

Abstract
There is a paramount need for expanding the drug armamentarium to counter the growing problem of drug-resistant tuberculosis. Salicyl-AMS, an inhibitor of salicylic acid adenylation enzymes, is a first-in-class antibacterial lead compound for the development of tuberculosis drugs targeting the biosynthesis of salicylic acid-derived siderophores. In this study, we determined the Ki of salicyl-AMS for inhibition of the salicylic acid adenylation enzyme MbtA from Mycobacterium tuberculosis (MbtAtb), designed and synthesized two new salicyl-AMS analogues to probe structure–activity relationships (SAR), and characterized these two analogues alongside salicyl-AMS and six previously reported analogues in biochemical and cell-based studies. The biochemical studies included determination of kinetic parameters and analysis of the mechanism of inhibition. For these studies, we optimized production and purification of recombinant MbtAtb, for which Km and kcat values were determined, and used the enzyme in conjunction with an MbtAtb-optimized, continuous, spectrophotometric assay for MbtA activity and inhibition. The cell-based studies provided an assessment of the antimycobacterial activity and post-antibiotic effect of the nine MbtAtb inhibitors. The antimycobacterial properties were evaluated using a strain of non-pathogenic, fast-growing Mycobacterium smegmatis that was genetically engineered for MbtAtb-dependent susceptibility to inhibitors. This convenient model system greatly facilitated the cell-based studies by bypassing the methodological complexities associated with the use of pathogenic, slow-growing M. tuberculosis. Collectively, these studies provide new information on the mechanism of inhibition of MbtAtb by salicyl-AMS and eight analogues, afford new SAR insights for these inhibitors, and highlight several suitable candidates for future preclinical evaluation.
Graphical Abstract
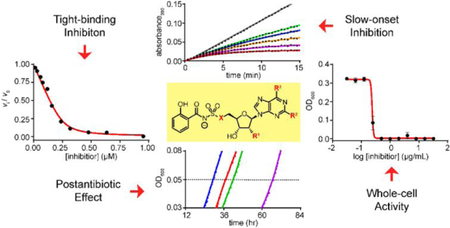
INTRODUCTION
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, is a resilient, obligate bacterial pathogen with a devastating impact on global public health.1 The intrinsic clinical resistance of Mtb to many antimicrobial drugs is one of the challenges at the center of the problematic chemotherapy and global control of tuberculosis.2 Standard tuberculosis treatment requires prolonged and expensive chemotherapy with multiple drugs, and is associated with adverse side effects and compliance challenges.3,4 The cumbersome chemotherapy regimens against tuberculosis result in high frequency of suboptimal or incomplete drug treatment courses,4,5 a situation that over the decades has led to the rise of tuberculosis cases produced by multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains of Mtb.1 The rise of these resistant strains compounds the already challenging problem of tuberculosis chemotherapy and presents a growing threat to global tuberculosis control and eradication efforts. This grim scenario underscores the need for expanding the antituberculosis drug armamentarium.
Towards this end, we previously developed the first-in-class nucleoside antibiotic salicyl-AMS (5´-O-[N-salicylsulfamoyl]adenosine) (1, Figure 1A).6 We rationally-designed 1 as a salicyl-AMP intermediate mimetic inhibitor of the enzyme MbtA of Mtb (MbtAtb, Figure 1B).7 MbtA has no human homologues and is required for the biosynthesis of salicylic acid-derived mycobactin (MBT) siderophores, which are high-affinity Fe3+ chelators involved in the scavenging and uptake of iron (Fe),7,8 a micronutrient essential for Mtb growth and pathogenesis (Figure 1C).9–11 The realization of the critical role of the MBT siderophore system in Mtb biology emerges from multiple studies demonstrating that Mtb mutant strains with gene knockouts in the siderophore biosynthesis or transport systems have impaired survival in macrophages12–14 and various degrees of attenuation in guinea pig14 and mouse13,15–17 models of tuberculosis. Thus, MBT biosynthesis is considered an attractive potential target for developing tuberculosis drugs with novel mechanisms of action.18–20
Figure 1.

(A) Nucleoside antibiotic salicyl-AMS and salicyl-AMP intermediate synthesized by the salicylate adenylation enzyme activity of MbtAtb. (B) Reactions catalyzed by MbtAtb during mycobactin (MBT) biosynthesis. MbtAtb catalyzes formation of the first covalent acyl-enzyme intermediate during MBT acyl-chain assembly through a mechanism involving two-half reactions. The first half reaction is the ATP-dependent adenylation of salicylic acid to generate a salicyl-AMP intermediate that remains non-covalently bound to the active site. The second half-reaction is the transfer of the salicyl moiety of the adenylate onto the phosphopantetheinyl group of the carrier protein domain of the peptide synthetase MbtB. (C) Representative mycobactin siderophores of M. tuberculosis. R represents variable fatty acyl groups (mycobactin variants) or acyl substituents terminating in a carboxylate or a methyl ester (carboxymycobactin variants). All of these variants are herein collectively referred to as MBTs.
Our previous in vitro studies on salicyl-AMS (1) demonstrated that it is a potent, selective, tight-binding inhibitor (TBI) of MbtAtb, as well as other salicylate adenylation enzymes from pathogenic bacteria,6 including YbtE from Yersinia pestis21 and PchD from Pseudomonas aeruginosa.22 Moreover, we have shown that salicyl-AMS (1) inhibits the biosynthesis of MBTs in Mtb and, as expected, restricts the growth of the pathogen with much greater potency under Fe-limiting conditions,6 in which production of MBTs is crucial for Fe acquisition. In all, this early work provided proof of principle for the druggability of salicylate adenylation enzymes, validated pharmacological inhibition of siderophore biosynthesis as a new mechanism of antibiotic action, and established salicyl-AMS (1) as a first-in-class antibacterial lead compound for the development of tuberculosis drugs targeting siderophore biosynthesis. Subsequent studies by Aldrich and coworkers confirmed the inhibitory activity of salicyl-AMS (1) against MtbAtb and Mtb, demonstrated that the inhibitor is not cytotoxic against mammalian cells, and provided extensive in vitro structure–activity relationship (SAR) analysis for inhibition of MtbAtb biochemical activity and Mtb growth using a wide range of salicyl-AMS analogues.23–32
More recently, we have also reported studies on the in vivo efficacy of salicyl-AMS (1) in a mouse model of tuberculosis.33 Importantly, this work showed that monotherapy with salicyl-AMS (1) at 5.6 or 16.7 mg/kg correlated with a significant reduction of Mtb growth in the mouse lung, thus supporting MbtAtb as a promising target for the development of novel tuberculosis drugs blocking siderophore biosynthesis. However, we also observed in vivo toxicity at ≥16.7 mg/kg doses, precluding further dose escalation to improve efficacy. Thus, there is a continued need to develop new salicyl-AMS analogues with the long-term goal of improving pharmacokinetics, efficacy, and toxicity profiles.
Towards this end, we report herein detailed in vitro evaluation of salicyl-AMS (1), two novel salicyl-AMS analogues designed to probe new SAR regions, and six of the most potent analogues reported previously by Aldrich and coworkers.28 We determined intrinsic Ki values for salicyl-AMS (1), biochemical residence times and other kinetic parameters of these tight-binding inhibitors, and assessed them for antibacterial and post-antibiotic effects in cell culture. In the course of this work, we also optimized production and purification of recombinant MbtAtb and developed new cellular models using the non-pathogenic, fast-growing Mycobacterium smegmatis (Msm) species. Collectively, these studies provide new information on the mechanism of inhibition of MbtAtb by salicyl-AMS (1) and eight analogues, provide new SAR insights for MbtAtb inhibitors, and highlight several MbtAtb inhibitors as suitable candidates for further preclinical evaluation.
MATERIALS AND METHODS
Synthesis of MbtA inhibitors.
Salicyl-AMS (1)6 was synthesized by WuXi AppTec (Shanghai, China). Salicyl-2´-dAMS (2),25 salicyl-2-Ph-AMS (3),23 salicyl-2-PhNH-AMS (4),23 salicyl-AMSN (4a),24 salicyl-6-N-Me-AMS (5a),23 and salicyl-6-N-c-Pr-AMS (5b),23 were synthesized as previously described. Salicyl-AMSNMe (4b) was synthesized in seven steps from 6-N-Boc-2´,3´-isopropylideneadenosine (Figure S1). Salicyl-6-MeO-AMSN (6) was synthesized in 10 steps from inosine (Figure S2). Compounds (except 6) were converted to the corresponding sodium salts by ion exchange (see Supporting Information).
Overexpression and purification of H10MbtAopt.
Mtb MbtA (UniProtKB P71716), codon-optimized for expression in Escherichia coli with an N-terminal His10 tag (H10MbtAopt), was overproduced in E. coli BL21(DE3)pLysS carrying plasmid pH10MbtAopt (see Supporting Information for codon optimization and Tables S1 and S2 for strains and plasmids used in this study, respectively). The strain was cultured in Luria-Bertani broth34 in Fernsbach baffled flasks (wide-mouth, 2.8-L capacity) under rotary agitation (220 rpm) at 25 °C to OD600 = 0.3. The temperature was then reduced to 20 °C and incubation was continued. Protein overproduction was induced by addition of 1 mM IPTG (isopropyl β-d-1-thiogalactopyranoside) at OD600 ≈ 0.6. After 16 h of additional incubation, the cultures were chilled on ice and the cells were harvested by centrifugation. The cells were resuspended in 20 mL of lysis buffer per liter of culture (50 mM Tris·HCl, pH 8; 10 mM imidazole; 0.5 M NaCl; 20% sucrose; 1 mM -mercaptoethanol; 1 mM PMSF; 0.1% IGEPAL CA-630). Lysozyme (300 µg/ml), DNase I (100 µg/ml), and MgCl2 (25 mM) were added to the cell suspension, which was then incubated at 0 °C for 30 min and subsequently subjected to a freeze/thaw cycle for lysis. The lysate was then sonicated (Branson Ultrasonics Digital Sonifier; 2 × 30 sec, 90% intensity), diluted 1.3 times in lysis buffer, subjected to high-speed centrifugation (1 h, 20,000 g), filtered (Whatman filter paper, 2.7 µm pore size), and degassed under reduced pressure. H10MbtAopt was purified from the cleared lysate by Ni2+-column chromatography using Ni–NTA Superflow resin according to the manufacturer’s instructions (Qiagen) and an ÄKTA Purifier UPC10 FPLC System (GE Healthcare). The loaded column (7 mL) was washed with 5 column volumes of wash buffer (75 mM Tris·HCl, pH 8; 20 mM imidazole; 0.5 M NaCl; 5% glycerol), and proteins were eluted using an imidazole gradient [solvent A: wash buffer; solvent B: elution buffer (75 mM Tris·HCl, pH 8; 0.8 M imidazole; 0.2 M NaCl; 5% glycerol)]. H10MbtAopt eluted at ≈0.36 M imidazole. Fractions containing H10MbtAopt were then dialyzed (Slide-A-Lyzer Dialysis cassettes; Pierce) into dialysis buffer (25 mM Tris·HCl, pH 8; 0.2 M NaCl; 2 mM DTT; 5% glycerol), aliquoted, flash-frozen, and stored at –80 °C. Protein fraction quality and protein concentration were determined by SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) analysis34 and Bio-Rad Protein Assay (Bio-Rad Laboratories, Inc.), respectively.
Assay for MbtAtb activity and inhibition.
The adenylation activity of H10MbtAopt and its inhibition were evaluated using a H10MbtAopt-optimized version of the hydroxylamine–7-methyl-6-thioguanosine (HA–MesG) continuous, spectrophotometric assay.35 The assay was carried out in a 96-well plate format as we have recently reported.36 The assay reaction mixture was optimized for H10MbtAopt activity. Optimization experiments included evaluation of various concentrations of Tris·HCl (and pH), hydroxylamine, MesG, ATP, NaCl, MgCl2, glycerol, reducing agents (DTT and TCEP), and detergent [IGEPAL CA-630, Triton-X100, and CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate)]. Unless otherwise indicated for specific experiments, the optimized assay reaction mixture contained the following: 50 mM Tris·HCl, pH 8.0; 3 mM MgCl2; 0.5 mM DTT; 0.1 U purine nucleoside phosphorylase (PNP); 0.04 U inorganic pyrophosphatase (PPT); 450 mM hydroxylamine; 0.4 mM MesG; 1 mM ATP; 300 µM salicylic acid; 0.01% CHAPS buffer; 7.5% ultrapure glycerol; and H10MbtAopt at concentrations noted for specific experiments. When needed, MbtA inhibitors were added from 10% DMSO stock solutions, with a final DMSO concentration of 1% in both inhibitor-containing reactions and control reactions (no inhibitor). Reactions were preincubated for 10 min at 25 °C before being initiated by the addition of either salicylic acid for steady state kinetic analysis or H10MbtAopt for progress curve analysis. The phosphorolysis of MesG was measured continuously at either regular 1-min intervals (for steady state kinetic analysis) or 25-sec intervals (for progress curve analysis) for up to 45 min, at 360 nm and 25°C in a DTX 880 multimode detector microplate reader (Beckman Coulter, Inc.). The concentration of active H10MbtAopt was validated by active-site titration37 using salicyl-AMS (1) as the reference inhibitor. The calculated concentration of total H10MbtAopt used in the assays was essentially indistinguishable from the concentration of active H10MbtAopt determined by active-site titration (not shown).
Determination of Km and kcat.
Experiments to determine the Km value for ATP were carried out with ATP in the 1–67.5 µM range (1.5-fold dilution series) and a saturating salicylic acid concentration of 300 µM. Experiments to determine the Km value for salicylic acid were done with salicylic acid in the 0.027–1.3 µM range (1.5-fold dilution series) and a saturating ATP concentration of 1 mM. H10MbtAopt and MgCl2 were used at 250 nM and 5 mM, respectively. The spectrophotometric data were analyzed to calculate initial velocity (v0) data. Km and kcat values were then determined by nonlinear regression analysis of background-corrected v0 versus substrate concentration ([S]) datasets using the Henri–Michaelis–Menten equation with Vmax replaced by enzyme concentration ([E]) × kcat38 (Eq. 1). Prism v6.01 (GraphPad Software, Inc.) was used for curve-fitting, regression analyses, and statistical analyses of these datasets and all other experimental datasets generated in this study. The Km and kcat values reported are averages of two independent experiments.
| Eq. 1 |
Determination of Ki.
The Ki of salicyl-AMS (1) for H10MbtAopt was determined by Morrison analysis for TBIs.37,39 Two Ki values were determined using two experimental modalities with respect to substrates. One modality used variable, saturating concentrations of ATP (0.86–6.5 mM range) along with a constant, saturating concentration of salicylic acid (300 µM). The other modality used variable, saturating concentrations of salicylic acid (30.6–310 µM range) along with a constant, saturating concentration of ATP (6.5 mM). H10MbtAopt and MgCl2 were used in the assays at 250 nM and 15 mM, respectively. Salicyl-AMS (1) was tested using a 50–1,040 nM range. The range was covered using a 2-fold dilution series format for sections A and C of the dose-response curve and a 1.2-fold dilution series format for section B (“elbow”) of the curve as recommended.37,40 The spectrophotometric data were used to determine fractional initial velocities (vi/v0), where vi is the initial velocity with inhibitor and v0 is the initial velocity with no inhibitor (DMSO controls). values were calculated by Morrison analysis of background-corrected vi/v0 versus logarithm of inhibitor concentration ([I]) datasets using Eq. 2, as described37. A Ki value was then obtained from each of the two datasets of values vs. [S] as the y-intercept of the linear regression of the data fitted to Eq. 3.39 Each of the two Ki values reported was calculated from the average of datasets generated from three independent experiments.
| Eq. 2 |
| Eq. 3 |
Progress curves and determination of kinetic parameters , , koff, and tR.
Reactions were pre-incubated for 10 min before being initiated by the addition of H10MbtAopt (1 µM). The concentration range at which each inhibitor was tested was selected empirically by pilot experiments (not shown). The ranges were as follows: 4,000–1,041 nM range (1.4-fold dilution series) for 1, 4a, 4b, 5a, 5b, and 6; 2,000–1,157 nM range (1.2- and 1.3-fold dilution series combination) for 3a; 3,000–1,366 nM range (1.3- and 1.5-fold dilution series combination) for 3b; and 2,857–1,041 nM range (2.2- and 1.4-fold dilution series combination) for 2. As done in similar studies,41,42 the background-corrected spectrophotometric data were fitted to Eq. 4.43 In Eq. 4, A is the absorbance at time t, vps is the pre-steady state initial velocity, vs is the steady-state velocity at equilibrium, and kobs is the rate constant for progression to steady state. The datasets of vs versus [I] derived from the progress curves were fitted to Eq. 2 to calculate the values. The kobs values were determined with Eq. 4 for each inhibitor concentration and then used to calculate the dissociation rate constant (koff) values using Eq. 5,44 as previously reported.45,46 Subsequently, values were determined using Eq. 6 and the calculated koff and the values.44 Residence time (tR) values were calculated as the reciprocal of the koff value. To assess the inhibition mechanisms of the inhibitors, kobs values versus [I] datasets were plotted and fitted to Eq. 7.44 Each kinetic parameter reported is the average derived from a minimum of five independent experiments. Pearson correlation analysis between kinetic parameters was carried out using the statistical analysis package in Prism v6.01. Pearson correlation coefficients (PCCs) with Student’s t-test p values ≤0.05 were considered statistically significant.
| Eq. 4 |
| Eq. 5 |
| Eq. 6 |
| Eq. 7 |
Bacterial culturing and recombinant DNA manipulations.
Msm mc2155 (ATCC 700084)47 and its derivatives were regularly cultured under standard conditions in Middlebrook 7H9 or 7H11 (Difco) supplemented as reported.8 Msm strains were cultured in Fe-limiting GASTD medium or GASTD supplemented with 100 µM FeCl3 (GASTD+Fe medium)6,48 for specific experiments as noted below. Routine culturing of E. coli strains was done under standard conditions in Luria-Bertani media.34 When required, kanamycin (30 μg/ml), chloramphenicol (34 μg/ml), ampicillin (100 μg/ml), sucrose (2%), and/or 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal, 70 μg/ml) were added to the growth media. DNA manipulations were carried out using established protocols and E. coli DH5α as the primary cloning host.34 PCR-generated DNA fragments used in plasmid constructions were sequenced to verify fidelity. The oligonucleotides used in this study are shown in Table S3. Genomic DNA isolation, plasmid electroporation into Msm, and selection of Msm transformants were carried out as reported.8 Unless otherwise indicated, molecular biology, biochemical, and microbiology reagents were purchased from Sigma-Aldrich Inc., Thermo Fisher Scientific Inc., New England Biolabs Inc., QIAGEN Inc., or Integrated DNA Technologies Inc.
Generation of M. smegmatis mutants ΔE, ΔEM, and ΔEM-pMbtAtb.
Mutants were generated using the p2NIL/pGOAL19-based flexible cassette method,49 as reported.8 Msm ΔE carried an unmarked, in-frame deletion of MSMEG_0019, encoding the peptide synthetase (7,523 amino acids, the largest protein in Msm50) predicted to be essential for biosynthesis of exochelin (EXO) siderophores.51–53 A MSMEG_0019 deletion cassette-delivery, suicide plasmid (pΔ0019) was used to generate the chromosomal deletion, which eliminated codons 5 through 7,519 of the gene. The deletion cassette contained from 5´- to 3´-end: the 984-bp segment upstream of the gene, the gene’s first 4 codons, the gene’s last 4 codons, the stop codon, and the 1,029-bp segment downstream of the gene. To generate the cassette, primer pair OF0019 and IR0019soe and primer pair IF0019soe and OR0019 were first used to generate the 5’ fragment (1,011 bp) and the 3’ fragment (1,059 bp) for the cassette, respectively, from genomic DNA template. The fragments, which had a 30-bp overlap at the splice site embedded in IF0019soe and IR0019soe, were then used together as a template for PCR with primers OF0019 and OR0019 to fuse the fragments. The PCR-generated cassette was cloned into pCR2.1Topo (TOPO TA Cloning Kit, Invitrogen, Thermo Fisher Scientific Inc.), then excised from the pCR2.1Topo construct using HindIII and EcoRV, and religated into p2NIL49 linearized by HindIII-PmlI digestion. The resulting plasmid (p2NILΔ0019) and pGOAL1949 were digested with PacI, and then the PacI marker cassette of pGOAL19 was ligated to the linearized p2NILΔ0019 to create pΔ0019. Electroporation of pΔ0019 into Msm wild-type (WT) and selection of potential single- and double-crossover mutants were conducted as reported.8 The MSMEG_0019 deletion was screened for and confirmed by PCR using two primer pairs (OF0019 and OR0019: yielding an undetectable 24,585-bp amplicon for WT and a 2,040-bp amplicon for mutant; IF0019 and IR0019: yielding a 148-bp amplicon for WT and no amplicon for mutant) (not shown).
EXO/MBT-deficient Msm ΔEM carried the MSMEG_0019 deletion noted above and an unmarked, in-frame deletion of Msm mbtA (mbtAsm), which encodes the salicylate adenylation enzyme essential for MBT biosynthesis.8 The mbtAsm deletion left behind only the gene’s start codon followed by the stop codon, and it was created in Msm ΔE with the same approach we have previously reported for generation of the identical mbtAsm deletion in Msm WT to generate the mutant referred to hereafter as Msm ΔM.8
Msm ΔEM was transformed with plasmid pMbtAtb (expressing mbtAtb) to generate strain Msm ΔEM-pMbtAtb. To construct pMbtAtb, a DNA fragment encompassing mbtA of Mtb (mbtAtb, Rv23848) was generated by PCR from genomic DNA template using primer pair mbtAtbF1 and mbtAtbR1. The PCR product, which included an optimized ribosome-binding site54 upstream of mbtAtb introduced by primer mbtAtbF1, was cloned into pCR2.1Topo. Subsequently, the insert was recovered from the pCR2.1Topo construct as a HpaI-NheI fragment and subcloned into the mycobacterial, low-copy number plasmid pCP055 linearized by HpaI-NheI digestion. This subcloning created pMbtAtb, in which mbtAtb is under the control of the constitutive mycobacterial hsp60 promoter located in pCP0.
Growth inhibition assays.
Dose-response experiments using microdilution assays in a 96-well plate platform were performed as reported.48,56 Msm strains were grown in GASTD or GASTD+Fe media. Cultures (200 µL/well) were started at OD600 = 0.0005 (≈9×104 CFU/well, as per concurrent CFU determination) from culture stocks prepared in GASTD medium as reported.6 Growth was assessed as OD600 after 4 days of incubation (37 °C, 170 rpm) using the DTX 880 microplate reader. Unless otherwise indicated in specific experiments, compounds were typically evaluated using a 0.031–64 µg/ml range (2-fold dilution series format). Inhibitors were added from 10% DMSO stock solutions, with a final DMSO concentration of 0.5% in both inhibitor-treated cultures and DMSO controls (no inhibitor). Minimum inhibitory concentration (MIC) values were calculated as the lowest concentration tested that inhibited growth by ≥95% relative to DMSO controls. Data presented are derived from three independent experiments.
Post-antibiotic effect (PAE) assays.
Cells from mid-log growth phase cultures of Msm ∆EM-pMbtAtb in GASTD medium (37 °C, 170 rpm, OD600 ≈ 0.75) were harvested by centrifugation and washed in GASTD medium (3 times with 1 culture volume). The washed cells were resuspended in GASTD medium and transferred to U-bottom 96-well cell culture plates (Corning, Inc.) for inhibitor exposure at a cell density corresponding to OD600 = 1.0 (50 µL/well). Cells were exposed to 5×, 50×, and 100× average MIC value, in line with reported studies with other antimycobacterial compounds.57 Inhibitors were added from 10% DMSO stock solutions, with a final DMSO concentration of 1% in both inhibitor-exposed cultures and DMSO controls (no inhibitor). After the exposure period (1 h, 37 °C, 170 rpm), the cultures were diluted in pre-warmed, inhibitor-free medium to OD600 = 0.001 (1,000-fold dilution, bringing inhibitor concentrations to 0.005×, 0.05×, and 0.1× average MIC values) and reseeded into flat-bottom, 96-well culture plates (Corning, Inc.) at 200 µl/well. The 96-well plates were incubated for culture growth at 37 °C in a FLUOstar Optima microplate reader (BMG Labtech, Inc.), and OD600 readings were taken every 30 min following plate shaking (5 min, 200 rpm) for 5 days. The growth vs. time datasets were analyzed to determine the time at which cultures reached an exponential growth phase threshold of OD600 = 0.05 (≈15% of maximal growth). The time-to-threshold data were used to calculate PAE as the difference between the time-to-threshold values of the inhibitor-exposed culture and the control cultures.58 Data presented are derived from three independent experiments. Pearson correlation analysis between PAE and tR datasets was carried out using Prism v6.01, and PCCs with p values ≤0.05 were considered statistically significant.
RESULTS AND DISCUSSION
Cloning, overexpression, and purification of recombinant MbtAtb.
MbtAtb catalyzes formation of the first covalent acyl-enzyme intermediate during MBT acyl-chain assembly7 and is the molecular target of the antibacterial lead compound salicyl-AMS (1)6 (Figure 1). Previous approaches for purification of recombinant MbtAtb expressed in E. coli have been characterized by low yields (0.1–2 mg/L) due to low expression, poor solubility, inefficient affinity tag removal, and/or the need for multiple purification steps.6,7,25 We sought to overcome this limitation and to make MbtAtb more readily available for our biochemical and inhibition studies. To this end, we explored codon optimization, alternative polyhistidine-tag fusion strategies, and changes in expression and purification conditions. We first carried out codon optimization, which led to changes in 322 of the 566 codons of mbtAtb (Figure S3). We then evaluated ten different polyhistidine affinity tag strategies (viz. alternative tag lengths and locations, double tags, and a tandem tag) for the codon-optimized MbtAtb (MbtAopt) (Figure S4A). This included unconventional tags that have been shown to be advantageous with other problematic recombinant proteins.59,60 Pilot experiments for assessment of protein expression, solubility, and binding to Ni2+-charged resin revealed that N-terminal deca-His tagged MbtAopt (H10MbtAopt) had the best properties overall (not shown). Thus, we advanced H10MbtAopt to larger-scale overproduction and purification experiments that ultimately led to the final methodology used to obtain the enzyme for the biochemical and inhibition studies described below. Overall, the optimizations and methodological improvements shortened the purification protocol by eliminating the need for tag cleavage and size exclusion chromatography, rendered purified H10MbtAopt with purity levels comparable to those reported for other recombinant MbtAtb variants (Figure S4B), and permitted final yields of up to ≈8 mg/L. This represents a 4-fold increase relative to the highest yield previously reported for MbtAtb.25
Validation of H10MbtAopt activity and inhibition by salicyl-AMS in the HA–MesG kinetic assay.
H10MbtAopt is a novel recombinant variant of MbtAtb with a relatively large His10 tag that could potentially hinder the salicylate adenylation activity of the enzyme and/or the ability of salicyl-AMS (1) to inhibit this activity with a potent, TBI modality.6 Therefore, we sought to validate the activity of H10MbtAopt and its inhibition by salicyl-AMS (1). We have previously used a traditional ATP–32PPi isotope exchange (ATP-PPi) assay to characterize the adenylation activity of MbtAtb7 and its inhibition by salicyl-AMS (1).6 Although this is a robust assay, it measures the reverse of the adenylation reaction and has several methodological disadvantages, including those arising from its end-point nature, the need for radiolabeled reagents, and a poor suitability for high-throughput analysis. Thus, we explored the adoption of a more convenient spectrophotometric HA–MesG kinetic assay that measures enzyme activity in the relevant forward direction of the biosynthetic pathway and has been recently validated with several adenylation enzymes, including MbtAtb.35 To this end, we used our recently reported study applying the HA–MesG assay to the characterization of a TBI of the cysteine adenylation domain of the Y. pestis siderophore synthetase HMWP236 as a methodological template. Encouragingly, results of the first assessment of the activity of H10MbtAopt and its inhibition by salicyl-AMS (1) using the HA–MesG assay demonstrated enzyme activity and the expected TBI behavior for 1 (i.e. IC50 ≈ ½ [E]) (Table 1, Figure S5). Out of an abundance of caution, we assessed whether salicyl-AMS (1) had any inhibitory effect on the PPT-PNP coupling system of the HA–MesG assay. We also evaluated whether any of four salicyl-AMS fragments that could possibly result from hydrolytic degradation of 1 and/or be present as trace contaminants (i.e.: AMS (7), salicyl-sulfamate (8), salicylamide (9), and N3-5´-cycloadenosine (10); Table S4) had a negative impact on the HA–MesG assay. We found that salicyl-AMS (1) did not affect the PPT-PNP coupling system when tested at up to 40 µM (10 times the maximum concentration of 1 used in the HA–MesG assay), and only salicyl-sulfamate (8) depressed the H10MbtAopt-dependent signal in the HA–MesG assay, however only to a negligible extent (IC50 ≈ 1 mM) (Table S4). Therefore, inhibition by an off-target effect of salicyl-AMS (1) or by potential trace amounts of fragments 7–10 is not an assay confounder.
Table 1.
Kinetic parameters for inhibitors of MbtAtb
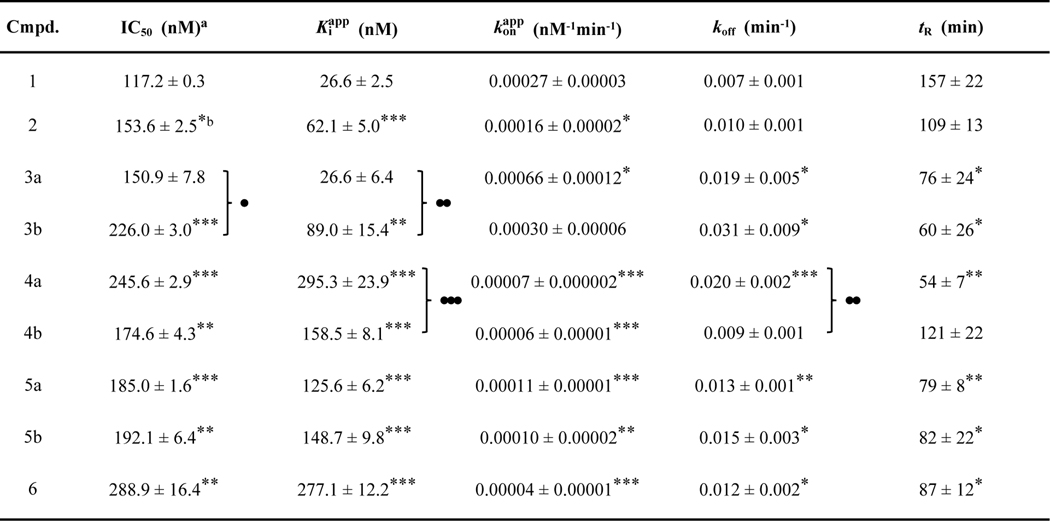 |
Values represent means ± SEM derived from two independent experiments for IC50 values (Figure S5) and at least 5 for other parameters.
Statistical significance as per Student’s t-test is indicated.
*, **, and *** represent p values of ≤ 0.05; ≤ 0.01, and ≤ 0.001, respectively, for analogues vs. 1. ●, ●●, and ●●● represent p values of ≤ 0.05; ≤ 0.01, and ≤ 0.001, respectively, for 3a vs. 3b, 4a vs. 4b, and 5a vs. 5b pairs. No dots or asterisks indicate no statistical significance.
The validation experiments noted above were carried out using the generic conditions of the HA–MesG assay originally reported.35 Thus, we optimized the assay for H10MbtAopt prior to its use in additional enzyme and inhibitor characterization studies. This optimization led to several changes in reaction composition and a ≈ 3-fold increase in H10MbtAopt activity (Figure S6). In all, our results ruled out the possibility of a detrimental effect of the His10 tag in H10MbtAopt and set the stage for the use of H10MbtAopt in conjunction with the HA–MesG assay for the studies reported here. To our knowledge, this is the first example of the use of the HA–MesG assay for the evaluation of inhibitors of a salicyl-AMP ligase.
Steady-state kinetic parameters of H10MbtAopt.
Prior to carrying out additional H10MbtAopt inhibition studies, we determined the Michaelis−Menten parameters for H10MbtAopt using the optimized HA–MesG assay (Figure 2). Kinetic analysis with salicylic acid as the variable substrate revealed and values of 0.31 ± 0.003 µM and 0.90 ± 0.06 min−1, respectively. With ATP as the variable substrate, the and values obtained were 13.0 ± 1.3 uM and 1.16 ± 0.04 min−1, respectively. The agreement of the two kcat values indicates consistency between the two evaluation modalities with respect to substrate and highlights the reliability of the experimental approach. Notably, the average kcat value of H10MbtAopt (≈1 min−1) is comparable to those of mechanistically related 2,3-dihydroxybenzoate adenylation enzymes involved in the biosynthesis of several aryl-capped siderophores,61 namely E. coli EntE (≈1 min−1), Acinetobacter baumannii BasE (≈1 min−1), and, to some extent Vibrio cholerae VibE ( 0.2 min−1), when using DHBA as the variable substrate in the HA–MesG assay.35 The relatively low kcat values of these ligases determined with the surrogate acyl-acceptor hydroxylamine in the HA–MesG assay might be tentatively attributed to the lack of interaction between the ligase and its phosphopantetheinylated carrier protein partner, which is thought to be needed to trigger the rotation of the C-terminal domain of the ligase and facilitate the acyl transfer step.62
Figure 2.

Michaelis−Menten kinetic parameters for H10MbtAopt. The v0 vs. [S] datasets were generated using salicylic acid (A) or ATP (B) as the variable substrate. Each plot shown is representative of two independent experiments. The datasets (●) were fitted to Eq. 1 to generate the solid line (R2 values ≥ 0.993). The kinetic parameters indicated are mean values ± SEM of n = 2 independent experiments.
This work provided and values for MbtAtb working in the relevant forward direction of the biosynthetic pathway. A previous study with a different MbtAtb recombinant variant and HA–MesG assay conditions not optimized for MbtAtb found a value identical to the one reported here, but a value ≈3-fold lower than the one determined by our analysis.35 The higher value found herein might be due to a more robust MbtAtb recombinant variant and/or our optimized assay conditions. Interestingly, the Km values derived from our analysis indicate that MbtAtb has ≈40-fold higher affinity for salicylic acid than for ATP. This finding is in agreement with the 65- and 32-fold preference for salicylic acid found with MbtAtb and the salicylate adenylation enzyme YbtE from Y. pestis (45% sequence identity with MbtAtb) for the reverse of the adenylation reaction in the ATP-PPi assay.6,25 It is worth noting, however, that the and values derived from the ATP-PPi assay were 10–20 times larger (i.e.: lower substrate affinities) than those we determined with H10MbtAopt and the optimized HA–MesG assay.
Determination of intrinsic Ki for salicyl-AMS (1) with H10MbtAopt.
The Ki value of salicyl-AMS (1) for inhibition of MbtAtb has not been reported, and thus we determined the Ki value for H10MbtAopt inhibition using the optimized HA–MesG assay. To this end, we followed the approach we previously applied to determine the Ki value of salicyl-AMS (1) for inhibition of the Y. pestis salicylate adenylation enzyme YbtE noted above.6 Due to the bisubstrate nature of the adenylation reaction catalyzed by MbtAtb, two Ki values were independently calculated using two approaches with respect to the substrates. One approach had variable saturating concentrations of ATP along with a constant, saturating concentration of salicylic acid, whereas the other had the opposite relationship of constant vs. variable substrate. For both approaches, values increased linearly with increasing [S] (Figure 3). This indicated a competitive inhibition mechanism for salicyl-AMS (1) with respect to both MbtAtb substrates,39 a property found also for a carbocyclic analogue of salicyl-AMS (1) with MbtAtb in an ATP-PPi assay-based study25 and suggestive of an equilibrium random mechanism for the adenylation reaction catalyzed by MbtAtb.63 The competitive mechanism determined for salicyl-AMS (1) allowed for the calculation of Ki values as the y-intercepts of the fit lines for the value vs. [S] datasets.39 The values determined were 0.76 ± 0.43 nM for and 1.04 ± 0.48 nM for (Figure 3). These Ki values are essentially indistinguishable from one another, demonstrating correspondence between the two evaluation approaches with respect to substrate and underlining the robustness of the methodological approach. Notably, the average Ki value of 0.9 nM found with MbtAtb is comparable to our previously reported Ki for the inhibition of Y. pestis YbtE using the ATP-PPi assay (0.7 nM).6 Overall, this indicates that salicyl-AMS (1) is a potent salicylate adenylation enzyme inhibitor with sub-nM Ki values.
Figure 3.

Ki value of salicyl-AMS for H10MbtAopt inhibition. Two Ki values were independently calculated using two approaches having opposite relationship of constant versus variable substrate. The value (A) and value (B) were calculated from datasets of values vs. ATP and salicylic acid concentration, respectively, as the y-intercept of the linear regression of the data (●) fitted to Eq. 3 (solid line; R2 values ≥ 0.6). The Ki values indicated were calculated from the average of vs. variable substrate datasets generated from three independent experiments. Error bars on data points represent SEM of n = 3.
Selection of salicyl-AMS analogues for further evaluation.
We selected six previously reported salicyl-AMS analogues for detailed in vitro analysis (Figure 4). These compounds are among the most potent analogues reported by Aldrich and coworkers, and represent modifications in different regions of the lead compound, including deletion of the ribose 2´-hydroxyl group (salicyl-2´-dAMS, 2),25 replacement of the ribose 5´-oxygen (salicyl-AMSN, 4a),24 and substitutions of the adenine ring at the C2 position (salicyl-2-Ph-AMS, 3a; salicyl-2-NHPh-AMS, 3b) and 6-amino group (salicyl-6-N-Me-AMS, 5a; salicyl-6-N-c-Pr-AMS, 5b).23
Figure 4.

Structural differences between salicyl-AMS (1) and its various analogues with modifications in the AMS moiety (dotted line box) evaluated herein. Me, methyl; Ph, phenyl; c-Pr, cyclopropyl.
We also designed two analogues to expand the SAR analysis (Figure 4). Salicylate adenylation enzymes are members of the ANL family (acyl-CoA synthetase, non-ribosomal peptide synthetase adenylation domain, luciferase)62 that bind their substrates in an unusual cisoid conformation (Figure 5a).64,65 In an effort to promote this pharmacophoric conformation, we introduced a 5´-N-methyl substituent in salicyl-AMSN as a turn element in salicyl-AMSNMe (4b). The compound was synthesized by sulfamoylation and salicylation of a protected 5´-methylamino-5´-deoxyadenosine (see Supporting Information Figure S1 for complete details).
Figure 5.

Design rationales for new salicyl-AMS analogues. (A) Cocrystal structure of 2,3-dihydroxybenzoyl-AMP (DHB-AMP, gray) bound in the active site of the DHBA adenylation enzyme Bacillus subtilis DhbE (cyan and white, PDB: 1MDB),64 illustrating pharmacophoric cisoid binding conformation.65 Salicyl-AMSNMe (4b) is designed to promote this cisoid binding conformation. (B) Structures of previously reported inosine analogue salicyl-IMS (11) and dimethylated analogue salicyl-6-N,N-diMe-AMS (12), both of which are weak MbtAtb inhibitors,23 presumably due to loss of hydrogen-bonding interaction with the main-chain carbonyl oxygen of MbtAtb V352 (DhbE V329). Salicyl-6-MeO-AMS (6) is designed to remove this hydrogen-bonding interaction without introducing a proton at N1 via tautomerization (cf. 11) and without introducing a potential steric clash with V352 (cf. 12).
In addition, we were intrigued by reports that small 6-amino substituents could be accommodated in salicyl-AMS analogues (5a, 5b).23,66,67 In contrast, however, the corresponding inosine analogue (11, Figure 5b) was a poor MbtAtb inhibitor and exhibited no antimicrobial activity (; Mtb MIC > 100 µM).23 As inosine adopts a tautomeric (6-oxo) form compared to adenine, it was proposed that a hydrogen-bond donor is required at the C6-substituent position, which could interact with the main-chain carbonyl oxygen of MbtAtb V352.67,68 This was further supported by the finding that the adenine 6-N,N-dimethyl analogue (12) was also a weak inhibitor (; Mtb MIC = 50 µM). However, the inosine tautomeric form also introduces a proton at N1 in analogue 11 and the 6-N,N-dimethyl modification introduces additional steric constraints in analogue 12. Thus, to probe this SAR further, we introduced a 6-methoxy substituent in salicyl-6-MeO-AMSN (6), which lacks the C6-substituent hydrogen-bond donor but also maintains the adenine tautomeric form (N1 lone pair) and is isosteric to the active 6-N-methyl analogue 5a. Notably, initial attempts to synthesize the corresponding sulfamate analogue, salicyl-6-MeO-AMS (not shown), were thwarted by product instability, necessitating replacement with the more stable sulfamide in 6.24 Thus, salicyl-6-MeO-AMSN (6) was synthesized from inosine by conversion of the 6-oxo group to a 6-methoxy group, followed by installation of the 5´-amino group, sulfamoylation, and salicylation (see Supporting Information Figure S2 for complete details).
Progress curve analysis of kinetics of MbtAtb inhibition by salicyl-AMS and analogues.
To evaluate the activity of the new analogues salicyl-AMSNMe (4b) and salicyl-6-MeO-AMSN (6), and to gain additional insight into the inhibition of MbtAtb by salicyl-AMS (1) and the six previously reported analogues (2, 3a, 3b, 4a, 5a, 5b), we investigated the time dependence of the onset of H10MbtAopt inhibition by each of the compounds. To this end, we determined and compared the kinetics of their H10MbtAopt inhibition by progress curve analysis. For each compound in the series, the progress curves displayed a nonlinear profile of H10MbtAopt inhibition with the characteristic three phases of a time-dependent, slow-onset mechanism of inhibition (Figure 6A and Figure S7A); i.e.: an initial linear phase that extrapolates to a slope corresponding to a pre-equilibrium initial velocity (vps) at t = 0; a final linear phase with a slope representing the equilibrium, steady-state velocity (vs); and an exponential phase that connects the two linear phases with a pseudo-first order rate constant (kobs) for the approach to the steady state.43,44 In contrast, the progress curves for uninhibited reactions (DMSO controls) showed the expected linear profile of the steady-state kinetics (Figure 6A and Figure S7A). Thus, our results demonstrate that salicyl-AMS (1) and the eight analogues are slow-onset inhibitors of H10MbtAopt. Encouragingly, the results also provide the first indication of the potent activity of salicyl-AMSNMe (4b) and salicyl-6-MeO-AMSN (6) against MbtAtb.
Figure 6.

Representative data for the kinetics of time-dependent inhibition of H10MbtAopt. (A) Progress curves for MbtAtb inhibition by different concentrations of 1, 4b, and 6. The inhibitor number is indicated on the upper left corner of the plot. Each plot is representative of at least five independent experiments. The solid lines represent curve fitting to the dataset (●) using Eq. 4 to extract kobs values. The R2 values for ≈270 progress curves generated were in the 0.789–0.999 range, with an average of 0.985 and a median of 0.995, demonstrating excellent fit for the large majority of the curves. (B) Datasets showing the dependence of the extracted kobs on the concentration of 1, 4b, and 6. The datasets (●) were fitted to Eq. 7 to generate the solid lines (R2 values ≥ 0.955). kobs datapoints represent mean values ± SEM of n ≥ 5 independent experiments.
To explore further the mechanism of MbtAtb inhibition, progress curves were fitted to Eq. 4 for slow-onset inhibitors, and vps, vs, and kobs values were obtained for each inhibitor concentration using nonlinear regression analysis (Figure 6A and Figure S7A).43,44 The vps, vs, and kobs values of each progress curve were then used to calculate the dissociation rate constant (koff) values using Eq. 5.44,45 Analysis of the relationship between kobs values and inhibitor concentration revealed a linear trend (Figure 6B and Figure S7B), which is a property indicative of a static, slow-onset mechanism of inhibition.44 To our knowledge, these results provide the first indication that salicyl-AMS (1) exhibits this type of mechanism of inhibition with its salicylate adenylation enzyme target. Moreover, our analysis demonstrates that all of the analogues also exhibited this mechanism of inhibition. Incidentally, salicyl-AMS (1) was recently shown to have a static, slow-onset inhibitory mechanism with the 2,3-dihydroxybenzoate adenylation enzyme EntE from E. coli (40% sequence identity with MbtAtb) in a study using a methodology not reliant on the HA–MesG assay41. This reinforces our overall conclusions as to the inhibitory mechanism of salicyl-AMS (1) with its MbtAtb target and supports our methodological approach, thus increasing the confidence in the inhibitory mechanism assignment made for the eight salicyl-AMS analogues analyzed.
Determination of kinetic parameters for inhibition of H10MbtAopt by salicyl-AMS and analogues.
To characterize further the kinetic underpinnings of the inhibition of MbtAtb by salicyl-AMS (1) and analogues (2–6), and to provide the first side-by-side comparison of these inhibitors, we sought to determine their , , koff, and tR kinetic parameters. Since none of the eight salicyl-AMS analogues had been previously evaluated using the HA–MesG assay, we first assessed whether any of them had an off-target effect on the PPT-PNP coupling system of the assay. We found that none of the analogues inhibited the coupling system when tested at up to ten times the maximum concentration used in the HA–MesG assay (Table S4), thus clearing the way for their evaluation with this assay. We then carried out pilot experiments to assess whether the TBI behavior of the previously reported analogues (2, 3a, 3b, 4a, 5a, 5b) as per the ATP-PPi assay23–26 was recapitulated under the conditions of the HA–MesG assay and whether the novel analogues salicyl-AMSNMe (4b) and salicyl-6-MeO-AMSN (6) were also TBIs. Encouragingly, all inhibitors displayed IC50 values ≈ ½ [E], thus indicating a TBI behavior (Table 1 and Figure S5). Kinetic parameters for each of the inhibitors were then derived from analysis of progress curve datasets using established methodologies.41,42 The results of this analysis are summarized in Table 1.
The analysis rendered values in the 27–295 nM range (≈11-fold relative spread), thus indicating that all the inhibitors have potent activity against H10MbtAopt. As expected for TBIs,37 the values had a better discrimination power of inhibitory potency than the IC50 values, which covered a narrower 117–289 nM range (≈2.5-fold relative spread). The values spanned a 0.00004–0.00066 nM−1min−1 range (≈17-fold relative spread), whereas the koff values and the tR values covered a 0.007–0.031 min−1 range and a 54–157 min range, respectively (4-fold and 3-fold relative spread, respectively). Collectively, these results indicate that the members of the inhibitor series are relatively similar to one another in terms of their kinetic parameters. In other words, none of the structural features of the analogues setting them apart from salicyl-AMS (1) had a drastic impact (i.e.: 50-fold change) on any kinetic parameter. Encouragingly, Pearson pairwise correlation analysis revealed the somewhat expected relatively strong correlations between IC50 and values [Pearson correlation coefficient (PCC) = 0.86, p value = 0.003] and between koff and tR values (PCC = −0.79, p value = 0.012). The correlation analysis also showed a weaker but statistically significant correlation between and values (PCC = −0.69, p value = 0.040).
A two-dimensional kinetic map representation of the kinetic parameters illustrated several SAR trends and inhibitor clustering patterns (Figure 7). We found that the of salicyl-AMS (1) (27 nM) was the lowest in the series and that the inhibitor had the best tR (157 min). This is perhaps not surprising considering that salicyl-AMS (1) has the most similar structure in the inhibitor series to that of the cognate salicyl-AMP reaction intermediate (Figure 1), which is thought to be retained tightly bound to the active site in the enzyme.6 The loss of the ribose 2´-hydroxyl in salicyl-2´-dAMS (2) was well tolerated across the board, leading to a very modest worsening of the kinetic parameters relative to salicyl-AMS (1) (ca. ≤2-fold change). In agreement with this, salicyl-2´-dAMS (2) has been reported to exhibit a marginal potency increase relative to salicyl-AMS (1) (0.5-fold decrease in ) using the ATP-PPi assay.25 The three analogues with modifications at the C6-substituent of the adenine ring (5a, 5b, 6) clustered with respect to tR and koff, showing a ≈0.5-fold decrease and ≈2-fold increase, respectively, compared with salicyl-AMS (1). Within this triad, the pair with the native sulfamate-based linker and substitutions on the adenine 6-amino group (5a, 5b) were also essentially indistinguishable from each other with respect to and values, which were in turn ≈5.5-fold higher and ≈2.5-fold lower, respectively, than those of salicyl-AMS (1). In contrast, replacement of the 6-amino group with a 6-methoxy group in salicyl-6-MeO-AMSN (6) led to a more drastic worsening of and values, which were 10-fold higher and 6-fold lower, respectively, than those of salicyl-AMS (1). However, this may be the result of a dominant negative effect arising from concurrent replacement of the sulfamate linker of salicyl-AMS (1) with a sulfamide in this analogue. Indeed, the presence of the sulfamide linker in analogues 4a, 4b, and 6 correlated with the worst and values in the series. This correlation contrasts with an earlier study reporting essentially indistinguishable values for salicyl-AMSN (4a) and salicyl-AMS (1) as determined using the ATP-PPi assay.26 This difference may be due to the different experimental conditions used in the studies (i.e.: different assay, recombinant proteins, batches of inhibitors, etc.). Notably, the comparison of the values of salicyl-AMSN (4a) and salicyl-AMSNMe (4b) indicated that addition of the 5´-N-methyl substituent led to a modest 0.5-fold improvement in value. This suggests that substitution of the sulfamide linker might be a strategy to mitigate the negative effect of this linker on MbtAtb inhibitory potency revealed by our analysis herein. Lastly, the two analogues with aryl substituents at the adenine C2 position (3a, 3b) clustered away from the analogues with modifications on the adenine C6 substituent (5a, 5b, 6) and displayed the worst koff and tR values in the series. Thus, although these analogues exhibited a modest decrease (3b) or no decrease (3a) in values and an increase in values compared to salicyl-AMS (1), fast koff rates are typically viewed as an undesired property.69,70 It is tempting to speculate that the bulky substituents in analogues 3a and 3b compromise some of the binding interactions in the MbtAtb active site, perhaps facilitating analogue dissociation and reducing the lifetime of the analogue-MbtAtb complexes. Interestingly, salicyl-6-MeO-AMSN (6) remains a fairly potent inhibitor of MbtAtb, despite the lack of a hydrogen-bond donor on the C6-substituent and the presence of the sulfamide linker. This result was unexpected based on previous SAR studies,23 and is not readily rationalized based on protein structure analysis.64,67 Nonetheless, it opens the door to further investigation of such C6-substituents.
Figure 7.

Two-dimensional kinetic map for salicyl-AMS and its analogues. The map represents the data summarized in Table 1 and facilitates visualization of relationships between inhibitors and the kinetic parameters koff, , tR, and (diagonal dashed lines) noted in the text. The clustering of analogues with sulfamide-based linkers (4a, 4b, 6; red oval), modifications at C2 (3a, 3b; green oval), and modifications at C6 (5a, 5b, 6; blue oval) is indicated.
Contrasting with our results, a previous study reported analogues 3a, 3b, 5a, and 5b to be ≈4– 24-fold more potent than salicyl-AMS (1) as per values determined using the ATP-PPi assay.23 It appears that this study referenced a value for salicyl-AMS (1) that was determined separately,23 and this may explain the overall discrepancy with the findings reported herein. Because values are highly dependent on experimental conditions, we believe that our side-by-side comparison, derived from multiple independent experimental replicates, is likely to offer a more reliable comparison of the values of this group of inhibitors.
Construction of a M. smegmatis strain with MbtAtb-dependent susceptibility to salicyl-AMS.
The study of the antimicrobial properties of tuberculosis lead compounds using Mtb is challenging due to the need for biosafety level 3 procedures and the bacterium’s slow growth rate. Thus, to simplify the analysis of the antimicrobial properties of MbtAtb inhibitors, we sought to use the nonpathogenic, fast-replicating Msm model system. Msm has the MBT siderophore system, and we have shown that the MbtAtb orthologue in Msm (MbtAsm) (71% sequence identity) is essential for MBT biosynthesis.8 However, Msm also has a second siderophore system that accounts for 90–95% of siderophore activity in the bacterium (i.e. the EXO system noted above).71,72 We hypothesized that production of EXOs would render Msm resistant to salicyl-AMS (1), thus impeding the use of Msm for evaluation of MbtA inhibitors. To explore this view, we first investigated the susceptibility of Msm WT, Msm E (EXO−, Figure S8A), and Msm M (MBT−, Figure S8B) to salicyl-AMS (1) in Fe-limiting and Fe-rich growth media (Table 2). As expected, among these three strains, only Msm ∆E was susceptible, and the potent activity of salicyl-AMS (1) (MIC = 0.5–1 µg/mL) was exquisitely conditional to the Fe-limiting condition, a property consistent with its mechanism of action in inhibiting MBT siderophore biosynthesis. Under this condition, Msm ∆E relied on MBT production to acquire the Fe needed for growth, as indicated by the failure of Msm EM (EXO−/MBT−) to grow in the Fe-limiting medium (Table 2). This lack-of-growth phenotype, which is viewed as a reference for the effect of 100% pharmacological inhibition of MbtA in Msm ∆E, was corrected in the complementation control strain with episomal expression of mbtAsm (Figure S8C), thus ruling out confounding mutations or polar effects in the double mutant. The lack of growth of the siderophore-deficient Msm ∆EM mutant in the Fe-limiting medium is in agreement with previous observations73,74 and mimics the phenotype seen for Mtb MBT− mutants.12,15,17,74–76 In accordance with previous reports,73,74 the growth defect of Msm ∆EM was corrected by addition of Fe to the culture medium (Figure S7C).
Table 2.
Susceptibility of M. smegmatis strains to salicyl-AMS
| Msm strain (EXO / MBT)a | MIC (µg/ml)b |
|
|---|---|---|
| GASTD medium | GASTD+Fe medium | |
| Msm WT (+ / +) | >1000 | >1000 |
| Msm ΔE (− / +) | 0.5 – 1.0 | >1000 |
| Msm ΔM (+ / −) | >1000 | >1000 |
| Msm ΔEM (− / −) | no growth | >1000 |
| Msm ΔEM-pMbtAtb (− / +) | 0.5 – 1.0 | >1000 |
The siderophore production phenotype of each strain is indicated between brackets. EXO, exochelins; MBT, mycobactins; +, production; -, deficiency.
MIC data shown is derived from three independent experiments.
Notably, the lack of susceptibility seen with the EXO+ strains under the Fe-limiting condition and with all strains under the Fe-rich condition indicated that salicyl-AMS (1) (at up to 1 mM, ≈1,000 × MIC; Table 2) does not have significant off-target effects in Msm. This finding contrasts with the observation that salicyl-AMS (1) (Table 2) and analogues 3a, 3b, 5a, and 5b (Table 3) have significant antimycobacterial activity against Mtb even in Fe-rich media (MIC values increased by only 4–64 fold relative to low-Fe medium).23 The results obtained with Mtb have been attributed to off-target effects.23 Alternatively, our review of the literature revealed that MBT-deficient mutants have been shown to display either a significant growth defect17,74–77 or no defect12,15 compared with WT when grown in iron-rich media. This difference is study-dependent, and suggests that MBT production might be needed for WT growth even in Fe-rich media, but only under some experimental conditions. This precludes determination of whether the Mtb growth inhibition resulting from MbtA inhibitors in the Fe-rich media is due to MBT inhibition, off-target effects, or a combination of both. Thus, the possibility of off-target effect confounders in Mtb-based SAR analyses of MbtA inhibitors cannot be ruled out, a disadvantage not found with the Msm model developed herein.
Table 3.
Antimicrobial activity and post-antibiotic effect of MbtAtb inhibitors in M. smegmatis ∆EM-pMbtAtb
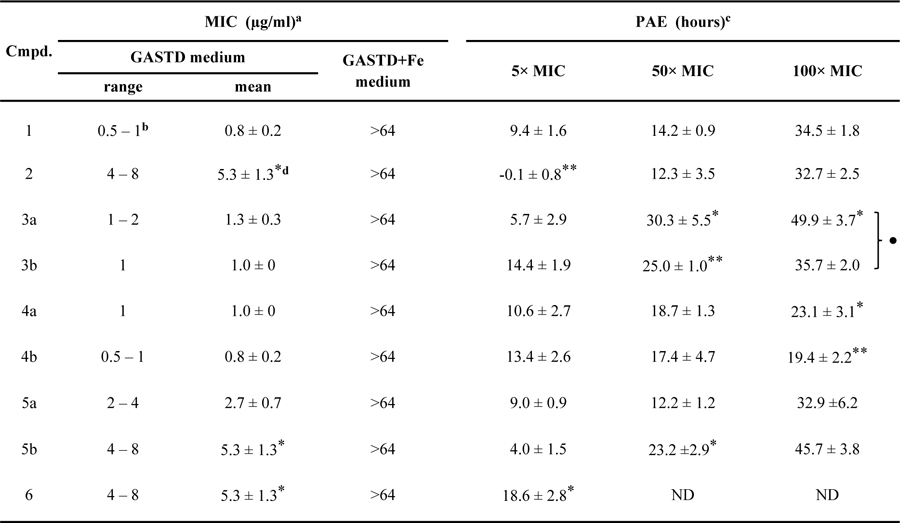 |
MIC data against Msm ∆EM-pMbtAtb shown represent ranges and mean values ± SEM of n = 3 independent experiments.
The MIC value for 1 was derived from experiments independent from those that rendered the MIC value shown in Table 2.
PAE was calculated as the difference between the time-to-threshold values of the inhibitor-exposed culture and the control cultures (Figure S9). The PAE data in Msm ∆EM-pMbtAtb represent mean values ± SEM of n = 3 independent experiments.
Statistical significance as per Student’s t-test is indicated.
*, *, and *** represent p values of ≤0.05; ≤0.01, and ≤0.001, respectively, for analogues vs. 1. ● represents p value of ≤0.05 for 3a vs. 3b, 4a vs. 4b, and 5a vs. 5b pairs. No dots or asterisks indicate no statistical significance. ND, not determined.
Collectively, the above results demonstrated that salicyl-AMS (1) has potent and selective antimicrobial activity against Msm ∆E. Encouraged by these results, we next explored the possibility of developing a Msm EXO− strain that is dependent upon MbtAtb, the primary intended target of salicyl-AMS (1). To this end, we transformed the Msm ∆EM double mutant with pMbtAtb to enable heterologous expression of mbtAtb. We found that the transformant (Msm ∆EM-pMbtAtb) regained MBT production (Figure S8B) and had the same pattern of susceptibility to salicyl-AMS (1) seen for Msm ∆E (Table 2 and Figure S8D). Moreover, salicyl-AMS (1) inhibited MBT production in Msm ∆EM-pMbtAtb (Figure S8B), a result paralleling that seen with Mtb.6 Taken together, these results established that Msm ∆EM-pMbtAtb has high, MbtAtb-dependent salicyl-AMS susceptibility, and thus the strain represents a convenient model system to assess and compare the antimycobacterial properties of the MbtAtb inhibitors.
Antimicrobial activity and post-antibiotic effect of salicyl-AMS and analogues.
To assess the antimycobacterial activity of our two novel MbtAtb inhibitors (4b, 6) and to gain additional insight into the antibacterial properties of salicyl-AMS (1) and the six previously reported analogues (2, 3a, 3b, 4a, 5a, 5b), we determined MIC values and investigated in vitro PAE using Msm EM-pMbtAtb (Table 3). Notably, despite the relevance of PAE information to the lead optimization and prioritization phases of antibiotic development,78–80 to our knowledge PAE studies have not been undertaken previously for salicyl-AMS (1) or any of its analogues.
Evaluation of antimicrobial activity in Fe-limiting media revealed MIC values in the 0.8–5.3 µg/mL range (≈6-fold relative spread), indicating that all of the MtbAtb inhibitors had potent activity against Msm ∆EM-pMbtAtb. This narrow range parallels the modest, 11-fold relative spread observed for the range (Table 1). The MIC value for salicyl-AMS (1) (0.8 µg/mL) was at the lowest end of the range, and none of the analogues displayed improved potency (Table 3), an outcome mirroring the results of the analysis of values (Table 1). In this light, it is perhaps not surprising that Pearson correlation analysis revealed no statistically significant correlation between the antibacterial potency captured by the MIC dataset and the MbtAtb inhibitory potency informed by the dataset (PCC < 0.5). The relatively small differences seen in in vitro potency are probably overpowered by the numerous cellular factors known to impact its translation into cell-based assay potency (e.g., drug penetration/efflux/inactivation, non-specific off-target binding, target vulnerability/turnover, etc.),79,81 and thus it is likely that any existing -MIC correlation would have been confounded. On the other hand, lack of correlation between biochemical activity and whole-cell antimycobacterial activity is a well-known challenge in the tuberculosis drug development field, a phenomenon thought to arise from a mostly unpredictable combination of factors (e.g. penetration through the mycomembrane, intracellular metabolism, and extrusion by efflux pumps) differentially affecting each compound.82–84
Encouragingly, the results demonstrated potent antimicrobial activity for the novel analogues salicyl-AMSNMe (4b) and salicyl-6-MeO-AMSN (6). While N-methylsulfamide analogue 4b had an MIC value indistinguishable from that of salicyl-AMS (1), 6-methoxy analogue 6 displayed a 6-fold decrease in potency compared to 1. Among the previously reported analogues, we found no statistically significant difference between the MIC values of salicyl-AMS (1), salicyl-2-Ph-AMS (3a), salicyl-2-NHPh-AMS (3b), salicyl-AMSN (4a), and salicyl-6-N-Me-AMS (5a), whereas salicyl-2´-dAMS (2) and salicyl-6-N-c-Pr-AMS (5b) showed a significant 6-fold decrease in potency compared to 1 (Table 3). Notably, neither salicyl-AMS (1) nor the analogues reached MIC when tested at up to 1,000 and 64 µg/mL, respectively, in Fe-rich medium (Tables 2 and 3). This selectivity is consistent with their expected mechanism of action in inhibition of MBT siderophore biosynthesis, and revealed that the structural features of the analogues setting them apart from the lead compound 1 did not lead to unintended off-target effects of significance in Msm.
Notably, the MIC of 0.5–1 µg/ml found for salicyl-AMS (1) against Msm ∆EM-pMbtAtb is in reasonable agreement with the MIC of ≈0.1–0.5 µg/ml reported previously for Mtb in Fe-limiting medium, when using a bacterial inoculum level comparable to that herein.23,24,33 The similarity between the MIC values of salicyl-AMS (1), salicyl-6-N-Me-AMS (5a), and salicyl-AMSN (4a) determined herein against Msm ∆EM-pMbtAtb is also in reasonable agreement with the MIC data reported with Mtb, considering the experimental variability of MIC assessments.23,24 Notably, salicyl-2´-dAMS (2) displayed reduced activity in both bacterial species, but much more so in Mtb. This might be a consequence of the longer incubation period of the MIC assay for Mtb, which could lead to some degradation of the less-stable 2´-deoxynucleoside structure.28,85
On the other hand, we found no statistically significant difference between the MIC value of salicyl-AMS (1) and those of adenine C2-substituted analogues 3a and 3b against Msm ∆EM-pMbtAtb, in contrast to a previous report that these analogues are ten times more potent than 1 against Mtb. Moreover, we found salicyl-6-N-c-Pr-AMS (5b) to be six times less potent than salicyl-AMS (1) against Msm EM-pMbtAtb, while this analogue was previously reported to be four times more potent than 1 against Mtb. Thus, there is only partial agreement between the MIC profiles of the inhibitor series emerging from testing against Msm and Mtb. The possibility that the increase in potency of analogues 3a, 3b, and 5b relative to salicyl-AMS (1) seen in Mtb is due to an off-target effect cannot be ruled out. Alternatively, other species-specific factors and/or variations in the experimental conditions might have contributed to the differences.
Encouragingly, the assessment of the in vitro PAE, using washout experiments, revealed that all the inhibitors had a substantially delayed regrowth following inhibitor exposure (Table 3). A concentration-dependent PAE trend was found for the eight inhibitors tested at different concentrations, showing 0–14 h, 12–30 h, and 19–50 h ranges for the 5×, 50×, and 100× MIC exposures, respectively. For reference, drugs commonly used to treat tuberculosis have reported PAEs in the 2–68 h range86 and drugs to treat infections with the fast-growing Mycobacterium fortuitum have PAEs in the 5–15 h range.87 Within each inhibitor concentration evaluated, the majority of the PAE values of the analogues had no statistically significant difference from that of salicyl-AMS (1) (Table 3). A correlation between in vitro PAE and tR datasets is occasionally seen for inhibitor series,88 and thus we explored this possibility. Pearson pairwise correlation analysis revealed no significant correlation between the PAE and tR datasets (PCC < 0.5). This is not surprising considering that the relatively modest tR differences between the inhibitors are probably eclipsed by the various cellular factors influencing the extent of intracellular target engagement (e.g., inhibitor penetration and efflux, target turnover, etc.),79,81 and thus it is likely that any existing tR–PAE correlation would have been confounded. In all, to our knowledge, these results provide the first demonstration of PAE for MbtAtb inhibitors. Moreover, they validate Msm ∆EM-pMbtAtb as a convenient model system for analyses of inhibitors of MbtAtb.
CONCLUSIONS
Our analyses of salicyl-AMS and its analogues as MbtAtb inhibitors indicate that they all are potent, competitive TBIs, with a static, slow-onset inhibition mechanism, and a considerable tR. The inhibitors are relatively similar to one another in terms of kinetic parameters. The fairly potent MbtAtb inhibition activity of the new analogue salicyl-6-MeO-AMSN (6) is unexpected based on previously published SAR and modeling analyses. Further SAR exploration of C6-substituents is therefore warranted. The second new analogue salicyl-AMSNMe (4b), which was designed to promote a favorable pharmacophoric cisoid conformation, has improved MbtAtb inhibitory potency compared with the unsubstituted counterpart. This observation highlights substitutions promoting a cisoid conformation as a topic deserving further investigation. Our evaluation of the antimicrobial properties of the MtbAtb inhibitors were greatly facilitated by using a convenient Msm-based model system. This model is free of off-target effect confounders, whereas such confounders cannot be ruled out in SAR analyses of MbtAtb inhibitors using Mtb. All of these inhibitors have potent antimycobacterial activity and are similar to one another in terms of MIC values, a finding that parallels their relatively narrow range of values. All of the inhibitors have also a substantial PAE, a clinically significant property shared with drugs commonly used against mycobacterial infections. This property, along with the long tR of the inhibitors, may explain the efficacy of salicyl-AMS monotherapy in the murine model of tuberculosis using daily dosing (5.6 or 16.7 mg/kg), despite its relatively rapid clearance (lung half-life = 13 min after 50 mg/kg intraperitoneal administration).33 The PAE and long tR of salicyl-AMS suggest retention of the compound at the target site during the efficacy studies, which would not be detected in the pharmacokinetic studies where there are no bacteria to maintain the compound in the lung. In all, the insights gained in the present study support the advancement of the new analogues to testing in Mtb strains and set the stage for further preclinical evaluation of pharmacological and toxicological properties to identify the most promising candidates for evaluation of in vivo efficacy in mouse models of tuberculosis.
Supplementary Material
ACKNOWLEDGMENT
We thank Omar Omar (L.E.N.Q. laboratory) for assistance with mutant constructions and Lukman Solola (L.E.N.Q. laboratory) for help with CAS-Q assays. We thank Yasutomi Asano (Takeda), Makoto Fushimi (Takeda), Naoyoshi Noguchi (Takeda), and Michael A. Foley (Tri-Institutional Therapeutics Discovery Institute) for assistance with the synthesis of salicyl-AMS.
Funding Sources
This work was supported in part by the National Institutes of Health (R01 AI118224 to D.S.T. and L.E.N.Q., F31 AI129244 to L.C.S., CCSG P30 CA008748 to C. B. Thompson), the Tri-Institutional Therapeutics Discovery Institute, and endowment support from Carol and Larry Zicklin to L.E.N.Q.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The following files are available free of charge.
Supplemental materials and methods, figures, and tables (PDF)
Accession Codes
MbtA: UniProtKB P71716
Notes
The authors declare competing financial interest. D.S.T. and L.E.N.Q. are inventors on U.S. Patents 8,461,128 and 8,946,188. D.S.T., L.E.N.Q, L.C.S., and G.V.B. are inventors on a patent application based on this work.
REFERENCES
- (1).Global tuberculosis report 2017 Geneva: World Health Organization (2017). Licence: CC BY-NCSA 3.0 IGO. [Google Scholar]
- (2).Barry CE 3rd, and Mdluli K (1996) Drug sensitivity and environmental adaptation of mycobacterial cell wall components. Trends Microbiol 4, 275–281. [DOI] [PubMed] [Google Scholar]
- (3).Nahid P, Dorman SE, Alipanah N, Barry PM, Brozek JL, Cattamanchi A, Chaisson LH, Chaisson RE, Daley CL, Grzemska M, Higashi JM, Ho CS, Hopewell PC, Keshavjee SA, Lienhardt C, et al. (2016) Official american thoracic society/centers for disease control and prevention/infectious diseases society of america clinical practice guidelines: Treatment of drug-susceptible tuberculosis. Clin. Infect. Dis 63, e147–e195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Alipanah N, Jarlsberg L, Miller C, Linh NN, Falzon D, Jaramillo E, and Nahid P (2018) Adherence interventions and outcomes of tuberculosis treatment: A systematic review and meta-analysis of trials and observational studies. PLoS Med 15, e1002595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Awofeso N Anti-tuberculosis medication side-effects constitute major factor for poor adherence to tuberculosis treatment, Bull. World Health Organ 2008. March;86(3):B-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ferreras JA, Ryu JS, Di Lello F, Tan DS, and Quadri LE (2005) Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol 1, 29–32. [DOI] [PubMed] [Google Scholar]
- (7).Quadri LE, Sello J, Keating TA, Weinreb PH, and Walsh CT (1998) Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol 5, 631–645. [DOI] [PubMed] [Google Scholar]
- (8).Chavadi SS, Stirrett KL, Edupuganti UR, Sadhanandan G, Vergnolle O, Schumacher E, Martin C, Qiu WG, Soll CE, and Quadri LEN (2011) Mutational and phylogenetic analyses of the mycobacterial mbt gene cluster. J. Bacteriol 193, 5905–5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Quadri LEN, and Ratledge C (2005) Iron metabolism in the tubercle bacillus and other mycobacteria, In Tuberculosis and the Tubercle Bacillus (Cole ST, Eisenach KD, McMurray DN, et al. , Eds.), pp 341–357, ASM Press, Washington, DC. [Google Scholar]
- (10).Ratledge C (2004) Iron, mycobacteria and tuberculosis. Tuberculosis (Edinb) 84, 110–130. [DOI] [PubMed] [Google Scholar]
- (11).De Voss JJ, Rutter K, Schroeder BG, and Barry CE 3rd. (1999) Iron acquisition and metabolism by mycobacteria. J. Bacteriol 181, 4443–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, and Barry CE 3rd. (2000) The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. U.S.A 97, 1252–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rodriguez GM, and Smith I (2006) Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J. Bacteriol 188, 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Reddy PV, Puri RV, Chauhan P, Kar R, Rohilla A, Khera A, and Tyagi AK (2013) Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. J. Infect. Dis 208, 1255–1265. [DOI] [PubMed] [Google Scholar]
- (15).Madigan CA, Martinot AJ, Wei JR, Madduri A, Cheng TY, Young DC, Layre E, Murry JP, Rubin EJ, and Moody DB (2015) Lipidomic analysis links mycobactin synthase K to iron uptake and virulence in M. tuberculosis. PLoS Pathog 11, e1004792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wells RM, Jones CM, Xi Z, Speer A, Danilchanka O, Doornbos KS, Sun P, Wu F, Tian C, and Niederweis M (2013) Discovery of a siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog 9, e1003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tufariello JM, Chapman JR, Kerantzas CA, Wong KW, Vilcheze C, Jones CM, Cole LE, Tinaztepe E, Thompson V, Fenyo D, Niederweis M, Ueberheide B, Philips JA, and Jacobs WR Jr. (2016) Separable roles for Mycobacterium tuberculosis ESX-3 effectors in iron acquisition and virulence. Proc. Natl. Acad. Sci. U. S. A 113, E348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Quadri LE (2007) Strategic paradigm shifts in the antimicrobial drug discovery process of the 21st century. Infect. Disord. Drug Targets 7, 230–237. [DOI] [PubMed] [Google Scholar]
- (19).Meneghetti F, Villa S, Gelain A, Barlocco D, Chiarelli LR, Pasca MR, and Costantino L (2016) Iron acquisition pathways as targets for antitubercular drugs. Curr. Med. Chem 23, 4009–4026. [DOI] [PubMed] [Google Scholar]
- (20).Monfeli RR, and Beeson C (2007) Targeting iron acquisition by Mycobacterium tuberculosis. Infect. Disord. Drug Targets 7, 213–220. [DOI] [PubMed] [Google Scholar]
- (21).Gehring AM, Mori I, Perry RD, and Walsh CT (1998) The nonribosomal peptide synthetase HMWP2 forms a thiazoline ring during biogenesis of yersiniabactin, an iron-chelating virulence factor of Yersinia pestis. Biochemistry 37, 11637–11650. [DOI] [PubMed] [Google Scholar]
- (22).Quadri LE, Keating TA, Patel HM, and Walsh CT (1999) Assembly of the Pseudomonas aeruginosa nonribosomal peptide siderophore pyochelin: in vitro reconstitution of aryl-4,2-bis-thiazoline synthetase activity from PchD, PchE, and PchF. Biochemistry 38, 14941–14954. [DOI] [PubMed] [Google Scholar]
- (23).Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE 3rd, Bennett EM, and Aldrich CC (2008) Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure-activity relationships of the nucleobase domain of 5’-O-[N-(salicyl)sulfamoyl]adenosine. J. Med. Chem 51, 5349–5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE 3rd, and Aldrich CC (2006) Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem 49, 31–34. [DOI] [PubMed] [Google Scholar]
- (25).Somu RV, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, Barry CE 3rd, and Aldrich CC (2006) Antitubercular nucleosides that inhibit siderophore biosynthesis: SAR of the glycosyl domain. J. Med. Chem 49, 7623–7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Vannada J, Bennett EM, Wilson DJ, Boshoff HI, Barry CE 3rd, and Aldrich CC (2006) Design, synthesis, and biological evaluation of β-ketosulfonamide adenylation inhibitors as potential antitubercular agents. Org. Lett 8, 4707–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Nelson KM, Viswanathan K, Dawadi S, Duckworth BP, Boshoff HI, Barry CE 3rd, and Aldrich CC (2015) Synthesis and pharmacokinetic evaluation of siderophore biosynthesis inhibitors for Mycobacterium tuberculosis. J. Med. Chem 58, 5459–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Duckworth BP, Nelson KM, and Aldrich CC (2012) Adenylating enzymes in Mycobacterium tuberculosis as drug targets. Curr. Top. Med. Chem 12, 766–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dawadi S, Boshoff HIM, Park SW, Schnappinger D, and Aldrich CC (2018) Conformationally constrained cinnolinone nucleoside analogues as siderophore biosynthesis inhibitors for tuberculosis. ACS Med. Chem. Lett 9, 386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Engelhart CA, and Aldrich CC (2013) Synthesis of chromone, quinolone, and benzoxazinone sulfonamide nucleosides as conformationally constrained inhibitors of adenylating enzymes required for siderophore biosynthesis. J. Org. Chem 78, 7470–7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dawadi S, Kawamura S, Rubenstein A, Remmel R, and Aldrich CC (2016) Synthesis and pharmacological evaluation of nucleoside prodrugs designed to target siderophore biosynthesis in Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett 24, 1314–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Krajczyk A, Zeidler J, Januszczyk P, Dawadi S, Boshoff HI, Barry CE 3rd, Ostrowski T, and Aldrich CC (2016) 2-Aryl-8-aza-3-deazaadenosine analogues of 5’-O-[N-(salicyl)sulfamoyl]adenosine: Nucleoside antibiotics that block siderophore biosynthesis in Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett 24, 3133–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lun S, Guo H, Adamson J, Cisar JS, Davis TD, Chavadi SS, Warren JD, Quadri LE, Tan DS, and Bishai WR (2013) Pharmacokinetic and in vivo efficacy studies of the mycobactin biosynthesis inhibitor salicyl-AMS in mice. Antimicrob. Agents Chemother 57, 5138–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sambrook J, Russell DW (2001) Molecular cloning: A laboratory manual, 3rd ed., Cold Spring Harbor Press, Cold Spring Harbor, NY. [Google Scholar]
- (35).Wilson DJ, and Aldrich CC (2010) A continuous kinetic assay for adenylation enzyme activity and inhibition. Anal. Biochem 404, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Davis TD, Mohandas P, Chiriac MI, Bythrow GV, Quadri LE, and Tan DS (2016) Design, synthesis, and biological evaluation of -hydroxyacyl-AMS inhibitors of amino acid adenylation enzymes. Bioorg. Med. Chem. Lett 26, 5340–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Copeland RA (2013) Tight binding inhibition, In Evaluation of enzyme inhibitors in drug discovery, pp 245–285, John Wiley & Sons, Inc. [Google Scholar]
- (38).Brooks HB, Geeganage S, Kahl SD, Montrose C, Sittampalam S, Smith MC, and Weidner JR (2004) Basics of enzymatic assays for HTS, In Assay guidance manual (Sittampalam GS, Coussens NP, Brimacombe K, et al. , Eds.), Bethesda (MD). [Google Scholar]
- (39).Copeland RA (2002) Tight binding inhibitors, In Enzymes, pp 305–317, John Wiley & Sons, Inc. [Google Scholar]
- (40).Murphy DJ (2004) Determination of accurate KI values for tight-binding enzyme inhibitors: An in silico study of experimental error and assay design. Anal. Biochem 327, 61–67. [DOI] [PubMed] [Google Scholar]
- (41).Sikora AL, Wilson DJ, Aldrich CC, and Blanchard JS (2010) Kinetic and inhibition studies of dihydroxybenzoate-AMP ligase (EntE) from Escherichia coli. Biochemistry 49, 3648–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan Z, Rudolph J, and Raetz CR (2005) A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciprofloxacin. Biochemistry 44, 16574–16583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Morrison JF, and Walsh CT (1988) The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol 61, 201–301. [DOI] [PubMed] [Google Scholar]
- (44).Copeland RA (2013) Slow binding inhibitors, In Evaluation of enzyme inhibitors in drug discovery, pp 203–244, John Wiley & Sons, Inc. [Google Scholar]
- (45).Chang A, Schiebel J, Yu W, Bommineni GR, Pan P, Baxter MV, Khanna A, Sotriffer CA, Kisker C, and Tonge PJ (2013) Rational optimization of drug-target residence time: Insights from inhibitor binding to the Staphylococcus aureus FabI enzyme-product complex. Biochemistry 52, 4217–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Larionova NI, Gladysheva IP, and Gladyshev DP (1997) Human leukocyte elastase inhibition by Bowman-Birk soybean inhibitor. Discrimination of the inhibition mechanisms. FEBS Lett 404, 245–248. [DOI] [PubMed] [Google Scholar]
- (47).Snapper SB, Melton RE, Mustafa S, Kieser T, and Jacobs WR Jr. (1990) Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol 4, 1911–1919. [DOI] [PubMed] [Google Scholar]
- (48).Ferreras JA, Gupta A, Amin ND, Basu A, Sinha BN, Worgall S, Jayaprakash V, and Quadri LE (2011) Chemical scaffolds with structural similarities to siderophores of nonribosomal peptide-polyketide origin as novel antimicrobials against Mycobacterium tuberculosis and Yersinia pestis. Bioorg. Med. Chem. Lett 21, 6533–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Parish T, and Stoker NG (2000) Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146, 1969–1975. [DOI] [PubMed] [Google Scholar]
- (50).Mohan A, Padiadpu J, Baloni P, and Chandra N (2015) Complete genome sequences of a Mycobacterium smegmatis laboratory strain (MC2 155) and isoniazid-resistant (4XR1/R2) mutant strains. Genome Announc 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Fiss EH, Yu S, and Jacobs WR Jr. (1994) Identification of genes involved in the sequestration of iron in mycobacteria: The ferric exochelin biosynthetic and uptake pathways. Mol. Microbiol 14, 557–569. [DOI] [PubMed] [Google Scholar]
- (52).Yu S, Fiss E, and Jacobs WR Jr. (1998) Analysis of the exochelin locus in Mycobacterium smegmatis: Biosynthesis genes have homology with genes of the peptide synthetase family. J. Bacteriol 180, 4676–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Zhu W, Arceneaux JE, Beggs ML, Byers BR, Eisenach KD, and Lundrigan MD (1998) Exochelin genes in Mycobacterium smegmatis: Identification of an ABC transporter and two non-ribosomal peptide synthetase genes. Mol. Microbiol 29, 629–639. [DOI] [PubMed] [Google Scholar]
- (54).Ma J, Campbell A, and Karlin S (2002) Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J. Bacteriol 184, 5733–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ferreras JA, Stirrett KL, Lu X, Ryu JS, Soll CE, Tan DS, and Quadri LE (2008) Mycobacterial phenolic glycolipid virulence factor biosynthesis: Mechanism and small-molecule inhibition of polyketide chain initiation. Chem. Biol 15, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Stirrett KL, Ferreras JA, Jayaprakash V, Sinha BN, Ren T, and Quadri LE (2008) Small molecules with structural similarities to siderophores as novel antimicrobials against Mycobacterium tuberculosis and Yersinia pestis. Bioorg. Med. Chem. Lett 18, 2662–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Chan CY, Au-Yeang C, Yew WW, Leung CC, and Cheng AF (2004) In vitro postantibiotic effects of rifapentine, isoniazid, and moxifloxacin against Mycobacterium tuberculosis. Antimicrob. Agents Chemother 48, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Stubbings WJ, Bostock JM, Ingham E, and Chopra I (2004) Assessment of a microplate method for determining the post-antibiotic effect in Staphylococcus aureus and Escherichia coli. J. Antimicrob. Chemother 54, 139–143. [DOI] [PubMed] [Google Scholar]
- (59).Khan F, He M, and Taussig MJ (2006) Double-hexahistidine tag with high-affinity binding for protein immobilization, purification, and detection on Ni-nitrilotriacetic acid surfaces. Anal. Chem 78, 3072–3079. [DOI] [PubMed] [Google Scholar]
- (60).Lee J, and Kim SH (2009) High-throughput T7 LIC vector for introducing C-terminal poly-histidine tags with variable lengths without extra sequences. Protein Expr. Purif 63, 58–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Quadri LE (2000) Assembly of aryl-capped siderophores by modular peptide synthetases and polyketide synthases. Mol. Microbiol 37, 1–12. [DOI] [PubMed] [Google Scholar]
- (62).Gulick AM (2009) Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol 4, 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Fromm HJ (1995) Reversible enzyme inhibitors as mechanistic probes. Methods Enzymol 249, 123–143. [DOI] [PubMed] [Google Scholar]
- (64).May JJ, Kessler N, Marahiel MA, and Stubbs MT (2002) Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. U. S. A 99, 12120–12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Cisar JS, Ferreras JA, Soni RK, Quadri LE, and Tan DS (2007) Exploiting ligand conformation in selective inhibition of non-ribosomal peptide synthetase amino acid adenylation with designed macrocyclic small molecules. J. Am. Chem. Soc 129, 7752–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Tawari NR, and Degani MS (2011) Predictive models for nucleoside bisubstrate analogs as inhibitors of siderophore biosynthesis in Mycobacterium tuberculosis: pharmacophore mapping and chemometric QSAR study. Mol. Divers 15, 435–444. [DOI] [PubMed] [Google Scholar]
- (67).Labello NP, Bennett EM, Ferguson DM, and Aldrich CC (2008) Quantitative three dimensional structure linear interaction energy model of 5’-O-[N-(salicyl)sulfamoyl]adenosine and the aryl acid adenylating enzyme MbtA. J. Med. Chem 51, 7154–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Maganti L, Open Source Drug Discovery, C., and Ghoshal N. (2014) Probing the structure of Mycobacterium tuberculosis MbtA: model validation using molecular dynamics simulations and docking studies. J. Biomol. Struct. Dyn 32, 273–288. [DOI] [PubMed] [Google Scholar]
- (69).Copeland RA (2013) Drug–target residence time, In Evaluation of enzyme inhibitors in drug discovery, pp 287–344, John Wiley & Sons, Inc. [Google Scholar]
- (70).Nunez S, Venhorst J, and Kruse CG (2012) Target-drug interactions: First principles and their application to drug discovery. Drug Discov. Today 17, 10–22. [DOI] [PubMed] [Google Scholar]
- (71).Ratledge C, and Ewing M (1996) The occurrence of carboxymycobactin, the siderophore of pathogenic mycobacteria, as a second extracellular siderophore in Mycobacterium smegmatis. Microbiology 142, 2207–2212. [DOI] [PubMed] [Google Scholar]
- (72).Sharman GJ, Williams DH, Ewing DF, and Ratledge C (1995) Isolation, purification and structure of exochelin MS, the extracellular siderophore from Mycobacterium smegmatis. Biochem. J 305, 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Siegrist MS, Unnikrishnan M, McConnell MJ, Borowsky M, Cheng TY, Siddiqi N, Fortune SM, Moody DB, and Rubin EJ (2009) Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc. Natl. Acad. Sci. U. S. A 106, 18792–18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Jones CM, Wells RM, Madduri AV, Renfrow MB, Ratledge C, Moody DB, and Niederweis M (2014) Self-poisoning of Mycobacterium tuberculosis by interrupting siderophore recycling. Proc. Natl. Acad. Sci. U. S. A 111, 1945–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Jones CM, and Niederweis M (2011) Mycobacterium tuberculosis can utilize heme as an iron source. J. Bacteriol 193, 1767–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Madigan CA, Cheng TY, Layre E, Young DC, McConnell MJ, Debono CA, Murry JP, Wei JR, Barry CE 3rd, Rodriguez GM, Matsunaga I, Rubin EJ, and Moody DB (2012) Lipidomic discovery of deoxysiderophores reveals a revised mycobactin biosynthesis pathway in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A 109, 1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Sassetti CM, Boyd DH, and Rubin EJ (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol 48, 77–84. [DOI] [PubMed] [Google Scholar]
- (78).Craig WA (2007) Pharmacodynamics of antimicrobials: General concepts and applications, In Antimicrobial pharmacodynamics in theory and clinical practice (Nightingale CH, Ambrose PG, Drusano GL, et al. , Eds.) 2nd ed., pp 1–22, Informa Healthcare USA, Inc., New York, NY. [Google Scholar]
- (79).Tonge PJ (2018) Drug-target kinetics in drug discovery. ACS Chem. Neurosci 9, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).MacKenzie FM, and Gould IM (1993) The post-antibiotic effect. J. Antimicrob. Chemother 32, 519–537. [DOI] [PubMed] [Google Scholar]
- (81).Davis TD, Gerry CJ, and Tan DS (2014) General platform for systematic quantitative evaluation of small-molecule permeability in bacteria. ACS Chem. Biol 9, 2535–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Singh V, and Mizrahi V (2017) Identification and validation of novel drug targets in Mycobacterium tuberculosis. Drug Discov. Today 22, 503–509. [DOI] [PubMed] [Google Scholar]
- (83).Sharma U (2011) Current possibilities and unresolved issues of drug target validation in Mycobacterium tuberculosis. Expert Opin. Drug Discov 6, 1171–1186. [DOI] [PubMed] [Google Scholar]
- (84).Machado D, Girardini M, Viveiros M, and Pieroni M (2018) Challenging the drug-likeness dogma for new drug discovery in tuberculosis. Front. Microbiol 9, 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715. [DOI] [PubMed] [Google Scholar]
- (86).Chan CY, Au-Yeang C, Yew WW, Hui M, and Cheng AF (2001) Postantibiotic effects of antituberculosis agents alone and in combination. Antimicrob. Agents Chemother 45, 3631–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Tsui SY, Yew WW, Li MS, Chan CY, and Cheng AF (1993) Postantibiotic effects of amikacin and ofloxacin on Mycobacterium fortuitum. Antimicrob. Agents Chemother 37, 1001–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Walkup GK, You Z, Ross PL, Allen EK, Daryaee F, Hale MR, O’Donnell J, Ehmann DE, Schuck VJ, Buurman ET, Choy AL, Hajec L, Murphy-Benenato K, Marone V, Patey SA, et al. (2015) Translating slow-binding inhibition kinetics into cellular and in vivo effects. Nat. Chem. Biol 11, 416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.