B) mRNA polyplexes in HCT116 cells. (C) and (D): Transfection efficiency of the (C) DNA and (D) mRNA polyplexes in NHDF cells. The weight ratios of PBAP:PEG–PBAP–PEG:mRNA or CLPBAP:PEG–CLPBAP–PEG:mRNA in the PBAP and CLPBAP polyplexes, respectively,were fixed at 48:24:1. The AD:β-CD molar ratio in the crosslinked PBAP polyplexes was fixed as 4:3*: p < 0.05; **: p < 0.01; ****: p < 0.0001; n = 3.
Genome Editing Using Polyplexes
Cas9 is an endonuclease that is able to cleave double-stranded DNA at a target genomic locus guided by a sgRNA sequence. After double-stranded DNA cleavage occurs by the RNP, gene knockout can be achieved by the nonhomologous end joining (NHEJ) DNA repair pathway 13, 44. Moreover, by introducing a donor DNA template along with the RNP, precise sequences from a donor template can be incorporated into the genome through the homology-directed repair (HDR) pathway 16. Compared to nucleic acids such as DNA and mRNA, genome editing payloads such as Cas9/sgRNA RNP and S1mplex, have a net negative surface charge and more sophisticated structures. To investigate whether non-crosslinked PBAP and crosslinked CLPBAP polyplexes can efficiently deliver RNP payload, we designed a sgRNA targeting the mCherry transgene in a transgenic HEK 293 cell line. To enhance the nuclear transportation, Cas9 protein with two NLS peptides (sNLS–Cas9–sNLS) was next used to form the RNP complexes. Both non-crosslinked and crosslinked RNP polyplexes were prepared by varying the polymer and RNP weight ratios. As shown in Figure 7(A), both non-crosslinked and crosslinked polyplexes exhibited the highest genome editing efficiencies for mCherry gene disruption at a PBAP:PEG-PBAP-PEG:RNP or CLPBAP:PEG–CLPBAP–PEG:RNP weight ratio of 12:7:1 (the corresponding PBAP (or CLPBAP) and PEG-PBAP (or CLPBAP)-PEG concentrations were 18.7 μg/ml and 10.9 μg/ml, respectively). Furthermore, both non-crosslinked and crosslinked polyplexes possessed similar targeted gene disruption efficiencies to Lipo 2000.
Figure 7. Efficient delivery of CRISPR-Cas9 gene editing machinery by polyplexes.

(A) Genome editing efficiency of non-crosslinked and crosslinked RNP polyplexes in mCherry-expressing HEK 293 cells. The RNP polyplexes were prepared by varying the polymer-to-RNP weight ratios (i.e., PBAP: PEG–PBAP–PEG: RNP for non-crosslinked polyplexes, or CLPBAP: PEG–CLPBAP–PEG: RNP for crosslinked polyplexes). mCherry knock-out efficiency was assayed by flow cytometry for loss of mCherry fluorescence. (B) Precise gene correction efficiency of the non-crosslinked and crosslinked S1mplex polyplexes in BFP-expressing HEK 293 cells. S1mplex polyplexes were prepared by varying the polymer-to-S1mplex weight ratios (i.e., PBAP: PEG–PBAP–PEG: S1mplex for non-crosslinked polyplexes, or CLPBAP: PEG–CLPBAP–PEG: S1mplex for crosslinked polyplexes). Precise gene correction efficiency of the BFP to the GFP from the S1mplex repair ssODN repair template was assayed by flow cytometry for gain of GFP fluorescence. NS: not significant; n = 3.
In addition to gene disruption, many gene editing applications require precise gene correction using a repair nucleic acid template. The S1mplex payload is capable of precise gene correction of a target gene when previously delivered to cells through nucleofection 16..The gene correction efficiencies of both non-crosslinked and crosslinked S1mplex polyplexes were investigated using a BFP-expressing HEK 293 cell line. The S1mplex was designed to modify the BFP transgene to GFP through a 3 nucleotide switch 45. The precise gene correction efficiencies of the S1mplex polyplexes were monitored by flow cytometry by gating for GFP-positive cells. As shown in Figure 7(B), both non-crosslinked and crosslinked polyplexes induced precise gene correction as indicated by significant GFP expression. Furthermore, at the PBAP:PEG-PBAP-PEG:RNP or CLPBAP:PEG-CLPBAP-PEG:RNP weight ratio of 24:14:1 (the corresponding PBAP (or CLPBAP) and PEG-PBAP (or CLPBAP)-PEG concentrations were 56.4 μg/ml and 32.9 μg/ml, respectively), the two types of polyplexes exhibited similar genome correction efficiencies to Lipo 2000. These studies indicate that the non-crosslinked PBAP and crosslinked CLPBAP polyplexes are suitable for the delivery of not only nucleic acids, but also complex CRISPR genome editing payloads.
Cytotoxicity within human cells
The cell viability of the optimal polyplex formulations were studied using an MTT assay at the same payload concentration used in the transfection or genome editing studies. For plasmid DNA and mRNA, 200 ng/well were used at a PBAP (or CLPBAP):PEG-PBAP (or CLPBAP)-PEG:RNP weight ratio of 48:28:1. The corresponding PBAP (or CLPBAP) and PEG-PBAP (or CLPBAP)-PEG concentrations were 96 μg/ml and 56 μg/ml, respectively. For RNP, 156 ng/well were used at a weight ratio of 12:7:1. The corresponding PBAP (or CLPBAP) and PEG-PBAP (or CLPBAP)-PEG concentrations were 18.7 μg/ml and 10.9 μg/ml, respectively. For S1mplex, 235 ng/well were used at a weight ratio of 24:14:1. The corresponding PBAP (or CLPBAP) and PEG-PBAP (or CLPBAP)-PEG concentrations were 56.4 μg/ml and 32.9 μg/ml, respectively. As shown in Figure 8, no significant cytotoxicity was observed for both non-crosslinked PBAP and crosslinked CLPBAP polyplex-treated groups. In contrast, at the same payload, Lipo 2000 (0.5 μl/well, as shown in Figure S9) induced significant cell death, indicating that Lipo 2000 had a much higher cytotoxicity than the polyplexes used to deliver the same amount of payload in this study.
Figure 8. Cytotoxicity of polyplexes in vitro.

Cell viability study of the non-crosslinked and crosslinked complexes loaded with (A) DNA, (B) mRNA, (C) RNP, and (D) S1mplex. Cytotoxicity was studied using the polymer/payload formulation yielding the highest transfection efficiency for DNA or RNA payloads or genome editing efficiency for RNP or S1mplex payloads. NS: not significant; *: p < 0.05; **: p < 0.01; ***: p < 0.005; n = 5.
DISCUSSION
The cytotoxicity data, in combination with the transfection/genome editing efficiency studies, indicate that both types of polyplexes outperform the commercially available Lipo 2000 for delivery of nucleic acids and CRISPR-Cas9 genome editing payloads. Furthermore, the crosslinked polyplexes also possess superior stability in the presence of polyanions such as BSA suggesting good in vivo stability due to its crosslinked structure. DNA, mRNA, Cas9 RNP, and S1mplex are very different payloads both in terms of structure and functionality. Therefore, it is not surprising to see some of the reports suggesting that minor changes in the chemical structure of the cationic polymers can affect the transfection efficiency of DNA and mRNA differently 46–47. For example, the odd-even effects of the repeating aminoethylene units in the side chain of N-substituted polyaspartamides, had contradictory effects to DNA and mRNA transfection efficiencies owing to their different endo/lysosomal escape capabilities 46–47. Furthermore, studies also found that minor changes in the chemical structure of the cationic polymers can also dramatically affect the transfection efficiency of the same payload (e.g., DNA) 48–49. We did not observe such sensitive changes in transfection efficiency with our nanoplatform, as it had broad versatility for several different payloads. This versatility is likely to facilitate the delivery of other complex CRISPR payloads, such as epigenomic editors, RNA editors and base editors. Further, other gene editing systems such as zinc finger nucleases and TAL-like effector nucleases maybe loaded into this nanoplatform 50.
One key advantage of the nanoplatform is the ease of formulation once the PBAP polymers have been synthesized. The PBAP polymers could be off-the-shelf reagents that can be mixed with many payloads within an hour and easily optimized for each particular payload and clinical application. Incorporation of targeting ligands onto the PBAP polymers would further add versatility and potential cell-targeting capability of the nanoplatform. Overall, we anticipate the nanoplatform to be easily customized by researchers without synthetic chemistry expertise, such as engineers, biologists and clinicians, for potent gene editing in vitro and in vivo.
CONCLUSIONS
Two types of redox-responsive polyplexes, namely non-crosslinked PBAP and crosslinked CLPBAP polyplexes were designed for the efficient delivery of various genetic materials including DNA, mRNA, Cas9 RNP, and S1mplex. These polyplexes were made of a family of PBAP polymers containing disulfide bond in their backbones and bearing imidazole groups, thus enabling the efficient encapsulation, endo/lysosomal escape, and cytosolic release of various negatively charged payloads. To further enhance the stability of the non-crosslinked polyplexes, PBAP-based polymers were conjugated with either AD or β-CD which can form crosslinks via the host–guest interactions between β-CD and AD. Unlike non-crosslinked polyplexes, the crosslinked CLPBAP polyplexes exhibited excellent stability against polyanions, a feature desirable for in vivo applications. Both non-crosslinked and crosslinked polyplexes demonstrated similar or better transfection/genome editing efficiencies in comparison with Lipo 2000, a commercially available delivery agent. While Lipo 2000 consistently exhibited severe cytotoxicity, the two types of polyplexes did not induce any cytotoxicity at the same dosage studied. Furthermore, the surface of these PEGylated polyplexes can also be easily conjugated with desirable moieties such as targeting ligands or imaging probes. Taken together, these versatile GSH-responsive polyplexes constitute a highly promising off-the-shelf platform to deliver a variety of payloads for a broad range of applications in vitro and in vivo.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge the financial support from the NIH (1K25CA166178 and R01 HL129785 to SG and 1R35GM119644–01 to KS).
Footnotes
SUPPORTING INFORMATION
Synthesis scheme of the PBAP polymers for non-crosslinked polyplexes. NMR spectra of the CLPBAP polymers. NMR analysis of the host–guest interaction between the CLPBAP polymers. MFI analysis of DNA, mRNA, RNP, and S1mplex delivery by non-crosslinked and crosslinked polyplexes. Optimization of the PBAP-based polymers and mRNA weight ratios for mRNA transfection. Intracellular trafficking of the non-crosslinked DNA polyplexes with and without NLS. Intracellular trafficking of the non-crosslinked and crosslinked mRNA polyplexes. Molecular weight measurement of PBAP and CLPBAP polymers by GPC. Size and zeta-potential of non-crosslinked PBAP and crosslinked CLPBAP polyplexes.
REFERENCES
- 1.Yin H; Kanasty RL; Eltoukhy AA; Vegas AJ; Dorkin JR; Anderson DG, Non-Viral Vectors for Gene-Based Therapy. Nat Rev Genet 2014, 15 (8), 541–555. [DOI] [PubMed] [Google Scholar]
- 2.Jin L; Zeng X; Liu M; Deng Y; He N, Current Progress in Gene Delivery Technology Based on Chemical Methods and Nano-Carriers. Theranostics 2014, 4 (3), 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gill S; Kalos M, T Cell-Based Gene Therapy of Cancer. Translational Research 2013, 161 (4), 365–379. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H; Lee M-Y; Hogg MG; Dordick JS; Sharfstein ST, Gene Delivery in Three-Dimensional Cell Cultures by Superparamagnetic Nanoparticles. ACS nano 2010, 4 (8), 4733–4743. [DOI] [PubMed] [Google Scholar]
- 5.Guan S; Rosenecker J, Nanotechnologies in Delivery of Mrna Therapeutics Using Nonviral Vector-Based Delivery Systems. Gene Ther 2017, 24 (3), 133–143. [DOI] [PubMed] [Google Scholar]
- 6.Islam MA; Reesor EKG; Xu Y; Zope HR; Zetter BR; Shi J, Biomaterials for Mrna Delivery. Biomaterials science 2015, 3 (12), 1519–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song H; Yu M; Lu Y; Gu Z; Yang Y; Zhang M; Fu J; Yu C, Plasmid DNA Delivery: Nanotopography Matters. Journal of the American Chemical Society 2017, 139 (50), 18247–18254. [DOI] [PubMed] [Google Scholar]
- 8.Sahay G; Alakhova DY; Kabanov AV, Endocytosis of Nanomedicines. Journal of controlled release 2010, 145 (3), 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J; Wang W; He Y; Li Y; Yan EZ; Zhang K; Irvine DJ; Hammond PT, Structurally Programmed Assembly of Translation Initiation Nanoplex for Superior Mrna Delivery. ACS nano 2017, 11 (3), 2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y; Su H.-h.; Yang Y; Hu Y; Zhang L; Blancafort P; Huang L, Systemic Delivery of Modified Mrna Encoding Herpes Simplex Virus 1 Thymidine Kinase for Targeted Cancer Gene Therapy. Mol Ther 2013, 21 (2), 358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaczmarek JC; Kowalski PS; Anderson DG, Advances in the Delivery of Rna Therapeutics: From Concept to Clinical Reality. Genome medicine 2017, 9 (1), 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lächelt U; Wagner E, Nucleic Acid Therapeutics Using Polyplexes: A Journey of 50 Years (and Beyond). Chemical reviews 2015, 115 (19), 11043–11078. [DOI] [PubMed] [Google Scholar]
- 13.Doudna JA; Charpentier E, The New Frontier of Genome Engineering with Crispr-Cas9. Science 2014, 346 (6213), 1258096. [DOI] [PubMed] [Google Scholar]
- 14.Hsu PD; Lander ES; Zhang F, Development and Applications of Crispr-Cas9 for Genome Engineering. Cell 2014, 157 (6), 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glass Z; Li Y; Xu Q, Nanoparticles for Crispr–Cas9 Delivery. Nature Biomedical Engineering 2017, 1 (11), 854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlson-Stevermer J; Abdeen AA; Kohlenberg L; Goedland M; Molugu K; Lou M; Saha K, Assembly of Crispr Ribonucleoproteins with Biotinylated Oligonucleotides Via an Rna Aptamer for Precise Gene Editing. Nature communications 2017, 8 (1), 1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu Z; Biswas A; Zhao M; Tang Y, Tailoring Nanocarriers for Intracellular Protein Delivery. Chemical Society Reviews 2011, 40 (7), 3638–3655. [DOI] [PubMed] [Google Scholar]
- 18.Leader B; Baca QJ; Golan DE, Protein Therapeutics: A Summary and Pharmacological Classification. Nature reviews Drug discovery 2008, 7 (1), 21. [DOI] [PubMed] [Google Scholar]
- 19.Zuris JA; Thompson DB; Shu Y; Guilinger JP; Bessen JL; Hu JH; Maeder ML; Joung JK; Chen Z-Y; Liu DR, Cationic Lipid-Mediated Delivery of Proteins Enables Efficient Protein-Based Genome Editing in Vitro and in Vivo. Nat Biotech 2015, 33 (1), 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang M; Zuris JA; Meng F; Rees H; Sun S; Deng P; Han Y; Gao X; Pouli D; Wu Q, Efficient Delivery of Genome-Editing Proteins Using Bioreducible Lipid Nanoparticles. Proceedings of the National Academy of Sciences 2016, 113 (11), 2868–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nault J-C; Datta S; Imbeaud S; Franconi A; Mallet M; Couchy G; Letouzé E; Pilati C; Verret B; Blanc J-F, Recurrent Aav2-Related Insertional Mutagenesis in Human Hepatocellular Carcinomas. Nat Genet 2015, 47 (10), 1187–1193. [DOI] [PubMed] [Google Scholar]
- 22.Thomas CE; Ehrhardt A; Kay MA, Progress and Problems with the Use of Viral Vectors for Gene Therapy. Nat Rev Genet 2003, 4 (5), 346–358. [DOI] [PubMed] [Google Scholar]
- 23.Dong Y; Eltoukhy AA; Alabi CA; Khan OF; Veiseh O; Dorkin JR; Sirirungruang S; Yin H; Tang BC; Pelet JM, Lipid ‐ Like Nanomaterials for Simultaneous Gene Expression and Silencing in Vivo. Advanced healthcare materials 2014, 3 (9), 1392–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B; Luo X; Deng B; Wang J; McComb DW; Shi Y; Gaensler KML; Tan X; Dunn AL; Kerlin BA, An Orthogonal Array Optimization of Lipid-Like Nanoparticles for Mrna Delivery in Vivo. Nano letters 2015, 15 (12), 8099–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steyer B; Carlson-Stevermer J; Angenent-Mari N; Khalil A; Harkness T; Saha K, High Content Analysis Platform for Optimization of Lipid Mediated Crispr-Cas9 Delivery Strategies in Human Cells. Acta biomaterialia 2016, 34, 143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao J-F; Lee J-C; Lin C-K; Wei K-C; Chen P-Y; Yang H-W, Self-Assembly DNA Polyplex Vaccine inside Dissolving Microneedles for High-Potency Intradermal Vaccination. Theranostics 2017, 7 (10), 2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nochi T; Yuki Y; Takahashi H; Sawada S.-i.; Mejima M; Kohda T; Harada N; Kong IG; Sato A; Kataoka N, Nanogel Antigenic Protein-Delivery System for Adjuvant-Free Intranasal Vaccines. Nature materials 2010, 9 (7), 572–578. [DOI] [PubMed] [Google Scholar]
- 28.Wang H-X; Li M; Lee CM; Chakraborty S; Kim H-W; Bao G; Leong KW, Crispr/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem Rev 2017, 117 (15), 9874–9906. [DOI] [PubMed] [Google Scholar]
- 29.Wang P; Zhang L; Zheng W; Cong L; Guo Z; Xie Y; Wang L; Tang R; Feng Q; Hamada Y, Thermo‐Triggered Release of Crispr‐Cas9 System by Lipid‐Encapsulated Gold Nanoparticles for Tumor Therapy. Angewandte Chemie International Edition 2017. [DOI] [PubMed] [Google Scholar]
- 30.Pissuwan D; Niidome T; Cortie MB, The Forthcoming Applications of Gold Nanoparticles in Drug and Gene Delivery Systems. Journal of controlled release 2011, 149 (1), 65–71. [DOI] [PubMed] [Google Scholar]
- 31.Lee K; Conboy M; Park HM; Jiang F; Kim HJ; Dewitt MA; Mackley VA; Chang K; Rao A; Skinner C, Nanoparticle Delivery of Cas9 Ribonucleoprotein and Donor DNA in Vivo Induces Homology-Directed DNA Repair. Nature Biomedical Engineering 2017, 1 (11), 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oba M; Miyata K; Osada K; Christie RJ; Sanjoh M; Li W; Fukushima S; Ishii T; Kano MR; Nishiyama N, Polyplex Micelles Prepared from Ω-Cholesteryl Peg-Polycation Block Copolymers for Systemic Gene Delivery. Biomaterials 2011, 32 (2), 652–663. [DOI] [PubMed] [Google Scholar]
- 33.Sarett SM; Werfel TA; Chandra I; Jackson MA; Kavanaugh TE; Hattaway ME; Giorgio TD; Duvall CL, Hydrophobic Interactions between Polymeric Carrier and Palmitic Acid-Conjugated Sirna Improve Pegylated Polyplex Stability and Enhance in Vivo Pharmacokinetics and Tumor Gene Silencing. Biomaterials 2016, 97, 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin H; Kauffman KJ; Anderson DG, Delivery Technologies for Genome Editing. Nature Reviews Drug Discovery 2017, 16 (6), 387. [DOI] [PubMed] [Google Scholar]
- 35.Suk JS; Xu Q; Kim N; Hanes J; Ensign LM, Pegylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Advanced drug delivery reviews 2016, 99, 28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shim MS; Xia Y, A Reactive Oxygen Species (Ros)‐Responsive Polymer for Safe, Efficient, and Targeted Gene Delivery in Cancer Cells. Angewandte Chemie International Edition 2013, 52 (27), 6926–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlson-Stevermer J; Goedland M; Steyer B; Movaghar A; Lou M; Kohlenberg L; Prestil R; Saha K, High-Content Analysis of Crispr-Cas9 Gene-Edited Human Embryonic Stem Cells. Stem cell reports 2016, 6 (1), 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodell CB; Kaminski AL; Burdick JA, Rational Design of Network Properties in Guest–Host Assembled and Shear-Thinning Hyaluronic Acid Hydrogels. Biomacromolecules 2013, 14 (11), 4125–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moret I; Peris JE; Guillem VM; Benet M; Revert F; Dasí F; Crespo A; Aliño SF, Stability of Pei–DNA and Dotap–DNA Complexes: Effect of Alkaline Ph, Heparin and Serum. Journal of Controlled Release 2001, 76 (1–2), 169–181. [DOI] [PubMed] [Google Scholar]
- 40.Lee Y; Miyata K; Oba M; Ishii T; Fukushima S; Han M; Koyama H; Nishiyama N; Kataoka K, Charge‐Conversion Ternary Polyplex with Endosome Disruption Moiety: A Technique for Efficient and Safe Gene Delivery. Angewandte Chemie 2008, 120 (28), 5241–5244. [DOI] [PubMed] [Google Scholar]
- 41.Wang RE; Tian L; Chang Y-H, A Homogeneous Fluorescent Sensor for Human Serum Albumin. Journal of pharmaceutical and biomedical analysis 2012, 63, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arvizo RR; Miranda OR; Thompson MA; Pabelick CM; Bhattacharya R; Robertson JD; Rotello VM; Prakash YS; Mukherjee P, Effect of Nanoparticle Surface Charge at the Plasma Membrane and Beyond. Nano letters 2010, 10 (7), 2543–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fröhlich E, The Role of Surface Charge in Cellular Uptake and Cytotoxicity of Medical Nanoparticles. International journal of nanomedicine 2012, 7, 5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cong L; Ran FA; Cox D; Lin S; Barretto R; Habib N; Hsu PD; Wu X; Jiang W; Marraffini L, Multiplex Genome Engineering Using Crispr/Cas Systems. Science 2013, 1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson CD; Ray GJ; DeWitt MA; Curie GL; Corn JE, Enhancing Homology-Directed Genome Editing by Catalytically Active and Inactive Crispr-Cas9 Using Asymmetric Donor DNA. Nat Biotechnol 2016, 34 (3), 339. [DOI] [PubMed] [Google Scholar]
- 46.Uchida H; Itaka K; Nomoto T; Ishii T; Suma T; Ikegami M; Miyata K; Oba M; Nishiyama N; Kataoka K, Modulated Protonation of Side Chain Aminoethylene Repeats in N-Substituted Polyaspartamides Promotes Mrna Transfection. Journal of the American Chemical Society 2014, 136 (35), 12396–12405. [DOI] [PubMed] [Google Scholar]
- 47.Uchida H; Miyata K; Oba M; Ishii T; Suma T; Itaka K; Nishiyama N; Kataoka K, Odd–Even Effect of Repeating Aminoethylene Units in the Side Chain of N-Substituted Polyaspartamides on Gene Transfection Profiles. Journal of the American Chemical Society 2011, 133 (39), 15524–15532. [DOI] [PubMed] [Google Scholar]
- 48.Lin C; Blaauboer C-J; Timoneda MM; Lok MC; van Steenbergen M; Hennink WE; Zhong Z; Feijen J; Engbersen JFJ, Bioreducible Poly (Amido Amine) S with Oligoamine Side Chains: Synthesis, Characterization, and Structural Effects on Gene Delivery. Journal of Controlled Release 2008, 126 (2), 166–174. [DOI] [PubMed] [Google Scholar]
- 49.Green JJ; Langer R; Anderson DG, A Combinatorial Polymer Library Approach Yields Insight into Nonviral Gene Delivery. Accounts Chem Res 2008, 41 (6), 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H; La Russa M; Qi LS, Crispr/Cas9 in Genome Editing and Beyond. Annu Rev Biochem 2016, 85, 227–264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.