

ARID1A is important for genome folding, spatial partitioning of interphase chromosomes, and intermixing of small chromosomes.
Abstract
ARID1A, a subunit of the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin-remodeling complex, localizes to both promoters and enhancers to influence transcription. However, the role of ARID1A in higher-order spatial chromosome partitioning and genome organization is unknown. Here, we show that ARID1A spatially partitions interphase chromosomes and regulates higher-order genome organization. The SWI/SNF complex interacts with condensin II, and they display significant colocalizations at enhancers. ARID1A knockout drives the redistribution of condensin II preferentially at enhancers, which positively correlates with changes in transcription. ARID1A and condensin II contribute to transcriptionally inactive B-compartment formation, while ARID1A weakens the border strength of topologically associated domains. Condensin II redistribution induced by ARID1A knockout positively correlates with chromosome sizes, which negatively correlates with interchromosomal interactions. ARID1A loss increases the trans interactions of small chromosomes, which was validated by three-dimensional interphase chromosome painting. These results demonstrate that ARID1A is important for large-scale genome folding and spatially partitions interphase chromosomes.
INTRODUCTION
ARID1A is a subunit of the SWI/SNF chromatin-remodeling complex (1, 2). ARID1A is among the most frequently mutated genes across human cancers (1, 3). For example, ARID1A is mutated in up to 62% of ovarian clear cell carcinoma (OCCC) (4, 5). More than 90% of ARID1A mutations in OCCC leads to loss of protein expression (4, 5). Although ARID1A localizes to both promoters and enhancers, there is evidence suggesting that enhancer-mediated gene regulation plays a dominant role in its tumor suppressor function (1, 6–8).
Genome topological mapping reveals that interphase chromosomes are spatially organized by a complex collection of chromosome interactions (9, 10). For example, enhancers, in particular, require chromatin looping to regulate transcription. Interphase chromosomes alternate between euchromatic, actively transcribed A-compartments and heterochromatic, transcriptionally inactive B-compartments (9–13). On a finer scale, topologically associated domains (TADs) are discrete intrachromosomal units of chromatin that facilitate intra-TAD interactions between regulatory elements and promoters but insulate inter-TAD interactions (9–13). Although ARID1A’s tumor suppressor function is linked to enhancer-mediated gene regulation (6) and chromosome organization is thought to coordinate gene transcription (14), the role of ARID1A in higher-order spatial genome organization is unknown.
Condensin is a multisubunit complex that belongs to the family of structural maintenance of chromosome (SMC) complexes (15). Condensin regulates chromosome structure in a wide range of processes, including chromosome segregation and gene regulation (15, 16). The main molecular activity of condensin is to form chromosomal loops through their conserved SMC ring structure (15). Mammalian cells contain condensin I and II complexes, which include unique rate-limiting NCAPH and NCAPH2 subunits, respectively (17). Condensin II, but not condensin I, plays a role in interphase genome organization (15). For example, in Drosophila melanogaster, condensin II promotes axial compaction of chromosomes to form distinct chromosomal territories (18–21). In the present study, we demonstrate that ARID1A spatially partitions interphase chromosomes, which correlates with ARID1A-regulated distribution of condensin II, preferentially at enhancers.
RESULTS
SWI/SNF complex interacts with condensin II
To identify new interacting proteins of the SWI/SNF complex, we purified the SWI/SNF complex by pulling down the essential core subunit BAF155 in both ARID1A wild-type OVCAR429 and ARID1A-mutated TOV21G OCCC cells. In addition, we pulled down endogenously FLAG-tagged ARID1A generated by CRISPR from ARID1A wild-type RMG1 OCCC cells. Liquid chromatography–tandem mass spectrometry (LC-MS/MS) of the pulldown revealed the presence of SMC core subunits SMC2 and SMC4 in all three analyses (fig. S1A). Co-immunoprecipitation (IP) analysis by pulling down FLAG-ARID1A showed that ARID1A interacted with condensin II regulatory subunit NCAPH2 but not with condensin I regulatory subunit NCAPH (Fig. 1A). Notably, ARID1A knockout does not affect expression of condensin II subunits such as NCAPH2 and NCAPD3 (Fig. 1B). The interaction between subunits of the SWI/SNF and condensin II complexes such as BAF155 and NCAPH2 is DNA independent, because the deoxyribonuclease I (DNase I) digest did not affect the interaction (Fig. 1C).
Fig. 1. The SWI/SNF complex interacts with the condensin II complex.
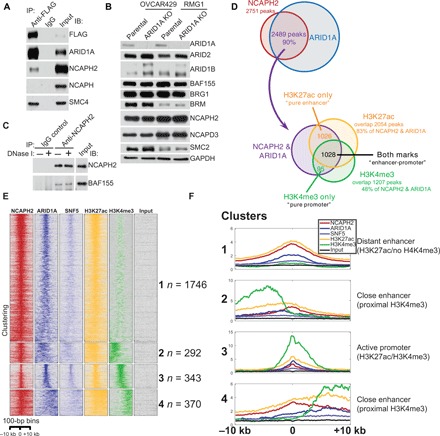
(A) Validation of the interaction between the SWI/SNF and condensin II complexes by co-IP in RMG1 cells with endogenously FLAG-tagged ARID1A. Nuclear fractions were subjected to IP using an anti-FLAG antibody and immunoblotting using the indicated antibodies. (B) Expression of the indicated SWI/SNF and condensin II subunits in ARID1A wild-type OVCAR429 and RMG1 cells with or without ARID1A knockout determined by immunoblotting. (C) Co-IP analysis between NCAPH2 and BAF155 using an anti-NCAPH2 antibody from nuclear extracts prepared from RMG1 cells treated with or without DNase I. (D) Venn diagram showing the overlap of ChIP-seq peaks among NCAPH2, ARID1A, H3K27ac, and H3K4me3 in RMG1 cells. (E) Heatmap clustering of ChIP-seq profiles of NCAPH2, ARID1A, SNF5, H3K27ac, and H3K4me3 in RMG1 cells. The number of binding sites in each of the four clusters is indicated. (F) Average profiles of the ChIP-seq signal for the indicated antibodies within four distinct clusters found by k-means clustering. Immunoglobulin G, IgG.
To explore the genome-wide distribution of ARID1A and NCAPH2, we performed chromatin IP followed by next-generation sequencing (ChIP-seq) analysis for NCAPH2, ARID1A, core SWI/SNF subunit SNF5, enhancer epigenetic mark H3K27ac, and promoter epigenetic mark H3K4me3 in ARID1A wild-type RMG1 cells. The analysis revealed that ~90% of NCAPH2 peaks overlap with ARID1A peaks (Fig. 1D and fig. S1, B and C). Eighty three percent of NCAPH2/ARID1A peaks overlap with H3K27ac peaks, while 48% of them overlap with H3K4me3 peaks (Fig. 1D). Notably, only ~4% of NCAPH2/ARID1A peaks overlap with H3K4me3 peaks without H3K27Ac (Fig. 1D). This was further confirmed by the unbiased clustering of NCAPH2 binding sites, which revealed that NCAPH2 and ARID1A colocalize at distant and proximal enhancers and at enhancers colocalized promoters (Fig. 1, E and F). Therefore, we conclude that NCAPH2 and ARID1A show significant overlap on enhancers marked by H3K27ac in ARID1A wild-type cells.
ARID1A loss does not affect the interaction between SWI/SNF complex and condensin II
Next, we sought to determine whether or not ARID1A affects the interaction between the SWI/SNF and condensin II complexes. Toward this goal, we performed a co-IP analysis for the essential SWI/SNF core subunit BAF155 and the condensin II regulatory subunit NCAPH2 in ARID1A wild-type and the matched isogenic ARID1A knockout RMG1 cells. ARID1A knockout did not affect the interaction between the SWI/SNF and condensin II complexes (Fig. 2A and fig. S2A). Similar results were also obtained in ARID1A wild-type and the matched isogenic ARID1A knockout OVCAR429 cells (fig. S2, B and C). In addition, based on glycerol gradient analysis (22, 23), compared with ARID1A wild-type RMG1 controls, ARID1A knockout did not affect the cosegregation of the condensin II and SWI/SNF complexes (Fig. 2B). Similar observations were also made using sucrose gradient analysis and in both RMG1 and OVCAR429 ARID1A wild-type cells (fig. S2, D and E). Consistently, the SWI/SNF and condensin II complexes showed the same segregation pattern in ARID1A-mutated TOV21G cells as those in ARID1A wild-type cells (Fig. 2C). Together, we conclude that ARID1A status does not affect expression of condensin II complex subunits or the interaction between condensin II and SWI/SNF complexes.
Fig. 2. ARID1A loss does not affect the interaction between the SWI/SNF complex and condensin II.

(A) Parental control and ARID1A knockout (KO) RMG1 cells were subjected to co-IP analysis using an antibody against a core SWI/SNF subunit BAF155 and examined for interactions with the indicated subunits of the SWI/SNF and condensin I and II complexes by immunoblotting. Asterisks indicate the nonspecific bands in the immunoblotting. (B) Glycerol sedimentation (10 to 30%) assay of the SWI/SNF and condensin II complexes from parental control and ARID1A knockout RMG1 cells. The red box indicates the cosegregation of the SWI/SNF and condensin II complexes. (C) Sucrose sedimentation (10 to 50%) assay of the SWI/SNF and condensin II complexes in ARID1A-mutated TOV21G OCCC cells. The red box indicates the cofractionation of SWI/SNF and condensin II complexes.
Condensin II redistribution at enhancers driven by ARID1A loss correlates with changes in gene expression
We next determined whether ARID1A affects condensin II genome-wide distribution by ChIP-seq analysis of NCAPH2 in ARID1A wild-type and knockout RMG1 cells. Notably, ARID1A knockout does not affect cell proliferation (24, 25). An unbiased subclassification of all NCAPH2 binding sites in wild-type and ARID1A knockout cells revealed that there was an overall decrease of NCAPH2 binding upon ARID1A knockout (51% of the binding sites) (Fig. 3, A and B). ARID1A knockout increased NCAPH2 binding on 12% of the existing or de novo NCAPH2 binding sites (Fig. 3, A and B). We performed function and pathway enrichment analysis for the genes whose NCAPH2 occupancy changed at least twofold in ARID1A knockout compared to wild-type RMG1 cells. While there was no significant enrichment among genes that showed increased NCAPH2 occupancy, the genes with decreased NCAPH2 occupancy showed significant enrichments for a number of functions and pathways, including angiogenesis and the adenosine 5′ monophosphate-activated protein kinase (AMPK) signaling pathway (fig. S3A).
Fig. 3. ARID1A loss redistributes the condensin II complex preferentially at enhancers.
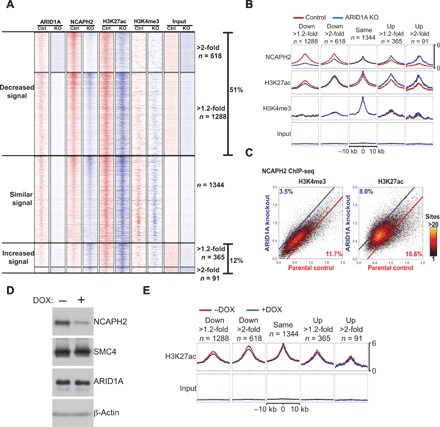
(A and B) Heatmap clustering of ChIP-seq profiles of ARID1A, NCAPH2, H3K27ac, and H3K4me3 in the indicated control (Ctrl) and ARID1A knockout RMG1 cells based on changes in NCAPH2 binding. The number of binding sites in each of the five clusters was indicated (A). Average profiles of the ChIP-seq signal for the indicated antibodies were generated on the basis of the five clusters (B). (C) Changes in NCAPH2 binding signal in parental control and ARID1A knockout RMG1 cells in regions with H3K4me3 or H3K27ac marks. Percentages represent proportion of binding sites that have at least a 1.2-fold change in binding signal cutoff indicated by blue and red lines. (D) Validation of NCAPH2 knockdown. RMG1 cells with inducible short hairpin RNA against the human NCAPH2 gene (shNCAPH2). Expression of NCAPH2, SMC4, ARID1A, and β-actin was examined by immunoblotting. DOX, doxycycline. (E) Same as (B) but for the H3K27ac mark in the indicated control and shNCAPH2 RMG1 cells. Note that the input is the same as those used in (B).
In addition, the subclassification of peaks revealed that the unaffected NCAPH2 binding was associated with the active promoter H3K4me3 mark, while the gain or loss of NCAPH2 binding correlated with the H3K27ac enhancer mark (Fig. 3B). Compared with changes in NCAPH2 binding in H3K4me3-enriched promoter regions, changes in NCAPH2 binding in H3K27ac-enriched enhancer regions were ~2-fold higher (Fig. 3C). In contrast, NCAPH2 knockdown did not overtly affect H3K27ac levels at these loci (Fig. 3, D and E). This suggests that ARID1A-driven changes in H3K27ac are upstream of NCAPH2 binding. Given that ARID1A knockout does not affect NCAPH2 expression (Fig. 1B), we conclude that ARID1A knockout preferentially affects the binding of NCAPH2 at H3K27ac-marked enhancers genome-wide.
We next determined the changes in gene expression induced by ARID1A knockout or inducible NCAPH2 knockdown using RNA sequencing (RNA-seq) analysis in RMG1 cells. There was a significant overlap of genes affected by ARID1A knockout and NCAPH2 knockdown (Fig. 4A), supporting the notion that ARID1A and NCAPH2 function in the same pathway to regulate gene expression. Consistent with an overall decrease in NCAPH2 chromatin binding when ARID1A was knocked out (Fig. 3A) and the fact that NCAPH2 is typically associated with transcriptionally active genes (26), most of the overlapped genes affected by ARID1A knockout or NCAPH2 knockdown were down-regulated, resulting in the overlap that is more than 10-fold over that expected by chance (P = 10−247 by hypergeometric test) (Fig. 4A).
Fig. 4. ARID1A-driven condensin II redistribution correlates with changes in gene expression.
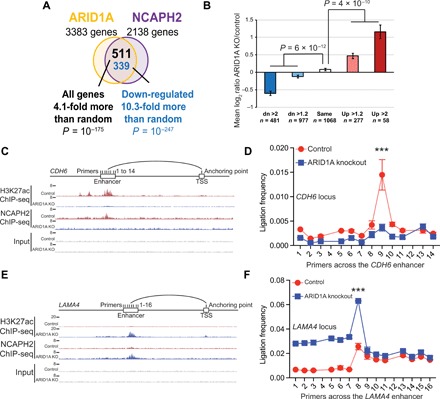
(A) Venn diagram showing the overlap of changes in gene expression induced by ARID1A knockout or NCAPH2 knockdown in RMG1 cells determined by RNA-seq analysis. (B) Average gene expression changes show positive correlation with changes in NCAPH2 binding and gene expression induced by ARID1A knockout. (C and D) Schematic of 3C primers covering the enhancer and proximal promoter anchoring of the down-regulated CDH6 gene locus aligned with ChIP-seq tracks for H3K27ac and NCAPH2 in control and ARID1A knockout RMG1 cells (C). 3C reveals that ARID1A knockout in RMG1 cells elicits a significant reduction of the interaction frequency between the enhancer and the promoter of the CDH6 gene locus (D). (E and F) Same as (C) and (D) but for the up-regulated LAMA4 gene. Error bars represent means ± SD. ***P < 0.0001.
Further supporting the role of NCAPH2 in driving the changes in the expression of the overlapped genes, NCAPH2 redistribution pattern positively correlated with the changes in gene expression (Fig. 4B). We validated six down-regulated and four up-regulated randomly selected NCAPH2 direct target genes in ARID1A knockout cells by quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis (fig. S3, B to D). ChIP-qPCR analysis revealed that changes in gene expression positively correlated with changes in the association of NCAPH2 and H3K27ac for both up-regulated and down-regulated genes in ARID1A knockout cells, which can be rescued by ectopic ARID1A expression (fig. S3, E to G). Chromosome conformation capture (3C) on the enhancers of a down-regulated gene locus CDH6 (Fig. 4, C and D) and an up-regulated gene locus LAMA4 (Fig. 4, E and F) validated that changes in interactions between the enhancers and proximal promoters correlated with changes in their expression. We conclude that the ARID1A-regulated redistribution of NCAPH2 at enhancers positively correlates with changes in gene expression.
ARID1A loss enhances TAD border strength and can drive compartment switching
Since ARID1A affects the condensin II genome-wide distribution and the role of ARID1A in regulating genome organization has not been explored, we performed a Hi-C analysis in ARID1A wild-type, ARID1A knockout, and NCAPH2 knockdown RMG1 cells. We first analyzed TADs using a resolution of 40 kb (Fig. 5A and fig. S4A). On the basis of ChIP-seq signal distribution relative to TAD borders (TBs), H3K4me3 showed the highest enrichment at the TBs, followed by H3K27ac, ARID1A, and NCAPH2 (Fig. 5B and fig. S4B). In addition, an insulation score–based analysis of the strength of TBs revealed that NCAPH2 knockdown only had minimal effects on TB strength (~1.4% of TB changed), while ARID1A knockout resulted in changes in TB strength at ~15% of TBs (Fig. 5A). This could be due to the fact that there may still be residual NCAPH2 in the NCAPH2 knockdown cells, while ARID1A is completely knocked out. Regardless, there was a statistically significant overlap in the pool of TBs affected by ARID1A knockout and NCAPH2 knockdown (Fig. 5A).
Fig. 5. ARID1A loss enhances TAD border strength and can drive compartment switching.
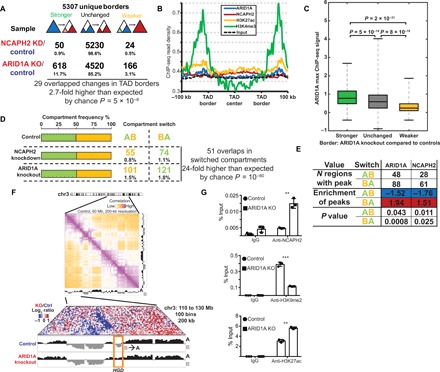
(A) Changes in the strength of TAD borders induced by NCAPH2 knockdown or ARID1A knockout in RMG1 cells. (B) Average ChIP-seq peak intensity of ARID1A, NCAPH2, H3K27ac, and H3K4me3 across the TADs. (C) ARID1A ChIP-seq signal intensity at the TBs divided on the basis of the strength of TBs, which become stronger, unchanged, or weaker after ARID1A knockout. (D) Overall distribution of the indicated euchromatic A-compartments and heterochromatic B-compartment and compartments switch induced by NCAPH2 knockdown or ARID1A knockout. (E) Distribution of ARID1A and NCAPH2 ChIP-seq signal in regions with compartments switch compared to those without compartments switch induced by ARID1A knockout in RMG1 cells. (F) An example of a region on chromosome 3 (chr3) showing a B-to-A compartment switching that is associated with the HGD gene locus, as indicated by the box. (G) Validation of an increase in NCAPH2, a decrease in H3K9me2, and an increase in H3K27ac on the HGD locus that showed a B-to-A compartment switch by ChIP analysis using the indicated antibodies or an IgG control. ***P < 0.0001 and **P < 0.001.
Notably, loss of ARID1A led to increased insulation (border strengthening) at most affected TBs (~12% increased versus 3% decreased insulation score) (Fig. 5A). This is also consistent with the increased number of TADs called after ARID1A knockout compared to controls (fig. S4A). Consistently, ARID1A ChIP-seq analysis showed that higher levels of ARID1A at the border in control cells correlated with the strengthening of TBs upon ARID1A knockout (Fig. 5C). This result indicates that ARID1A normally antagonizes the insulation of TADs.
We next examined the effects of ARID1A loss on genome compartmentalization. Compartmentalization refers to the fact that the genome is partitioned into broadly active (A-compartment) and inactive (B-compartment) chromatin domains, where domains of the same type show an enhanced contact frequency with one another (10). We assigned genomic intervals into euchromatic A-compartments and heterochromatic B-compartments using a resolution of 200 kb (a total of 13,535 bins with defined compartments). We found an equal proportion of A- and B-compartments in control, ARID1A knockout, and NCAPH2 knockdown RMG1 cells (Fig. 5D). Although less than 4% of regions showed a significant switch between compartments (A to B or B to A) across all samples, ARID1A knockout cells showed approximately twice as much compartment switching as NCAPH2 knockdown (Fig. 5D).
Notably, there was a significant overlap of the switched compartments induced by ARID1A knockout or NCAPH2 knockdown (Fig. 5D). To determine whether the compartment switch is associated with ARID1A and NCAPH2 binding within these compartments, we analyzed the regions with the most significant ARID1A and NCAPH2 binding peaks. Compared to regions without compartment switching, ARID1A and NCAPH2 binding signal in wild-type control cells had a lower number of significant peaks in the regions that showed that A-to-B compartments switch upon ARID1A knockout (Fig. 5E). In contrast, the number of significant ARID1A and NCAPH2 binding peaks in wild-type control cells was higher in regions that showed B-to-A compartment switching upon ARID1A knockout (Fig. 5E). This suggests that binding of ARID1A contributes to B-compartment formation in wild-type cells. After ARID1A knockout, there were 57 B-to-A switched compartments that had both ARID1A and NCAPH2 binding in RMG1 control cells. Together, this compartment switching after ARID1A knockout affected the expression of 38 genes (fig. S4C). HGD is one of the identified genes whose up-regulation is associated with B-to-A compartment switching after ARID1A knockout (Fig. 5D). We validated that the compartment switch correlated with an increase in NCAPH2, a decrease in the heterochromatin mark H3K9me2, and an increase in the euchromatin mark H3K27ac following ARID1A knockout (Fig. 5G). Together, these findings suggest that ARID1A loss can result in compartment switching.
ARID1A partitions interphase chromosome territory and promotes intermixing of small chromosomes
We next analyzed intra- and interchromosomal interactions at a resolution of 1 Mb. Both ARID1A knockout and NCAPH2 knockdown reduced the frequency of intrachromosomal interactions greater than 1 Mb and especially at genomic distances greater than 10 Mb (fig. S5A). Compared with ARID1A knockout cells, intra- and interchromosomal interactions were affected to a lesser extent by NCAPH2 knockdown (fig. S5B).
While ARID1A knockout reduced overall interchromosomal interactions, the interaction among small chromosomes [such as chromosome 19 (chr19) and chr22] was increased, with the exception of chr18 (Fig. 6A and fig. S5, B to D). Notably, average ARID1A binding was typically higher in small chromosomes (fig. S5E), and ARID1A binding positively correlated with changes in interchromosomal interactions (Pearson r = 0.77, P = 2 × 10−5) (Fig. 6B). Consistent with the idea that ARID1A colocalizes with NCAPH2 and ARID1A knockout predominantly redistributes NCAPH2 away from its binding sites, NCAPH2 redistribution significantly correlated with the chromosome size (Pearson r = 0.66, P = 0.0009) (Fig. 6C). There was a significant difference (P = 4 × 10−19) in the relative ratio of the number of NCAPH2 peaks between large (chr1 to chr13 and chrX) and small (chr14 to chr22) chromosomes (fig. S5F). This result indicates that NCAPH2 binding tends to increase on larger chromosomes and decrease on smaller chromosomes after ARID1A knockout. NCAPH2 redistribution significantly, negatively correlated with increased interchromosomal interactions observed in the Hi-C analysis (Fig. 6D).
Fig. 6. ARID1A loss promotes intermixing of small chromosomes.
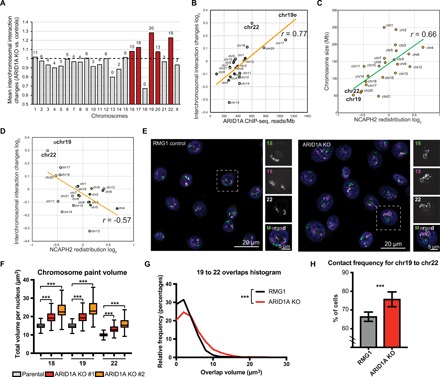
(A) Mean interchromosomal interaction changes across chromosomes. Number of chromosome pairs that showed significant [false discovery rate (FDR) < 5%] increase of interaction for each chromosome are indicated on the top of bars. Red bars highlight chromosomes with >50% significantly increased interchromosomal interactions. (B) Correlation between ARID1A ChIP-seq signal reads within significant peaks per megabase of chromosome length and changes in interchromosomal interaction. (C) NCAPH2 redistribution (relative change in number of NCAPH2 peaks in ARID1A knockout versus control RMG1 cells) across chromosomes. (D) Correlation between NCAPH2 redistribution and interchromosomal interactions. (E) Representative images of 3D chromosomal painting showing chr18 (green), chr19 (purple), and chr22 (white) in parental control and ARID1A knockout RMG1 cells. (F) Volumes of chr18, chr19, and chr22 (calculated on the basis of 3D chromosome painting from at least 790 nuclei) in parental control and two independent ARID1A knockout RMG1 clones. Error bars = mean with SD. (G and H) Distribution of 3D chromosome overlap area (G) or contact frequency (H) (from at least 747 nuclei) between chr19 and chr22 in parental control and ARID1A knockout RMG1 cells. Error bars = mean with SEM. P value was calculated by Mann-Whitney U test. Note that chr18 was excluded from the analysis in (B) to (D). Correlation was calculated by Pearson analysis. ***P < 0.001.
We next sought to validate the Hi-C results using three-dimensional (3D) chromosome painting analysis. We generated highly specific human chromosome paints using Oligopaints targeting three small chromosomes: chr18, chr19, and chr22 (Fig. 6E). The overall volume of all three chromosomes increased upon ARID1A knockout (Fig. 6F). In addition, the nuclear volume was increased upon ARID1A knockout (fig. S6A). The nuclear area is larger in a panel of ARID1A-mutated compared to wild-type OCCC cells (fig. S6B). Notably, the percentage of monosomies for chr18 was significantly higher in ARID1A knockout cells (fig. S5C), which is consistent with the observed decrease in interactions between chr18 and all other small chromosomes (fig. S5D). Consistent with the Hi-C analysis, we observed a significant increase in the intermixing volumes and interchromosomal contact frequencies between chr19 and chr22 in ARID1A knockout cells compared to control cells (Fig. 6, G and H). Similar results were also obtained using an independent ARID1A knockout RMG1 clone (Fig. 6F and fig. S6, C and D). Together, we conclude that ARID1A partitions interphase chromosome territories with its loss, increasing the intermixing between small chromosomes such as chr19 and chr22.
DISCUSSION
Here, we show that ARID1A loss drives the redistribution of NCAPH2 among H3K27ac-marked enhancers, with the predominant effect being loss of NCAPH2 in H3K27ac-positive ARID1A binding sites. ARID1A loss also increased NCAPH2 and H3K27ac levels at a small percentage of both existing and de novo binding sites. Our data support a model whereby ARID1A functions upstream of H3K27ac, which drives redistribution of NCAPH2. Consistently, NCAPH2 does not affect H3K27ac distribution. Thus, these data indicate that NCAPH2 functions downstream of ARID1A-regulated H3K27ac recruitment. Notably, compared with NCAPH2 knockdown, ARID1A loss has a greater effect on both TB strength and compartment switching. These findings support a model whereby loss of NCAPH2 contributes to, instead of recapitulates, the effects of ARID1A loss.
Although ARID1A loss predominantly led to loss of NCAPH2 and H3K27ac at enhancers, and ARID1A suppresses insulation of TADs, ARID1A contributes to heterochromatic B-compartment formation. This suggests that ARID1A may indirectly contribute to B-compartment formation through organizing A-compartments. Notably, several subunits of the SWI/SNF complexes, including ARID1A, appear to contribute to nuclear positioning of gene loci (27). This is consistent with our findings that ARID1A regulates chromosomal partitioning. In addition, knockdown of SMARCA4, one of the two mutually exclusive catalytic subunits of the SWI/SNF complexes (1), was reported to weaken TAD boundary strength in human mammary epithelial MCF-10A cells (28). ARID1A knockout weakens 3.1% of TBs while strengthening 11.7% (Fig. 5A). This highlights the context-dependent complexity and potential subunit-specific regulation of TAD structure by the SWI/SNF complexes.
ARID1A knockout increases nuclear and chromosome volume and increases the intermixing volume between small chromosomes such as chr19 and chr22. Notably, this correlates with a higher average ARID1A binding to these small chromosomes and a decrease in NCAPH2 binding to these small chromosomes induced by ARID1A knockout. CAP-H2 inactivation induces similar changes in interphase Drosophila nuclei (21). However, the overall decrease in interchromosomal interactions suggests that both NCAPH2 loss from ARID1A binding sites and de novo gain of binding sites contribute to the changes in spatial chromosome partitioning induced by ARID1A inactivation. In summary, our study shows that ARID1A spatially partitions interphase chromosomes, which correlates with its role in regulating the binding of condensin II at enhancers genome-wide.
MATERIALS AND METHODS
Cell lines, culture conditions, and transfection
OCCC cell line RMG1 cells were cultured in 1:1 Dulbecco’s modified Eagle’s medium (DMEM)/F12 supplemented with 10% fetal bovine serum (FBS). OCCC cell line TOV21G and OVCAR429 cells were cultured in RPMI 1640 with 10% FBS and 1% penicillin/streptomycin at 37°C, supplied with 5% CO2. Viral packing cells 293FT and Phoenix were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin at 37°C, supplied with 5% CO2. TOV21G OCCC cells harbor a frameshift 1645insC mutation that leads to a complete loss of ARID1A protein expression (5). All the cell lines were authenticated at The Wistar Institute Genomics Facility using short tandem repeat DNA profiling. Mycoplasma testing was performed using LookOut Mycoplasma PCR detection (Sigma-Aldrich) every month. Transfection was performed using Lipofectamine 2000 (Life Technologies) following the manufacturer’s specifications.
Reagents, plasmids, and antibodies
DNase I was purchased from Sigma-Aldrich (4536282001). Inducible NCAPH2 knockdown was performed using the pTRIPZ-shNCAPH2 lentiviral construct from K.-i.N.’s laboratory and as previously published (29). To induce NCAPH2 knockdown, cells infected with the lentivirus expressing shNCAPH2 were treated with doxycycline (1 μg/ml) for 72 hours. For ARID1A CRISPR knockout, PX458 (Addgene plasmid no. 48138) and pFETCh-donor (Addgene plasmid no. 63934) constructs were obtained from Addgene. Guide RNA sequence (TGTCCCACGGCTGTCATGAC) targeting the terminal codon of ARID1A was inserted into PX458. About 500 base pairs (bp) of homologous arms at both sides of the guide RNA targeting site were cloned and inserted into pFETCh-donor. ARID1A knockout clones were isolated after puromycin (1 μg/ml) selection and validated by immunoblotting.
The following antibodies were used: rabbit anti-ARID1A (1:1000 for Western blot; Cell Signaling Technology, catalog no. 12354), rabbit anti-ARID1A (5 μg per IP for ChIP; Abcam, catalog no. ab182560), mouse anti-ARID1B (1:1000 for Western blot; Abgent, catalog no. AT1189a), rabbit anti-ARID2 (1:1000 for Western blot; Bethyl, catalog no. A302-230A), rabbit anti-BRG1 (1:1000 for Western blot; Cell Signaling Technology, catalog no. 49360), rabbit anti-brahma (BRM) (1:1000 for Western blot; Cell Signaling Technology, catalog no. 11966), rabbit anti-BAF155 (2 μg per IP for IP; Abcam, catalog no. ab172638), rabbit anti-BAF155 (1:1000 for Western blot; Cell Signaling Technology, catalog no. 11956), rabbit anti–CAP-D3 (1:1000 for Western blot; Bethyl, catalog no. A300-604A), rabbit anti–CAP-H (1:1000 for Western blot; Bethyl, catalog no. A300-603A), rabbit anti–CAP-H2 (2 μg per IP for IP and 5 μg per IP for ChIP or ChIP-seq; Bethyl, catalog no. A302-276A), mouse anti–CAP-H2 (1:1000 for Western blot; Santa Cruz Biotechnology, catalog no. sc-393333), rabbit anti-H3K9me2 (5 μg per IP for ChIP; Cell Signaling Technology, catalog no. 4568), rabbit anti-H3K9me3 (5 μg per IP for ChIP; Active Motif, catalog no. 39161), rabbit anti-H3K27ac (3 μg per IP for ChIP and 5 μg per IP for ChIP-seq; Abcam, catalog no. ab4729), rabbit anti-H3K27me3 (5 μg per IP for ChIP; Cell Signaling Technology, catalog no. 9733), rabbit anti-RNA polymerase II (Pol II) (5 μg per IP for ChIP-seq; Santa Cruz Biotechnology, catalog no. sc-47701), rabbit anti-RAD21 (1:1000 for Western blot; Bethyl, catalog no. A300-080A), rabbit anti-SMC2 (1:1000 for Western blot; Cell Signaling Technology, catalog no. 5329), rabbit anti-SMC4 (1:1000 for Western blot; Cell Signaling Technology, catalog no. 5547), rabbit anti-SNF5 (2 μg per IP for IP and 1:1000 for Western blot; Bethyl, catalog no. A301-087A), mouse anti-Flag M2 (2 μg per IP for IP and 1:2000 for Western blot; Sigma-Aldrich, catalog no. F1804), mouse anti–μ-actin (1:5000 for Western blot; Sigma-Aldrich, catalog no. A5316), and mouse anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (1:1000 for Western blot; Millipore, MAB374).
Western blot and IP
Whole-cell protein was extracted using radioimmunoprecipitation assay lysis buffer [50 mM tris (pH 8.0), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 1 mM phenylmethylsulfonyl fluoride (PMSF)]. Proteins were separated by SDS-PAGE (polyacrylamide gel electrophoresis) and transferred to a polyvinylidene difluoride membrane (Millipore). Membranes were blocked with 5% nonfat milk and then incubated with primary antibodies and secondary antibodies.
Nuclear fractions for IP were prepared by ammonium sulfate precipitation. Briefly, cells were resuspended in buffer A [10 mM Hepes (pH 7.6), 10 mM KCl, 25 mM 10% glycerol, 1 mM dithiothreitol (DTT), and 1 mM PMSF] for 5 min on ice. Nuclei were harvested by centrifugation (1300g for 4 min) and lysated by 0.3 M ammonium sulfate in buffer C [10 mM Hepes (pH 7.6), 3 mM MgCl2, 100 mM KCl, 0.1 mM EDTA, 10% glycerol, 1 mM DTT, and 1 mM PMSF]. Soluble nuclear proteins were separated by ultracentrifugation (100,000g for 30 min) and precipitated with ammonium sulfate (0.3 g/ml) for 30 min on ice. Protein precipitate was isolated by ultracentrifugation (100,000g for 30 min) and resuspended in IP lysis buffer [50 mM tris-HCl (pH 8.0), 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 1 mM DTT, and 1 mM PMSF] for IP or gradient sedimentation.
Mass spectrometry
Endogenous complexes for LC-MS/MS were affinity-purified following the IP protocol. Quality of the samples was determined by silver staining and Western blot. Briefly, the SDS-PAGE gel was fixed in fixation buffer 1 (50% methanol and 10% acetic acid) for at least 15 min and fixation buffer 2 (10% methanol and 7% acetic acid) for 2 hours. The gel was washed with gluteraldehyde (25%):water solution (1:10) for 15 min and then three times with deionized water for 15 min. The staining solution was prepared by dropping solution B (1 g of AgNO3 in 5 ml of deionized water) into solution A (0.185 ml of 10 M NaOH, 2.8 ml of NH4OH, and 22.5 ml of deionized water). The final volume was brought to 100 ml by adding 70 ml of deionized water. The gel was stained for 15 min and washed three times with deionized water for 2 min. The stain was developed with developing solution (0.5 ml of 1% citric acid and 0.05 ml of 38% formaldehyde in 100 ml of deionized water) to appropriate signal and then stopped by stop solution (50% methanol and 5% acetic acid) for 10 min.
LC-MS/MS was performed by The Wistar Proteomic Facility. MS/MS spectra generated from the LC-MS/MS runs were searched using full tryptic specificity against the UniProt human database using the MaxQuant 1.5.2.8 program. Protein quantification was performed using unique and razor peptides. Razor peptides are shared (nonunique) peptides assigned to the protein group with the most other peptides (Occam's razor principle). False discovery rates (FDR) for protein and peptide identifications were set at 1%.
Sucrose and glycerol gradient sedimentation assay
Nuclear fractions were prepared following the IP protocol. The nuclear protein precipitate was resuspended in HEMG-0 buffer [25 mM Hepes (pH7.9), 0.1 M EDTA, 12.5 mM MgCl2, and 100 mM KCl]. One milligram of nuclear protein was carefully overlaid onto a 5-ml 20 to 50% sucrose gradient (in HEMG-0 buffer) or 10 to 30% glycerol gradient prepared in a 5-ml 13 mm × 51 mm polyallomer centrifuge tube (Beckman Coulter, 326819). Tubes were centrifuged in an SW-55 Ti swing-bucket rotor at 4°C for 16 hours at 100,000g. Fractions (0.4 ml) were collected for immunoblotting analyses.
Quantitative reverse transcription PCR
Total RNA was isolated using Trizol (Invitrogen) according to the manufacturer's instruction. Extracted RNAs were used for RT-PCR with the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). qPCR was performed using QuantStudio 3 Real-Time PCR System. The primer sequences used for qRT-PCR were as follows: CDH6, 5′-GCCAGTGGCTCAAACTTTAC-3′ (forward) and 5′-GGTGCTCATCTCGTGTCTATTA-3′ (reverse); KCNK5, 5′-CATCATCTTCTACCTGGCCATC-3′ (forward) and 5′-GATGCAGCTTCTGTGTGTAGTA-3′ (reverse); PROM1, 5′-CATCTCTCAATGACCCTCTGTG-3′ (forward) and 5′-CCTCAGTTCAGGGTTGCTATT-3′ (reverse); AGPAT4, 5′-CAGCTTCATCCTCGTCTTCTT-3′ (forward) and 5′-TTCTGCTTGCTGTCAGAGTT-3′ (reverse); LAMA4, 5′-GACATCACAAGGAGAGGGAAAT-3′ (forward) and 5′-TCCATTGACCATCAGGAGAATAAG-3′ (reverse); NCAPH2, 5′-CCAAGCAGCTCTCTTCGGTG-3′ (forward) and 5′-CATCCAGCGACAGGAACTCA-3′ (reverse); DEPDC1B, 5′-GGAAATTCTGAAAGTCCCTTTGG-3′ (forward) and 5′-CCATATCAGCTCCTGGGTATTT-3′ (reverse); HSD17B2, 5′-CACCAGTGACAAGTGGGAAA-3′ (forward) and 5′-CCGCTGTGCTAAGATGTAGTC-3′ (reverse); MAP1B, 5′-CCCGTCAGGATGTCGATTTAT-3′ (forward) and 5′-CCCGTCAGGATGTCGATTTAT-3′ (reverse); PTPRC, 5′-AACTCTTGGCATTTGGCTTTG-3′ (forward) and 5′-CTGGGCATCTTTGCTGTAGT-3′ (reverse); UGT8, 5′-CTACAGTCCAAGAGCGGAATA-3′ (forward) and 5′-CCATCAGGTCACAGTTCTTAGT-3′ (reverse); and B2M, 5′-GGCATTCCTGAAGCTGACA-3′ (forward) and 5′-CTTCAATGTCGGATGGATGAAAC-3′ (reverse). B2M was used as an internal control.
Chromatin IP
Cells were cross-linked with 1% formaldehyde for 10 min at room temperature. The reaction was quenched by 0.125 M glycine for 5 min. Fixed cells were lysated with ChIP lysis buffer 1 [50 mM Hepes-KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA (pH 8.0), 1% Triton X-100, and 0.1% deoxycholate] on ice and lysis buffer 2 [10 mM tris (pH 8.0), 200 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA] at room temperature. Chromatin was digested with MNase (Cell Signaling Technology) in digestion buffer [10 mM tris (pH 8.0), 1 mM CaCl2, and 0.2% Triton X-100] at 37°C for 15 min. The nucleus was broken down by one pulse of bioruptor with high output. The following antibodies were used for ChIP or ChIP-seq: rabbit anti-ARID1A (Abcam, catalog no. ab182560), rabbit anti-NCAPH2 (Bethyl, catalog no. A302-276A), rabbit anti-H3K4me1 (Abcam, catalog no. ab8895), rabbit anti-H3K4me3 (Active Motif, catalog no. 39159), rabbit anti-H3K9me2 (Cell Signaling Technology, catalog no. 4568), rabbit anti-H3K27ac (Abcam, catalog no. ab4729), rabbit anti–Pol II (Santa Cruz, catalog no. sc-47701), and rabbit anti-SNF5 (Bethyl, catalog no. A301-087A). Chromatin was incubated overnight at 4°C, and Protein A/G Dynabeads were added to the reaction for another 1.5 hours. Magnetic beads were washed, and chromatin was eluted. Eluted DNA was treated with proteinase K at 55°C for 45 min and decross-linked at 65°C overnight. A Zymo ChIP DNA clean and concentrator kit (Zymo Research, catalog no. D5205) was used to purify the ChIP DNA. ChIP DNA was used for ChIP-qPCR or ChIP-seq.
For ChIP-qPCR, the following primers were used: CDH6 locus, 5′-TCCCATGAAAGTCTCAGGAATG-3′ (forward) and 5′-AGACACTGGGTTTCCTCTCTA-3′ (reverse); KCNK5 locus, 5′-AGGGCCTGAGTTAGCATTTC-3′ (forward) and 5′-TCAGGGTCTTAGGTCTCAGATT-3′ (reverse); PROM1 locus, 5′-GCATCCACTTGGCATGATATTG-3′ (forward) and 5′-GAAGCTGGTCTCATACAGGATTTA-3′ (reverse); AGPAT4 locus, 5′-CCCAAAGTCACTCCAGTGATAG-3′ (forward) and 5′-GAACGGGTCTCTGTTGCTATT-3′ (reverse); LAMA4 locus, 5′-CACTGTGAGTACCATCCCTTT-3′ (forward) and 5′-GAGTTTCAGTCCCATCCTTCTT-3′ (reverse); and HGD locus, 5′-CATCCAGATCCAAGACCAAGAC-3′ (forward) and 5′-CCTGGTGTCTTGTGGCTAAA-3′.
In situ Hi-C
In situ Hi-C was performed as described in a previously published protocol (30) with a few modifications. Five million cells were fixed with 1% (v/v) formaldehyde in fresh medium at a concentration of 1 × 106 cells/ml. The reaction was quenched by 0.2 M glycine at room temperature for 5 min. Fixed cells were lysed in cold Hi-C lysis buffer [10 mM tris (pH 8.0) and 10 mM NaCl, 0.2% IGEPAL CA-630 with proteinase inhibitor] for 15 min, followed by a wash with 500 μl of cold Hi-C lysis buffer once. The pellet was gently resuspended in 50 μl of 0.5% SDS and incubated at 62°C for 10 min. Permeabilization was quenched by adding 145 μl of water and 25 μl of 10% Triton X-100 and incubated at 37°C for 15 min. Twenty-five microliters of 10× NEBuffer 2 and 100 U of Mbo I restriction enzyme were added, and chromatin was digested overnight or for at least 2 hours at 37°C, with rotation. Following inactivation of Mbo I at 62°C for 20 min, biotin 2′-deoxyadenosine 5′-triphosphate (dATP) fill-in was performed at 37°C for 1.5 hours with rotation by adding 15 nM biotin dATP, 3′-deoxythymidine 5′-triphosphate, 2′-deoxycytidine 5′-triphosphate, and 2′-deoxyguanosine 5′-triphosphate; 40 U of DNA Pol I; and a large Klenow fragment. In situ ligation was performed at room temperature for 4 hours with slow rotation by adding 900 μl of ligation master mix [120 μl of 10× NEB (New England Biolabs) T4 DNA ligase buffer, 100 μl of 10% Triton X-100, 12 μl of bovine serum albumin (BSA; 10 mg/ml), 5 μl of T4 DNA ligase (400 U/μl), and 663 μl of water]. Following centrifugation at 10,000 rpm for 5 min, the pellet was resuspended in 200 μl of decross-link buffer [50 mM tris-HCl (pH 8.0), 10 mM EDTA, and 1% SDS]. Eight microliters of 5 M sodium chloride and 8 μl of proteinase K (10 mg/ml) were added, and reverse cross-linking was performed at 68°C for at least 1.5 hours. Decross-linked DNA was purified by phenol/chloroform extraction. The pellet was dissolved in 200 μl of 1× tris buffer [10 mM tris-HCl (pH 8)] and incubated at 37°C for 15 min to fully dissolve the DNA. The DNA was sheared to a size of 300 to 500 bp by bioruptor with the following setting: high output, 30 s on/30 s off, five times. Sheared DNA was purified by Ampure beads and quantified by Qubit. One hundred fifty microliters of Dynabeads MyOne Streptavidin T1 (10 mg/ml) was used per sample to capture the biotin-labeled fragments. Following biotin pulldown, the DNA was used for library construction.
Chromatin conformation capture
Chromatin conformation capture (3C) was performed as described in the in situ Hi-C protocol, with a few modifications. Following inactivation of Mbo I at 62°C for 20 min, the ligation was performed at room temperature for 4 hours with slow rotation by adding 750 μl of ligation master mix [100 μl of 10× NEB T4 DNA ligase buffer, 80 μl of 10% Triton X-100, 10 μl of BSA (10 mg/ml), 5 μl of T4 DNA ligase (400 U/μl), and 555 μl of water]. Following centrifugation at 10,000 rpm for 5 min, the pellet was resuspended in 200 μl of reverse cross-link buffer with sodium chloride and proteinase K, and reverse cross-linking was performed at 68°C for at least 1.5 hours. Reverse cross-linked DNA was purified by phenol/chloroform extraction and dissolved in 200 μl of 1× tris buffer [10 mM tris-HCl (pH 8)]. Primer design was done following the qPCR primer design standard. Only primers no more than 100 bp close to Mbo I sites were used.
Chromosome paints for 3D Oligopaint FISH in interphase cells
Oligopaint libraries were designed as previously described, using the Oligoarray 2.1 software (31) and the hg19 genome build, and purchased from CustomArray. Chromosome paints were designed to have 42 bases of homology with a density of 1.3 probes/kb measured with a 50-kb sliding window. Coordinates for all paints can be found below (Table 1). Oligopaints were synthesized, as previously described (21).
Table 1. Coordinates for the indicated paints for chr18, chr19, and chr22 used in the current study.
| Chromosome | Start | End | Total oligos |
| 18 | 14,921,486 | 78,016,661 | 88,197 |
| 19 | 238,392 | 59,093,250 | 49,788 |
| 22 | 16,054,663 | 51,220,472 | 38,360 |
Fluorescence in situ hybridization (FISH) was performed as previously described, with a few modifications to the protocol (21). In short, cells from log-phase cultures were settled on poly-l-lysine–treated glass slides for 30 to 60 min and subsequently fixed for 10 min with 4% formaldehyde in PBS-T [1× phosphate-buffered saline (PBS) with 0.1% Triton X-100] at room temperature, followed by three 5-min washes in PBS-T at room temperature. Slides were then permeabilized by washing with PBS-T 0.3% for 15 min at room temperature. Next, slides were subjected to an ethanol row for drying: 70, 90, and 100% for 5 min each at −20°C. Slides were then allowed to dry for 10 min at room temperature. After drying, slides were rehydrated by washing once for 5 min in 2× SSCT (0.3 M NaCl, 0.03 M sodium citrate, and 0.1% Tween-20) at room temperature, followed by one wash in 2× SSCT/50% formamide for 5 min at room temperature. Slides were then predenatured in 2× SSCT/50% formamide at 92°C for 2.5 min and then in 2× SSCT/50% formamide at 60°C for 20 min. Primary Oligopaint probes in hybridization buffer (10% dextran sulfate/2× SSCT/50% formamide/4% polyvinylsulfonic acid) were then added to the slides, covered with a coverslip, and sealed with rubber cement. Slides were denatured on a heat block in a water bath set to 92°C for 2.5 min, after which slides were transferred to a humidified chamber and incubated overnight at 37°C. Each Oligopaint probe (100 pmol) was used per slide in a final volume of 25 μl. Approximately 16 to 18 hours later, coverslips were removed with a razor blade, and slides were washed in 2× SSCT at 60°C for 15 min, 2× SSCT at room temperature for 15 min and in 0.2× SSC at room temperature for 5 min. Secondary probes (10 pmol per 25 μl) containing fluorophores were added to slides, again resuspended in hybridization buffer, and covered with a coverslip sealed with rubber cement. Slides were incubated at 37°C for 2 hours in a humidified chamber, followed by washes in 2× SSCT at 60°C for 15 min, 2× SSCT at room temperature for 15 min, and 0.2× SSC at room temperature for 5 min. All slides were washed with Hoechst DNA stain (1:10,000 in PBS) for 5 min, followed by two 5-min washes in PBS before mounting in Slowfade (Invitrogen).
Images were acquired on a Leica wide-field fluorescence microscope, using a 1.4–numerical aperture 63× oil-immersion objective (Leica) and Andor iXon Ultra emCCD camera. All images were processed using the Leica LAS-X 3.3 software, deconvolved using Huygens Essential deconvolution software, and saved as leica image file (LIF) files. Images were segmented and measured using a modified version of the TANGO 3D-segmentation plugin for ImageJ (5). Chromosome paints were segmented using a hysteresis-based segmentation algorithm. Statistical tests were performed using Prism 7 software by GraphPad.
RNA-seq and ChIP-seq
For RNA-seq, extracted RNAs were digested with DNase I (Qiagen, 79254) and purified using the RNeasy MinElute Cleanup Kit (Qiagen, 74106). RNA-seq libraries were prepared using the TruSeq Total RNA Sample Prep Kit (Illumina) and sequenced with Illumina NextSeq 500 using a 75-bp paired-end run. For ChIP-seq, 10 ng of ChIP DNA was used for library construction. The NEBNext Ultra DNA Library Prep Kit (NEB, E7645) was used to prepare the sequencing library. The libraries were sequenced in a 75-bp single-end run using Illumina NextSeq 500.
Bioinformatics analysis
RNA-seq data were aligned using bowtie2 (32) against the hg19 version of the human genome, and RSEM v1.2.12 software (33) was used to estimate raw read counts and Reads Per Kilobase of transcript, per Million (RPKM) using the Ensemble transcriptome. DESeq2 (34) was used to estimate the significance of differential expression between group pairs. Overall gene expression changes were considered significant if they passed FDR thresholds of <5%. Significance of overlap between genes was estimated using hypergeometric test.
ChIP-seq data were aligned using bowtie (35) against hg19 version of the human genome, and HOMER (36) was used to generate bigWig files and call significant peaks versus input and between pairs of samples using the –style histone option. Peaks that passed FDR threshold of <5% were considered. Normalized signals for significant peaks were derived from bigWig files using the bigWigAverageOverBed tool from the University of California, Santa Cruz toolbox (37) with mean0 option. Fold differences between samples were then calculated with the average input signal 0.4 used as a floor for the minimum allowed signal. Only peaks with a FDR of <5% that passed the additional cutoff of 4-fold versus input or 1.2-fold between wild-type and knockout conditions were considered significant. Genes that had the closest transcript’s transcription starting site (TSS) were assigned to each peak. ChIP-seq profile groups were generated using k-means clustering with Pearson correlation distance. All peaks with an FDR of <5% were used for global chromosome CAPH2 redistribution analysis. A number of CAPH2 peaks in ARID1A wild-type and knockout conditions per 1-MB bin were used to calculate log2 ratios (knockout/wild type) for bins with at least one peak. All ratios were then mean-centered, and the average for each chromosome was used as a measure of CAPH2 redistribution.
For in situ Hi-C data analysis, 76-bp paired-end reads were separately aligned to the human genome (hg19) using bowtie2 (32) with iterative alignment strategy. Redundant paired reads derived from a PCR bias, reads aligned to repetitive sequences, and reads with low mapping quality (MapQ < 30) were removed. Reads potentially derived from self-ligation and undigested products were also discarded. Hi-C biases in contact maps were corrected using the iterative correction and eigenvector (ICE) method (38). The ICE normalization was repeated 30 times. For compartment calculation, the ratio of observed and expected score of ICE-normalized contact matrices were calculated using a resolution of 200 kb. Converted matrices were subjected to the principal components analysis (PCA). The signs of PCA score were determined by gene density. A PCA score with positive values were defined as A-compartment, and negative values were defined as B-compartment, as previously described (10). PCA values were upper quartile–normalized and used to estimate the significance of compartment A-to-B or B-to-A switch with Bioconductor limma package (39). Enrichment of peaks within significantly switched compartments was estimated by Fisher’s exact test. TADs were identified according to Van et al. (40) on ICE-normalized contact matrices at a resolution of 40 kb, and the significance of change of boundary strength determined by insulation score was tested using limma package (39). Significance of difference of interchromosomal interactions was estimated using DESeq2 on raw counts, and P values were adjusted for multiple testing using Bonferroni correction. An FDR of <5% was used as a significance threshold. Average interchromosomal interaction values for each chromosome were used for analysis of association with chromosome sizes and NCAPH2 redistribution. Gene set enrichment analysis for enrichment of gene ontology biological processes, molecular functions, and pathways (KEGG and BIOCARTA) was done using DAVID software, and enrichments of at least twofold that passed the FDR cutoff of <5% were considered significant.
Statistical analysis and reproducibility
Statistical analysis was performed using GraphPad Prism 7 (GraphPad) for macOS. Experiments were repeated three times unless otherwise stated. The representative images were shown unless otherwise stated. Quantitative data were expressed as means ± SEM unless otherwise stated. Analysis of variance (ANOVA) with Fisher’s least significant difference was used to identify significant differences in multiple comparisons.
Supplementary Material
Acknowledgments
Funding: This work was supported by NIH grants (R01CA160331, R01CA163377, and R01CA202919 to R.Z.; P01AG031862 to R.Z. and K.-i.N.; R35GM128903 to E.F.J.; R50CA221838 to H.-Y.T.; and R50CA211199 to A.V.K.) and the U.S. Department of Defense (OC140632P1 and OC150446 to R.Z.). Support of core facilities was provided by Cancer Centre Support Grant (CCSG) CA010815 to The Wistar Institute. This work was supported by the NIH/NIMGS grant R01GM124195 to K.-i.N. Author contributions: S.W., N.F., L.R., J.M.L., O.I., and H.-Y.T. performed the experiments and analyzed data. S.W., N.F., and R.Z. designed the experiments. A.G., K.-i.N., and D.W.S. participated in the experimental design. H.T., A.V.K., and H.Y.T. performed the bioinformatics analysis. E.F.J. and R.Z. supervised the studies. S.W., E.F.J., and R.Z. wrote the first draft of the manuscript. R.Z. conceived the study. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All sequencing data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE120060. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaaw5294/DC1
Fig. S1. The SWI/SNF complex colocalizes with the condensin II complex at enhancers.
Fig. S2. ARID1A loss does not affect the interaction between the SWI/SNF and condensin II complexes.
Fig. S3. ARID1A loss redistributes condensin II complex binding at enhancers to regulate gene expression.
Fig. S4. ARID1A suppresses insulation of TADs.
Fig. S5. ARID1A partitions chromosomal territories.
Fig. S6. ARID1A loss increases chromosome and nuclei volume and promotes small chromosome intermixing in trans.
REFERENCES AND NOTES
- 1.Kadoch C., Crabtree G. R., Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 1, e1500447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu J. N., Roberts C. W. M., ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 3, 35–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence M. S., Stojanov P., Mermel C. H., Robinson J. T., Garraway L. A., Golub T. R., Meyerson M., Gabriel S. B., Lander E. S., Getz G., Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murakami R., Matsumura N., Brown J. B., Higasa K., Tsutsumi T., Kamada M., Abou-Taleb H., Hosoe Y., Kitamura S., Yamaguchi K., Abiko K., Hamanishi J., Baba T., Koshiyama M., Okuno Y., Yamada R., Matsuda F., Konishi I., Mandai M., Exome sequencing landscape analysis in ovarian clear cell carcinoma shed light on key chromosomal regions and mutation gene networks. Am. J. Pathol. 187, 2246–2258 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Wiegand K. C., Shah S. P., Al-Agha O. M., Zhao Y., Tse K., Zeng T., Senz J., McConechy M. K., Anglesio M. S., Kalloger S. E., Yang W., Heravi-Moussavi A., Giuliany R., Chow C., Fee J., Zayed A., Prentice L., Melnyk N., Turashvili G., Delaney A. D., Madore J., Yip S., McPherson A. W., Ha G., Bell L., Fereday S., Tam A., Galletta L., Tonin P. N., Provencher D., Miller D., Jones S. J. M., Moore R. A., Morin G. B., Oloumi A., Boyd N., Aparicio S. A., Shih I.-M., Mes-Masson A.-M., Bowtell D. D., Hirst M., Gilks B., Marra M. A., Huntsman D. G., ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 363, 1532–1543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathur R., Alver B. H., San Roman A. K., Wilson B. G., Wang X., Agoston A. T., Park P. J., Shivdasani R. A., Roberts C. W. M., ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 49, 296–302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson B. G., Roberts C. W. M., SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11, 481–492 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Kelso T. W. R., Porter D. K., Amaral M. L., Shokhirev M. N., Benner C., Hargreaves D. C., Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife 6, e30506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bickmore W. A., van Steensel B., Genome architecture: Domain organization of interphase chromosomes. Cell 152, 1270–1284 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Lieberman-Aiden E., van Berkum N. L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B. R., Sabo P. J., Dorschner M. O., Sandstrom R., Bernstein B., Bender M. A., Groudine M., Gnirke A., Stamatoyannopoulos J., Mirny L. A., Lander E. S., Dekker J., Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciabrelli F., Cavalli G., Chromatin-driven behavior of topologically associating domains. J. Mol. Biol. 427, 608–625 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Parada L. A., Misteli T., Chromosome positioning in the interphase nucleus. Trends Cell Biol. 12, 425–432 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Dixon J. R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J. S., Ren B., Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dekker J., Mirny L., The 3D genome as moderator of chromosomal communication. Cell 164, 1110–1121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noma K.-i., The yeast genomes in three dimensions: Mechanisms and functions. Annu. Rev. Genet. 51, 23–44 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Iwasaki O., Noma K.-i., Condensin-mediated chromosome organization in fission yeast. Curr. Genet. 62, 739–743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buster D. W., Daniel S. G., Nguyen H. Q., Windler S. L., Skwarek L. C., Peterson M., Roberts M., Meserve J. H., Hartl T., Klebba J. E., Bilder D., Bosco G., Rogers G. C., SCFSlimb ubiquitin ligase suppresses condensin II–mediated nuclear reorganization by degrading Cap-H2. J. Cell Biol. 201, 49–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauer C. R., Hartl T. A., Bosco G., Condensin II promotes the formation of chromosome territories by inducing axial compaction of polyploid interphase chromosomes. PLOS Genet. 8, e1002873 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.George C. M., Bozler J., Nguyen H. Q., Bosco G., Condensins are required for maintenance of nuclear architecture. Cell 3, 865–882 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith H. F., Roberts M. A., Nguyen H. Q., Peterson M., Hartl T. A., Wang X.-J., Klebba J. E., Rogers G. C., Bosco G., Maintenance of interphase chromosome compaction and homolog pairing in Drosophila is regulated by the condensin cap-h2 and its partner Mrg15. Genetics 195, 127–146 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosin L. F., Nguyen S. C., Joyce E. F., Condensin II drives large-scale folding and spatial partitioning of interphase chromosomes in Drosophila nuclei. PLOS Genet. 14, e1007393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadoch C., Hargreaves D. C., Hodges C., Elias L., Ho L., Ranish J., Crabtree G. R., Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 45, 592–601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helming K. C., Wang X., Wilson B. G., Vazquez F., Haswell J. R., Manchester H. E., Kim Y., Kryukov G. V., Ghandi M., Aguirre A. J., Jagani Z., Wang Z., Garraway L. A., Hahn W. C., Roberts C. W. M., ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 20, 251–254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bitler B. G., Aird K. M., Garipov A., Li H., Amatangelo M., Kossenkov A. V., Schultz D. C., Liu Q., Shih I.-M., Conejo-Garcia J. R., Speicher D. W., Zhang R., Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 21, 231–238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bitler B. G., Wu S., Park P. H., Hai Y., Aird K. M., Wang Y., Zhai Y., Kossenkov A. V., Vara-Ailor A., Rauscher F. J. III, Zou W., Speicher D. W., Huntsman D. G., Conejo-Garcia J. R., Cho K. R., Christianson D. W., Zhang R., ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 19, 962–973 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dowen J. M., Bilodeau S., Orlando D. A., Hübner M. R., Abraham B. J., Spector D. L., Young R. A., Multiple structural maintenance of chromosome complexes at transcriptional regulatory elements. Stem Cell Reports 1, 371–378 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shachar S., Voss T. C., Pegoraro G., Sciascia N., Misteli T., Identification of gene positioning factors using high-throughput imaging mapping. Cell 162, 911–923 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barutcu A. R., Lajoie B. R., Fritz A. J., McCord R. P., Nickerson J. A., van Wijnen A. J., Lian J. B., Stein J. L., Dekker J., Stein G. S., Imbalzano A. N., SMARCA4 regulates gene expression and higher-order chromatin structure in proliferating mammary epithelial cells. Genome Res. 26, 1188–1201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yokoyama Y., Zhu H., Zhang R., Noma K.-i., A novel role for the condensin II complex in cellular senescence. Cell Cycle 14, 2160–2170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rao S. S. P., Huntley M. H., Durand N. C., Stamenova E. K., Bochkov I. D., Robinson J. T., Sanborn A. L., Machol I., Omer A. D., Lander E. S., Aiden E. L., A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beliveau B. J., Joyce E. F., Apostolopoulos N., Yilmaz F., Fonseka C. Y., McCole R. B., Chang Y., Li J. B., Senaratne T. N., Williams B. R., Rouillard J.-M., Wu C.-t., Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci. U.S.A. 109, 21301–21306 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kent W. J., Zweig A. S., Barber G., Hinrichs A. S., Karolchik D., BigWig and BigBed: Enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imakaev M., Fudenberg G., McCord R. P., Naumova N., Goloborodko A., Lajoie B. R., Dekker J., Mirny L. A., Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K., limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Bortle K., Nichols M. H., Li L., Ong C.-T., Takenaka N., Qin Z. S., Corces V. G., Insulator function and topological domain border strength scale with architectural protein occupancy. Genome Biol. 15, R82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaaw5294/DC1
Fig. S1. The SWI/SNF complex colocalizes with the condensin II complex at enhancers.
Fig. S2. ARID1A loss does not affect the interaction between the SWI/SNF and condensin II complexes.
Fig. S3. ARID1A loss redistributes condensin II complex binding at enhancers to regulate gene expression.
Fig. S4. ARID1A suppresses insulation of TADs.
Fig. S5. ARID1A partitions chromosomal territories.
Fig. S6. ARID1A loss increases chromosome and nuclei volume and promotes small chromosome intermixing in trans.