

Abstract
Excessive connective tissue deposition in skin and various internal organs is characteristic of systemic sclerosis (SSc). The profibrotic growth factor TGF-β plays a crucial role in SSc pathogenesis. The expression of NADPH oxidase 4 (NOX4), a critical mediator of oxidative stress, is potently stimulated by TGF-β. Here, we evaluated the effect of NOX4 on the development of TGF-β-induced tissue fibrosis. C57BL6/J control mice and Nox4 knockout mice were implanted subcutaneously with osmotic pumps containing either saline or 2.5 μg TGF-β1. After 28 days, skin and lung samples were isolated for histopathologic analysis, measurement of hydroxyproline content and gene expression analysis. Histopathology of skin and lungs from normal C57BL6/J mice treated with TGF-β1 showed profound dermal fibrosis and peribronchial and diffuse interstitial lung fibrosis. In contrast, TGF-β-treated Nox4 knockout mice showed normal skin and lung histology. Hydroxyproline levels in TGF-β-treated C57BL6/J mice skin and lungs demonstrated significant increases, however, hydroxyproline content of TGF-β-treated Nox4 knockout mice tissues was not changed. Expression of various profibrotic and fibrosis-associated genes was upregulated in skin and lungs of TGF-β1-treated C57BL6/J mice but was not significantly changed in TGF-β1-treated Nox4 knockout mice. The induction of skin and lung tissue fibrosis by TGF-β1 parenteral administration in mice was abrogated by the genetic deletion of Nox4 confirming that NOX4 is an essential mediator of the profibrotic effects of TGF-β. These results suggest Nox4 inhibition as a potential therapeutic target for SSc and other fibroproliferative disorders.
Introduction
The generation of reactive oxygen species (ROS) is frequently correlated with various progressive and chronic inflammatory disorders [1]. ROS, including superoxide (O2−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH−) are continuously produced intracellularly as byproducts of several enzymatic systems, including the mitochondrial respiratory chain [2], lipid peroxidation [3], endothelial nitric oxide (NO) synthesis [4], xanthine oxidase [5], and cytochrome P450 enzymes [6]. The cellular roles of ROS are complex and paradoxical. At low levels, ROS function as an antioxidant system critical for the maintenance of redox homeostasis and for the regulation of cellular proliferation, angiogenesis, apoptosis, and migration [7–10]. However, abnormal ROS regulation resulting in elevated intracellular ROS levels induces cellular stress and other molecular events causing damage to proteins, lipids, and DNA [11, 12]. The resulting molecular alterations and structural damage can trigger cellular senescence, a state of permanently arrested cellular division accompanied by the acquisition of a senescence-associated secretory phenotype that is characterized by the production of numerous senescence-associated factors and secreted molecules including proinflammatory chemokines, cytokines and growth factors [13–15].
The production of ROS is the primary function of the NADPH oxidase (NOX) family of proteins [16–18]. The NOX family consists of seven distinct NOX isoforms of membrane-associated enzymes that catalyze the reduction of O2 to generate ROS. NOX isoforms display remarkable differences in their tissue distribution [19, 20]. NADPH oxidase 4 (NOX4) is unique among the NOX family members because its activation is not dependent on the binding of other protein subunits but is determined by its expression levels [21]. Recently, the role of NOX-mediated generation of ROS in the pathogenesis of fibroproliferative disorders including idiopathic pulmonary fibrosis (IPF), liver fibrosis, renal fibrosis, and systemic sclerosis (SSc) has become an area of intense interest [22–24].
SSc is a systemic and often progressive autoimmune disorder of unknown etiology characterized by small vessel vasculopathy, numerous immunologic alterations including the appearance of autoantibodies, and the exaggerated synthesis and deposition of extracellular matrix macromolecules including the fibrillary collagens and fibronectin [25–29]. Although the most prominent manifestations of the disorder occur in the skin, visceral organs such as the lungs, kidneys, heart, and gastrointestinal tract are also frequently affected.
Transforming growth factor-β (TGF-β) causes marked increases in the expression and production of various molecules involved in tissue fibrotic processes [30, 31]. TGF-β signaling plays a crucial role in SSc pathogenesis [32–34] and increased TGF-β production and elevated expression of TGF-β-regulated genes, including the TGF-β receptor I (TRBI), have been demonstrated in affected tissues from SSc patients [35, 36]. TGF-β triggers the trans-differentiation of quiescent tissue fibroblasts into activated myofibroblasts, α-smooth muscle actin (α-SMA)-expressing cells responsible for the increased synthesis and exaggerated deposition of ECM macromolecules in the skin and affected organs in fibroproliferative disorders [37–40]. Although myofibroblast transdifferentiation was considered a terminal differentiation process [41], several recent studies have suggested that this process may be partially or totally reversible [42–44]. Recently, lineage tracing studies have demonstrated that mesenchymal cells are a heterogeneous population comprising a spectrum of subpopulations with varying phenotypes, marker expression, and tissue distribution. Indeed, a recent study that examined mesenchymal cell phenotypes present in the normal and fibrotic lungs of mice identified six distinct subpopulations in normal lungs and seven distinct subpopulations in fibrotic lungs [45]. In agreement with these observations, other studies have shown that during the wound healing process, some populations of fibroblasts display certain features characteristic of activated myofibroblasts, such as increased synthesis of ECM macromolecules but remarkably, lack other important features such as α-SMA expression [46, 47].
There has been strong recent interest in the study of the interactions of TGF-β with ROS in the development of fibrotic pathologies. These studies have shown that TGF-β signaling can generate a redox imbalance by increasing ROS production and decreasing antioxidant enzyme levels [48–50]. On the other hand, ROS can induce TGF-β signaling and regulate TGF-β-mediated fibrosis [48–51]. Intriguingly, several growth factors important in the induction and progression of pathologic tissue fibrosis including TGF-β, platelet-derived growth factor (PDGF), and endothelin induce and stimulate the expression of NOX4 [52–55]. Fibroblasts isolated from numerous organs including skin, lung, and heart produce NOX4 in vitro following stimulation with TGF-β, resulting in their activation and transdifferentiation into myofibroblasts [56–59]. Upregulation of NOX4 expression has also been reported in various fibrotic processes including patients with SSc [53, 60–67]. One study showed that NOX4 expression is upregulated in the lungs of IPF patients and siRNA-mediated knockdown of NOX4 transcript levels ameliorates TGF-β1-induced collagen expression and deposition in the lungs of mice in response to bleomycin injury [61]. Several other studies have investigated the role of NOX4 in various forms of tissue fibrosis. One study showed that small molecule inhibition of NOX4 activity blocked bleomycin-induced pulmonary fibrosis [62] and Nox4 knockout mice display attenuated fibrosis following bleomycin treatment [63]. NOX4 has been reported to mediate TGF-β-induced kidney myofibroblast activation [64] and NOX4-mediated ROS production activates profibrotic gene expression and matrix deposition in diabetic nephropathy [65]. NOX4 is upregulated in patients with hepatitis C-associated liver fibrosis and in patients with non-alcoholic steatohepatitis [66] and stimulation of hepatic stellate cells with TGF-β1 triggers NOX4-dependent ROS formation [67]. Furthermore, it has recently been shown that targeting NOX4/Nox4 expression in fibrotic human and murine dermal fibroblasts in vitro suppressed the pro-fibrotic phenotype of these cells and blocked bleomycin-induced fibrosis in vivo [68]. In agreement with these observations it has recently been reported that a selective inhibitor of NOX1/NOX4 abrogates the profibrotic gene expression and protein production in dermal fibroblasts isolated from lesional SSc skin biopsies [69].
Here, we examined the role of NOX4 in TGF-β-induced dermal and pulmonary fibrosis in vivo by assessing the ability of TGF-β administered employing subdermal osmotic pumps to induce dermal and pulmonary fibrosis in mice carrying a genetic deletion of the Nox4 allele. We report that genetic deletion of NOX4 in mice abrogated the ability of TGF-β to induce skin and lung tissue fibrosis in vivo, confirming the role for NOX4 as an important mediator of the profibrotic effects of TGF-β and highlighting the potential of NOX4 inhibition as a novel therapeutic strategy for SSc and other fibroproliferative disorders.
Materials and methods
Induction of TGF-β-mediated skin and pulmonary fibrosis in mice employing subcutaneously implanted osmotic pumps
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of Thomas Jefferson University (Protocol Number 01629). All surgery was performed under ketamine/xylazine anesthesia. B6.129-Nox4tm1Kkr/J mice (Nox4 knockout mice) have a neomycin cassette in place of exon four of the endogenous Nox4 gene resulting in abolition of Nox4 expression and NOX4 production [63]. B6.129-Nox4tm1Kkr/J and C57BL6/J normal control mice were purchased from The Jackson Laboratory and maintained as breeding colonies. Eight-week-old normal male and female C57BL6/J and Nox4 knockout mice were anesthetized and implanted subcutaneously in the inter-scapular region with Alzet osmotic pumps containing either saline or 2.5 μg TGF-β1. Five male and six female C57BL6/J and two male and two female Nox4 knockout mice were implanted with saline-containing pumps and four male and female animals of each strain were implanted with TGF-β-containing pumps. Mice were sacrificed 28 days post-implantation.
Histopathologic tissue analysis
Following sacrifice full thickness skin samples were excised from the dorsum of each mouse and isolation of both lungs was performed as previously described [70]. A portion of the tissue samples was fixed in 10% buffered formalin overnight and then stored in 70% ethanol. Subsequently, the fixed tissues were embedded in paraffin, sectioned (5 μm thickness) and stained with hematoxylin and eosin (H&E) or with Masson’s trichrome. Dermal thickness was measured in H&E-stained sections viewed under ×40 microscopic examination by measuring the distance between the epidermal–dermal junction and the dermal–adipose layer junction at five randomly selected fields from one sample from each animal.
Analysis of endothelial-to-mesenchymal transition (EndoMT) in lung tissues
Paraffin-embedded sections (5 μm thickness) of lungs from control and TGF-β-treated animals of each strain were deparaffinized and dehydrated following antigen retrieval with a citric acid buffer, as described previously [70]. Slides were first incubated with blocking IgG solution for 1 h, and then overnight with anti-α-SMA (Abcam; 1:100 dilution) and anti-von Willebrand (anti-vWF; Dako; 1:50 dilution) antibodies. IgG binding was revealed following incubation with an F(ab′) sheep anti-rabbit Cy3 antibody and an F(ab′) sheep anti-mouse fluorescein isothiocyanate-conjugated antibody (Sigma) for 1 h. Nuclei were counterstained with DAPI (Jackson ImmunoResearch). Samples were examined with a Zeiss 51 confocal laser microscope to evaluate the colocalization of immunoreactivity.
Analysis of oxidative stress-induced DNA damage in lung tissues
Paraffin-embedded sections (5 μm thickness) of lungs from control and TGF-β-treated animals of each strain were deparaffinized as described previously [70]. Slides were first incubated with blocking IgG solution for 1 h, and then overnight with anti-8-oxo-dG antibody (Abcam; 1:100 dilution). IgG binding was revealed following incubation with an F(ab′) sheep anti-mouse fluorescein isothiocyanate–conjugated antibody (Sigma) for 1 h.
Determination of tissue hydroxyproline content
A portion of the skin and lung samples isolated from each animal was weighed and acid-hydrolyzed overnight in 6 N HCl at 107 °C and assayed for their total hydroxyproline content as described [71]. The total content of hydroxyproline in the hydrolyzed samples was determined from a standard curve prepared by dissolving chemically pure hydroxyproline in distilled H2O and was expressed as μg hydroxyproline per mg of tissue wet weight.
RNA isolation and real-time polymerase chain reaction (PCR)
Total RNA was extracted from a portion of skin and lung samples using Trizol and was reverse-transcribed using SuperScript II Reverse Transcriptase (Invitrogen) to generate first-strand cDNA. Transcript levels of genes associated with fibrosis and with myofibroblast differentiation and activation, and of genes encoding downstream TGF-β transcription factors were determined using SYBR Green real-time PCR as previously described [70]. Primers were designed using Primer Quest (Integrated DNA Technologies, Coralville, IA) and were validated for specificity. The sequence of the primers employed is shown in Table 1. The differences in the number of mRNA copies in each PCR were corrected for 18S RNA endogenous control transcript levels; levels in control mice were set at 100% and all other values were expressed as normalized multiples of the control values.
Table 1.
Primers employed for quantitative real-time PCR
| Gene | Forward primer (5′−3′) | Reverse primer (5′−3′) |
|---|---|---|
| 18S | ACCAGAGCGGAAAGCATTTGCCA | TCGGCATCGTTTATGGTCGGAA |
| Acta2 | GACTCTCTTCCAGCCATCTTTC | GACAGGACGTTGTTAGCATAGA |
| Col1a1 | GCATGGCCAAGAAGACATCG | TCCACGTCTCAGCATTGGG |
| Col3a1 | AGCTTTGTGCAAAGTGGAACCTGG | CAAGGTGGCTGCATCCCAATTCAT |
| Col4a1 | CTCACTGTGGATCGGCTATTC | CGCTTCTAAACTCTTCCAGACA |
| Comp | GGAGCTGGTGGAGAAGAATATC | AGGAACTGTCTGCTACCTTTG |
| Fn1 | TCCAGGACAACAGCATCAGTGTCA | CCACAGTGGGTTGCAAACCTTCAA |
| Fn-Eda | TAAAGGACTGGCATTCACTGA | GTGCAAGGCAACCACACTGAC |
| Mrtfa | CGTGGGCTGGAAGGATAAA | TACTAGCTCAGGACCCTCATAG |
| Postn | GGTGTCCTAGAAAGGATCATGG | CAGAGCACTGGAGGGTATTTAG |
| Snai1 | TTGTGTCTGCACGACCTGTGGAAA | TCTTCACATCCGAGTGGGTTTTGCA |
| Snai2 | AAAGGCCACTGGGTAAAGGAGAGT | AAAGGCCACTGGGTAAAGGAGAGT |
| Twist | ATGTCCGCGTCCCACTA | CCAGAGTCTCTAGACTGTCCAT |
Statistical analysis
Values reflect the mean ± SD from skin or lung isolated from five male and six female saline-treated C57BL6/J or two male and two female Nox4 knockout mice and four male and female TGF-β1-treated C57BL6/J or Nox4 knockout mice performed in triplicate. The statistical significance of all the data was assessed by a two-tailed Student’s t-test. A p < 0.05 was considered statistically significant.
Results
Nox4 knockout mice are protected from TGF-β-induced dermal fibrosis
Histopathologic analysis of H&E-stained full thickness skin sections from four male C57BL6/J mice treated with TGF-β1 displayed a marked increase in dermal thickness compared to five male saline-treated C57BL6/J control mice (Fig. 1a, upper panel). Analysis of H&E-stained skin sections from four female mice treated with TGF-β1 exhibited comparable increases in dermal thickness compared to six female saline-treated control mice (Supplementary Figure 1A, upper panel). Sections stained with Masson’s trichrome demonstrated a striking accumulation of irregularly arranged, thickened collagen bundles in TGF-β1-treated C57BL6/J mice throughout the upper and lower dermis with decreased amounts of subcutaneous adipose tissue (Fig. 1a). Trichrome-stained sections from female mice showed similarly marked increases in thickened collagen bundle deposition but with little apparent change in subcutaneous adipose tissue (Supplementary Figure 1A, upper panel). Remarkably, no fibroproliferative response was noted in the TGF-β1-treated Nox4 mice of either sex, in contrast to the robust response induced by TGF-β1 in their C57BL6/J counterparts (Fig. 1a, lower panel; Supplementary Figure 1A, lower panel). The skin thickness quantification showed a 1.7-fold increase in skin thickness in TGF-β-treated C57BL6/J animals of both genders whereas no change was observed in the skin thickness of TGF-β-treated Nox4 knockout mice compared to saline-treated control Nox4 knockout mice (Fig. 1a, Supplementary Figure 1A). Male mice demonstrated greater baseline levels of collagen content levels in both the C57BL6/J and Nox4 knockout strain and male mice of the C57BL6/J strain displayed a more pronounced tissue fibrotic reaction in response to TGF-β1. These remarkable gender differences in extent and severity of tissue fibrotic reactions are most likely related to the effects of the androgen receptor [72].
Fig. 1.

a Histopathology of skin from male C57BL6/J and Nox4 knockout mice implanted with subcutaneous osmotic pumps containing either saline or TGF-β1. Representative sections of skin tissues isolated from male C57BL6/J control mice (left panels) and B6.129-Nox4tm1Kkr/J (Nox4 knockout) mice (right panels) implanted with subcutaneous pumps containing either saline (upper rows) or 2.5 μg TGF-β1 (lower rows) stained with H&E or Masson’s trichrome. TGF-β1-treated male C57BL6/J mice display increased dermal thickness and collagen accumulation compared to saline-treated controls. The induction of a fibrotic response following treatment with TGF-β1 was abrogated in skin samples isolated from Nox4 knockout mice. b Relative thickness of skin from male C57BL6/J and Nox4 knockout mice implanted with subcutaneous osmotic pumps containing either saline or TGF-β1. The shortest distance between the epidermal–dermal junction and the dermal–adipose layer junction was measured at five randomly selected fields from one sample from each animal. The results were normalized to the mean skin thickness in saline-treated C57BL6/J control mice and represent the mean relative thickness ± SD of five measurements per mouse in each experimental group
Nox4 knockout mice are protected from TGF-β1-induced pulmonary fibrosis
H&E staining of the lungs isolated from male TGF-β1-treated C57BL6/J mice displayed severe alterations in the alveolar structure with extensive thickening of the alveolar septae and pronounced parenchymal consolidation (Fig. 2, upper panel). H&E-stained sections from female mice displayed similar levels of structural changes (Supplementary Figure 2). Trichrome-stained sections showed increased accumulation of collagen in the interstitium and surrounding the small vessels and peribronchiolar regions in both sexes. In contrast, minimal changes in the alveolar architecture were observed in the lungs of TGF-β1-treated Nox4 knockout mice in H&E-stained sections and no increases in collagen deposition were evident in trichrome-stained lung sections (Fig. 2, lower panel; Supplementary Figure 2, lower panel).
Fig. 2.

Histopathology of lungs from male C57BL6/J and Nox4 knockout mice implanted with subcutaneous osmotic pumps containing either saline or TGF-β1. Representative sections of lungs isolated from male C57BL6/J control mice (left panels) and B6.129-Nox4tm1Kkr/J (Nox4 knockout) mice (right panels) implanted with subcutaneous pumps containing either saline (upper rows) or 2.5 μg TGF-β1 (lower rows) stained with H&E or Masson’s trichrome. TGF-β1-treated C57BL6/J mice display marked loss of normal alveolar morphology with tissue consolidation, thickening of alveolar septae and perivascular and interstitial collagen. The induction of a fibrotic response following treatment with TGF-β1 was abrogated in lung samples isolated from Nox4 knockout mice
Hydroxyproline content of skin and lungs of TGF-β1-treated and control C57BL6/J and Nox4 knockout mice
Samples of skin and lungs isolated from male and female saline-treated and TGF-β1-treated C57BL6/J and Nox4 knockout mice were assayed for hydroxyproline content to quantitatively analyze the extent of TGF-β1-mediated collagen deposition. The results are displayed in Fig. 3 and Supplementary Figure 3, respectively. Skin samples from TGF-β1-treated C57BL6/J mice showed a 1.7-fold increase in hydroxyproline levels. In contrast, there was no increase in the hydroxyproline content the skin of TGF-β1-treated Nox4 knockout mice (Fig. 3a, Supplementary Figure 3A). A similar pattern was observed in the hydroxyproline content of lungs from these animals. Lungs isolated from TGF-β-treated C57BL6/J mice exhibited a 2.4-fold increase in hydroxyproline levels. In contrast, lungs from TGF-β-treated Nox4 knockout mice showed no significant increase in the amount of hydroxyproline measured (Fig. 3b, Supplementary Figure 3B).
Fig. 3.

Hydroxyproline content of skin and lungs from male C57BL6/J and Nox4 knockout mice implanted with subcutaneous osmotic pumps containing either saline or TGF-β1. Hydroxyproline content of skin (a) and lung (b) tissues isolated from male control mice treated with either saline or with TGF-β1 and from Nox4 knockout mice receiving the same treatments. (N = 5 for saline-treated C57BL6/J mice and N = 2 for saline-treated Nox4 knockout mice of each strain; N = 4 for TGF-β1-treated groups.) Samples were hydrolyzed and analyzed for hydroxyproline content (expressed as μg/mg of tissue wet weight) in triplicate. Significance determined by Student’s two-tailed t-test. ***p < 0.001
TGF-β1-mediated increased expression of extracellular matrix genes associated with fibrosis is abrogated in Nox4 knockout mice
The expression of the genes encoding the fibrillar collagens Col1a1 and Col3a1 were upregulated in the skin by an average of 4.5 and 6.0 fold, respectively in male TGF-β1-treated C57BL6/J mice. In marked contrast, TGF-β1 treatment of male Nox4 knockout mice failed to induce increased expression of Col1a1 or Col3a1 transcripts in the skin samples (Fig. 4a). The assessment of gene expression in the lung tissues displayed a similar pattern. In the lungs of male C57BL6/J mice treated with TGF-β1, expression levels of Col1a1 were upregulated by 5.4 fold and those of Col3a1 were upregulated by 5.1 fold (Fig. 4b). In contrast, treatment of male Nox4 knockout mice with TGF-β1 did not significantly increase the expression of either gene in the lungs. Expression of Col4a1 increased by 2.3 fold in the skin and by 2.8 fold in the lungs of male TGF-β1-treated C57BL6/J mice, whereas treatment of male Nox4 knockout mice with TGF-β1 did not significantly affect the expression levels of Col4a1 in either the skin or lung of these mice. Total Fn1 expression in male C57BL6/J mice increased by 7.1 fold in the skin and by 6.6 fold in the lungs following TGF-β1-treatment whereas, as observed for the other extracellular matrix genes, treatment of male Nox4 knockout mice with TGF-β1 did not significantly increase the expression levels of Fn1 in either the skin or lung of these mice (Fig. 4a, b). Analysis of the expression of these genes in skin and lung tissues isolated from female mice displayed similar results to those observed in male mice with TGF-β1 inducing increased expression in C57BL6/J mice but not in Nox4 knockout female animals (Supplementary Figures 4A and 4B).
Fig. 4.

Expression of genes encoding extracellular matrix components in the skin and lungs of male saline-treated and TGF-β1-treated C57BL6/J and Nox4 knockout mice. Expression of Col1a1, Col3a1, Col4a1, and Fn in skin (a) and lung (b). The values shown are the mean (±SD) fold change levels of gene expression from each treatment group performed in triplicate for each tissue. Gene expression levels were normalized to 18S levels. Values for other samples are expressed relative to the saline-treated C57BL6/J control group which was arbitrarily set at the 100% expression level. Significance was determined by Student’s t-test. Statistical significance: ***p < 0.001
TGF-β1-induced fibroblast to myofibroblast transdifferentiation is abrogated in the skin and lungs of Nox4 knockout mice
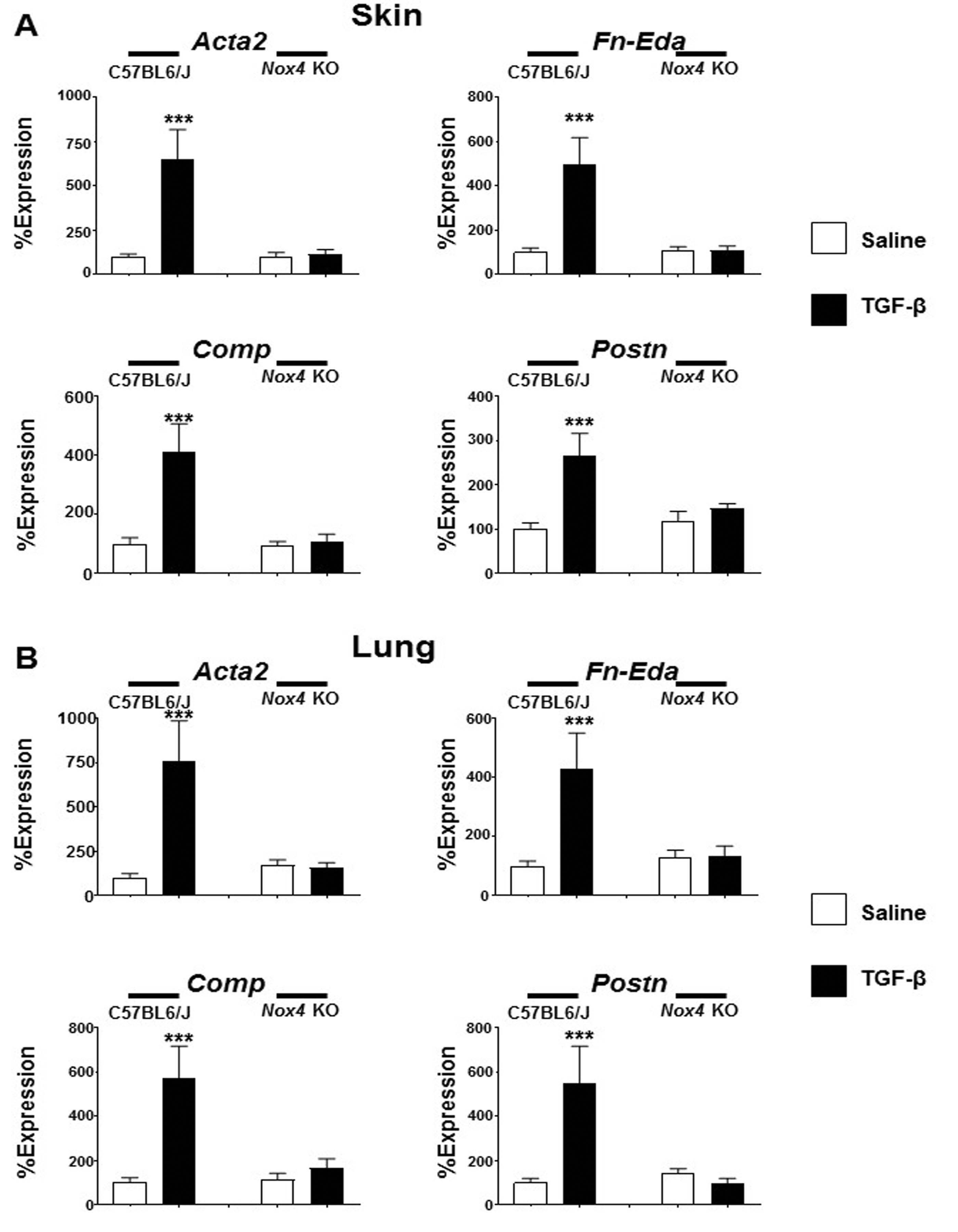
The expression of Acta2, the gene encoding the myofibroblast marker α-SMA was upregulated by an average of 8.8 fold in the skin of male TGF-β1-treated C57BL6/J mice (Fig. 5a) whereas in the lungs of male TGF-β1-treated C57BL6/J mice Acta2 was upregulated by 6.7 fold. In contrast, TGF-β1 treatment of male Nox4 knockout mice failed to induce any increase in Acta2 expression in either tissue (Fig. 5b). Expression levels of cartilage oligomeric matrix protein (Comp) increased by 2.7 fold in the skin and by 4.0 fold in the lungs of male TGF-β1-treated C57BL6/J mice. Treatment of male Nox4 knockout mice with TGF-β1 did not significantly increase the expression levels of Comp in either the skin or lung of these mice. Following TGF-β1-treatment, the expression levels of the EDA alternative splice variant of fibronectin (Fn-Eda) increased by 4.3 fold in the skin and by 6.8 fold in the lungs in male C57BL6/J mice. In contrast, TGF-β1 treatment of male Nox4 knockout mice failed to induce a significant change in the expression levels of Fn-Eda compared with the levels measured in saline-treated male Nox4 knockout mice (Fig. 5a, b). Finally, expression of periostin (Postn) in TGF-β-treated C57NL6/J male mice increased by 4.9 fold in the skin and 3.3 fold in the lungs compared to saline-treated control mice whereas no change in expression was measured in TGF-β-treated Nox4 knockout male mice compared to saline-treated control animals. Expression levels of these genes in the skin and lung isolated from female mice displayed a similar pattern of change to that observed in male mice with TGF-β1 inducing increased expression in C57BL6/J mice but not in Nox4 knockout female animals (Supplementary Figure 5A, B).
Fig. 5.

Expression of genes associated with myofibroblast differentiation in the skin and lungs of male saline-treated and TGF-β1-treated C57BL6/J and Nox4 knockout mice. Expression of Acta2, Fn-Eda, Comp, and Postn in skin (a) and lung (b). The values shown are the mean (±SD) fold change levels of gene expression from each treatment group performed in triplicate for each tissue. Significance was determined by Student’s t-test. Statistical significance: ***p < 0.001
TGF-β1-mediated increases in the expression of mesenchymal cell-specific profibrotic transcription factors are abrogated in Nox4 knockout mice
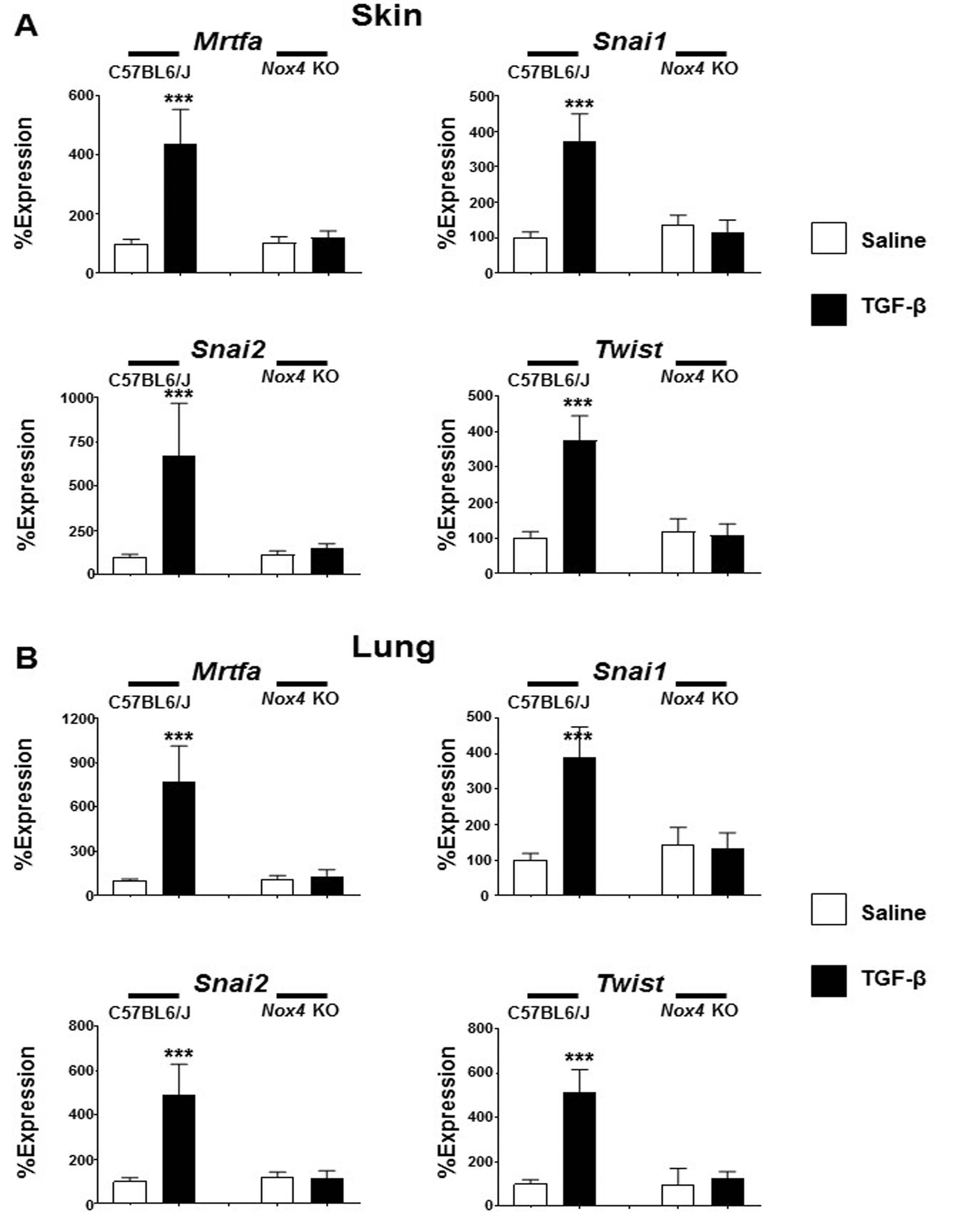
In the skin of male TGF-β1-treated C57BL6/J mice the expression levels of myocardin-related transcription factor A (Mrtfa) increased by 6.1 fold (Fig. 6a), whereas in the lungs of male TGF-β1-treated C57BL6/J mice Mrtfa increased by 4.7 fold (Fig. 6b). The Snail1 zinc finger protein transcription factor (Snai1) increased by 10.9 fold and 3.0 fold in the skin and lungs of male TGF-β-treated C57BL6/J mice, respectively, and expression levels of the Snail2 transcription factor (Snai2) increased by 3.3 fold in the skin of TGF-β1-treated male C57BL6/J mice and by 4.8 fold in the lungs of TGF-β1-treated C57BL6/J mice. Finally, expression levels of the Twist-related protein (Twist) transcription factor increased by 4.0 fold in the skin of male TGF-β1-treated C57BL6/J mice and by 6.9 fold in the lungs of male TGF-β1-treated C57BL6/J mice. In contrast to this TGF-β1-mediated increase in gene expression of relevant transcription factors involved in the fibro-blast to myofibroblast phenotypic transition observed in C57BL6/J mice, no significant changes in the expression of the genes encoding any of these factors was demonstrated in Nox4 knockout mice in response to treatment with TGF-β1 (Fig. 6a, b). The changes in gene expression in the skin and lung isolated from female mice were similar to those observed in male mice. In both male and female mice, TGF-β1-induced increased expression in C57BL6/J mice but not in Nox4 knockout female animals (Supplementary Figures 6A and 6B).
Fig. 6.

Expression of genes encoding profibrotic TGF-β-regulated transcription factors in the skin and lungs of male saline-treated and TGF-β1-treated C57BL6/J and Nox4 knockout mice. Expression of Mrtfa, Snai1, Snai2, and Twist from skin (a) and lung (b). The values shown are the mean (±SD) fold change levels of gene expression from each treatment group performed in triplicate for each tissue. Significance was determined by Student’s t-test. Statistical significance: ***p < 0.001
TGF-β1-mediated EndoMT is attenuated in Nox4 knockout animals
Immunofluorescence staining of paraffin-embedded sections of lung of saline-treated control C57BL6/J mice for α-SMA and vWF showed a clear cellular segregation in the expression of each of these proteins in the tissue with vWF staining confined to the vascular endothelial cell layer and the α-SMA-staining confined to sub-endothelial/medial smooth muscle cells, with no detectable overlap of expression (Fig. 7a). In TGF-β1-treated C57BL6/J mice, there was a striking increase in the number of α-SMA-positive cells, particularly in the subendothelial region and surrounding the vasculature of the small vessels and numerous vWF-positive cells simultaneously displayed the expression of α-SMA (Fig. 7b). In stark contrast, staining of lung sections isolated from saline-treated and TGF-β1-treated Nox4 knockout mice did not display an increased level of α-SMA and vWF colocalization in response to TGF-β1 (Fig. 7c, d).
Fig. 7.

Immunohistology of endothelial cells in the lungs of male saline-treated and TGF-β1-treated C57BL6/J and Nox4 knockout mice. Confocal microscopy staining for von Willebrand factor (red) and α-smooth muscle actin (green) in the lungs from male saline-treated C57BL6/J (a) or Nox4 knockout mice (c), or TGF-β-treated C57BL6/J (b) or Nox4 knockout mice (d). DAPI was used for counterstaining of nuclei. Magnification: ×20 (a); ×40 (b–d). Endothelial cells expressing vWF (red) are seen lining the large and small vessels of the lung. Activated myofibroblasts expressing α-SMA (green) are seen surrounding the vessels and in the interstitium. Cells co-staining for vWF and α-SMA (yellow; white arrows) in the small vessels representing cells in the process of endothelial-to-mesenchymal transition are observed in the TGF-β-treated C57BL6/J animals (b) whereas none are observed in saline-treated C57BL6/J animals. A single cell with co-staining for vWF and α-SMA was found in a similar section of lungs from saline-treated and TGF-β-treated Nox4 knockout mice, indicating that treatment with TGF-β failed to induce EndoMT in these mice (c and d)
TGF-β1-mediated increase in stress-induced DNA damage is abrogated in Nox4 knockout animals
Immunofluorescence staining of paraffin-embedded sections of lung of TGF-β1-treated C57BL6/J mice for the marker of oxidative stress-induced DNA damage 8-hydroxydeoxyguanosine (8-oxo-dG) showed a clear increase in the tissue compared with saline-treated control C57BL6/J lungs (compare Fig. 8b to Fig. 8a). In contrast, in tissue samples from TGF-β1-treated Nox4 knockout mice, there was no detectable increase in 8-oxo-dG-positive cells (Fig. 8d) compared with saline-treated Nox4 knockout animals.
Fig. 8.

Immunohistology of oxidative stress-induced DNA damage in the lungs of male saline-treate and TGF-β1-treated C57BL6/J and Nox4 knockout mice. Confocal microscopy staining for 8-hydroxydeoxy guanosine (green) in the lungs from male saline-treated C57BL6/J (a) or Nox4 knockout mice (c), or TGF-β-treated C57BL6/J (b) or Nox4 knockout mice (d). Magnification: ×20. Saline-treated male C57BL6/J (a) and Nox4 knockout animals show little or no staining for 8-oxo-dG (green) indicative of low levels of oxidative stress-induced DNA damage. Treatment with TGF-β dramatically increases cells positive for 8-oxo-dG in C57BL6/J mice (c) whereas no comparable increase is detected in the Nox4 knockout mouse lung (d)
Discussion
Disruption of the normal balance between the production of ROS and the antioxidant cellular response results in oxidative stress [16–18]. The NOX enzyme family are the major producers of ROS [19, 20]. At low concentrations, ROS serve as important signaling molecules involved in the maintenance and regulation of cellular proliferation, migration, and apoptosis. NOX-produced ROS are a primary source of oxidative stress that promotes crucial events in the development of skin fibrosis, IPF, liver fibrosis, and kidney fibrosis. Although several studies have examined the effect of Nox4 on bleomycin-induced fibrosis [61–63], no in vivo studies have analyzed the effect of NOX4 on TGF-β-mediated tissue fibrosis.
We report here the results from in vivo studies demonstrating that genetic deletion of the Nox4 gene in mice abrogates TGF-β1-mediated skin and lung fibrosis, as well as TGF-β-induced stimulation of expression of numerous genes associated with the fibrotic process. Treatment of C57BL6/J mice with TGF-β1 delivered continuously over 28 days in subcutaneous osmotic pumps induced a robust fibrotic response in the skin and lungs of these animals. Histopathologic analysis of the skin demonstrated a marked increase in dermal thickness accompanied by abnormally increased deposition of irregularly arrayed collagen fibers in the upper and lower dermis. In the lungs, TGF-β1 caused loss of normal tissue architecture with alveolar septae thickening and parenchymal consolidation with striking interstitial perivascular and peribronchial collagen deposition. In contrast, treatment of Nox4 knockout mice with TGF-β failed to produce significant changes in either tissue architecture or in levels of collagen deposition.
These observations were confirmed by measurement of the hydroxyproline content of the skin and lungs. The levels of hydroxyproline in the skin and lungs displayed a significant increase in TGF-β1-treated C57BL6/J mice compared to saline-treated mice of the same strain whereas hydroxyproline levels in the skin and lungs isolated from Nox4 knockout mice were unchanged in response to treatment with TGF-β1. Consistent with these analyses, expression levels of relevant profibrotic and fibrosis-associated genes in skin and lung samples demonstrated marked increases in the expression of genes encoding extracellular matrix macromolecules, such as Col1a1, Col3a1, Col4a1, and Fn1, as well as in genes regulating the differentiation and activation of quiescent fibroblast to profibrotic-activated myofibroblasts, such as Acta2, Comp, the fibrosis-associated Fn1 splice variant, Fn-Eda, and Postn. Expression of the TGF-β-induced gene periostin recently reported to promote liver fibrogenesis by stimulation of the matrix cross-linking protein lysyl oxidase [73] and the expression of several mesenchymal-specific transcription factors were upregulated in TGF-β1-treated C57BL6/J mice. One remarkable observation of our studies is that TGF-β1 treatment failed to increase the expression levels of any of these genes in Nox4 knockout mice.
These data confirm an important role for NOX4 in mediating the development of tissue fibrosis as demonstrated by the resistance of mice carrying a genetic deletion of Nox4 to the induction of tissue fibrosis. Furthermore, the attenuation of TGF-β1-mediated EndoMT in Nox4 knockout animals suggests a role for NOX4-mediated ROS production in this process. In support of these results are our observations that induction of constitutive TGF-β signaling in mouse microvascular endothelial cells induces EndoMT and tissue fibrosis in skin and several visceral organs including the lung [70]. Collectively, these results provide a mechanism for the previously reported ability of hypoxia to induce EndoMT in human coronary endothelial cells [74].
These data provide strong in vivo demonstration of the important role of NOX4 in mediating the profibrotic effects of TGF-β1 signaling, since the induction of skin and lung fibrosis observed and the increased expression of numerous profibrotic genes in C57BL6/J mice following treatment with TGF-β1 was not observed in Nox4 knockout mice. The results reported here emphasize the potential for inhibition of NOX4 activity to be employed as a novel therapeutic intervention for fibroproliferative diseases, such as SSc and other human systemic and organ-specific fibrotic diseases.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Supported by NIH grant AR 19616 to SAJ. We thank the expert assistance of Alana Pagano in the preparation of the manuscript.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41374-018-0161-1) contains supplementary material, which is available to authorized users.
References
- 1.Wingler K, Hermans JJ, Schiffers P, et al. NOX 1, 2, 4, 5: counting out oxidative stress. Br J Pharmacol. 2011;164:866–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy MP. Understanding and preventing mitochondrial oxidative damage. Biochem Soc Trans. 2016;44:1219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang P, Xie K, Wang C, et al. Oxidative stress induced by lipid peroxidation is related with inflammation of demyelination and neurodegeneration in multiple sclerosis. Eur Neurol. 2014;72: 249–54. [DOI] [PubMed] [Google Scholar]
- 4.Rochette L, Lorin J, Zeller M, et al. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: possible therapeutic targets? Pharmacol Ther. 2013;140:239–57. [DOI] [PubMed] [Google Scholar]
- 5.McNally JS, Davis ME, Giddons DP, et al. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol. 2003;285:H2290–H2297. [DOI] [PubMed] [Google Scholar]
- 6.Zangar RC, Davydov DR, Verma S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol Appl Pharmacol. 2004;199:316–31. [DOI] [PubMed] [Google Scholar]
- 7.D’Autreaux B, Toledano MB. ROS as signaling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–24. [DOI] [PubMed] [Google Scholar]
- 8.Valko M, Leibfritz D, Moncol J, et al. Free radicals and anti-oxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. [DOI] [PubMed] [Google Scholar]
- 9.Halliwell B The antioxidant paradox: less paradoxical now? Br J Clin Pharmacol. 2013;75:637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res. 2015;116:531–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–91. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki M, Kajiya H, Ozeki S, et al. Reactive oxygen species promotes cellular senescence in normal human epidermal keratinocytes through epigenetic regulation of p16(INK4a). Biochem Biophys Res Commun. 2014;452:622–8. [DOI] [PubMed] [Google Scholar]
- 14.Barascu A, Le Chalony C, Pennarun G. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J. 2012;31: 1080–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurundar A, Thannickal VJ. Redox mechanisms in age-related lung fibrosis. Redox Biol. 2016;9:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiszt M NADPH oxidases: new kids on the block. Cardiovasc Res. 2006;71:289–99. [DOI] [PubMed] [Google Scholar]
- 17.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. [DOI] [PubMed] [Google Scholar]
- 18.Brandes RP, Weissmann N, Schroder K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med. 2014;76:208–26. [DOI] [PubMed] [Google Scholar]
- 19.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–97. [DOI] [PubMed] [Google Scholar]
- 20.Cheng G, Cao Z, Xu X, et al. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001; 269:131–40. [DOI] [PubMed] [Google Scholar]
- 21.Serrander L, Cartier L, Bedard K, et al. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406:105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bargagli E, Olivieri C, Bennett D, et al. Oxidative stress in the pathogenesis of diffuse lung diseases: a review. Respir Med. 2009;103:1245–56. [DOI] [PubMed] [Google Scholar]
- 23.Richter K, Kietzmann T. Reactive oxygen species and fibrosis: further evidence of a significant liason. Cell Tissue Res. 2016;365: 591–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piera-Velazquez S, Jimenez SA. Role of cellular senescence and NOX4-mediated oxidative stress in Systemic Sclerosis pathogenesis. Curr Rheumatol Rep. 2015;17:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360:1989–2003. [DOI] [PubMed] [Google Scholar]
- 26.Allanore Y, Simms R, Distler O, et al. Systemic sclerosis. Nat Rev Dis Primers. 2015;1:15002. [DOI] [PubMed] [Google Scholar]
- 27.McCray CJ, Mayes MD. Update on systemic sclerosis. Curr Allergy Asthma Rep. 2015;15:25. [DOI] [PubMed] [Google Scholar]
- 28.Barsotti S, Stagnaro C, Della Rossa A. Systemic sclerosis: a critical digest of the recent literature. Clin Exp Rheumatol. 2015;33 (4 Suppl 91):S3–S14. [PubMed] [Google Scholar]
- 29.Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390: 1685–99. [DOI] [PubMed] [Google Scholar]
- 30.Sporn MB, Roberts AB, Wakefield LM, et al. Transforming growth factor-beta: biological function and chemical structure. Science. 1986;233:532–4. [DOI] [PubMed] [Google Scholar]
- 31.Varga J, Rosenbloom J, Jimenez SA. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem J. 1987; 247:597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lafyatis R Transforming growth factor β--at the centre of systemic sclerosis. Nat Rev Rheumatol. 2014;10:706–19. [DOI] [PubMed] [Google Scholar]
- 33.Varga J, Whitfield ML. Transforming growth factor-beta in systemic sclerosis (scleroderma). Front Biosci. 2009;1:226–35. [DOI] [PubMed] [Google Scholar]
- 34.Denton CP, Abraham DJ. Transforming growth factor-beta and connective tissue growth factor: key cytokines in scleroderma pathogenesis. Curr Opin Rheumatol. 2001;13:505–11. [DOI] [PubMed] [Google Scholar]
- 35.Kawakami T, Ihn H, Xu W, et al. Increased expression of TGF-beta receptors by scleroderma fibroblasts: evidence for contribution of autocrine TGF-beta signaling to scleroderma phenotype. J Invest Dermatol. 1998;110:47–51. [DOI] [PubMed] [Google Scholar]
- 36.Pannu J, Gore-Hyer E, Yamanaka M, et al. An increased transforming growth factor beta receptor type I:type II ratio contributes to elevated collagen protein synthesis that is resistant to inhibition via a kinase-deficient transforming growth factor beta receptor type II in scleroderma. Arthritis Rheum. 2004;50:1566–77. [DOI] [PubMed] [Google Scholar]
- 37.Hinz B, Phan SH, Thannickal VJ, et al. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. 2007;39: 666–71. [DOI] [PubMed] [Google Scholar]
- 39.Hinz B, Phan SH, Thannickal VJ, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kramann R, DiRocco DP, Humphreys BD. Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J Pathol. 2013;231: 273–89. [DOI] [PubMed] [Google Scholar]
- 41.Evans RA, Tian YC, Steadman R, et al. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation—the role of Smad proteins. Exp Cell Res. 2003;282:90–100. [DOI] [PubMed] [Google Scholar]
- 42.Garrison G, Huang SK, Okunishi K, et al. Reversal of myofibroblast differentiation by prostaglandin E(2). Am J Respir Cell Mol Biol. 2013;48:550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Driesen RB, Nagaraju CK, Abi-Char J, et al. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc Res. 2014;101:411–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desai VD, Hsia HC, Schwarzbauer JE. Reversible modulation of myofibroblast differentiation in adipose-derived mesenchymal stem cells. PLoS ONE. 2014;9:e86865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie T, Wang Y, Deng N, et al. Single-cell deconvolution of fibroblast heterogeneity in mouse pulmonary fibrosis. Cell Rep. 2018;22:3625–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu X, Khalil H, Kanisicak O, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. 2018;128:2127–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Macarak EJ, Lotto CE, Koganti D, et al. Trametinib prevents mesothelial-mesenchymal transition and ameliorates abdominal adhesion formation. J Surg Res. 2018;227:198–210. [DOI] [PubMed] [Google Scholar]
- 48.Thannickal VJ, Day RM, Klinz SG, et al. Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and TGF-beta 1. FASEB J. 2000;14: 1741–8. [DOI] [PubMed] [Google Scholar]
- 49.Kayanoki Y, Fujii J, Suzuki K, et al. Suppression of antioxidative enzyme expression by transforming growth factor-beta1 in rat hepatocytes. J Biol Chem. 1994;269:15488–92. [PubMed] [Google Scholar]
- 50.Liu R-M, Desai LP. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015;6:565–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rhyu DY, Yang Y, Ha H, et al. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16:667–75. [DOI] [PubMed] [Google Scholar]
- 52.Sturrock A, Cahill B, Norman K, et al. Transforming growth factor-beta 1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscles cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L661–L673. [DOI] [PubMed] [Google Scholar]
- 53.Piera-Velazquez S, Makul A, Jimenez SA. Increased expression of NADPH oxidase 4 in systemic sclerosis dermal fibroblasts: regulation by transforming growth factor β. Arthritis Rheumatol. 2015;67:2749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wagner B, Ricono JM, Gorin Y, et al. Mitogenic signaling via platelet-derived growth factor beta in metanephric mesenchymal cells. J Am Soc Nephrol. 2007;18:2903–11. [DOI] [PubMed] [Google Scholar]
- 55.Datla SR, Peshavariya H, Dusting GJ. Important role of Nox4 type NADPH oxidase in angiogenic responses in human micro-vascular endothelial cells in vitro. Arterioscler Thromb Vasc Biol. 2007;27:2319–24. [DOI] [PubMed] [Google Scholar]
- 56.Bondi CD, Manickham N, Lee DY, et al. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. 2010;21:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cucoranu I, Clempus R, Dikalova A, et al. NAD(P)H oxidase-4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97: 900–7. [DOI] [PubMed] [Google Scholar]
- 58.Ghatak S, Hascall VC, Markwald RR, et al. Transforming growth factor β1 (TGFβ1)-induced CD44v6-NOX4 signaling in pathogenesis of idiopathic pulmonary fibrosis. J Biol Chem. 2017;292: 10490–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alili L, Sack M, Puschmann L, et al. Fibroblast-to-myofibroblast switch is mediated by NAD(P)H oxidase generated reactive oxygen species. Biosci Rep 2014;34:pii e00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spadoni T, Svegliati Baroni S, Amico D, et al. A reactive oxygen species-mediated loop maintains increased expression of NADPH oxidases 2 and 4 in skin fibroblasts from patients with systemic sclerosis. Arthritis Rheumatol. 2015;67:1611–22. [DOI] [PubMed] [Google Scholar]
- 61.Hecker L, Vittal R, Jones T, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15:1077–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jarman ER, Khambata VS, Cope C, et al. An inhibitor of NADH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am J Respir Cell Mol Biol. 2014;50:158–69. [DOI] [PubMed] [Google Scholar]
- 63.Carnesecchi S, Deffert C, Donati Y, et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal. 2011;15:607–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manicakam N, Patel M, Griendling KK, et al. RhoA/Rho kinase mediates TGF-β1-induced kidney myofibroblast activation through Poldip/Nox4-derived reactive oxygen species. Am H Physiol Ren Physiol. 2014;307:F159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sedeek M, Callera G, Montezano A, et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Renal Physiol. 2010;299:F1348–58. [DOI] [PubMed] [Google Scholar]
- 66.Bettaieb A, Jiang JX, Sasaki Y, et al. Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology. 2015;149: 468–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Proell V, Carmona-Cuenca I, Murillo MM, et al. TGF-beta dependent regulation of oxygen radicals during transdifferentiation of activated hepatic stellate cells to myofibroblastoid cells. Comp Hepatol. 2007;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dosoki H, Stegemann A, Taha M, et al. Targeting of NADPH oxidase in vitro and in vivo suppresses fibroblast activation and experimental skin fibrosis. Exp Dermatol. 2017;26: 73–81. [DOI] [PubMed] [Google Scholar]
- 69.Murphy-Marshman H, Quensel K, Shi-Wen X. Antioxidants and NOX1/NOX4 inhibition blocks TGFβ1-induced CCN2 and α-SMA expression in dermal and gingival fibroblasts. PLoS ONE. 2017;12:e0186740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wermuth PJ, Carney KR, Mendoza FA, et al. Endothelial cell-specific activation of transforming growth factor-β signaling in mice induces cutaneous, visceral, and microvascular fibrosis. Lab Invest. 2017;97:806–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Del Galdo F, Sotgia F, de Almeida CJ, et al. Decreased expression of caveolin 1 in patients with systemic sclerosis: crucial role in the pathogenesis of tissue fibrosis. Arthritis Rheum. 2008;58: 2854–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Manne J, Markova M, Siracusa LD, et al. Collagen content in skin and internal organs of the tight skin mouse: an animal model of scleroderma. Biochem Res Int. 2013;2013:436053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kumar P, Smith T, Raeman R, et al. Periostin promotes liver fibrogenesis by activating lysyl oxidase in hepatic stellate cells. J Biol Chem. 2018;293:12781–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu X, Tan X, Tampe B, et al. Snail is a direct target of hypoxia-induced factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition in human coronary endothelial cells. J Biol Chem. 2015;290:16653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.