

Abstract
Purpose of review
Senescent cells have recently been identified as key players in the development of metabolic dysfunction. In this review, we will highlight recent developments in this field and discuss the concept of targeting these cells to prevent or treat cardiometabolic diseases.
Recent findings
Evidence is accumulating that cellular senescence contributes to adipose tissue dysfunction, presumably through induction of low-grade inflammation and inhibition of adipogenic differentiation leading to insulin resistance and dyslipidaemia. Senescent cells modulate their surroundings through their bioactive secretome and only a relatively small number of senescent cells is sufficient to cause persistent physical dysfunction even in young mice. Proof-of-principle studies showed that selective elimination of senescent cells can prevent or delay the development of cardiometabolic diseases in mice.
Summary
The metabolic consequences of senescent cell accumulation in various tissues are now unravelling and point to new therapeutic opportunities for the treatment of cardiometabolic diseases.
Keywords: cardiometabolic disease, cellular senescence, insulin resistance, metabolic syndrome, senolytics
INTRODUCTION
Cellular or replicative senescence is a protective response against endogenous and exogenous stressors in which cells permanently arrest their cell-cycle and undergo phenotypic alterations. The term senescence was initially introduced in 1961 by Hayflick and Moorhead [1] who observed that fibroblasts in culture were only able to divide a limited number of times before entering a state of permanent growth arrest. Compelling in-vivo evidence for this concept had long been lacking, until the discovery that cellular senescence can act as a potent cancer defence mechanism that prevents the proliferation of preneoplastic cells [2]. Furthermore, cellular senescence is a fundamental player in a range of physiological and pathophysiological processes, including wound healing, embryogenesis, ageing and the development of age-related diseases [3–10]. A variety of stimuli can induce cellular senescence, including telomere shortening (replicative senescence), oncogenic activity (oncogene-induced senescence) and stressors such as DNA damage, oxidative stress, inflammatory mediators and metabolites (stress-induced premature senescence) that induce growth arrest via activation of the p53-p21 and p16Ink4a/Rb tumour suppressor pathways [11–13]. Senescent cells display several hallmarks, including upregulation of the cyclin-dependent kinase inhibitor p16Ink4a, increased senescence-associated lysosomal β-galactosidase activity (SA-β-gal) and a characteristic secretome consisting of pro-inflammatory cytokines, chemokines, growth factors and proteases, the so-called senescence-associated secretory phenotype (SASP) [14–19]. The SASP is enriched for components that can attract immune cells and are thought to mediate natural elimination of senescent cells. However, during biological ageing, senescent cells accumulate within tissues, presumably due to increased cellular stress with ageing and deterioration of the immune system [20–26]. Remarkably, only a relatively small number of senescent cells seemingly is sufficient to drive tissue dysfunction throughout the body. Persistent SASP secretion may trigger senescence in distantly located cells [8,27,28,29▪▪], thereby perhaps promoting chronic tissue inflammation and degeneration. In this review, we delineate what is currently known about the role of senescent cells in metabolic and cardiovascular disorders.
Box 1.

no caption available
CELLULAR SENESCENCE AS A CAUSE OF METABOLIC DYSFUNCTION
Ageing is the major risk factor for the development of multiple chronic diseases and decline in physical functions. The first evidence that senescent cells are causally involved in ageing came from studies by Baker et al.[30,31], who examined the budding uninhibited by benomyl related-1 hypomorphic mice (BubR1H/H), which show accelerated ageing due to low levels of the core mitotic checkpoint protein BubR1 (Table 1) [30,31]. Inducible elimination of p16Ink4a-positive cells from BubR1H/H mice delayed the onset of age-related diseases such as sarcopenia, cataract and lipodystrophy (Table 1). Later studies in naturally aged mice confirmed this relationship and demonstrated that elimination of senescent cells extended life span [8]. Although senescent cells accumulate with ageing in multiple tissues, recent studies have raised the possibility that cellular senescence in adipose tissue may promote age-related diseases and frailty [34].
Table 1.
Clarification of the mouse models used to study the relationship between senescence and cardiometabolic diseases
| Mouse model | Description | Major findings | References |
| BubR1 hypomorphic mouse (BubR1H/H) | BubR1 is a core protein of the spindle assembly checkpoint, a safeguard that ensures correct chromosome segregation. BubR1 hypomorphic mice produce 10% of the BubR1 protein. | BubR1H/H is a model of accelerated ageing, as mice show markedly shortened lifespan and display several age-related diseases, including sarcopenia, cataracts, fat loss, arterial wall stiffening and impaired wound healing. BubR1H/H mice accumulate p16Ink4a-positive cells in several tissues, including adipose tissue, skeletal muscle and eye. | [30,31] |
| INK-ATTAC naturally aged mouse | The INK-ATTAC transgene allows for selective elimination of p16Ink4a-positive cells upon administration of the synthetic drug AP20187, which induces dimerization of a membrane-bound myristoylated FK506-binding-protein-caspase 8 (FKBP-Casp8) fusion protein expressed specifically in senescent cells via the p16Ink4a promoter. Furthermore, an internal ribosome entry site (IRES) followed by an open reading frame (ORF) coding for enhanced green fluorescence protein (EGFP), which allows detection and collection of p16Ink4a-positive senescent cells is incorporated in the construct. | Clearance of p16Ink4a-positive cells resulted in increased lifespan in male and female mice, delayed tumorigenesis and attenuated age-related diseases, including lipodystrophy, kidney dysfunction and cardiac dysfunction. Mechanistically, elimination of p16Ink4a-positive cells enhanced adipogenic transcription factors, reduced circulating levels of activin A and reduced fat accumulation in the liver of aged mice. | [8,32,33▪▪] |
| INK-ATTAC BubR1H/H mouse | Incorporation of the INK-ATTAC transgene in the progeroid BubR1H/H mouse. | Removal of p16Ink4a-positive cells in a model of accelerated ageing resulted in delayed onset of age-associated features, including sarcopenia, cataracts and lipodystrophy. | [30] |
Senescence as inducer of adipose tissue dysfunction
Adipose tissue is an active and dynamic endocrine organ that apart from its primary function to store energy in the form of fats also regulates systemic metabolism in response to nutrient intake, lifestyle and environmental changes [35]. With ageing, the distribution and function of adipose tissue changes significantly [35]. Old age is associated with a marked reduction in subcutaneous white adipose tissue (sWAT) and brown adipose tissue (BAT) and increased presence of visceral WAT (vWAT), accompanied by diminished lipid handling, altered secretion of adipokines, low-grade inflammation, defective thermogenesis and de-novo adipogenesis, which combined contributes to the development of insulin resistance and dyslipidaemia [35–39]. Senescent preadipocytes were shown to accumulate in BubR1 progeroid mice wherein they were first shown to cause lipodystrophy [31,40], a finding that was later confirmed in naturally aged mice [8]. Removal of senescent cells in mice resulted in a reduction of the pro-inflammatory SASP factor interleukin 6 (IL-6). Senescent cell accumulation can be accelerated in mice by excessive calorie intake and genomic instability [28,41]. In humans, obesity and diabetes is associated with senescent cell accumulation in adipose tissue, which correlated with adipose tissue dysfunction [28,35,41].
Senescent preadipocytes that cease to divide may limit the ability of the adipose tissue to expand, a process essential for storage of excess nutrients and to maintain metabolic health during obesity. During adipogenesis, preadipocytes can differentiate into insulin-responsive white adipocytes that store fats or into beige adipocytes that control thermogenesis by converting glucose and fats into heat [42,43]. The potential to form white and beige adipocytes declines with age [44–47]. Senescent adipocyte progenitors from fat pads of elderly human donors displayed markedly reduced levels of adipogenic transcription factors [peroxisome proliferator activated receptor gamma 2 (PPARγ2) and CCAAT/enhancer-binding protein alpha (C/EBPα)] and mature adipocyte markers (leptin, adiponectin, fatty acid-binding protein 4) as well as reduced adipogenic capacity of preadipocytes in culture [32,48]. Activin A, a member of the transforming growth factor beta superfamily and a critical inhibitor of proliferation and differentiation of preadipocytes, was identified as an important component of the senescent cell secretome and impaired adipogenesis in neighbouring, nonsenescent progenitors [32,49].
A study by Berry et al.[50▪] targeting the main regulators of cellular senescence p21 and p16Ink4a revealed that upregulation of p21 disrupted the potential of beige progenitors to differentiate into cold-induced beige adipocytes in mice. Both deletion and pharmacological inhibition of the p38/MAPK-p16Ink4a pathway were able to reverse this phenotype and resulted in improved glucose sensitivity [50▪]. Similarly, adipocyte-specific deletion of p53 or inhibition of p53 using pifithrin-α resulted in enhanced beige adipocyte formation upon cold exposure, increased energy expenditure and improved glucose clearance. Mechanistically, increased expression of p53 in aged adipose tissue appears to prevent adipocyte beiging through stimulation of mitophagy and prevention of increase in mitochondrial mass necessary for white-to-beige adipocyte conversion [51]. AP20187 treatment of naturally aged mice carrying the INK-ATTAC transgene, which allows for selective elimination of p16Ink4a-positive cells upon administration of AP20187 (Table 1), resulted in reduced circulating levels of activin A, enhanced expression of adipogenic transcription factors and reduced fat loss, although it should be noted that clearance of senescent cells was not demonstrated [32]. Nevertheless, these data are consistent with the idea that cellular senescence impairs adipogenesis, which can result in fatty acid spill over and ectopic lipid deposition in other organs promoting insulin resistance, nonalcoholic fatty liver disease (NAFLD) and atherosclerosis [52].
In addition to adipocytes, endothelial cells lining the microvasculature of the adipose tissue determine adipose tissue mass. Previous work by Kanda et al.[53] demonstrated a critical role for endothelial PPARγ in adipose tissue expansion in response to a high fat diet. Deletion of PPARγ from endothelial cells resulted in reduced adipose tissue mass and adipocyte size [53]. Accumulating evidence suggests that cellular senescence can affect the adipose tissue endothelial cells thereby impairing fatty acid handling and enhancing immune cell infiltration [54]. Interestingly, visceral adipose tissue depots isolated from obese individuals showed enhanced expression of pro-inflammatory mediators and senescence markers and reduced expression of metabolism-related genes as compared to subcutaneous adipose tissue [54,55]. Recently, Briot et al.[54] demonstrated that activation of PPARγ using its agonist rosiglitazone stimulated fatty acid uptake through expression of fatty acid transporters FATP1, FATP4 and CD36 in endothelial cells isolated from human adipose tissue. Remarkably, after induction of replicative senescence, activation of PPARγ by rosiglitazone promoted expression of pro-inflammatory mediators instead of fatty acid transporters [54]. The molecular events behind this surprising shift in PPARγ transcriptional activity remain to be elucidated. PPARγ was recently identified to act upstream of methyltransferase SETD8, which catalyses methylation of histone H4 at lysine 20 (H4K20me) and thereby silences expression of p16Ink4a and p21[56,57]. It remains to be investigated whether this mechanism also plays a role in endothelial cells.
Senescent cells that accumulate in adipose tissue during ageing, obesity and diabetes can disrupt the adipose tissue microenvironment via secretion of SASP components, thereby promoting adipose tissue inflammation and insulin resistance [35]. Recent studies highlight the murine double minute 2 (MDM2)-p53 axis as essential player in senescence-associated adipocyte dysfunction. Adipocyte-specific ablation of p53 in a mouse model of type 2 diabetes mellitus (T2DM) resulted in reduced senescent cell accumulation, reduced adipose tissue inflammation and improved insulin resistance. Conversely, p53 overexpression induced adipocyte senescence together with a pro-inflammatory environment causing impaired insulin sensitivity [41]. Ageing reduces the expression of Mdm2, an upstream inhibitor of p53 in WAT and BAT [58▪]. Adipocyte-specific deletion of Mdm2 resulted in age-dependent lipodystrophy caused by p53-dependent induction of apoptosis and senescence, which was associated with development of T2DM, NAFLD and hyperlipidaemia [58▪]. Inhibition of p53 attenuated senescence in adipose tissue, improved adipose function together with insulin sensitivity and glucose tolerance in a mouse model with elevated DNA damage due to Polh gene ablation [59]. Suppression of the janus kinase (JAK)-signal transducer and activator of transcription pathway, known for its role in regulating cytokine production, in aged mice reduced both adipose tissue and systemic inflammation, preserved fat mass, increased insulin sensitivity and reduced lipotoxicity [32]. Although these studies indicate that reduction of adipose inflammation resolves systemic dysfunction, they do not provide direct evidence for involvement of senescent cells in adipose tissue, as p53 or JAK inhibitors have pleiotropic effects on multiple tissues.
Very recently, adverse systemic effects of senescent adipose tissue cells were revealed via transplantation experiments of senescent adipocyte progenitors into fat tissue of healthy young mice. Senescent cells conferred senescence induction of host cells, not only locally but also in other tissues such as skeletal muscle, resulting in long-lasting physical dysfunction [29▪▪]. Transplantation of small numbers of senescent cells into aged or metabolically stressed mice resulted in more severe systemic dysfunction, accompanied with reduced survival of older recipients. Interestingly, replacement of senescent adipose tissue from adipocyte-specific Mdm2 knockout mice by healthy adipose tissue from wild-type mice largely reversed glucose intolerance, insulin resistance and hyperlipidaemia and partially reduced senescence markers in liver and skeletal muscle [58▪]. Collectively, these findings indicate adverse effects of senescent cells on adipose tissue function via induction of low-grade inflammation and insulin resistance, which could be accompanied by effects on distant tissues important for metabolic control possibly through secretion of SASP factors (Fig. 1).
FIGURE 1.
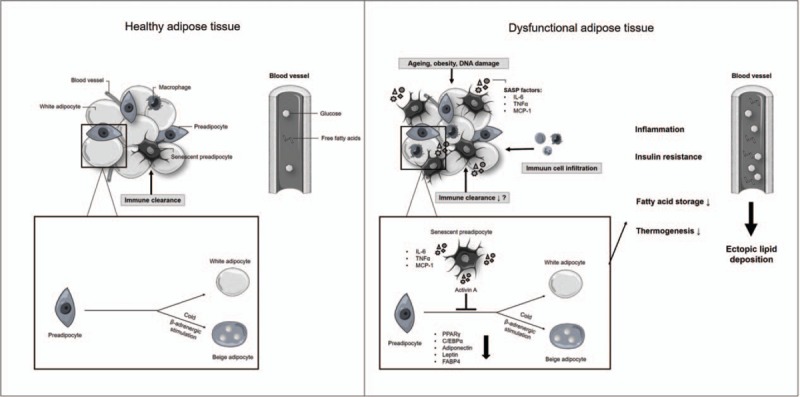
Potential mechanisms by which senescent cells contribute to adipose tissue dysfunction. Healthy adipose tissue is able to adapt to nutrient availability and environmental changes through adipogenesis providing metabolic flexibility. Senescent cell accumulation leads to increased secretion of SASP factors which can attract immune cells leading to low grade inflammation, insulin resistance and decreased formation of white and beige adipocytes. These changes can disturb systemic metabolic homeostasis. This figure was created using Servier Medical Art (http://smart.servier.com/).
THE ROLE OF CELLULAR SENESCENCE IN METABOLIC SYNDROME RELATED COMPLICATIONS
Accumulation of senescent cells in adipose tissue seems to play an important causal role in accelerated development of metabolic syndrome with age. Insulin resistance and dyslipidaemia are important inducers of metabolic diseases, such as T2DM, NAFLD and cardiovascular diseases. Here, we will discuss the latest research regarding the role of senescence in the pathophysiology of NAFLD and atherosclerosis.
Role of senescence in nonalcoholic fatty liver disease
Metabolic syndrome is a major risk factor for the development and progression of NAFLD, the most common liver disease worldwide and one of the most serious diseases associated with obesity with an estimated worldwide prevalence of 25% [60]. NAFLD is characterized by accumulation of excess fat within hepatocytes (steatosis). Although in itself relatively benign, it progresses in approximately 25% of all patients into nonalcoholic steatohepatitis (NASH), which can eventually develop into more serious conditions such as fibrosis, cirrhosis and hepatocellular carcinoma (HCC) [61]. Senescent cell accumulation has been reported in human livers, which correlated with T2DM, hepatic steatosis progression and fibrosis stage [22,62–64]. Excessive calorie intake in mice resulted in upregulation of senescence markers in hepatocytes, which was closely correlated with lipid deposition in the liver and was ameliorated by both dietary restriction and exercise [28,33▪▪,41,65,66]. A disturbed metabolic homeostasis can also trigger senescent cell accumulation associated with liver steatosis as seen in adipose-specific Mdm2-knockout mice [58▪] or muscle-specific mitochondrial fusion protein optic atrophy 1 (Opa1) deficient mice [67]. Induction of senescence in isolated primary hepatocytes promoted steatosis due to mitochondrial dysfunction followed by reduced fatty acid oxidation capacity [33▪▪]. One of the pathways involved in the development of age-associated hepatic steatosis might be the Cdk4-C/EBPα-p300 axis, as inhibition of cyclin dependent kinase 4 (Cdk4) reversed hepatic steatosis via reduction in C/EBPα-p300 complexes, resulting in reduction of senescent cells and alterations of chromatin structures in hepatocytes [68]. Importantly, senescent cells seem to contribute to NAFLD, as elimination of senescent cells, using the INK-ATTAC transgene reduced fat accumulation in the liver of aged, obese and diabetic mice [33▪▪] (Table 1) (Fig. 2).
FIGURE 2.
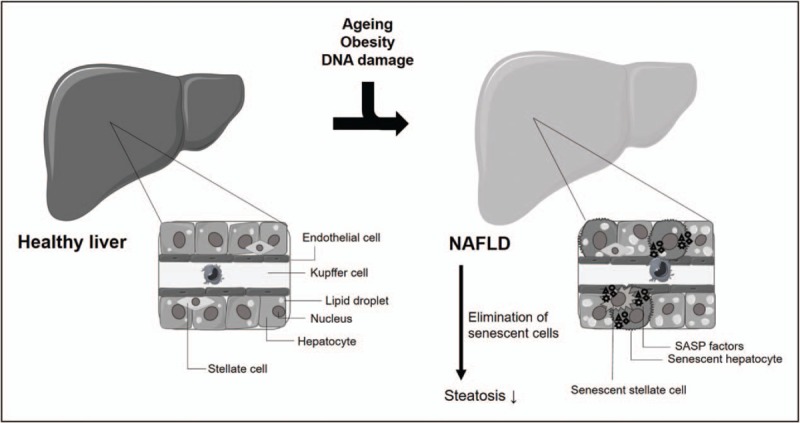
Potential role of senescent cells in the development of hepatic steatosis. Senescent hepatocytes induce lipid accumulation. Senescent stellate cells secrete SASP factors, which can trigger activation of immune cells such as Kupffer cells leading to NAFLD progression. This figure was created using Servier Medical Art (http://smart.servier.com/).
A key factor in the transition of NAFLD to NASH is the activation of innate immune cells, which initiates and amplifies hepatic inflammation. Senescent cells in the liver can promote inflammation by secretion of SASP factors. The innate immune-sensing mechanism cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING) are important SASP regulators [69▪–72▪]. Loss of the cGAS-STING pathway in senescent cells greatly compromises SASP factor secretion [69▪,71▪,72▪]. Consistent with this, NAFLD patients show increased expression of STING in nonparenchymal liver cells [73▪]. Interestingly, STING activation induces liver steatosis and inflammation [74▪] and STING deficiency attenuated steatosis, fibrosis and inflammation in murine models of NASH [73▪,74▪]. It should be noted that in those studies, the effects were ascribed to activated macrophages and Kupffer cells, while senescence was not addressed. Future studies are required to reveal whether the interplay between STING activity, SASP and hepatic lipid accumulation accelerates NAFLD development and progression.
Together, these studies suggest that senescent cells in the liver may contribute to the progression of NAFLD. However, senescence might also have beneficial roles in NAFLD under certain circumstances. In a toxin-induced liver damage model, senescence of activated stellate cells limited the progression of fibrosis [75]. In addition, the role of senescent cells and cGAS-STING in the development of HCC may be context-dependent. For example, in a mouse model of HCC with persistent overexpression of oncogenic Ras, deficiency of STING resulted in intrahepatic tumour formation due to loss of immune-mediated clearance of premalignant hepatocytes [69▪]. However, HCC development was diminished in the same Sting knockout mice under conditions of a single dose of carcinogen treatment followed by 30 weeks of high fat diet [70▪]. Therefore, it is possible that cGAS-STING activation and SASP from acutely generated senescent cells promote immune-surveillance, whereas long-term exposure to SASP factors of obesity-induced senescent hepatic stellate cells acts detrimental.
Role of cellular senescence in atherosclerosis
Ageing is accompanied by proatherogenic changes in the vasculature, including arterial stiffness, calcification and increased arterial permeability [76]. The presence of metabolic derangements, such as dyslipidaemia, initiates and accelerates atherosclerotic plaque formation. Interestingly, genome-wide association studies (GWAS) revealed that polymorphisms at the chromosome 9p21 locus are the most robust genetic markers for atherogenesis [77]. These associations were independent of established cardiovascular risk factors, such as blood lipid levels. Although studies have suggested that this locus owes its functional relevance to the long coding RNA ANRIL (antisense noncoding RNA in the INK4 locus) [78,79], it also encodes the cyclin-dependent kinase inhibitors and major regulators of senescence p16INK4A, p15INK4B and the p53 regulatory protein p14ARF. Vascular smooth muscle cells (VSMCs) and vascular endothelial cells derived from human atherosclerotic plaques display features of senescence, including SA-β-gal activity, increased expression of p16INK4A and p21 and hypophosphorylation of the retinoblastoma tumour suppressor protein Rb [80–83]. However, it remains unclear whether cellular senescence also contributes to atherosclerosis development. Mice deficient in p19Arf, p21 and p53 display accelerated atherosclerosis development and, although these cell cycle regulators are involved in many processes and findings of these studies were mainly attributed to effects on apoptosis [84–86], a role of cellular senescence in atherogenesis cannot be excluded. Whether cellular senescence of VSMCs is beneficial or adverse for plaque development is controversial. Gizard et al.[87] previously demonstrated protective effects of cellular senescence due to limiting proliferation and accumulation of VSMCs in the tunica intima. However, VSMC proliferation is protective in early and advanced atherosclerosis [88]. Wang et al.[89] reported that VSMC senescence promoted atherosclerosis development and features of plaque vulnerability. Mechanistically, senescent cells might promote plaque formation and vulnerability via pro-inflammatory SASP cytokines that could facilitate macrophage influx as well as matrix-degrading SASPs that could trigger plaque rupture [81,90]. The only conclusive evidence for a role of cellular senescence in atherosclerosis comes from a study by Childs et al.[91▪] that used both pharmacological and INK-ATTAC mediated clearance of senescent cells in atherosclerosis prone LDL receptor knockout mice and convincingly demonstrated that removal of p16Ink4a-positive foamy macrophages blocks lesion growth and promotes plaque remodelling associated with plaque stability. Overall, these findings demonstrate that the role of VSMC senescence in atherosclerotic plaque development is inconclusive, whereas accumulation of p16Ink4a-positive foamy macrophages might be detrimental for lesion progression and stability.
CONCLUSION
Cellular senescence has a causal role in adipose tissue dysfunction, presumably through induction of low-grade inflammation and inhibition of adipogenic differentiation resulting in insulin resistance, dyslipidaemia and ultimately development of cardiometabolic disease. Removal of senescent cells can potentially play an important role in treatment or prevention of these diseases. Current senolytic agents interfere with the pro-survival pathways, on which senescent cell survival depends. An alternative approach might be to target the secretion or activity of SASP factors, as these are likely to facilitate systemic tissue dysfunction.
Acknowledgements
None.
Financial support and sponsorship
J.K.K. was supported by grant 40-45900-98-152 from The Netherlands Organisation for Health Research and Development. J.W.J. was supported by grants from The Netherlands Organization for Scientific Research (VICI grant 016.176.640 to JWJ) and European Foundation for the Study of Diabetes (award supported by EFSD/Novo Nordisk). B.v.d.S was supported by the Noaber foundation. J.v.D. was supported by the Glenn Foundation for Medical Research and grants R01CA096985 (NIH) and R01AG057493 (NIH).
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25:585–621. [DOI] [PubMed] [Google Scholar]
- 2.Sieben CJ, Sturmlechner I, van de Sluis B, van Deursen JM. Two-step senescence-focused cancer therapies. Trends Cell Biol 2018; 28:723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muñoz-Espin D, Cañamero M, Maraver A, et al. Programmed cell senescence during mammalian embryonic development. Cell 2013; 155:1104–1118. [DOI] [PubMed] [Google Scholar]
- 4.Muñoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15:482–496. [DOI] [PubMed] [Google Scholar]
- 5.Storer M, Mas A, Robert-Moreno A, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013; 155:1119–1130. [DOI] [PubMed] [Google Scholar]
- 6.Childs BG, Durik M, Baker DJ, Van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 2015; 21:1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 2014; 31:722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016; 530:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeon OH, Kim C, Laberge RM, et al. Local clearance of senescent cells attenuates the development of posttraumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 2017; 23:775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bussian TJ, Aziz A, Meyer CF, et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018; 562:578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serrano M, Lin AW, McCurrach ME, et al. Oncogenic Ras provokes premature cell senescence associated with accumulation of p53 and p16Ink4a. Cell 1997; 88:593–602. [DOI] [PubMed] [Google Scholar]
- 12.Aguilera A, Garcia-Muse T. Causes of genome instability. Annu Rev Genet 2013; 47:1–32. [DOI] [PubMed] [Google Scholar]
- 13.Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010; 141:81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coppé JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008; 6:e301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodier F, Coppé JP, Patil K, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 2009; 11:973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodier F, Muñoz DP, Teachenor R, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci 2011; 124:68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuilman T, Michaloglou C, Vredeveld LCW, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008; 133:1019–1031. [DOI] [PubMed] [Google Scholar]
- 19.Acosta JC, O’Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008; 133:1006–1018. [DOI] [PubMed] [Google Scholar]
- 20.Herbig U, Ferreira M, Condel L, et al. Cellular senescence in aging primates. Science 2006; 311:1257–11257. [DOI] [PubMed] [Google Scholar]
- 21.Lawless C, Wang C, Jurk D, et al. Quantitative assessment of markers for cell senescence. Exp Gerontol 2010; 45:772–780. [DOI] [PubMed] [Google Scholar]
- 22.Wang C, Jurk D, Maddick M, et al. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009; 8:311–323. [DOI] [PubMed] [Google Scholar]
- 23.Krishnamurthy J, Torrice C, Ramsey MR, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest 2004; 114:1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16ink4a induces an age-dependent decline in islet regenerative potential. Nature 2006; 443:453–470. [DOI] [PubMed] [Google Scholar]
- 25.Jeyapalan JC, Ferreira M, Sedivy JM, et al. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev 2007; 128:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Deursen JM. The role of senescent cells in ageing. Nature 2014; 509:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acosta J, Banito A, Wuestefeld T, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013; 15:978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schafer MJ, White TA, Evans G, et al. Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes 2016; 65:1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29▪▪.Xu M, Pirtskhalava T, Farr JN, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med 2018; 24:1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, the driving force of senescent cells on age-related disease was addressed by transplanting a relative small number of senescent preadipocytes in young and old mice.
- 30.Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011; 479:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker DJ, Perez-Terzic C, Jin F, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol 2008; 10:825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu M, Palmer AK, Ding H, et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 2015; 4:e12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33▪▪.Ogrodnik M, Miwa S, Tchkonia T, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 2017; 8:15691. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that hepatic senescent cells can induce a fatty liver in mice. Furthermore, they show that markers of hepatocyte senescence correlate with the severity of NAFLD in patients.
- 34.Stout MB, Justice JN, Nicklas BJ, Kirkland JL. Physiological aging: links among adipose tissue dysfunction, diabetes, and frailty. Physiology 2017; 32:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tchkonia T, Morbeck DE, Von Zglinicki T, et al. Fat tissue, aging and cellular senescence. Aging Cell 2010; 9:667–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kyle UG, Genton L, Hans D, et al. Age-related differences in fat-free mass, skeletal muscle, body cell mass and fat mass between 18 and 94 years. Eur J Clin Nutr 2001; 55:663–672. [DOI] [PubMed] [Google Scholar]
- 37.Raguso CA, Kyle U, Kossovsky MP, et al. A 3-year longitudinal study on body composition changes in the elderly: role of physical exercise. Clin Nutr 2006; 25:573–580. [DOI] [PubMed] [Google Scholar]
- 38.Garg A, Agarwal AK. Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta 2009; 1791:507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuk JL, Saunders TJ, Davidson LE, et al. Age-related changes in total and regional fat distribution. Ageing Res Rev 2009; 8:339–348. [DOI] [PubMed] [Google Scholar]
- 40.Baker DJ, Weaver RL, van Deursen JM. p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep 2013; 3:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minamino T, Orimo M, Shimizu I, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med 2009; 15:1082–1087. [DOI] [PubMed] [Google Scholar]
- 42.Hamm JK, el Jack AK, Pilch PF, Farmer SR. Role of PPAR gamma in regulating adipocyte differentiation and insulin-responsive glucose uptake. Ann N Y Acad Sci 1999; 892:134–145. [DOI] [PubMed] [Google Scholar]
- 43.Wu J, Boström P, Sparks LM, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012; 150:366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogers NH, Landa A, Park S, Smith RG. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging Cell 2012; 11:1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoneshiro T, Aita S, Matsushita M, et al. Age-related decrease in cold-activated brown adipose tissue and accumulation of body fat in healthy humans. Obesity 2011; 19:1755–1760. [DOI] [PubMed] [Google Scholar]
- 46.Kirkland JL, Hollenberg CH, Gillon WS. Age, anatomic site, and the replication and differentiation of adipocyte precursors. Am J Physiol 1990; 258:C206–C210. [DOI] [PubMed] [Google Scholar]
- 47.Khanh VC, Zulkifli AF, Tokunaga C, et al. Aging impairs beige adipocyte differentiation of mesenchymal stem cells via the reduced expression of Sirtuin 1. Biochem Biophys Res Commun 2018; 500:682–690. [DOI] [PubMed] [Google Scholar]
- 48.Mitterberger MC, Lechner S, Mattesich M, Zwerschke W. Adipogenic differentiation is impaired in replicative senescent human subcutaneous adipose-derived stromal/progenitor cells. J Gerontol Ser A Biol Sci Med Sci 2014; 69:13–24. [DOI] [PubMed] [Google Scholar]
- 49.Zaragosi LE, Wdziekonski B, Villageois P, et al. Activin A plays a critical role in proliferation and differentiation of human adipose progenitors. Diabetes 2010; 59:2513–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50▪.Berry DC, Jiang Y, Arpke RW, et al. Cellular aging contributes to failure of cold-induced beige adipocyte formation in old mice and humans. Cell Metab 2017; 25:166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]; Adaptive thermogenesis is decreased with age. This study shows that senescent cells can block the potential to form cold-induced beige adipocytes.
- 51.Fu W, Liu Y, Sun C, Yin H. Transient p53 inhibition sensitizes aged white adipose tissue for beige adipocyte recruitment by blocking mitophagy. FASEB J 2019; 33:844–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Byrne CD, Targher G. Ectopic fat, insulin resistance, and nonalcoholic fatty liver disease. Arterioscler Thromb Vasc Biol 2014; 34:1155–1161. [DOI] [PubMed] [Google Scholar]
- 53.Kanda T, Brown JD, Orasanu G, et al. PPAR( in the endothelium regulates metabolic responses to high-fat diet in mice. J Clin Invest 2009; 119:110–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Briot A, Decaunes P, Volat F, et al. Senescence alters PPAR((peroxisome proliferator-activated receptor gamma)-dependent fatty acid handling in human adipose tissue microvascular endothelial cells and favors inflammation. Arterioscler Thromb Vasc Biol 2018; 38:1134–1146. [DOI] [PubMed] [Google Scholar]
- 55.Villaret A, Galitzky J, Decaunes P, et al. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes 2010; 59:2755–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shih CT, Chang YF, Chen YT, et al. The PPAR(-SETD8 axis constitutes an epigenetic, p53-independent checkpoint on p21-mediated cellular senescence. Aging Cell 2017; 16:797–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka H, Takebayashi SI, Sakamoto A, et al. The SETD8/PR-Set7 Methyltransferase functions as a barrier to prevent senescence-associated metabolic remodeling. Cell Rep 2017; 18:2148–2161. [DOI] [PubMed] [Google Scholar]
- 58▪.Liu Z, Jin L, Yang JK, et al. The dysfunctional MDM2-p53 axis in adipocytes contributes to aging-related metabolic complications by induction of lipodystrophy. Diabetes 2018; 67:2397–2409. [DOI] [PubMed] [Google Scholar]; This study looked into the role of the MDM2-p53 axis in the regulation of adipose tissue ageing using adipocyte-specific MDM2-deficient mice.
- 59.Chen YW, Harris RA, Hatahet Z, Chou K. Ablation of XP-V gene causes adipose tissue senescence and metabolic abnormalities. Proc Natl Acad Sci U S A 2015; 112:E4556–E4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2017; 15:11–20. [DOI] [PubMed] [Google Scholar]
- 61.Perumpail BJ, Khan MA, Yoo ER, et al. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol 2017; 23:8263–8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wiemann SU, Satyanarayana A, Tsahuridu M, et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J 2002; 16:935–942. [DOI] [PubMed] [Google Scholar]
- 63.Aravinthan A, Scarpini C, Tachtatzis P, et al. Hepatocyte senescence predicts progression in nonalcohol-related fatty liver disease. J Hepatol 2013; 58:549–556. [DOI] [PubMed] [Google Scholar]
- 64.Aravinthan A, Shannon N, Heaney J, et al. The senescent hepatocyte gene signature in chronic liver disease. Exp Gerontol 2014; 60:37–45. [DOI] [PubMed] [Google Scholar]
- 65.Zhang X, Zhou D, Strakovsky R, et al. Hepatic cellular senescence pathway genes are induced through histone modifications in a diet-induced obese rat model. Am J Physiol Gastrointest Liver Physiol 2012; 302:G558–G564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jurk D, Wilson C, Passos JF, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun 2014; 2:4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tezze C, Romanello V, Desbats MA, et al. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab 2017; 25:1374–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nguyen P, Valanejad L, Cast A, et al. Eliminaton of age-associated hepatic steatosis and correction of aging phenotype by inhibition of cdk4-C/EBP(-p300 axis. Cell Rep 2018; 24:1597–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69▪.Dou Z, Ghosh K, Vizioli MG, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017; 550:402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that the cytosolic DNA sensor cGAS and STING regulate senescence and the SASP phenotype. To investigate the role of SASP in vivo, using STING-deficient mice, this study showed that SASP are important for inflammation and immunosurveillance.
- 70▪.Takahashi A, Loo TM, Okada R, et al. Downregulation of cytoplasmic DNAses is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 2018; 9:1249. [DOI] [PMC free article] [PubMed] [Google Scholar]; Senescent cells affect their surrounding through their SASP phenotype. This study shows that cytoplasmic accumulation of nuclear DNA plays a key role in the onset of SASP. Cytoplasmic DNA is sensed by STING and loss of STING in mice decreases obesity-induced SASP and liver tumor formation.
- 71▪.Yang H, Wang H, Ren J, et al. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 2017; 114:E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that cGAS is essential for cellular senescence during spontaneous immortalization or in response to DNA-damaging agents and that deletion of cGAS abolished expression of SASP factors.
- 72▪.Glück S, Guey B, Gulen MF, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 2017; 19:1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that cGAS promotes the production of SASP factors via STING upon sensing of cytosolic chromatin fragments in senescent cells thereby promoting paracrine senescence.
- 73▪.Luo X, Li H, Ma L, et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology 2018; 155:1971–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that STING is increased in liver tissue from patients with NAFLD and used mice to determine the role of STING in NAFLD development and progression.
- 74▪.Yu Y, Liu Y, An W, et al. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest 2019; 129:546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors investigated the role of STING in the progression of NASH in mice.
- 75.Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell 2008; 134:657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Head T, Daunert S, Goldschmidt-Clermont PJ. The aging risk and atherosclerosis: a fresh look on arterial homeostasis. Front Genet 2017; 8:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arterioscler Thromb Vasc Biol 2012; 32:196–206. [DOI] [PubMed] [Google Scholar]
- 78.Holdt LM, Stahringer A, Sass K, et al. Circular noncoding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun 2016; 7:12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lo Sardo V, Chubukov P, Ferguson W, et al. Unveiling the role of the most impactful cardiovascular risk locus through haplotype editing. Cell 2018; 175:1796–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bennett MR, Macdonald K, Chan SW, et al. Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ Res 1998; 82:704–712. [DOI] [PubMed] [Google Scholar]
- 81.Minamino T, Yoshida T, Tateno K, et al. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation 2003; 108:2264–2269. [DOI] [PubMed] [Google Scholar]
- 82.Matthews C, Gorenne I, Scott S, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis. Circ Res 2006; 99:156–164. [DOI] [PubMed] [Google Scholar]
- 83.Gorenne I, Kavurma M, Scott S, Bennett M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res 2006; 72:9–17. [DOI] [PubMed] [Google Scholar]
- 84.Khanna AK. Enhanced susceptibility of cyclin kinase inhibitor p21 knockout mice to high fat diet induced atherosclerosis. J Biomed Sci 2009; 16:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mercer J, Figg N, Stoneman V, et al. Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ Res 2005; 96:667–674. [DOI] [PubMed] [Google Scholar]
- 86.González-Navarro H, Abu Nabah YN, Vinué Á, et al. p19ARF deficiency reduces macrophage and vascular smooth muscle cell apoptosis and aggravates atherosclerosis. J Am Coll Cardiol 2010; 55:2258–2268. [DOI] [PubMed] [Google Scholar]
- 87.Gizard F, Amant C, Barbier O, et al. PPAR alpha inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16Ink4a. J Clin Invest 2005; 115:3228–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res 2016; 118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang J, Uryga AK, Reinhold J, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation 2015; 132:1909–1919. [DOI] [PubMed] [Google Scholar]
- 90.Gardner SE, Humphry M, Bennett MR, Clarke MCH. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1(-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol 2015; 35:1963–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91▪.Childs BG, Baker DJ, Wijshake T, et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016; 354:472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using atherosclerotic-prone Ldlr knockout mice, the authors show that senescent intimal foam cells are key drivers of atheroma formation and maturation.