

Abstract
Advances in design and use of light-sensitive and light-emitting sensors have facilitated observation, measurement, and control of neuronal activities. Viruses are effective vectors for delivery of these valuable research tools to mammalian brains. Recombinant viruses are optimized to mediate regulatable, long-term, and cell-specific gene expression. Here, we describe production methods for three of the most commonly used types of recombinant viruses in neurobiology research: adeno-associated virus (AAV), retro/lentivirus, and glycoprotein deleted rabies virus. These viral constructs are frequently used for calcium imaging or to deliver neural tracers and optogenetic tools. Popular constructs are readily obtained commercially; however, customized virus production through commercial sources is time consuming and costly. This article aims to provide readers with detailed technical information for rapid production and validation of high quality viral particles in a laboratory setting while highlighting advantages and limitations of each viral type.
Keywords: Recombinant virus, Viral production, AAVs, Retrovirus, Lentivirus, Rabies delta-G
INTRODUCTION
The ability to deliver genes to living organisms has significantly advanced our understanding of complex organs such as mammalian brains. Rapid development of viral technology provides molecular tools for researchers to dissect, observe, and manipulate the nervous system in vivo. Over the last few decades, multiple daises have been developed for design and production of recombinant viruses. Three of the most common types of viruses used for gene delivery in neurobiology are adeno-associated viruses (AAVs), retro/lentiviruses, and surface glycoprotein deleted rabies viruses (Rabies dG).
The current AAV production platforms are mostly based on methods generated and validated by teams led by Jude Samulski, Xiao Xiao, and James Wilson (Lock et al., 2010; Xiao, 2010; Zolotukhin et al., 1999). A tremendous body of work was published by Didier Trono and Patrick Salmon describing details of lentiviral packaging and titering methods (Salmon & Trono, 2007). The extensive retrograde activity of rabies dG, its characteristics, and production method were first described by the Ed Callaway team and developed further by laboratories of Thomas Jessell, Marco Tripodi, and Ian Wickersham (Chatterjee et al., 2018; Ciabatti, Gonzalez-Rueda, Mariotti, Morgese, & Tripodi, 2017; Osakada & Callaway, 2013; Reardon et al., 2016). Protocols described in this unit are adapted from their published work to provide a side-by-side comparison between production efforts and effective use of each viral type.
This unit aims to provide a brief overview of viral properties, production process, and capabilities of each viral type – refer to the accompanying overview “Recombinant Viral Vectors as Neuroscience Tools” for additional details. Basic Protocol 1 contains comprehensive steps for packaging and validation of recombinant AAVs and provides two alternative methods for small and large-scale productions. The retro/lentiviral protocol (Basic Protocol 2) includes variations in packaging for retroviruses, lentiviruses, integrase deficient lentiviruses (IDLVs) and concentration methods for VSV-G and Env A pseudotyped lentiviruses. In the rabies dG protocol (Basic Protocol 3), we describe the differences between SADB19-Rabies dG, SADB19-Rabies dGL, SADB19 Self-Inactivating Rabies (SiR), CVS-N2c Rabies dG and necessary modifications to the protocol for production of each type of rabies dG virus.
Many commercial sources manufacture and provide recombinant viruses rapidly and at a reasonable cost. Here, we describe small-scale production methods for creation of customized vectors used for mutational analysis and development of molecular toolbox in a laboratory setting.
STRATEGIC PLANNING
Considerations for the use of AAVs in transducing mammalian neurons
AAVs are non-enveloped resilient viruses that can withstand multiple freeze-thaw cycles, changes in pH, varying salt concentrations, and high centripetal force during ultracentrifugation (Lock et al., 2010). Moreover, there is no known disease associated with AAVs; their capsids can be engineered to alter viral tropism; and they offer a lasting expression in non-dividing cells such as neurons. Thus, AAVs are versatile and innocuous vectors for delivering molecular tools to mammalian brains in vivo.
AAV genes necessary for production of viral particles are delivered by three plasmids: (1) AAV transfer plasmid, (2) the Helper plasmid, and (3) the Rep/Cap plasmid (Figure 1). Genes encoding serotypes are delivered by the Rep/Cap plasmid during the transfection of host cells. For example, Rep2/Cap9 plasmid is used to package AAV2/9. Since most AAVs are produced from Rep2 genes, the nomenclature is often shortened from AAV2/9 to AAV9. The Helper plasmid delivers additional genes VA RNAs (Viral Associated non-coding RNAs), E2A, and E4OEF6 that are necessary for packaging AAVs (Clement and Grieger 2016) and the transfer plasmid supplies the gene of interest flanked by Inverted Terminal Repeats (ITRs) in an AAV transfer vector.
Figure 1. Reconstitution of recombinant viruses from plasmids.

AAVs are reconstituted from transfer vector, Rep/Cap, and Helper plasmids. Serotypes are determined by the Cap genes. 2nd generation packaging plasmids for a VSV-G pseudotyped lentivirus include transfer vectors, psPax2, and pMD2.G. Rabies dG transfer vector and viral proteins N, P, L, and G are necessary for formation of initial viral particles.
The major disadvantage of using AAVs, is the limited size of the genetic load in transfer plasmids. The production yield decreases significantly for vector genomes greater than 3.5 kilobases (kb) and never exceeds 5.2 kb (Wu, Yang et al. 2010). The genome can still accommodate regulatable promoters and recombinase-dependent expression of genes (Castle, Gershenson, Giles, Holzbaur, & Wolfe, 2014). Elements such as self-cleaving P2A and T2A are often used to link gene expression from a single promoter to reduce the size of delivered genes in a transfer vector. A myriad of AAV transfer vector plasmids carrying optogenetics tools and sensors are available from non-profit plasmid repositories such as Addgene.
AAVs are ideal for delivery of small genes to a population of neurons. Although AAV transduction cannot be limited within the targeted population, the expression of delivered genes can be controlled by specialized promoters or recombinase technology (e.g. Cre-lox). Small-scale production of AAV is accomplished within 10 days (Figure 2A) and genes delivered by AAVs are expressed 2–3 weeks after in vivo transduction. AAVs offer lasting expression in neurons.
Figure 2A. Workflow of recombinant virus production.

General steps and length of time for production of Adeno-associated viruses.
Recombinant AAVs are non-propagating viruses with Biosafety Level 2 designation. Please plan all experiments accordingly to your institute’s health and safety guidelines and use personal protective clothing and gear to protect yourself and your coworkers.
Considerations for use and production of retro and lentiviruses
Retro and Lentiviruses are members of the Retroviridae family of viruses. The lentiviral genome is a single-stranded RNA that expresses gag, pro, pol and env genes that encode viral components such as capsid proteins, reverse transcriptase, protease, integrase and the envelope glycoproteins, respectively (Salmon & Trono, 2007). The lentiviral genome is flanked by long terminal repeats (LTRs) that are required for viral genome replication and integration. During the development of recombinant lentiviral vectors, the viral genes were divided among multiple plasmids to create packaging generations. Each subsequent generation of lentiviral vector was designed to enhance safety features for use in research laboratories.
The lentiviral packaging generations dispense several accessory genes and/or splits the lentiviral genome into more plasmids to reduce the potential for recombination and production of replication-competent lentiviruses. The 2nd generation packaging system (Figure 1), described in Protocol 2, can be used to produce lentiviruses using 2nd, 3rd, or higher generation lentiviral transfer vectors. Higher generations offer higher safety features but at the cost of lowered yields since host cells are transfected with multiple plasmids to reconstitute the viral genome, and therefore, not all cells will contain all the necessary components for packaging the viruses.
Lentiviruses are readily pseudotyped with membrane proteins that are expressed on the surface of the packaging cell line. Membrane protein VSV-G is commonly used for pseudotyping lentiviruses since it binds to low-density lipoprotein receptors that are ubiquitously expressed on the surface of most mammalian cells. VSV-G broadens lentiviral tropism to include neurons and glia (Finkelshtein, Werman, Novick, Barak, & Rubinstein, 2013). VSV-G also stabilizes lentiviral virion structure and facilitates concentration of the particles by ultracentrifugation at high speed in 2 hours. Lentiviruses pseudotyped with other envelope proteins or the wild type lentiviral Env protein are concentrated at much lower speeds and for an extended length of time (typically 16–20 hours).
In a lentiviral transfer plasmid, LTRs flank the desired transgene sequences and their promoters. The genomic sequences inserted between the LTRs are randomly integrated into the host genome after viral transduction and become a permeant part of their host cell genome. Random integration could lead to insertional mutations and aberrant function in host cells. The lentiviral transfer vectors accommodate inserts of up to 9 kb (Dull et al., 1998; Parr-Brownlie et al., 2015).
Retroviral and lentiviral vectors are used less extensively than AAVs in neurobiological research since their delivered genes do not express robustly. A common application for retroviruses is to identify and manipulate neuronal stem/progenitor cells since retroviruses only transduce dividing cells (Beier, Samson, Matsuda, & Cepko, 2011; Jessberger, Toni, Clemenson, Ray, & Gage, 2008). As an alternative to using recombinant pseudotyped rabies dG viruses, lentiviruses are also utilized as neural tracers when pseudotyped with rabies virus glycoprotein (Rb-G) (Mazarakis et al., 2001) or the chimera of both VSV-G and Rb-G (Kato, Kobayashi, et al., 2011; Kato, Kuramochi, et al., 2011). Lentiviruses are easier and take less time to produce than recombinant pseudotyped rabies viruses. A lentiviral transfer vector can be packaged into viral particles in 5 days (Figure 2B).
Figure 2B. Workflow of recombinant virus production.

General steps and length of time for production of Lentiviruses.
Recombinant retro and lentiviruses are non-propagating viruses with Biosafety Level 2 designation. Depending on their pseudotypes, they are capable of infecting mammalian cells and randomly incorporating into chromosomes which can lead to insertional mutations. Please plan all experiments accordingly to your institute’s health and safety guidelines and use personal protective clothing and gear to protect yourself and your coworkers.
Considerations for use of Rabies dG vectors
Rabies is a neurotropic virus with pathogenic effects in humans. Initial symptoms of infected patients are fever, upper respiratory illness, and gastrointestinal tract disorders. The virus destroys the nervous system as it spreads from the peripheral motor neurons to the central nervous system (Fooks et al., 2017). Rabies is a member of the Rhabdoviridae family of viruses and similar to other members of this family has a single negative-strand RNA genome.
Recombinant SADB19 and CVS-N2c rabies are the most commonly used strains for trans-synaptic gene delivery in retrograde studies (Reardon et al., 2016). To make rabies virus safe for use in research laboratories, the surface glycoprotein G in recombinant rabies dG virus is deleted from the genome. Avian envelope proteins (e.g. Env A, Env B., etc) are often used to pseudotype rabies dG. The resulting virus is incapable of infecting neurons, propagation, or retrograde transfer. A secondary virus (typically an AAV) is needed to deliver the corresponding avian protein receptor (e.g TVA, TVB, etc.) and glycoprotein G to neurons for retrograde transmission to occur. The retrograde transmission is limited to one synaptic jump since the second neuron does not contain glycoprotein G. Importantly, rabies transmission is restricted to synaptic junctions and does not spread to adjacent neurons (Callaway & Luo, 2015).
Rabies dG viruses are a powerful tool for retrograde studies and can deliver up to 8 kb of genetic material. However, there are three major disadvantages in using rabies dG viruses for gene delivery: lack of control over transgene expression, high toxicity, and a lengthy production process (Figure 2c). Since recombinant rabies genome is maintained as an RNA in infected cells, delivered genes cannot contain any promoters or recombinase sensitive sequences to regulate expression. Rabies genes, and the inserted transgene, are expressed sequentially and based on the order of genes. Hence, genes closer to the start of the genome express more robustly than genes farther away.
Figure 2C. Workflow of recombinant virus production.

General steps and length of time for production of Pseudotyped rabies dG viruses.
Rabies dG viruses are also highly toxic and cause damage to neurons within a week or two after infection. Additional modifications to SADB19 rabies dG genome have given rise to less toxic viruses for prolonged expression. Rabies dGL (deletion of G and L genes) and self-inactivating rabies viruses (SiR dG) are promising new tools for studying neural network organizations (Chatterjee et al., 2018; Ciabatti et al., 2017). Rabies L protein is essential for viral genome transcription and protein production. Without L, rabies virus does not replicate, yet small amounts of dGL virus travel retrograde to infect projection neurons (Chatterjee et al., 2018). Neurons infected with SADB19 dGL appear intact and functional for many months. The Self-inactivating (SiR dG) rabies genome is engineered to contain a PEST protein sequence that targets viral protein for destruction (Ciabatti et al., 2017). Degradation of viral proteins removes toxicity and allows for long-term expression of genes for neuroanatomical studies. Since SADB19 dGL and SiR are less efficient in retrograde transmission than rabies dG, they are ideal for delivery of genes that are effective in trace amounts such as recombinases (e.g. Cre, or Flp).
Please note that packaging and pseudotyping cell lines and plasmids for SADB19 Rabies SiR dG and CVS-N2C rabies dG can be obtained from laboratories of Dr. Marco Tripodi and Dr. Andrew Murray that developed the protocols and created the cell lines, respectively (Ciabatti et al., 2017; Reardon et al., 2016). Necessary transfer vectors and packaging plasmids are generously provided by authors and deposited in Addgene.
Production and validation of pseudotyped recombinant SADB19 rabies dG, dGL, and dG SiR viruses can be accomplished in 4–6 weeks (Figure 2C). However, packaging and pseudotyping of recombinant CVS-N2c rabies dG will require several months.
Recombinant rabies dG viruses have Biosafety Level 2 designation. Depending on their pseudotypes, they are capable of infecting and inflicting severe damage to mammalian cells. Please plan all experiments accordingly to your institute’s health and safety guidelines and use personal protective clothing and gear to protect yourself and your coworkers.
BASIC PROTOCOL 1
Production of AAVs
The protocol described in this section is a step-by-step production method for AAV particles. This method was adapted from previously published protocols by Dr. Xiao Xiao, Dr. Jude Samulski, and Dr. James Wilson laboratories. Any AAV transfer vector or Rep/Cap plasmid can be substituted to modify delivered genes and/or serotypes.
Materials
All material and reagents used in this protocol are prepared and maintained with sterile techniques.
Packaging cell line:
293-AAV cells (Cell Biolabs Inc., catalog # AAV-100, RRID: CVCL_KA64)
Cell culture solutions:
DMEM, Dulbecco’s Modified Eagle Medium (Thermo Fisher Scientific, catalog # 11965–118)
L-Glutamine (Thermo Fisher Scientific, catalog # 25030081)
Trypsin-EDTA (Thermo Fisher Scientific, catalog # 25200–056, purchased as 0.25%)
Sodium pyruvate (Thermo Fisher Scientific, catalog # 11360070)
PBS (Phosphate Buffered Saline)
See REAGENT AND SOLUTIONS for Growth and AAV Packaging media recipe
Plasmids:
AAV transfer vector (or AAV shuttle vector)
pHelper plasmid (Cell Biolabs Inc., catalog # 340202)
Rep/Cap plasmid, pAAV2/1 (Cell Biolabs Inc., catalog # VPK-421), pAAV2/2 (Cell Biolabs Inc., catalog # VPK-422), pAAV2/5 (Cell Biolabs Inc., catalog # VPK-425), pAAV2/6 (Cell Biolabs Inc., catalog # VPK-426), pAAV2/8 (Cell Biolabs Inc., catalog # VPK-428), pAAV2/9 (UPenn Vector Core), or pAAV2/RETRO (Addgene catalog # 81070)
Plasmids can be resuspended in sterile water, TE, or EB (See REAGENT AND SOLUTIONS). Any plasmid concentration above 100 ng/ul is acceptable. The above plasmids are also available from other commercial sources, Addgene, and laboratories with published protocols.)
Oligos:
ITR II primers Forward 5’ – GGAACCCCTAGTGATGGAGTT
Reverse 5’ - CGGCCTCAGTGAGCGA
Transfection reagents:
150 mM NaCl
5 M NaCl
PEI, 16 mg/ml in H20 (Polyethylenimine, PEI “MAX”, Polysciences, Warrington, PA, USA)
Purification solutions:
Turbonuclease (Eton Bioscience, catalog # 1400010050)
40% sucrose in TNE (See REAGENT AND SOLUTIONS)
Consumables
T175 tissue culture flask
15 cm tissue culture dishes
14 ml screw top centrifuge tubes
50 ml screw top centrifuge tubes
30 ml conical ultracentrifuge tubes (Beckman Coulter, catalog # 358126, or equivalent)
1.5 ml conical screw top centrifuge tubes with o-rings
Cell scrapers
Sterile cotton tip applicators
0.45 um steriflip filters (Millipore catalog # SCHVU02RE or equivalent)
100-kDa molecular weight cutoff (MWCO) protein concentrator (Pierce, Rockford, IL, USA)
70% Ethanol
10% Bleach
Vortex mixer
Nutating shaker
Centrifuge (Beckman Coulter Allegra X-22R and SX4250 rotor, max speed 4,500 rpm, or equivalent)
Ultracentrifuge (Beckman Coulter and SW32Ti rotor, capable of 50,000 x g speed for centrifugation of 35ml samples, max speed 32,000 rpm, or equivalent)
Water bath (37° C), or equivalent
Tissue culture incubator (37°C, 5% CO2)
Laminar flow hood
Cell Culture
-
1
Maintain cells in growth media without antibiotics in T-175 flasks in a tissue culture incubator at 37°C, 5% CO2.
-
2Split cells using trypsin-EDTA at a ratio of 1:4 to 1:6 every two to three days.Maintain cells at less than 60% confluency and for less than 10 passages.
-
3
For transfection, seed 15cm dishes with 4e6 cells in 20 ml of growth media and incubate overnight. For small-scale preparations use 6 × 15cm dishes, for large-scale preparations use minimum of 20 × 15cm dishes.
Transfection to reassemble recombinant viral genes
-
4Prepare transfection solution. For 6 × 15cm dishes:
- Add 12 ml of 150 mM NaCl to a 14 ml screw top centrifuge tube
- Add 200ug of each plasmid (Transfer vector, Helper, and Rep/Cap, Figure 1).
- Vortex at low speed for a few seconds.
- Add 75 ul of PEI Max stock.
- Vortex at low speed for a few seconds.
- Incubate in a rack at room temperature for exactly 10 minutes.
-
5
Add 2 ml of transfection solution to each 15 cm dish and incubate at 37°C, 5% CO2.
-
6
24 hours post transfection, replace the media with 15 ml of pre-warmed packaging media and incubate at 37°C, 5% CO2 for an additional 96 hours.
Harvesting AAV particles
-
7120 hours post transfection, add 2 ml of 5 M NaCl to release AAV from cells into solution.At this time, AAV particles are both within cells and released into the culture media.
-
8
Incubate plates at 37°C, 5% CO2, for 2 additional hours.
-
9
Collect media and/or cells based on serotype.
-
10For AAV particles with serotypes 1, 5, 8, 9, RETRO, DJ, and PHP.B most viral particles are released into the supernatant:
- Collect supernatant and either proceed to purification step or store at −80°C.
-
11For serotypes 2 and 6, great portion of AAV particles remain with the cells:
- Collect supernatant.
- Scrape cells and add to collected media.
- Prepare dry ice-ethanol bath and perform 4 freeze-thaw cycles in dry ice-ethanol and 37°C water bath with vigorous vortexing for 30 s in between cycles to rupture cells and release AAV particles.
- Either proceed to purification step or store at −80°C.
For other/engineered serotypes use step 11 protocol or compare AAV levels in the supernatant and lysed cells to determine the optimal method for collection. Harvested AAVs can be stored at −80°C for an extended period of time (years).
AAV Purification
-
12
Add turbonuclease (50 units/ml) to the harvested AAV solution and incubate at 37°C in a water bath for 1 hour to digest remaining cellular and plasmid DNA.
-
13
Centrifuge the harvested AAV at 10,000 x g for 10 minutes at 4°C to remove most debris.
-
14
Collect as much supernatant as possible without disturbing the pellet.
-
15
Centrifuge the collected solution at 4,000 x g for 30 minutes at 4°C to remove additional debris.
-
16
Collect as much supernatant as possible without disturbing the pellet.
-
17
Filter AAV supernatant with 0.45 um steriflip filters.
-
18Load ultracentrifuge tubes with AAV solution (e.g. 6 × 15cm plates will yield approximately 6 × 17ml of AAV solution for loading 3 x ultracentrifuge tubes):
- Add 25 ml of filtered AAV solution onto each sterile 30 ml conical tube.
- Layer the bottom of the tube with 4 ml of 40% sucrose cushion in TNE.
- Fill to the top with the remainder of AAV solution (or PBS if not enough AAV) allowing a 2mm gap between the liquid and the rim of the tube.Tubes must be balanced to within 0.1 grams. At high speed, ultracentrifuge tubes will collapse if not filled close to the rim.
-
19
Ultracentrifuge for 16 hours at 50,000 x g (20,000 rpm in a SW32Ti rotor) at 4°C to pellet the AAV virus. For large-scale preparations, also see ALTERNATE PROTOCOL 1.
-
20
Remove and discard supernatant and sucrose cushion gently so the pellet is not disturbed.
-
21
Remove excess solution from ultracentrifuge tube walls with sterile cotton swabs.
-
22
Add 0.5 ml of PBS to each pellet.
-
23
Place tubes on a nutator at 4°C overnight.
-
24
After overnight mixing, pipet pellets up and down 50 times to resuspend in PBS.
-
25
Combine resuspended AAVs in one tube (14 ml or 50 ml depending on the size of the preparation).
-
26
Vortex AAV solution at max speed for 30 s.
-
27
Centrifuge at 500 x g at 4°C for 10 minutes to clear aggregates and debris.
-
28Load AAV supernatant onto a 100-kDa molecular weight cutoff (MWCO) protein concentrator.Multiple concentrator sizes are commercially available. Select the size appropriate for your preparation.
-
29
Centrifugation at 3,000 x g at room temperature for 10-minute intervals until the volume is reduced to 100–500 ul.
-
30Aliquot samples (3–5 ul/tube) in 1.5 ml sterile screw cap vials with O-ring seals and store at −80°C freezer. Avoid freeze-thawing of samples.There is no need to quick-freeze on dry ice, dry ice plus ethanol, or liquid nitrogen. Aliquoted samples stored at −80°C are effective for an extended period (years).
AAV titer and validation
AAV preparations are validated by determining their titer or number of genome copies per ml (GC/ml), identifying the amount of impurities in the sample solution, and their transduction efficiency in target cells.
-
31Perform qPCR or ddPCR, as previously described (Lock et al., 2010), on an aliquot of virus to determine the number of AAV genocopies per ml of solution.
- AAV samples are typically diluted from 10−6 to 10−8 fold
- If qPCR is used, prepare a standard curve using the transfer vector used for packaging. Please note that a plasmid copy is equivalent to two copies of AAV genome.
- The following primers have been validated:
- ITR forward primer 5’ GGAACCCCTAGTGATGGAGTT 3’
- ITR reverse primer 5’ CGGCCTCAGTGAGCGA 3’
-
32
Resolve an aliquot of AAV via polyacrylamide gel electrophoresis (PAGE) and silver stain the resulting gel. Sample results are discussed in Anticipated Results Section (Figure 3B).
-
33
Genes delivered by AAVs are expressed 2–3 weeks after in vivo transduction. To evaluate transduction efficiency of the selected serotypes in target cells rapidly and prior to in vivo transduction, infect cultured cells or ex vivo preparations of brain slices (Bastrikova, Gardner, Reece, Jeromin, & Dudek, 2008; Gu, Lamb, & Yakel, 2012; Stoppini, Buchs, & Muller, 1991). Transduced cells express genes delivered by AAVs within days in culture and expression in brain cultures is observed 1–2 week after transduction.
Figure 3. Validation of an AAV preparation for transduction efficiency and purity.
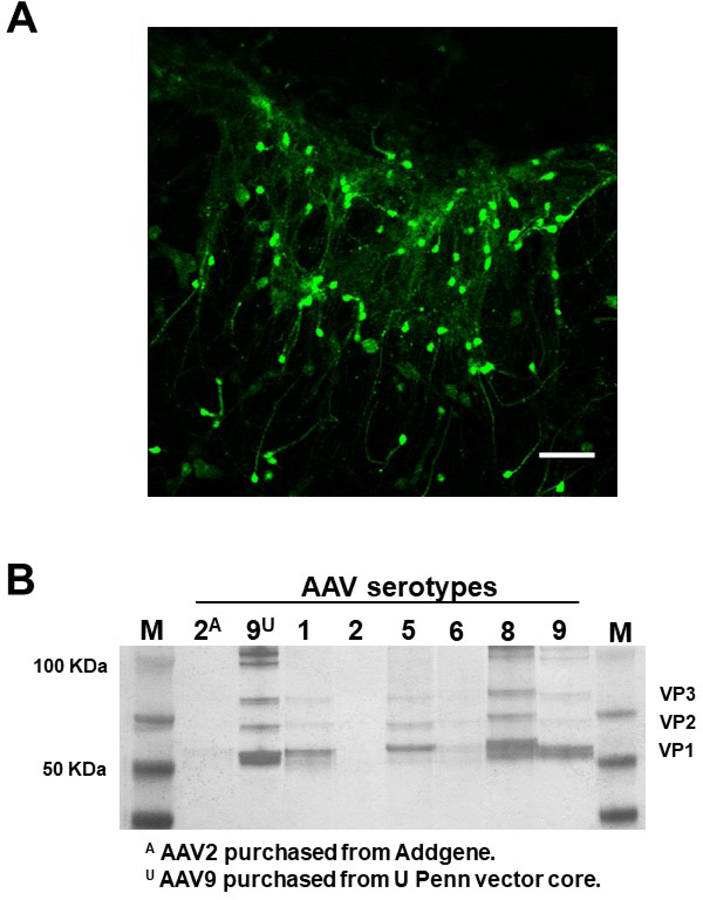
A. Organotypic mouse brain slice cultures were prepared as described previously from 6–8 days postnatal C57BL/6J mice (Bastrikova et al., 2008; Gu et al., 2012; Stoppini et al., 1991). Virus AAV9-hSyn-GFP was added on top of the culture 24 hours post dissection. GFP expression was observed 7 days after transduction and fluorescence images were captured on a Zeiss LSM710 (Carl Zeiss Inc, Oberkochen, Germany) using an EC Plan-Neofluar 10x/0.3 objective. AAV9-hSyn-GFP preparation was validated by ex vivo transduction. B. AAV samples were harvested from culture media (packaging cells were discarded) as described in Protocol 1. Samples were boiled in NuPage sample buffer, loaded onto a 4–12% NuPage Bis-Tris gels (Thermo Fisher Scientific, Rockford, IL, USA) and resolved via polyacrylamide gel electrophoresis (PAGE). Pierce Silver Staining kit (Thermo Fisher Scientific, Rockford, IL, USA) was used according to manufacturer’s protocols to stain the gel and reveal VP1, VP2, VP3 (AAV proteins) and impurities. AAV serotype 1, 5, 8, and 9 samples produced with small-scale protocol have suitable titers and comparable purities to commercially obtained samples. AAV packaged from the same vector backbones for serotypes 2 and 9 were purchased from Addgene and UPenn Vector Core and run on the gel as positive controls.
ALTERNATE PROTOCOL 1
Cesium Chloride and iodixanol gradient purification
An alternative method used by many commercial virus production laboratories is to concentrate large amounts of AAVs via tangential flow filtration (TFF) followed by cesium chloride or iodixanol gradient purification as described previously (Lock et al., 2010; Zolotukhin et al., 1999). AAV samples purified by cesium chloride gradients have been shown to have lowered transduction rates as compared to AAV samples purified by iodixanol gradients (Lock et al., 2010). Therefore, only iodixanol gradient purification is briefly discussed in the following section.
Materials
Harvested AAV particles from Basic Protocol 1
Optiprep (60% w/v sterile solution of iodixanol in water, Sigma-Aldrich, catalog # D1556–250ML)
Prepare 15%, 25%, and 40% iodixanol solutions as described in REAGENTS and SOLUTIONS section recipe
NovaSet™-LS LHV tangential flow filtration (TFF)
18-gauge needle
Quick-seal ultracentrifuge tubes (Beckman Quick-Seal Polypropylene Tube, 25×89mm, 39ml capacity, catalog # 342414 or equivalent)
Beckman Coulter Cordless Tube Topper (catalog # 358312) or equivalent
Ultracentrifuge (Beckman Coulter and 70Ti rotor, capable of 350,000 x g speed for centrifugation of 39ml samples, max speed 32,000 rpm, or equivalent)
Ultracentrifuge (Beckman Coulter and 70Ti rotor, or equivalent)
Large-Scale AAV preparation
If liters of AAV solution are harvested for preparation:
- Concentrate the clarified culture medium by tangential flow filtration (TFF).This can be accomplished by a NovaSet™-LS LHV TFF system or equivalent. Note that concentration of a large AAV preparation increases the amount of impurities in AAV sample several folds and the sample should be purified by cesium chloride or iodixanol gradients.
Prepare iodixanol gradient in quick-seal centrifuge tubes: 4ml of 15%, 9ml of 25%, 9ml of 40%, and 5ml of 54% iodixanol as described previously (Lock et al., 2010).
Apply concentrated AAV sample up to 12ml on top of the gradient or fill with PBS.
Seal the tube with Beckman Coulter Cordless Tube Topper or equivalent.
Centrifuge samples for 70min at 350,000 x g in Beckman 70Ti rotor or equivalent at 18°C.
Puncture the quick-seal tube with an 18-gauge needle on top and use a second needle to puncture and collect fractions at 1cm from the bottom.
Assay fractions as described in PROTOCOL 1 - AAV titer and validation section, steps 1 and 2 – to determine high quality fractions.
Pool high quality fractions, dilute in PBS, and dialyze in 35 mM NaCl-phosphate buffer to remove impurities.
OPTIONAL STEP: Depending on the volume of your resulting solution, use TFF or protein concentrator with 100-kDa molecular weight cutoff (MWCO) (Pierce, Rockford, IL, USA) to concentrate the AAV sample.
- Aliquot samples (3–5 ul/tube) in 1.5 ml sterile screw cap vials with O-ring seals and store at −80°C freezer. Avoid freeze-thawing of samples.There is no need to quick-freeze on dry ice, dry ice plus ethanol, or liquid nitrogen. Aliquoted samples stored at −80°C are effective for an extended period (years).
BASIC PROTOCOL 2
Production of Lentiviruses/Retroviruses
This protocol describes details of 2nd generation packaging system for VSV-G-pseudotyped lentiviral vectors. VSV-G is the most common protein used for pseudotyping lentiviruses due to its ability to transduce most mammalian cells. Other plasmids delivering plasma membrane proteins (e.g. Rabies G or avian envelope protein Env A) can be substituted in place of pMD2.G to pseudotype retro and lentiviruses. This protocol is adapted from the previously published protocol by Salmon and Trono (Salmon & Trono, 2006).
To package retroviral transfer vectors, helper packaging plasmid (pUMVC) is substituted for the lentiviral helper packaging plasmid (psPAX2). All other steps remain the same. Titers for retroviruses are approximately 10 times less than titers for lentiviruses.
HEK293 cells that stably express components of retro and lentiviral packaging genome are commercially available. Use of the HEK293 stable cell lines, eliminates the need to transduce cells with helper and/or envelope plasmids but increases the chance of recombination and creation of replication-competent viruses.
Materials
All material and reagents used in this protocol are prepared and maintained with sterile techniques.
Cell line:
HEK293T/17 cells (ATCC catalog # CRL-11268, RRID: CVCL_1926)
Tissue culture and transfection solutions:
DMEM, Dulbecco’s Modified Eagle Medium (Thermo Fisher Scientific, catalog # 11965–118)
L-Glutamine (Thermo Fisher Scientific, catalog # 25030081)
Trypsin- EDTA (Thermo Fisher Scientific, catalog # 25200–056, purchased as 0.25%)
Sodium pyruvate (Thermo Fisher Scientific, catalog # 11360070)
PBS (Phosphate Buffered Saline)
OPTI-MEM medium (Gibco, catalog # 31985–070)
100X Pen/Strep (Sigma-Aldrich, catalog # P0781)
Lipofectamine 2000 (Thermo Fisher Scientific, catalog # 11668019)
100x Penicillin-Streptomycin at 10,000 U/mL (Thermo Fisher Scientific, catalog # 15140122)
See REAGENT AND SOLUTIONS for Growth media recipe
Plasmids:
Lentiviral transfer vector (e.g. pWPXL, pLKO.1, pDEST673)
Helper plasmid psPAX2 (encoding gag, Pol, tat, and rev)
pMD2.G (encoding VSV-G envelope protein)
Plasmids can be resuspended in sterile water, TE, or EB (See REAGENT AND SOLUTIONS). Any plasmid concentration above 100 ng/ul is acceptable. The above plasmids are also available from commercial sources, Addgene, and laboratories with published protocols.)
Human genomic DNA (Promega catalog # G304A, or equivalent)
Oligos:
gag primers Forward 5’ - GGAGCTAGAACGATTCGCAGTTA
Reverse 5’ - GGTTGTAGCTGTCCCAGTATTTGTC
Human actin primers Forward 5’ - TCCGTGTGGATCGGCGGCTCCA
Reverse 5’ - CTGCTTGCTGATCCACATCTG
Psi (ψ) primer Forward 5’ - GCAGCATCGTTCTGTGTTGT
Reverse 5’ - GCTCGACATCTTTCCAGTGA
Consumables
T175 tissue culture flask
10 cm tissue culture dishes
14 ml screw top centrifuge tubes
50 ml screw top centrifuge tubes
30 ml conical ultracentrifuge tubes (Beckman Coulter, catalog # 358126) or equivalent
1.5 ml conical screw top centrifuge tubes with O-rings (Sarstedt, catalog # 72.692.005)
Cell scrapers
Sterile cotton tip applicators
0.45 um Durapore Striflip-GP PVDF filter (EMD Millipore, catalog # SE1M003M00) or equivalent
70% Ethanol
10% Bleach
Vortex mixer
Nutating shaker
Centrifuge (Beckman Coulter Allegra X-22R and SX4250 rotor, max speed 4,500 rpm, or equivalent)
Ultracentrifuge (Beckman Coulter and SW32Ti rotor, capable of 50,000 x g speed for centrifugation of 35ml samples, max speed 32,000 rpm, or equivalent)
Water bath (37° C)
Tissue culture incubator (37°C, 5% CO2)
Laminar flow hood
Cell Culture
-
1
Maintain HEK293T cells in growth media without antibiotics in T175 flasks in a tissue culture incubator at 37°C, 5% CO2.
-
2Split cells at a ratio of 1:4 to 1:8 using trypsin-EDTA three times per week.Discard cells after 10 passages. Frequent passages and maintaining the cells at less than 80% confluency helps ensure optimum yield.
-
3For transfection, split cells the day before and seed 10cm dishes with 6.5e6 cells per dish in 10ml of growth media without antibiotics.The cells should be 80–90% confluent at the time of transfection. One T175 flask at 60% confluency should provide ample cells to seed two to three 10cm dishes. One 10cm plate yields 10ml of unconcentrated lentivirus with titer of >1e6 TU/ml; for retroviruses a 10cm plate will yield 10ml of virus with titer of >1e5 TU/ml.
-
4
Incubate cells overnight in an incubator at 37°C, 5% CO2.
Transfection to reassemble recombinant viral genes
-
5
Replace media on 10cm plates with 6ml of growth media without antibiotics.
-
6
Three to four hours after replacing the media, transfect cell cultures. For each 10cm plate, mix plasmids in a sterile 15ml tube (transfer vector plasmid: 12 ug, helper plasmid psPAX2: 10.8 ug, and envelope plasmid pMD2.G: 1.2 ug, Figure 1) in 1.5 ml of Opti-MEM – Tube A.
-
7
Prepare a second 15ml tube containing 1.5 ml of Opti-MEM and add 72 ul of Lipofectamine 2000. Mix Lipofectamine 2000 gently. Incubate at room temperature for 5 minutes – Tube B.
-
8
Add the content of mixed plasmid solution (Tube A) to the diluted Lipofectamine 2000 (Tube B) for a final volume of 3 ml. Mix gently and incubate for 30 minutes at room temperature.
-
9
Add the transfection solution (3 ml per sample) dropwise on top of each 10cm plate. Mix gently by rocking the plate back and forth. Incubate the plate overnight at 37°C, 5% CO2 in a tissue culture incubator.
-
10
16–18 hours post-transfection, replace the medium with 10ml of pre-warmed growth media containing 1x Pen/Step antibiotics.
-
11
Collect the first virus-containing supernatant at 40–48 hours post-transfection and dispose of the cells after short incubation with 10% bleach.
-
12
Centrifuge supernatant at 2000 x g for 10 minutes at 4°C to remove cellular debris.
-
13Filter the viral supernatant through a 0.45 µm PVDF filter. Proceed to concentrate the virus by ultracentrifugation or aliquot and store unconcentrated viral supernatants at −80°C in screw cap vials with O-ring seals. Samples are typically stored as 0.5–1 ml aliquots.Frozen supernatant can be stored at −80°C for years with minimum effect on titer. Use 30 ml of supernatant for concentration of virus (supernatant from 3 × 10cm plates). Concentrated virus in PBS decreases in titer exponentially after 6–12 months. Each round of freeze thaw lowers the viral titer significantly.
Virus concentration by ultracentrifugation
-
14
Rinse ultracentrifuge tubes with 70% ethanol and allow to dry in the laminar flow hood.
-
15Carefully fill the tubes with lentiviral supernatant and layer the bottom of tube with 4ml of 20% sucrose cushion in Tris-sodium chloride-EDTA (TNE) buffer. Fill to the top allowing a 2mm gap between the liquid and the rim of the tube.Tubes must be balanced to within 0.1 grams. At high speed, ultracentrifuge tubes will collapse if not filled close to the rim.
-
16Centrifuge samples based on pseudotype as below.
- Viruses pseudotyped with VSV-G, centrifuge for 2 hours at 50,000 x g, 4°C (20,000 rpm in SW32Ti rotor).
- Centrifuge non-VSV-G retro and lentiviruses for 20 hours at 6,500 x g, 4°C (7,300 rpm in SW32Ti rotor).Viruses that are not pseudotyped with VSV-G, including retro and lentiviral Env proteins, will be mostly destroyed at high ultracentrifuge speeds.
-
17
Transfer tubes to a laminar flow hood and gently discard the supernatant into 10% bleach.
-
18
Allow the tube to drain in an inverted position. Tubes can rest up-side-down on a sterile 10cm plate for a few minutes. Do not allow the virus to dry.
-
19
Carefully wipe the inside of each tube with sterile cotton tip swabs to remove any remaining traces of liquid.
-
20
Resuspend the pellet in PBS (300 ul for 30 ml of concentrated viral supernatant) or desired volume/medium and allow the samples to resuspend overnight at 4°C with gentle rotation on a nutator. Pellets will not be visible to naked eye.
-
21
Mix the samples by pipetting up and down 50–100 times. Avoid creating bubbles when pipetting.
-
22Aliquot samples (25 ul/tube) in 1.5 ml sterile in screw cap vials with O-ring seals and store at −80°C freezer. Avoid freeze-thawing of samples.Do not snap-freeze using dry ice or dry ice plus ethanol; instead use liquid nitrogen to freeze viral aliquots quickly.
Retro lentiviral titer and validation – qPCR method
Retro and lentiviruses can be titered by a number of different methods. Three methods are discussed in this section: qPCR on chromosomal preparations from transduced cells, Flow cytometry on transduced cells with fluorescent lentiviruses, and p24 Assay on viral particles.
-
23qPCR on chromosomal preparations from transduced cells:
- Split HEK293T cells and seed 50,000 cells per well in a 6 well plate. Incubate overnight at 37°C, 5% CO2.
- Next day, infect cells with 10 ul of unconcentrated or 0.5 ul of concentrated lentiviral samples.
- 24 and 72 hours after infection, replace media with fresh growth media.
- 5 days after infection, harvest cells and isolate chromosomal DNA from cells using commercial kits (e.g. DNeasy Blood & Tissue Kit from Qiagen).
- Dilute chromosomal DNA by adding 5 uL of each DNA preparation to 195 uL of water. Use 5 uL of diluted DNA for qPCR.
- Prepare two sets of qPCR standards using lentiviral transfer vector plasmids and human genomic DNA (Promega catalog # G304A, or equivalent). Transfer plasmid range of 1e5–1e2 copies/5 ul and genomic DNA range of 2e4–1e2 copies/5 ul work well for chromosomal DNA dilutions from step e.
- Perform two qPCRs on 5 ul of diluted DNA from step e using gag primers (Forward 5’ - GGAGCTAGAACGATTCGCAGTTA and Reverse 5’ - GGTTGTAGCTGTCCCAGTATTTGTC) and human actin primers (Forward 5’ - TCCGTGTGGATCGGCGGCTCCA and Reverse 5’ - CTGCTTGCTGATCCACATCTG).
- Use standard curves to deduce the number of gag and actin copies per 5 ul of your sample.
- Number of gag copies per cell = (number of gag copies) / (number of actin copies/2)
- Lentiviral titer in transducing units per ml = starting number of cells at the time of transduction 100,000 x number of gag copies per cell x dilution factor (1000/ volume of virus used in ul)
The above protocol can be used to titer retroviruses with the following modifications: use retroviral transfer vectors for the standard curve and primers designed for the retroviral transfer vectors. For example, packaging sequence psi (ψ) is present in most retroviral transfer vectors and can be targeted for qPCR using the following primers: Forward 5’ - GCAGCATCGTTCTGTGTTGT, and Reverse 5’ - GCTCGACATCTTTCCAGTGA.
Retro lentiviral titer and validation – flow cytometry method
Flow cytometry on transduced cells with fluorescent retro or lentiviruses:
Split HEK293T cells and seed 50,000 cells per well in a 6 well plate. Incubate overnight at 37°C, 5% CO2.
Next day, infect cells by adding 50, 10, and 2 ul of unconcentrated or 0.5, 0.1, and 0.02 ul of concentrated lentiviral samples to the existing media.
24 and 72 hours after infection, replace media with fresh growth media.
5 days after infection, detach cells with trypsin-EDTA and fix in 1% formalin.
Use flow cytometry to determine the % fluorescent cells.
From your infection volumes in step b, select a lentiviral volume that produces 5–20% fluorescent cells. Populations with greater than 20% fluorescent cells have a greater chance of containing cells with multiple lentiviral incorporations.
- Lentiviral titer in transducing units per ml = starting number of cells at the time of transduction 100,000 x % fluorescent cell x dilution factor (1000/ volume of virus used for transduction)P24 assay is also able to convey the number of viral particles per volume. Since both retro and lentiviruses produce many non-transducing particles, p24 titers are usually 500–5000 times higher than actual transducing unit titers. This assay is commercially available as kits for retro and lentiviruses. Please follow manufactures recommendation for experimental procedure.
BASIC PROTOCOL 3
Production of Rabies dG (SADB19 dG, SiR, dGL, and CVS-N2c dG)
This protocol describes step by step production of SADB19 Rabies dG adapted from the previously published protocol by Dr. Callaway laboratory (Osakada & Callaway, 2013). Variations to the protocol for the production of SADB19 rabies dG SiR, dGL, and CVS-N2c dG are described in the strategic planning section.
Rabies virus is harvested at multiple stages to expand the amount of collected virus. Prior to each harvesting step, cultured cells should be checked for expression of rabies proteins or the gene of interest. Expression of fluorescent proteins is readily checked under a microscope. If the expressed protein is not fluorescent, cells should be harvested, divided into packaging and monitoring batches, and checked for expression with immunostaining. It may take weeks after the initial transfection of viral packaging plasmids for SADB19 rabies dG SiR, dGL, and CVS-N2c dG (in Neuro2a cells) to detect viral particles or the delivered gene of interest.
Materials
All material and reagents used in this protocol are prepared and maintained with sterile techniques.
Cell lines for packaging SADB19 dG and dGL:
HEK293T/17 cells, (ATCC catalog # CRL-11268, RRID: CVCL_1926)
The following cell lines are provided by Dr. Callaway’s laboratory and the Salk Institute Viral Vector Core (Osakada & Callaway, 2013).
B7GG: BHK-21 that stably express T7 RNA polymerase, rabies glycoprotein G, and histone-tagged GFP.
BHK-EnvA: BHK-21 cells stably express EnvA fused to the cytoplasmic domain of rabies glycoprotein G.
BHK-EnvB: BHK-21 cells stably express EnvB fused to the cytoplasmic domain of rabies glycoprotein G.
HEK 293T–TVA800: HEK293T cells that stably express TVA800.
HEK 293T–TVB cells: HEK293T cells that stably express TVB.
Cell lines for packaging SADB19 dG SiR:
The following cell lines are provided by Dr. Tripodi’s laboratory at Cambridge, UK (Ciabatti et al., 2017).
B7/ToGG: BHK-21 that stably express T7 RNA polymerase, TEV protease, rabies glycoprotein G, and histone-tagged GFP.
HEK/TGG: HEK293 cells that stably express T7 RNA polymerase, TEV protease, rabies glycoprotein G, and histone-tagged GFP.
BHK/EnvA-TEVp: BHK-21 cells stably express Tev protease, EnvA fused to the cytoplasmic domain of rabies glycoprotein G.
HEK/TVA-GFP: HEK293T cells that stably express TVA800.
Tissue culture and transfection solutions:
DMEM, Dulbecco’s Modified Eagle Medium (Thermo Fisher Scientific, catalog # 11965–118)
L-Glutamine (Thermo Fisher Scientific, catalog # 25030081)
Trypsin- EDTA (Thermo Fisher Scientific, catalog # 25200–056, purchased as 0.25%)
Sodium pyruvate (Thermo Fisher Scientific, catalog # 11360070)
PBS (Phosphate Buffered Saline)
OPTI-MEM medium (Gibco, catalog # 31985–070)
Lipofectamine 2000 (Thermo Fisher Scientific, catalog # 11668019)
HBSS (Thermo Fisher Scientific, catalog # 14175095, 1x, no calcium, no magnesium, no phenol red)
HBSS (Thermo Fisher Scientific, catalog # 14185052, 10x, no calcium, no magnesium, no phenol red)
20% sucrose in HBSS (See REAGENT AND SOLUTIONS)
Growth media (see REAGENT AND SOLUTIONS)
Plasmids:
Visit “Edward Callaway, Thomas Jessell, and Ian Wickersham Lab Plasmids” on Addgene for a generous collection of recombinant rabies plasmids:
pSADΔG-F3 (Addgene, catalog # 32634)
pcDNA-SADB19N (Addgene, catalog # 32630)
pcDNA-SADB19P (Addgene, catalog # 32631)
pcDNA-SADB19L (Addgene, catalog # 32632)
pcDNA-SADB19G (Addgene, catalog # 32633)
Plasmids can be resuspended in sterile water, TE, or EB (See REAGENT AND SOLUTIONS). Any plasmid concentration above 100 ng/ul is acceptable. The above plasmids are also available from commercial sources, Addgene, and laboratories with published protocols.)
Consumables
T175 tissue culture flask
15 cm tissue culture dishes
14 ml screw top centrifuge tubes
50 ml screw top centrifuge tubes
30 ml conical ultracentrifuge tubes (Beckman Coulter, catalog # 358126) or equivalent
1.5 ml conical screw top centrifuge tubes with o-rings
Cell scrapers
Sterile cotton tip applicators
0.45 um Durapore Striflip-GP PVDF filter (EMD Millipore, catalog # SE1M003M00) or equivalent
70% Ethanol
10% Bleach
Parafilm
Vortex mixer
Nutating shaker
Centrifuge (Beckman Coulter Allegra X-22R and SX4250 rotor, max speed 4,500 rpm, or equivalent)
Ultracentrifuge (Beckman Coulter and SW32Ti rotor, capable of 50,000 x g speed for centrifugation of 35ml samples, max speed 32,000 rpm, or equivalent)
Water bath (37° C)
Tissue culture incubator (37°C, 5% CO2)
Tissue culture incubator (35°C, 3% CO2)
Laminar flow hood
Cell Culture
-
1
Maintain (1) packaging cell line B7GG, (2) pseudotyping cell lines BHK-EnvA or BHK-EnvB, and (3) titering cell lines HEK293T, TVA800, or TVB in growth media without antibiotics in T175 flasks in tissue culture incubators at 37°C, 5% CO2.
-
2Split cells at a ratio of 1:4 to 1:8 using trypsin-EDTA three times per week.Discard cells after 10 passages. Frequent passages and maintaining the cells at less than 80% confluency helps ensure optimum yield.
-
3Prior to transfection, split B7GG cells the day before and seed 10cm dishes with 3.3e6 cells per dish in 10ml of growth without antibiotics.The cells should be about 40–50% confluent at the time of transfection. One T175 flask at 60% confluency should provide 1–3e7 cells.
-
4
Incubate cells overnight in an incubator at 37°C, 5% CO2.
Transfection to reassemble recombinant viral genes
-
5Replace media on 10cm plate(s) with 5.5ml of OPTI-MEM.For planning purposes: the transfection takes 6 hours to complete.
-
6
For each 10cm plate, mix plasmid and transfer vector in a sterile 15ml tube (rabies pSADΔG-F3 transfer vector plasmid carrying your gene of interest: 30 ug, pcDNA-SADB19N plasmid: 15 ug, pcDNA-SADB19P plasmid: 7.5 ug, pcDNA-SADB19L plasmid: 15 ug, and pcDNA-SADB19G plasmid: 5 ug) in 1.25 ml of Opti-MEM – Tube A.
-
7
Prepare a second 15ml tube containing 1.25 ml of Opti-MEM and add 112.5 ul of Lipofectamine 2000 to Opti-MEM. Mix Lipofectamine 2000 gently. Use Incubate at room temperature for 5 minutes – Tube B.
-
8
Add the content of mixed plasmid solution (Tube A) to the diluted Lipofectamine 2000 (Tube B) for a final volume of 2.5 ml. Mix gently and incubate for 30 minutes at room temperature.
-
9
Add the transfection solution (2.5 ml per sample) dropwise on top of each 10cm plate. Mix gently by rocking the plate back and forth. Incubate the plate for 6 hours at 37°C, 5% CO2 in a tissue culture incubator.
-
10
After 6 hours, wash the dish with 10 ml of pre-warmed growth media.
-
11
Replace the media with 10ml of pre-warmed growth media and incubate the plate overnight at 37°C, 5% CO2.
-
12
One day after transfection, move the plate to a 35°C, 3% CO2 tissue culture incubator. Lowered temperature and CO2 levels decelerate cell growth and improve virus yield.
-
13
Two days after transfection, wash cells with 5ml of PBS, and detach cells with 2ml of trypsin-EDTA for 5–6 minutes.
-
14
Collect detached cells in a 15ml tube containing 10ml of growth media.
-
15
Centrifuge cells at 300 x g for 3 minutes.
-
16
Discard supernatant. Gently resuspend cells in growth media and transfer to a 15cm plate. Add growth media to a final volume of 15ml and incubate overnight at 35°C, 3% CO2.
-
17
One day after re-plating the cells, replace media with 24ml of pre-warmed growth media and incubate the plate for two days at 35°C, 3% CO2.
-
18Add 5 ml of growth media to plate and incubate for two additional days.Do not allow the media to become acidic as you propagate the rabies virus in the following steps. Add/replace 5 ml of growth media to plates as needed to avoid media turning yellow.
-
19Seven days after transfection, check for expression of your gene of interest or viral particles. If expression is low, replace 5 ml of media and return to this step after two days. If most cells show expression, collect the first batch of supernatant:
- Collect supernatant and spin at 300 x g for 3 minutes to remove debris.
- Filter supernatant through a 0.45 um low protein binding PVDF filter.
- Store at 4°C for up to 48 hours or store for longer periods of time at −80°C.
-
20Add 24ml of pre-warmed growth media and incubate the plates for two days at 35°C, 3% CO2.During the two-day incubation in step 20, expand B7GG stock of cells for propagation of the unpseudotyped virus. Seed 2 x T175 flasks of B7GG with 4e6 cells and maintain as described in the Cell Culture section.
-
21
Add 5 ml of growth media to 15cm plates and incubate for two days.
-
22
Collect supernatant as described in step 19 and discard cells.
-
23
23. Mix all collected supernatants and store at 4°C for up to 48 hours or store for longer periods of time at −80°C.
Expanding unpseudotyped rabies dG
-
24
Seed 3 × 15cm plates with 4e6 B7GG cells in 25 ml of growth media and incubate overnight at 37°C, 5% CO2.
-
25
Next day, remove media, divide supernatant from step 23 (approximately 58ml) and add to 3 × 15 cm plates (approximately 19ml).
-
26
Add additional growth media to plates to bring the total volume to 24 ml and incubate for 6 hours at 35°C, 3% CO2.
-
27
After 6 hours, replace media with 24 ml of pre-warmed growth media and incubate the plate for 4 days at 35°C, 3% CO2.
-
28
After incubation, collect supernatant as described in step 19. Add 24 ml of pre-warmed growth media and incubate the plate for 2 days at 35°C, 3% CO2.
-
29
After incubation, collect supernatant as described in step 19 and discard cells.
-
30
Mix all collected supernatants and store at 4°C for up to 48 hours or store for longer periods of time at −80°C.
NOTE: Rabies supernatant after step 24 can be stored to seed plates for future viral preparations. To do so, add 4 ml of growth media to 15cm plates three days after step 24. Collect supernatant two days later as described in step 19. Store 4 ml aliquots of virus at −80°C to seed 15cm plates in step 24 in later preparations. Collection of seed viral stock eliminates the repeat of steps 1–23 for subsequent viral preparations (saves 11 days).
NOTE: To produce 100 ul of pseudotype rabies dG, a minimum of 100ml of rabies dG per sample is needed. Repeat steps 24–30 and seed 10 × 15cm (instead of 3) with collected supernatant to increase the amount of harvested virus. Do not repeat this expansion more than once. Chance of recombination events leading to creation of replication competent rabies virus increases after each expansion process.
Pseudotyping rabies dG
-
31
Seed 10 × 15cm plates with 4e6 pseudotyping cells (example used in this protocol: BHK-Env A) in 25 ml of growth media and incubate overnight at 37°C, 5% CO2.
-
32
One day after seeding the cells, remove media, apply 10 ml of supernatant collected in step 30 (or enough virus to reach MOI 0.5–0.6) to each 15 cm plates.
-
33
Add additional growth media to plates to bring the total volume to 20 ml and incubate for 6 hours at 35°C, 3% CO2.
-
34After 6 hours, remove unpseudotyped rabies dG as follows:
- Wash each plate thoroughly with PBS twice.
- Apply 5 ml of trypsin-EDTA to each plate and incubate at 37°C, 5% CO2 for 3–5 minutes to detach cells and destroy the remainder of unpseudotyped rabies dG.
- Collect cells in 10ml of growth media.
- Centrifuge at 300 x g for 3 minutes to pellet cells.
- Discard supernatant.
- Gently resuspend cells in growth media and plate in a new 15cm plate.
- Add growth media to a final volume of 23 ml per plate and incubate the plate overnight at 35°C, 3% CO2.
-
35
After incubation, replace media with 23 ml of pre-warmed growth media and incubate the plate for 2 days at 35°C, 3% CO2.
-
36
After incubation, collect supernatant as described in step 19.
-
37
Mix all collected supernatants and store at 4°C for up to 48 hours or store for longer periods of time at −80°C.
Virus concentration by ultracentrifugation
-
38
For each pseudotyped sample, collect a minimum of 180 ml of supernatant from step 37 for concentration.
-
39
Rinse 6 × 30–35 ml ultracentrifuge tubes with 70% ethanol and allow to dry in the laminar flow hood.
-
40Carefully fill the tubes with pseudotyped rabies dG supernatant. Fill to the top allowing a 2 mm gap between the liquid and the rim of the tube.Tubes must be balanced to within 0.1 grams. At high speed, ultracentrifuge tubes will collapse if not filled close to the rim.
-
41
Centrifuge samples for 2 hours at 70,000 x g, 4°C (24,000 rpm in SW32Ti rotor).
-
42
Transfer tubes to a laminar flow hood and gently discard the supernatant into 10% bleach.
-
43
Allow the tube to drain in an inverted position. Tubes can rest up-side-down on a sterile 10cm plate for a few minutes. Do not allow the virus to dry.
-
44
Carefully wipe the inside of each tube with sterile cotton tip swabs to remove any remaining traces of liquid.
-
45
Resuspend the pellet in 300 ul of HBSS by wrapping the top in parafilm and gently vortexing.
-
46
Prepare another ultracentrifuge tube by rinsing the tube with 70% ethanol and allowing it to dry in a laminar flow hood.
-
47Carefully combine resuspended pellets in the new ultracentrifuge tube and layer the bottom of the tube with 4 ml of 20% sucrose cushion in HBSS buffer. Fill to the top allowing a 2 mm gap between the liquid and the rim of the tube.Tubes must be balanced to within 0.1 grams. At high speed, ultracentrifuge tubes will collapse if not filled close to the rim.
-
48
Centrifuge samples for 2 hours at 50,000 x g, 4°C (20,000 rpm in SW32Ti rotor).
-
49
Transfer tubes to a laminar flow hood and gently discard the supernatant into 10% bleach.
-
50
Allow tubes to drain in an inverted position. Tubes can rest up-side-down on a sterile 10cm plate for a few minutes. Do not allow the virus to dry.
-
51
Carefully wipe the inside of each tube with sterile cotton tip swabs to remove any remaining traces of liquid.
-
52
Resuspend the pellet in 100 ul of 1x HBSS by gentle rotation on a nutator.
-
53Aliquot and store at −80°C.Freeze-thaw cycles damage virus. Aliquot in small volumes (3–5 ul) accordingly.
Pseudotyped rabies dG titer and validation
Pseudotyped rabies dG viruses should be titered in HEK293T cells lines with and without TVA (or other corresponding avian receptor) expression. If HEK293Ts without TVA are transduced with the virus, the preparation contains unpseudotyped rabies dG virus and should be discarded.
-
54To determine titer:
- Split HEK 293-TVA800 (or corresponding avian receptor) cells and seed 150,000 cells per well in a 24 well plate. Incubate overnight at 37°C, 5% CO2.
- Next day, infect cells with 250ul of rabies dG virus serial dilutions (1e-3 to 1e-11 dilutions in 1000 ul aliquots).
- Each well should contain 0.5ml of media + 0.25 ml of viral dilution.
- Incubate at 37°C, 5% CO2.
- If pseudotyped rabies dG is fluorescent, after 1–2 days, count the number of fluorescent cells in a dilution well that has 10–100 fluorescent cells.
- If delivered protein is not fluorescent, 2 days post infection, immunostain cells for the presence of protein of interest or viral proteins, and count infected cells.
- Rabies dG titer in transducing units per ml = number of fluorescent cells x fraction of volume added (1000ul/250ul) x dilution factor (1e-3 … 1e-11)
REAGENTS AND SOLUTIONS
Growth media: DMEM, 10% Fetal Bovine Serum, 2 mM L-Glutamine, 1mM Sodium Pyruvate
AAV Packaging media: DMEM, 4mM L-Glutamine
1X TE buffer: dilute 10X TE buffer (Sigma-Aldrich, catalog # T9285) with ddH2O to 1X, or prepare 10mM Tris, pH 8.0, and 0.1mM EDTA in water; Sterilize by passing through a 0.22um pore size filter
1X EB buffer: 10mM Tris, pH 8.0, and 0.1mM EDTA in water; Sterilize by passing through a 0.22um pore size filter.
40% sucrose in Tri-sodium chloride-EDTA (TNE) buffer: 40 g of sucrose dissolved in 100 ml 1X TNE buffer (50 mM of Tris-HCl, pH7.4, 100 mM of NaCl, and 0.5 mM of EDTA); Sterilize by passing through 0.22µm pore size filter.
20% sucrose in Tri-sodium chloride-EDTA (TNE) buffer: 20 g of sucrose dissolved in 100 ml 1X TNE buffer (50 mM of Tris-HCl, pH7.4, 100 mM of NaCl, and 0.5 mM of EDTA); Sterilize by passing through 0.22µm pore size filter.
20% sucrose in HBSS: 20 g of sucrose dissolved in 90 ml of water and 10ml of 10X HBSS; Sterilized by passing through 0.22µm pore size filter.
15%, 25%, and 40% iodixanol solutions are prepared by diluting 60% (w/v) solution of iodixanol in water (OptiPrep, Sigma-Aldrich, catalog # D1556–250ML) in PBS with final concentrations of 10 mM magnesium chloride and 25 mM potassium
35 mM NaCl-phosphate buffer: 35 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, in water, pH 7.2 and sterile filter.
COMMENTARY
Background Information
Recombinant viral vectors are effective tools for delivery of tracers, sensors, and actuators to cells in mammalian brains (Callaway & Luo, 2015; Castle et al., 2014; Tervo et al., 2016). The recombinant technology offers a versatile method for modification and safe production of viral vectors. Each viral genome is divided into several plasmids and is reconstituted via transfection in a packaging cell line to produce viral particles (Figure 1). Transgenes are often inserted among the viral genome for delivery in vivo. Genetic material responsible for viral replication are often removed to render recombinant viruses non-propagating and safe for use in laboratory research. Recombinant adeno-associated viruses (AAVs), retro/lentiviruses, and rabies dG viruses can deliver up to 5.2 kb, 9 kb, and 8.5 kb of genetic material to mammalian cells, respectively.
AAVs are often used to deliver shorter genomic sequences and can infect dividing and non-dividing cells. They are routinely used to deliver calcium sensors, fluorescent markers, and optogenetic tools to mammalian brains. Small genes such as Genetically Encoded Calcium Indicators (GECIs: GCaMP, jRGECO1), Genetically Encoded Voltage Indicators (GEVIs: ArcLight, ASAPs), dopamine sensors (e.g. dLIGHT), and optogenetic actuators (e.g. Chanelrhodopsins, eNpHRs) are readily packaged and delivered via AAVs. However, genetic sequences such as spCas9 or calcium channels are too large to fit onto an AAV transfer vector. Larger genes are occasionally delivered via AAVs as split modules that recombine in the host cell or delivered using lentiviruses.
Viral capsid proteins dictate AAV tropism or range of host cells. Interactions between host cell surface proteins and AAV capsids result in transduction. Therefore, AAV tropism is determined by variations in the amino acid sequences of its capsid proteins. Some AAV serotypes such as PHP.B cross the blood brain barrier while others manifest retrograde and anterograde activities (Castle et al., 2014; Chan et al., 2017; Tervo et al., 2016; Zingg et al., 2017). Genes delivered by AAVs do not incorporate into their host chromosomes and thus do not cause insertional mutations. The AAV genome is maintained innocuously as concatemeric circles with lasting expression in non-dividing cells. Therefore, AAVs are among favorites for gene delivery in research.
Retro/lentiviruses and rabies dG are more toxic and immunogenic than AAVs. However, they are enveloped viruses that offer superb targeting and pinpoint delivery of larger transgenes. Enveloped viruses such as retro/lentiviruses and rabies dG are easily pseudotyped by expression of desired membrane protein on the surface of the packaging cell lines. The viral envelope is assembled from the plasma membrane of the packaging cell line, and therefore any membrane protein expressed on the surface of the cell is incorporated onto the viral envelope. Natural and designer antigen-receptor pairs can be used to pseudotype retro/lentiviruses and rabies dG and direct them to transduce antigen-expressing cells. Pseudotyped rabies dG is a powerful vector for monosynaptic retrograde delivery of molecular tools in neurobiology.
A myriad of transfer vector plasmids carrying optogenetics tools and sensors are available from non-profit plasmid repositories such as Addgene – refer to INTERNET RESOURCES section.
Critical Parameters
Viral transfer vectors
Design and construction of viral transfer plasmids are the initial steps in production of viral vectors. High quality preparations and validation of each plasmid before use are imperative steps that ensure great yields and effective viral products. Check plasmid integrity with restriction enzyme digestion prior to use.
AAV viral genome is delivered as a single-stranded DNA that is converted and maintained in cells as a double-stranded DNA. Therefore, the insert in the AAV transfer vector could contain promoters, recombinase recognition sites (lox-P), self-inactivating sequences (P2A, T2A, genes of interest). Similarly, ssRNA genome of retro and lentiviruses are reverse transcribed to DNA and can accommodate up to 9 kilobases of promoters, recombinase recognition sites (lox-P), self-cleaving sequences (P2A, T2A, etc.), and/or genes of interest.
AAV, retro, and lentiviral transfer vector plasmids are unstable and readily recombine at the repeat sites. Therefore, researchers should use recA mutant E. coli strains (e.g. Stbl3, Invitrogen) for plasmid propagation to maintain plasmid integrity.
Rabies dG transfer vectors are maintained in the cytoplasm and viral proteins and genes of interest are transcribed from RNA by a specialized viral polymerase. Therefore, it cannot carry promoters or recombinase recognition sites. Also, molecular cloning requires the gene of interest to be inserted in frame with other viral proteins (Osakada & Callaway, 2013).
Maintaining cells
Packaging cell lines often stably express components of viral genome. Therefore, maintaining stock cells at low passage under optimal growth conditions are required for high yield viral preparations.
During AAV packaging, cells are incubated in serum-free packaging media for 5 days (steps 6–8). Cell media will turn yellow-orange during these steps. Do not replace the serum-free media with growth media which contain serum. Presence of serum in media increases the amounts of undesired proteins in AAV preparations.
In contrast to AAV, do not allow the media to become acidic as you propagate the rabies dG virus in the following steps. Acidic pH destroys rabies dG virus. Add/replace 5 ml of growth media to plates as needed to avoid media turning yellow.
Storing viruses
AAVs are durable viruses that can be stored under a variety of conditions. Established guidelines recommend aliquoting and storing AAVS in −80°C freezers. Allowing virus to dry would destroy the virus very quickly. AAV aliquots are effective for years when properly stored.
Rabies dG, retro, and lentiviruses are enveloped viruses that are sensitive to freeze-thaw cycles, pH, salt concentration, and drying. Aliquot these viruses in small volumes (enough for one experimental setting) and store in −80°C freezers. Frozen aliquots of unconcentrated lentiviruses and rabies dG are effective for years. Concentrated lentiviruses and retroviruses lose infectivity gradually after several months at −80°C.
Safety
All recombinant viruses described in the protocols are non-propagating and safe for use in Biosafety Level 2 laboratories. Please consult and follow your institute’s biosafety guidelines for safe handling and disposal of viruses. Also, use personal protective clothing and gear to protect yourself and your coworkers.
All material that comes in contact with viruses should be decontaminated with 10% bleach prior to disposal and all experimentation should be performed in a tissue culture hood.
Troubleshooting
The most common problem with packaging viruses is low yield. Viruses delivering fluorescent moieties can be monitored by microscopy at every step of packaging to ensure expression.
For low yield AAVs:
For low yield retro and lentiviruses:
For low yield rabies dG viruses:
Anticipated Results
AAVs
AAV yield is affected by the chosen serotype, size of insert, and the nature of expressed genes. For example, serotypes such as 9 and DJ are harvested at higher titers in packaging cell lines than serotypes 2 and 6. A virus carrying a small inert insert such as GFP produces a higher yield than a larger insert such as channelrhodopsin tethered to a GFP gene. Transfection of 6 × 15 cm plates as starting material for a 100ul preparation produces 1e12–1e13 genome copies/ml of purified AAV.
AAVs are delivered in vivo via stereotactic injections. Transduction of cultured mouse brain slices can be accomplished by injection or adding the virus dropwise on top of brain slices. In Figure 3A, an AAV preparation expressing GFP from a human synapsin promoter was used to transduce organotypic mouse brain cultures to validate efficacy (Addgene, catalog # 50465; RRID: addgene_50465). Animals used in this study were ordered from Charles River and Jackson Laboratories, USA. All animal procedures complied with NIH/NIEHS animal care guidelines and were approved by the Animal Care and Use Committee (ACUC) at NIH/NIEHS, animal Protocol # 2012–0004.
For Figure 3B, AAV samples with serotypes 1, 2, 5, 6, 8, and 9 were prepared from culture media as described in the small-scale (6 × 15cm plates) production method. AAV particles were only collected from the culture media. Prepared samples and commercially obtained AAV particles with serotypes 2 and 9 (from Addgene and UPenn vector core, respectively) were resolved on a polyacrylamide gel and silver stained to validate purity (Figure 3B). Pure samples should only contain the VP1, VP2, VP3 viral proteins. AAV preparations had suitable purities and titer (1e12–1e13GC/ml, except for AAV2 and 6 with titers below 1e12 GC/ml). Serotypes 2 and 6 had low yields. Isolation of AAV particles with serotypes 2 and 6 from packaging cells (scraped cells in Protocol 2, step 11) greatly improves the amount of harvested virus and titer. Due to AAV tropism, serotypes dictate transduction efficiency and the size of infected area.
Retro and lentiviruses
Lentiviral titers are also affected by size of insert and the nature of expressed genes. Lentiviral titer before concentration ranges from 1e6–1e7 TU/ml. The conical ultracentrifuge tubes used in this protocol accommodate 30 ml of virus. Therefore, resuspension of pellets in 300 ul of PBS will result in a concentrated titer of 1e8–1e9 TU/ml. After concentration, Env A pseudotyped lentiviral titer is slightly lower than the VSV-G pseudotyped virus. Similar preparations for retroviruses produce titers of 1e5–1e6 TU/ml and 1e7–1e8 TU/ml after concentration. In vitro cultures of primary rat cortical neurons were transduced with 5ul of a concentrated lentivirus prepared from the transfer vector FCK-ChR2-GFP (Addgene, catalog # 15814) (Boyden, Zhang, Bamberg, Nagel, & Deisseroth, 2005). Robust expression of ChR2-GFP moiety was observed within 48 hours (Figure 4).
Figure 4. Transduction of primary rat cortical neurons with a concentrated lentivirus.
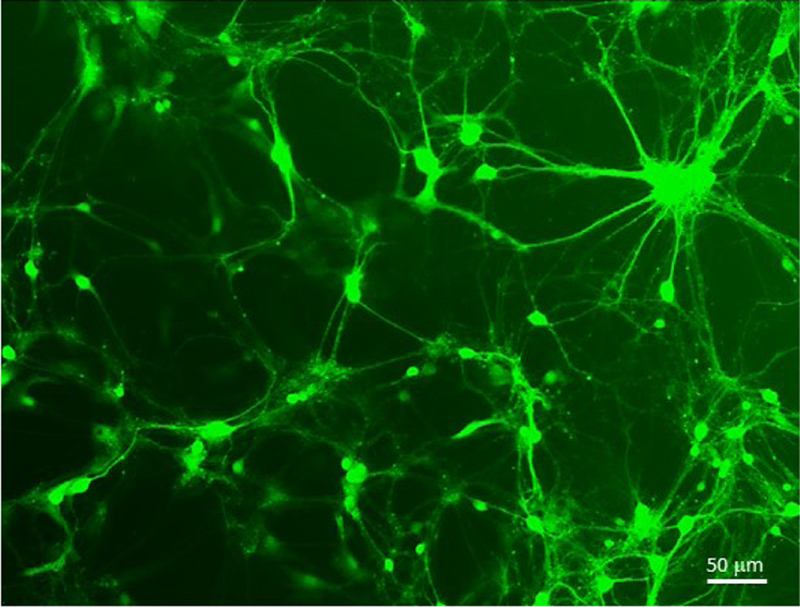
Lentiviral transfer vector FCK-ChR2-GFP delivering Channelrhodopsin-2 tethered to eGFP expressed from a cytomegalovirus (CMV) promoter was prepared as described in Protocol 2. The preparation had a yield of 1.2e8 TU/ml. 5 ul of virus was used to infect 1e6 cultured mouse primary neurons. eGFP expression was observed weakly at 24 hours and robustly after 48 hours.
Pseudotyped rabies dG
The described SADB19 rabies dG protocol can be used to produce 100 ul preparations with titers ranging from 1e7–1e9 TU/ml. Size of insert and the nature of expressed genes impact the resulting titer.
Self-inactivating SADB19 rabies dG expressing Cre preparations yield similar titers. However, titers for packaged SiR Flp and Channel Rhodopsin-mCherry were lowered significantly. Other laboratories report achieving higher titers for SiR rabies dG Flp (Ciabatti et al., 2017). This may be due to larger amounts of starting material and/or allowing packaging cell lines to express the virus for longer periods of time.
In Figure 5, avian receptor TVA and rabies glycoprotein G were delivered by AAV9-Camk2a-GFP-T2A-TVA-E2A-B19G to deep layers of entorhinal cortex via stereotactic injection. 6 weeks after AAV delivery, Env A pseudotyped SADB19 rabies dG expressing mCherry was delivered to the same region also via stereotactic injection. GFP-TVA-positive entorhinal neurons are depicted in green. Retrograde transmission of the rabies dG virus expressing mCherry was observed in the injection site and the CA1 pyramidal neurons. Expression of mCherry and absence of GFP demonstrates that CA1 pyramidal neurons synapse onto the neurons in the entorhinal cortex.
Figure 5. Env A pseudotyped SADB19 rabies dG retrograde transmission.
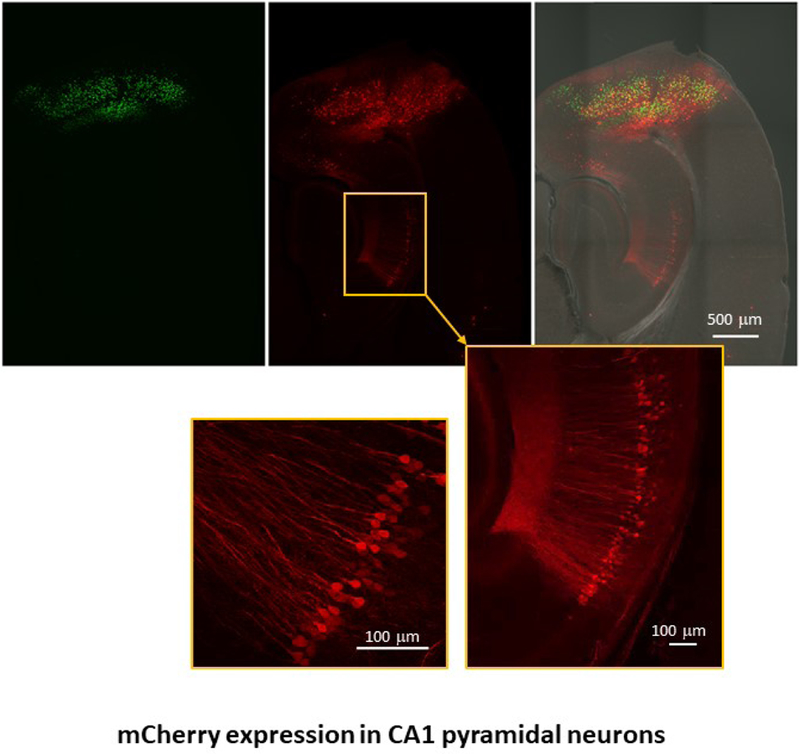
AAV9-Camk2a-T2A-TVA-E2A-B19G was delivered to deep layers of entorhinal cortex via stereotactic injection to mouse brain. The GFP expression marks the AAV tranduced cells. Env A pseudotyped SADB19 rabies dG expressing mCherry was injected into the same region after 2 weeks. Retrograde transmission of Env A-rabies dG to CA1 pyramidal neurons is indicated by mCherry expression.
Time Considerations
Design and construction of viral transfer vectors for each viral type to deliver genes of interest/promoters will vary depending on the complexity of desired genetic sequences.
AAV
Retro and lentiviruses
Pseudotyped SADB19 rabies dG
If seed virus is used to transduce and expand rabies dG, user can skip steps 1–29 and proceed to step 30. If a single plate is transfected with viral genome plasmids to reconstitute virus, minimum of 11 days is needed. Rabies dG SiR may take 2–3 weeks before ample expression is observed. Expansion of CVS-N2c Rabies dG in Neuro2a cells may take up to several months.
INTERNET RESOURCES
https://www.addgene.org/ Addgene is a non-profit plasmid repository and a copious source for obtaining viral transfer vector plasmids and packaged AAV particles – visit “Addgene Special Collections” section for categorized plasmid indexes. “Principal Investigator” section of Addgene contains recombinant rabies plasmids deposited by Edward Callaway, Thomas Jessell, and Ian Wickersham laboratories.
https://www.janelia.org/ The Janelia research campus website provides researchers with the latest news about the developed molecular and imaging tools for neurobiological research.
http://www.gensat.org/cre.jsp GENSAT is a gene expression nervous system atlas that contains a list of several BAC-Cre recombinase driver lines. Transfer vectors for AAVs, retro, and lentiviruses can be designed to include recombinase sensitive sequences and to target specific neuronal populations in these strains.
https://www.salk.edu/science/core-facilities/viral-vector-core/ The Salk Institute Viral Vector Core, SADB19 rabies dG packaging cell lines and plasmids
https://www2.mrc-lmb.cam.ac.uk/group-leaders/t-to-z/marco-tripodi/ Dr. Tripodi’s laboratory website, SiR - SADB19 rabies dG packaging cell lines and plasmids.
Table 1.
Troubleshooting Guide for Low-Yield AAVs
| Possible Source | Troubleshoot |
|---|---|
| Packaging plasmids and the transfer vector | Check plasmid integrity with restriction enzyme digestion and/or sequencing prior to use. |
| Length of insert | Insert (sequences between ITRS) should not exceed 5.2 Kb. Transfer vectors with >4.5 kb package poorly. Increase starting material. |
| Pelleted virus | Remove supernatant from ultracentrifuge tubes carefully to maintain an intact pellet. |
| Insert | Check if insert is toxic to cells by monitoring transfected packaging cells. |
| Serotype | AAVs with serotype 2 often yields low titers. Increase starting material or increase the centrifugation time at step 29 to reduce the volume of virus. |
Table 2.
Troubleshooting Guide for Low-Yield Retroviruses and Lentiviruses
| Possible Source | Troubleshoot |
|---|---|
| Packaging plasmids and the transfer vector | Check plasmid integrity with restriction enzyme digestion and/or sequencing prior to use. |
| Length of insert | Insert (sequences between LTRS) should not exceed 9 Kb. |
| Pelleted virus | Remove supernatant from ultracentrifuge tubes carefully to maintain an intact pellet. |
| Insert | Check if insert is toxic to cells by monitoring transfected packaging cells. |
| Virus type | Retroviruses have 10 times lower titers than lentiviruses. Increase starting material or decrease the amount of PBS used to resuspend the viral pellet in step 22. |
Table 3.
Troubleshooting Guide for Low-Yield Rabies dG Viruses
| Possible Source | Troubleshoot |
|---|---|
| Packaging plasmids and the transfer vector | Check plasmid integrity with restriction enzyme digestion and/or sequencing prior to use. |
| Length of insert | Rabies genome in transfer vector should not exceed 8.5 Kb. |
| Pelleted virus | Remove supernatant from ultracentrifuge tubes carefully to maintain an intact pellet. |
| Insert | Check if insert is toxic to cells by monitoring transfected packaging cells. |
| Transgene expression | Check Transgene expression before harvesting supernatant. A longer period of time or larger volumes for infection (higher MOI) may be necessary before harvesting supernatant. |
Table 4.
Time Considerations for AAV
| Process | Time |
|---|---|
| Transfection of cells with recombinant viral genome to collection of unconcentrated virus | 8 days |
| Ultracentrifugation and small-scale purification of virus | 2 days |
| Ultracentrifugation and large-scale purification of virus | 2 days |
| Titer by qPCR or ddPCR and silver stained gels | 1 day |
| In vitro and ex vivo titering method | 3 days to 2 weeks |
Table 5.
Time Considerations for Retroviruses and Lentiviruses
| Process | Time |
|---|---|
| Transfection of cells with recombinant viral genome to collection of unconcentrated virus | 4 days |
| Ultracentrifugation to concentrate virus | 1 day |
| Titer by qPCR or flow cytometry | 6 days |
Table 6.
Time Considerations for Pseudotyped SADB19 Rabies dG
| Process | Time |
|---|---|
| Transfection of cells with recombinant viral genome to collection of rabies dG | 0–11 days |
| Expansion of unpseudotyped rabies dG | 7–14 days |
| Pseudotyping of rabies dG | 5 days |
| Ultracentrifugation to concentrate virus | 1 day |
| Titer by transduction of HEK293T or pseudotype permissive cell lines | 2–3 day |
ACKNOWLEDGEMENT
This research was supported by the Intramural Research Program of the National Institute of Health (NIH), National Institute of Environmental Health and Sciences (NIEHS). We are grateful to Dr. Steve Wu, and Dr. Robert Petrovich for critical reading of the manuscript and helpful advice. We would also like to acknowledge and thank Dr. Charles Gerfen for intellectual input and initiating the concept for this paper. At the NIEHS, we would like to thank Dr. Jerrel Yakel, Dr. Bernd Gloss, Dr. Guohong Cui, Dr. Zhenglin Gu, Ms. Pattie lamb, and Ms. Amy Papaneri for intellectual and technical contributions. Many thanks to Dr. Ed Callaway, Dr. Marco Tripodi and Dr. Fabio Morgese for providing rabies dG and SiR rabies dG packaging components and Dr. Andrew Murray and Dr. Julia Sable for providing reagents and material for production of CVS-N2c rabies dG.
Footnotes
CONFLICT OF INTEREST
Authors declare that they have no conflict of interest.
REFERENCES
- Bastrikova N, Gardner GA, Reece JM, Jeromin A, & Dudek SM (2008). Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc Natl Acad Sci U S A, 105(8), 3123–3127. doi: 10.1073/pnas.0800027105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier KT, Samson ME, Matsuda T, & Cepko CL (2011). Conditional expression of the TVA receptor allows clonal analysis of descendents from Cre-expressing progenitor cells. Dev Biol, 353(2), 309–320. doi: 10.1016/j.ydbio.2011.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, & Deisseroth K (2005). Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci, 8(9), 1263–1268. doi: 10.1038/nn1525 [DOI] [PubMed] [Google Scholar]
- Callaway EM, & Luo L (2015). Monosynaptic Circuit Tracing with Glycoprotein-Deleted Rabies Viruses. J Neurosci, 35(24), 8979–8985. doi: 10.1523/JNEUROSCI.0409-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle MJ, Gershenson ZT, Giles AR, Holzbaur EL, & Wolfe JH (2014). Adeno-associated virus serotypes 1, 8, and 9 share conserved mechanisms for anterograde and retrograde axonal transport. Hum Gene Ther, 25(8), 705–720. doi: 10.1089/hum.2013.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, … Gradinaru V (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci, 20(8), 1172–1179. doi: 10.1038/nn.4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Sullivan HA, MacLennan BJ, Xu R, Hou Y, Lavin TK, … Wickersham IR (2018). Nontoxic, double-deletion-mutant rabies viral vectors for retrograde targeting of projection neurons. Nat Neurosci, 21(4), 638–646. doi: 10.1038/s41593-018-0091-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciabatti E, Gonzalez-Rueda A, Mariotti L, Morgese F, & Tripodi M (2017). Life-Long Genetic and Functional Access to Neural Circuits Using Self-Inactivating Rabies Virus. Cell, 170(2), 382–392 e314. doi: 10.1016/j.cell.2017.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, & Naldini L (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol, 72(11), 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelshtein D, Werman A, Novick D, Barak S, & Rubinstein M (2013). LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A, 110(18), 7306–7311. doi: 10.1073/pnas.1214441110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fooks AR, Cliquet F, Finke S, Freuling C, Hemachudha T, Mani RS, … Banyard AC (2017). Rabies. Nat Rev Dis Primers, 3, 17091. doi: 10.1038/nrdp.2017.91 [DOI] [PubMed] [Google Scholar]
- Gu Z, Lamb PW, & Yakel JL (2012). Cholinergic coordination of presynaptic and postsynaptic activity induces timing-dependent hippocampal synaptic plasticity. J Neurosci, 32(36), 12337–12348. doi: 10.1523/JNEUROSCI.2129-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Toni N, Clemenson GD Jr., Ray J, & Gage FH (2008). Directed differentiation of hippocampal stem/progenitor cells in the adult brain. Nat Neurosci, 11(8), 888–893. doi: 10.1038/nn.2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Kobayashi K, Inoue K, Kuramochi M, Okada T, Yaginuma H, … Kobayashi K (2011). A lentiviral strategy for highly efficient retrograde gene transfer by pseudotyping with fusion envelope glycoprotein. Hum Gene Ther, 22(2), 197–206. doi: 10.1089/hum.2009.179 [DOI] [PubMed] [Google Scholar]
- Kato S, Kuramochi M, Takasumi K, Kobayashi K, Inoue K, Takahara D, … Kobayashi K (2011). Neuron-specific gene transfer through retrograde transport of lentiviral vector pseudotyped with a novel type of fusion envelope glycoprotein. Hum Gene Ther, 22(12), 1511–1523. doi: 10.1089/hum.2011.111 [DOI] [PubMed] [Google Scholar]
- Lock M, Alvira M, Vandenberghe LH, Samanta A, Toelen J, Debyser Z, & Wilson JM (2010). Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther, 21(10), 1259–1271. doi: 10.1089/hum.2010.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarakis ND, Azzouz M, Rohll JB, Ellard FM, Wilkes FJ, Olsen AL, … Mitrophanous KA (2001). Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum Mol Genet, 10(19), 2109–2121. [DOI] [PubMed] [Google Scholar]
- Osakada F, & Callaway EM (2013). Design and generation of recombinant rabies virus vectors. Nat Protoc, 8(8), 1583–1601. doi: 10.1038/nprot.2013.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr-Brownlie LC, Bosch-Bouju C, Schoderboeck L, Sizemore RJ, Abraham WC, & Hughes SM (2015). Lentiviral vectors as tools to understand central nervous system biology in mammalian model organisms. Front Mol Neurosci, 8, 14. doi: 10.3389/fnmol.2015.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon TR, Murray AJ, Turi GF, Wirblich C, Croce KR, Schnell MJ, … Losonczy A (2016). Rabies Virus CVS-N2c(DeltaG) Strain Enhances Retrograde Synaptic Transfer and Neuronal Viability. Neuron, 89(4), 711–724. doi: 10.1016/j.neuron.2016.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon P, & Trono D (2006). Production and titration of lentiviral vectors. Curr Protoc Neurosci, Chapter 4, Unit 4 21. doi: 10.1002/0471142301.ns0421s37 [DOI] [PubMed] [Google Scholar]
- Salmon P, & Trono D (2007). Production and titration of lentiviral vectors. Curr Protoc Hum Genet, Chapter 12, Unit 12 10. doi: 10.1002/0471142905.hg1210s54 [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, & Muller D (1991). A simple method for organotypic cultures of nervous tissue. J Neurosci Methods, 37(2), 173–182. [DOI] [PubMed] [Google Scholar]
- Tervo DG, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, … Karpova AY (2016). A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron, 92(2), 372–382. doi: 10.1016/j.neuron.2016.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X (2010). Adeno-associated viral vectors found free in media. Hum Gene Ther, 21(10), 1221–1222. doi: 10.1089/hum.2010.921 [DOI] [PubMed] [Google Scholar]
- Zingg B, Chou XL, Zhang ZG, Mesik L, Liang F, Tao HW, & Zhang LI (2017). AAV-Mediated Anterograde Transsynaptic Tagging: Mapping Corticocollicular Input-Defined Neural Pathways for Defense Behaviors. Neuron, 93(1), 33–47. doi: 10.1016/j.neuron.2016.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, … Muzyczka N (1999). Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther, 6(6), 973–985. doi: 10.1038/sj.gt.3300938 [DOI] [PubMed] [Google Scholar]