

PDE1 inhibition enhances the proteasome and protects against proteinopathy and heart failure with preserved ejection fraction.
Abstract
No current treatment targets cardiac proteotoxicity or can reduce mortality of heart failure (HF) with preserved ejection fraction (HFpEF). Selective degradation of misfolded proteins by the ubiquitin-proteasome system (UPS) is vital to the cell. Proteasome impairment contributes to HF. Activation of cAMP-dependent protein kinase (PKA) or cGMP-dependent protein kinase (PKG) facilitates proteasome functioning. Phosphodiesterase 1 (PDE1) hydrolyzes both cyclic nucleotides and accounts for most PDE activities in human myocardium. We report that PDE1 inhibition (IC86430) increases myocardial 26S proteasome activities and UPS proteolytic function in mice. Mice with CryABR120G-based proteinopathy develop HFpEF and show increased myocardial PDE1A expression. PDE1 inhibition markedly attenuates HFpEF, improves mouse survival, increases PKA-mediated proteasome phosphorylation, and reduces myocardial misfolded CryAB. Therefore, PDE1 inhibition induces PKA- and PKG-mediated promotion of proteasomal degradation of misfolded proteins and treats HFpEF caused by CryABR120G, representing a potentially new therapeutic strategy for HFpEF and heart disease with increased proteotoxic stress.
INTRODUCTION
Heart failure (HF) is a clinical syndrome in which the heart fails to pump enough blood to meet the body’s need for oxygen and nutrients. It can be broadly divided into HF with reduced ejection fraction (HFrEF) and HF with preserved ejection fraction (HFpEF). Current guidelines classify HF with an ejection fraction (EF) of <40% as HFrEF. Conversely, patients with HFpEF, mainly characterized by diastolic malfunction, have a normal EF (>50%) (1). In the United States, approximately 50% of HF cases are HFpEF. Moreover, the prevalence of HFpEF relative to HFrEF has been increasing over time, and HFpEF is projected to become the most common type of HF (1). Despite tremendous therapeutic advances in improving HFrEF mortality in the past decades, currently, there is no effective pharmacological intervention available for HFpEF (2). Hence, HFpEF represents a large unmet need in cardiovascular medicine. Cardiac proteinopathy belongs to a family of cardiac diseases caused by increased expression of misfolded proteins and resultant proteotoxicity in cardiomyocytes. Emerging evidence suggests that increased proteotoxic stress (IPTS) plays an important role in the progression from a large subset of heart diseases to HF (3). As exemplified by desmin-related cardiomyopathy, restrictive cardiomyopathy or HFpEF is commonly seen in patients with cardiac proteinopathy (4). Similar to HFpEF, no evidence-based clinical strategy is currently available for treating cardiac proteinopathy or for targeting cardiac proteotoxicity.
Targeted clearance of misfolded proteins by the ubiquitin-proteasome system (UPS) is pivotal to protein quality control (PQC), which acts to minimize the amount and toxicity of misfolded proteins in the cell. UPS-mediated proteolysis begins with covalent attachment of a chain of ubiquitin molecules to a substrate protein molecule via a highly coordinated cascade of enzymatic reactions, a process known as ubiquitination; the polyubiquitinated protein molecule is subsequently degraded by the 26S proteasome (5). PQC inadequacy due to proteasome functional insufficiency (PFI) has been implicated in a large subset of human heart disease during their progression to HF (6), and studies using animal models have established a major pathogenic role for PFI in cardiac proteinopathy (7), ischemia-reperfusion (I/R) injury (7–9), and diabetic cardiomyopathy (10). Hence, improving cardiac proteasome functioning is implicated as a potential therapeutic strategy for HF treatment. Moreover, recent advances in cell biology excitingly unravel the proteasome as a nodal point for controlling UPS proteolysis (5). Our previous study has shown that guanosine 3′,5′-monophosphate (cGMP)–dependent protein kinase (PKG) stimulates proteasome proteolytic activities, improves cardiac UPS performance, and facilitates the degradation of misfolded proteins in cardiomyocytes (11). In addition, adenosine 3′,5′-monophosphate (cAMP)–dependent protein kinase (PKA) was also shown to stimulate proteasome activity and accelerate proteasomal degradation of misfolded proteins in cultured cells through directly phosphorylating the 19S subunit RPN6/PSMD11 at Ser14 (12, 13). Hence, it is conceivable, but remains to be formally tested, that duo-activation of PKG and PKA can facilitate proteasomal degradation of misfolded proteins in cardiomyocytes and thereby effectively treat heart disease with IPTS.
By breaking down cAMP and/or cGMP in a specific compartment of the cell, the cyclic nucleotide phosphodiesterase (PDE) plays a key role in cyclic nucleotide–mediated signaling amplitude, duration, and compartmentalization in the cell. The PDE superfamily consists of 11 family members, PDE1 through PDE11 (14). PDE1s are duo-substrate PDEs and account for most of the PDE activity in human myocardium (15). The PDE1 family is encoded by three distinct genes, PDE1A, PDE1B, and PDE1C, with PDE1A and PDE1C expressed in the heart and vessels. PDE1A is mostly expressed in rodents and displays a higher affinity for cGMP than for cAMP, whereas PDE1C is predominantly expressed in human myocardium and displays similar affinity to both cyclic nucleotides (14). Several studies have shown an important role of PDE1 in cardiovascular regulation. A PDE1-selective inhibitor (IC86340) was shown to attenuate isoproterenol (ISO)–induced cardiac hypertrophy and fibrosis in mice, with the mechanism involving stimulating the cGMP/PKG axis (16, 17). PDE1C depletion exhibits a protective effect on pressure overload–induced cardiac remodeling in a PKA-dependent manner (18). Given that both PKG and PKA positively regulate proteasome functioning, we sought to test the hypothesis that PDE1 inhibition augments proteasomal degradation of misfolded proteins and protects against cardiac proteinopathy, with the present study.
Here, we report that myocardial PDE1A expression was substantially increased in a mouse model of cardiac IPTS, including at its HFpEF stage; PDE1 inhibition increased proteasome activities and the degradation of a known UPS substrate in the heart; and chronic treatment of CryABR120G-based cardiac proteinopathy mice at their HFpEF stage with a PDE1-specific inhibitor (IC86430) improved cardiac diastolic function and remarkably delayed mouse premature death, which was associated with substantial increases in PKA-mediated proteasome phosphorylation and decreases in misfolded CryAB. In cultured cardiomyocytes, PDE1 inhibition increased 26S proteasome activities, enhanced degradation of a known UPS substrate in a PKA- and PKG-dependent manner, and promoted proteasomal degradation of CryABR120G, a misfolded protein known to cause human disease. Our data demonstrate that PDE1 inhibition, via activation of PKA and PKG, increases proteasome activities, promotes proteasomal degradation of misfolded proteins, and protects against cardiac proteinopathy-based HFpEF, representing a potential therapeutic strategy for HFpEF and heart disease with IPTS.
RESULTS
Up-regulated myocardial PDE1A in a mouse model of cardiac proteinopathy
We first determined changes in myocardial expression of PDE1A in a well-established transgenic (tg) mouse model of cardiac proteinopathy induced by cardiomyocyte-restricted expression of CryABR120G, a misfolded protein known to cause human disease (19). We examined non-tg (NTG), CryABR120G tg, and wild-type CryAB (CryABWT) tg mice at 4 and 6 months of age, when cardiac proteinopathy and cardiac malfunction are readily discernable in the CryABR120G tg mice. Western blot analyses with a validated anti-PDE1A antibody (fig. S1) revealed that myocardial PDE1A protein levels were significantly increased in CryABR120G tg mice compared with NTG littermates (P = 0.046 at 4 months and P = 0.017 at 6 months; Fig. 1, A and B). Myocardial PDE1A protein was not discernibly altered by overexpression of CryABWT (Fig. 1, C and D), suggesting that the up-regulation of myocardial PDE1A is specific to the CryABR120G-based proteinopathy. Moreover, consistent with the protein data, reverse transcription polymerase chain reaction (RT-PCR) analysis revealed that myocardial PDE1A mRNA levels were significantly increased in CryABR120G mice compared to NTG littermates (P < 0.05; Fig. 1, E and F), whereas no significant difference was detected between NTG and CryABWT tg mice (Fig. 1, E and F).
Fig. 1. Myocardial PDE1A expression in mouse hearts overexpressing CryABR120G or wild-type CryAB.
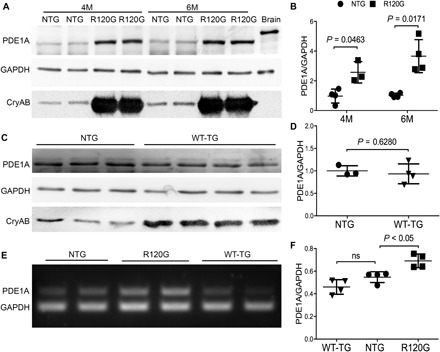
Total protein and total RNA from ventricular myocardial samples were used for Western blot analyses (A to D) and RT-PCR (E and F), respectively. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (A and B) Representative images (A) and pooled densitometry data (B) of Western blot analyses for PDE1A in 4-month-old (4M) or 6-month-old (6M) CryABR120G tg (R120G) and their NTG littermate mice. For each time point, four NTG (two males and two females) and four R120G (two males and two females) mice were used. (C and D) Western blot analysis for myocardial PDE1A in CryABWT tg (WT-TG) and NTG control mice at 6 months. Littermate NTG (one male and two females) and WT-TG (two males and two females) were used. (E and F) Representative image (E) and pooled densitometry data (F) of RT-PCR analyses for myocardial PDE1A mRNA levels in NTG, R120G, and WT-TG mice at 6 months. For each genotype, two males and two females were tested. P values are derived from two-tailed unpaired t test with Welch’s correction (B and D) or one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison tests (F). ns, not significant.
Decreasing GFPdgn protein levels in mouse hearts by PDE1 inhibition
Activation of PKG promotes UPS proteolytic function (11). Cell culture studies carried out with noncardiomyocyte cells have showed that cAMP/PKA also stimulates proteasome activities (12). Both PKA and protein phosphatase 2A were found in the proteasome complex isolated from cardiac muscle (20). Since PDE1 hydrolyzes both cAMP and cGMP, we sought to determine whether PDE1 inhibition in mice is capable of enhancing myocardial UPS degradation of GFPdgn, a well-established UPS reporter substrate and a slightly shorter version of GFPu, which is a green fluorescent protein (GFP) modified by carboxyl fusion of the degron CL1 (21). We examined changes in myocardial GFPdgn expression at 6 hours after administration of IC86430 [3 mg/kg, intraperitoneally (ip)], a PDE1-specific inhibitor (17). Myocardial GFPdgn protein levels were ~40% lower in the IC86430-treated mice than that in vehicle-treated mice (P = 0.0411; Fig. 2, A and B), with the GFPdgn reduction evidenced primarily in the cardiomyocyte compartment (Fig. 2C); however, GFPdgn mRNA levels were comparable between the two groups (P = 0.8980; Fig. 2, D and E), indicating that the cardiac reduction of GFPdgn proteins by PDE1 inhibition occurs at the posttranscriptional level, supporting the interpretation that the decreased GFPdgn protein level is indicative of enhanced UPS proteolysis. The 26S proteasome exists in two forms: doubly capped (i.e., a 20S core is capped at both ends by the 19S complex) and singly capped (i.e., only one end of the 20S core is capped by a 19S complex) (5). When the cytosolic fractions from the IC86430-treated GFPdgn mice were analyzed with native gel electrophoresis followed by an in-gel proteasome peptidase activity assay, they showed increased peptidase activities both in singly (P = 0.0006 versus vehicle; Fig. 2, F and G) and doubly capped species (P = 0.0026 versus vehicle; Fig. 2, F and G), whereas proteasome assembly was not discernibly altered (Fig. 2, F and H). Together, these results suggest that PDE1 inhibition by IC86430 increases myocardial 26S proteasome activities, stimulates UPS proteolytic function, and facilitates the clearance of a surrogate misfolded protein in vivo.
Fig. 2. Effects of IC86430 on GFPdgn expression and the assembly and activity of 26S proteasomes in mouse hearts.
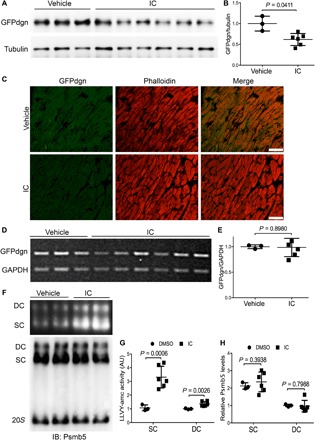
Adult GFPdgn tg mice were treated with IC86430 (IC; 3 mg/kg, ip; n = 6) or equivalent amount of vehicle (n = 3). Ventricular myocardium was sampled 6 hours after injection for the indicated assays. (A and B) Western blot analysis images (A) and pooled densitometry data (B) for the indicated proteins. (C) Representative fluorescence confocal micrographs of IC86430- or vehicle-treated GFPdgn mouse ventricular myocardium. Scale bars, 50 μm. (D and E) RT-PCR image (D) and densitometry data (E) of myocardial GFPdgn mRNA levels. Duplex RT-PCR for GFPdgn and GAPDH were performed. (F to H) Analyses for the activity and abundance of 26S proteasomes after native gel electrophoresis. Myocardial crude protein extracts were subject to native gel electrophoresis and then in-gel peptidase activity assays using Suc-LLVY-AMC as the fluorogenic substrate; the fractionated proteins in the native gel were then transferred to a polyvinylidene difluoride membrane and subjected to immunoblotting (IB) for Psmb5 to assess the abundance of the doubly capped 26S proteasomes (DC) and singly capped 26S proteasomes (SC). Shown are representative images of the in-gel peptidase activity assay (F, top) and corresponding Western blot (F, bottom), as well as the pooled densitometry data (G and H) of respective assays. All P values in this figure were derived from two-tailed unpaired t test with Welch’s correction. AU, arbitrary units.
Protection against HFpEF and premature death of CryABR120G mice by chronic PDE1 inhibition
Since PDE1 inhibition can enhance cardiac UPS proteolytic function and PFI has proven to be a major pathogenic factor in cardiac proteinopathy (7), we decided to test whether chronic PDE1 inhibition is able to mitigate CryABR120G-based cardiac proteinopathy in mice. Line 134 CryABR120G tg mice develop well-characterized cardiac proteinopathy (19). Briefly, the animals do not show any cardiac pathology at 1 month of age, but they do show restrictive cardiomyopathy at 3 months and HF at 6 months and die between 6 and 7 months of age (19). Hence, this model serves as an excellent cardiac proteinopathy model for studying pathogenesis and experimental intervention. We started the treatment at exactly 4 months of age, when cardiac hypertrophy and heart malfunction are readily discernible (fig. S2), but the mice are approximately 2 months younger than their median life span (7, 19). A cohort of line 134 CryABR120G tg mice was randomly assigned to either IC86430 (3 mg/kg per day; TG-IC) or vehicle control [equivalent amount of 60% dimethyl sulfoxide (DMSO) in saline; TG-Vehicle] treatment groups, and the treatment was designed to last for 4 weeks via osmotic minipumps. We carried out Kaplan-Meier survival analysis, in addition to serial echocardiography performed immediately before (baseline) and 4, 6, and 8 weeks after minipump implantation. As expected, no statistically significant difference in any of the echocardiographic parameters was detected between TG-IC and TG-Vehicle groups before initiation of the treatment (table S1). However, data from echocardiography performed at the day immediately before minipump implantation reveal a moderate HFpEF phenotype in CryABR120G tg mice, as reflected by significant decreases in left ventricular (LV) end diastolic volume (LVEDV), in stroke volume (SV), and in cardiac output (CO), as well as increases in LV end diastolic posterior wall thickness (LVPWd) along with unchanged EF, compared with the NTG littermate mice (fig. S2). These echocardiographic abnormities were substantially attenuated (CO and LVPWd) or even normalized to the NTG control levels (LVEDV and SV), without altering EF and the heart rate, at the end of the 4-week treatment in the TG-IC group compared with the TG-Vehicle group (fig. S3 and Fig. 3, A, C, E, G,and I). Although virtually all echocardiographic parameters became indistinguishable between the TG-IC and TG-Vehicle groups by 4 weeks after termination of the treatment (i.e., 8 weeks after initiation of treatment), analyses of the combined data of the entire 8 weeks of assessment period using the area under the curve (AUC) of each parameter as the index convincingly show significantly improvement in LVEDV, SV, and CO in the TG-IC group compared with the TG-Vehicle group, while EF and heart rate remained comparable between the two groups throughout (Fig. 3, B, D, F and J). The echocardiography-revealed differences in LVEDV, SV, CO, and EF among NTG, TG-Vehicle, and TG-IC groups were corroborated by findings from catheter-based LV pressure-volume assessments (fig. S4) performed on a parallel cohort at the end of 4 weeks of treatment. At this time point, the TG-Vehicle group also displayed significant increases in LV end diastolic pressure (LVEDP) and the time constant of relaxation (Tau), as well as reduced maximum rate of LV pressure decline (i.e., minimum dP/dt), compared with the NTG group. This confirms diastolic malfunction in TG-Vehicle mice. In addition, the increases in Tau and LVEDP were attenuated or nearly normalized by IC86430 treatment (fig. S4). Neither systolic nor diastolic blood pressure in the aorta was different between TG-Vehicle and TG-IC groups (fig. S5). Together, these data demonstrate that the IC86430 treatment improves cardiac function and effectively attenuates HFpEF in CryABR120G tg mice.
Fig. 3. Effects of PDE1 inhibition on CryABR120G tg mouse LV function and morphometry and life span.
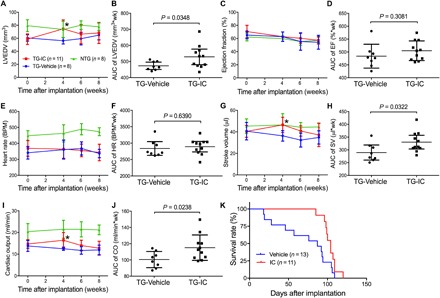
The treatment of CryABR120G tg mice with IC86430 (3 mg/kg per day, TG-IC) or vehicle control (TG-Vehicle) was initiated at exactly 4 months of age and lasted for 4 weeks via subcutaneous implantation of mini-osmotic pumps. Mice were observed daily for survival and subjected to serial echocardiography on the day before (baseline) and 4, 6, and 8 weeks after the minipump implantation. (A to J) LV morphometry and function parameters derived from the serial echocardiography of the TG-IC (four males and seven females) or TG-Vehicle (four males and four females) mice or of age-matched NTG littermates (four males and four females) without any intervention. The stacked line chart of each panel summarizes the time course of changes in the indicated parameter. Mean ± SD; *P < 0.05 TG-IC versus TG-Vehicle of the same time point (two-way repeated measures ANOVA followed by Bonferroni’s test). The scatter dot plot of each panel presents the AUC of the indicated parameter versus time obtained using the trapezoidal rule. Each dot represents a mouse; means ± SD are superimposed; two-tailed unpaired t test with Welch’s correction were used. wk, weeks; BPM, beats per minute. (K) Kaplan-Meier survival analysis. The median life spans of the TG-IC tg group (four males and seven females) and the TG-Vehicle (six males and seven females) groups are 224 days and 207 days, respectively. P = 0.0235, log-rank test.
Consistent with the echocardiography and hemodynamics data, the Kaplan-Meier survival analysis of the cohort revealed that IC86430 treatment remarkably delayed the premature death of CryABR120G tg mice (P = 0.0235; Fig. 3K). Notably, the TG-Vehicle group displayed a survival curve that is comparable to that was previously reported for this line of tg mice (7, 22, 23). During the 4-week IC86430 treatment and within the 6 weeks after termination of the IC86430 treatment, none of the TG-IC mice died, but ~50% of the TG-Vehicle mice died during this period (Fig. 3K), demonstrating a notable and long-lasting therapeutic benefit from the IC86430 treatment.
Decreasing oligomeric CryABR120G and increasing Ser14-phosphorylated Rpn6 in proteinopathic mouse hearts by PDE1 inhibition
Given that IC86430 treatment enhances degradation of a surrogate UPS substrate (GFPdgn) in mouse hearts (Fig. 2A), we next asked whether PDE1 inhibition would promote proteasomal degradation of misfolded CryABR120G proteins in the heart. A recent study developed a novel method to distinguish proteasomal degradation of misfolding-prone proteins from other cellular mechanisms that regulate total protein levels (24). In this assay, total protein lysate is separated into soluble (supernatant) and insoluble fractions (pellet, NI) using an NP-40 lysis buffer. The pellet is then suspended in an SDS buffer and is further divided into SDS-soluble (SS) and SDS-insoluble fractions. The SS fraction was rapidly increased by proteasome inhibition, suggesting that it derives from a misfolded species and can be readily prevented by the proteasome; therefore, changes in the level of a stably expressed misfolding-prone protein in the SS fraction inversely reflect proteasome proteolytic activity (24). Hence, we examined the SS fraction of CryAB at the end of the 4-week treatment. As expected, the protein level of the SS fraction of CryAB was barely detectable in NTG mouse hearts but was significantly increased in CryABR120G tg mice (fig. S6); the increase was remarkably attenuated by IC86430 treatment (P = 0.0014; Fig. 4, A and B). Furthermore, the protein level of Ser14-phosphorylated RPN6 (p-Rpn6), which has proven to be increased by PKA activation (12, 13), was significantly increased in TG-IC mice compared with TG-Vehicle mice (P = 0.0189; Fig. 4, C and D), indicating that myocardial PKA-mediated proteasome stimulation is enhanced by PDE1 inhibition. PDE4 specifically hydrolyzes cAMP (14). To test whether proteasome phosphoregulation and functioning can also be stimulated by increasing cAMP/PKA signaling alone in diseased hearts with IPTS, we treated a cohort of 4-month-old CryABR120G tg mice with piclamilast, a selective inhibitor of PDE4, at doses ranging from 0.03 to 1.0 mg/kg per injection (ip, twice daily for 3 days). Examination of the myocardial samples collected at 6 hours after the sixth injection revealed that myocardial p-Rnp6 (Fig. 4, E and F) and 26S proteasome chymotrypsin-like activities (Fig. 4G) were remarkably increased by piclamilast treatment in a dose-dependent manner. Together, these in vivo data highly support that PDE1 inhibition leads to PKA-mediated phosphorylation of Rpn6 and thereby increases proteasome activity and facilitates proteasomal degradation of misfolded proteins in the heart.
Fig. 4. Reduction of myocardial abundance of oligomeric CryAB by PDE1 inhibition is associated with PKA-mediated proteasome activation in CryABR1220G tg mice.
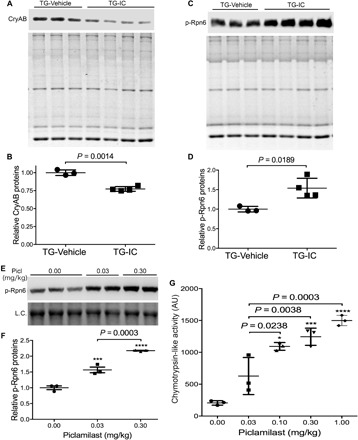
(A to D) A separate cohort of CryABR120G mice subject to the same treatment as described in Fig. 3 was euthanized immediately at the end of the 4-week treatment, and ventricular myocardium was sampled for protein analyses. Shown are the Western blot images (A) and summary of densitometry data (B) of the NP-40–insoluble but SS fraction of CryAB, as well as representative images (C) and pooled densitometry data (D) of Western blot analyses for p-Rpn6. The stain-free total protein imaging technology was used to visualize all proteins in the SDS-PAGE gel used for the Western blot analysis. The total protein signals of corresponding lane of the gel [the bottom image of (A) and (C)] are used as the in-lane loading control. Each lane (A and C) or each dot (B and D) represents an independent mouse. The difference between the TG-IC (three males and one female) and the TG-Vehicle (two males and one female) groups is evaluated statistically using two-tailed unpaired t test with Welch’s correction. (E to G) A cohort of 4-month-old CryABR120G tg mice were treated with a selective PDE4 inhibitor piclamilast at the indicated dosage (ip, twice daily × 3 days). Ventricular tissues collected at 6 hours after the sixth injection were used for Western blot analyses of myocardial p-Rnp6 (E and F) and for 26S proteasome chymotrypsin-like activity assays (G). Shown are a representative image (E) and the pooled densitometry data of Western blot for p-Rpn6, as well as the scatterplot of the individual slopes of 26S proteasome chymotrypsin-like activity assays (G). Each dot represents a mouse; means ± SD are superimposed. L.C., a segment of the in-lane loading control image derived from the stain-free total protein imaging technology. *P < 0.05, *** P < 0.005, and **** P < 0.0001 versus the 0 mg/kg (vehicle control) group; one-way ANOVA followed by Tukey’s test for pair-wise comparison.
Proteasome enhancement by PDE1 inhibition in cultured cardiomyocytes
Next, we tested the effect of PDE1 inhibition on proteasome proteolytic activities in cultured cardiomyocytes using both small peptide substrates and a full-length protein surrogate. In-gel proteasome peptidase activity assays using the cytosolic fractions revealed significantly higher peptidase activities in both singly capped and doubly capped proteasomes from IC86430-treated neonatal rat ventricular myocytes (NRVMs) than those from vehicle-treated cells (Fig. 5, A to C). To determine whether PDE1 inhibition enhances the degradation of a known substrate of the UPS in a cardiomyocyte-autonomous fashion, we tested the effect of IC86430 treatment on the stability of GFPu in cultured NRVMs. Adenoviral vectors expressing GFPu (Ad-GFPu) or a red fluorescent protein (Ad-RFP) were used. The RFP has a much longer half-life than GFPu and is not an efficient UPS substrate, whereas the GFPu is a proven UPS substrate (11). The Ad-RFP has exactly the same expression control elements for RFP as the Ad-GFPu for GFPu; hence, RFP protein levels can serve as a valid control for both viral infection efficiency and protein synthesis when a mixture of Ad-RFP and Ad-GFPu is used (11). In Ad-GFPu– and Ad-RFP–coinfected NRVMs, treatment with IC86430 dose-dependently decreased the ratio of GFPu to RFP, indicative of increased degradation of GFPu (Fig. 5, D and E). Moreover, a cycloheximide (CHX) chase assay showed that PDE1 inhibition significantly shortened the half-life of GFPu (P = 0.0026; Fig. 5, F and H), demonstrating that PDE1 inhibition promotes UPS proteolytic function in cultured cardiomyocytes. Together, these results strongly suggest that UPS proteolytic function enhancement by PDE1 inhibition is at least in part attributable to enhanced activities of the 26S proteasome.
Fig. 5. PDE1 inhibition by IC86430 increases proteasomal peptidase activities and proteasomal degradation of misfolded proteins in cultured neonatal rat ventricular myocytes (NRVMs).
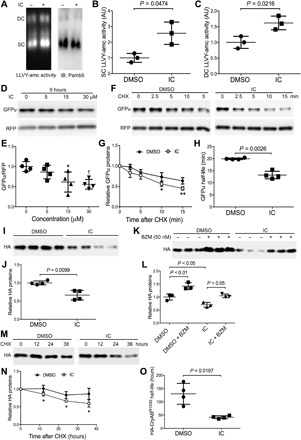
(A to C) In-gel proteasome peptidase activity assays followed by immunoblotting (IB) for Psmb5 were performed as described in Fig. 2F. NRVMs in culture were treated with IC86430 (15 μM) for 9 hours before being harvested for crude protein extraction. A fraction of each sample was used for the in-gel proteasomal peptidase assay (A to C). Representative images (A) and pooled data of three biological repeats (B and C) of the cleavage of the LLVY-AMC fluorogenic substrate are shown. Two-tailed unpaired t test with Welch’s correction. (D and E) Representative images (D) and pooled densitometry data (E) of Western blot analyses for the steady state protein levels of the co-overexpressed GFPu and RFP. Cultured NRVMs were coinfected with a mixture of recombinant adenoviruses expressing GFPu and RFP in serum-free medium for 6 hours. Cells were then cultured in DMEM containing 2% fetal bovine serum (FBS) for 48 hours before IC86430 treatment. Total cell lysates collected after 9 hours of IC86430 treatment at the indicated dosages were analyzed. *P < 0.05 and †P < 0.01 versus the 0 μM group; n = 4 repeats; one-way ANOVA followed by Dunnett’s test. (F to H) CHX chase assays for GFPu degradation. CHX treatment (100 μM) was started 5 hours after the treatment of IC86430 (15 μM) or DMSO. In each run of the CHX chase, the GFPu chase was started 15 min after addition of CHX to the culture dish, the GFPu image density at this time point was set as 1 arbitrary unit, and the GFPu levels of subsequent time points were calculated relative to it. Representative Western blot images (F), summarized GFPu decay curves (G), and the derived GFPu half-lives (H) are shown. *P < 0.05 and ** P < 0.01 versus DMSO group; two-tailed unpaired t test with Welch’s correction. (I to O) cultured NRVMs were infected with adenoviruses expressing HA-tagged CryABR120G (Ad-HA-CryABR120G) in serum-free medium for 6 hours. Cells were then cultured in DMEM containing 2% serum for 24 hours before treated with IC86430 (30 μM) or vehicle control (DMSO). (I and J) Representative image (I) and pooled densitometry data (J) of Western blot analyses for HA-CryABR120G in the NP-40–insoluble but SS fraction of NRVMs treated with IC86430 for 6 hours. (K and L) Reduction of a misfolded species of CryAB level by IC86430 is proteasome dependent. The proteasome inhibitor BZM (50 nM) or volume corrected DMSO was applied to NRVM cultures 10 hours after the initiation of IC86430 or DMSO treatment. Six hours later, the cells were harvested for extraction of the NP-40–insoluble but SS fraction of proteins. Representative images (K) and pooled densitometry data from three repeats (L) of Western blot analyses for HA-CryABR120G are shown. Two-way ANOVA followed by Tukey’s multiple comparisons test; n = 3 repeats. (M to O) CHX chase assays of HA-CryABR120G. CHX (50 μM) treatment was started after 16 hours of IC86430 or DMSO treatment. Total protein extracts from NRVMs harvested at the indicated time points after CHX administration were subject to Western blotting for HA-CryABR120G. Representative images (M), HA-CryABR120G decay curves (N), and the derived HA-CryABR120G half-lives (O) are presented. *P < 0.05 versus DMSO group, two-tailed unpaired t test with Welch’s correction; n = 4 repeats.
Accelerating proteasomal degradation of CryABR120G by PDE1 inhibition
To determine whether the PDE1 inhibition–induced acceleration of misfolded protein degradation in the heart is cardiomyocyte autonomous, we sought to further examine the effect of PDE1 inhibition on degradation of a human disease–linked misfolded protein, CryABR120G, in cultured NRVMs. Hemagglutinin epitope (HA)–tagged CryABR120G was overexpressed in NRVMs via adenoviral gene delivery. Consistent with in vivo data, PDE1 inhibition by IC86430 significantly reduced CryABR120G in the NP-40–insoluble but SS fraction (P = 0.0099; Fig. 5, I and J). The reduction of CryABR120G protein level by IC86430 is proteasome-dependent as the reduction was reversed in the presence of proteasome inhibitor bortezomib (BZM; Fig. 5, K and L). Furthermore, the CHX chase assay revealed that PDE1 inhibition significantly shortened CryABR120G protein half-life (P = 0.0197; Fig. 5, M to O). Together, these data further confirm that PDE1 inhibition enhances proteasome-mediated degradation of misfolded proteins in cardiomyocytes.
Attenuation of PDE1 inhibition’s proteasomal enhancement by inhibiting PKA or PKG
Experiments performed with noncardiac cells have demonstrated that stimulating the cAMP/PKA pathway increases 26S proteasome activities and promotes proteasomal degradation of several disease-linked misfolded proteins through phosphorylating RPN6 at its Ser14 (12). To test whether cAMP/PKA would do the same in cardiomyocytes, we treated cultured NRVMs with forskolin, a potent activator of adenylate cyclase, to increase cAMP, in the absence or the presence of a PKA inhibitor H89. CHX chase assays further showed that forskolin significantly shortened the half-life of GFPu in the cultured NRVMs (Fig. 6, A and B), confirming that elevation of cAMP improves UPS proteolytic function in cardiomyocytes. Western blot analyses revealed that forskolin remarkably increased the level of p-Rpn6, and this increase was completely blocked by H89 cotreatment (Fig. 6C), confirming that the proteasome phosphoregulation by cAMP/PKA does occur in cardiomyocytes. Previously, we have reported that cGMP/PKG positively regulates 26S proteasome activities and the proteasomal degradation of misfolded proteins in cardiomyocytes (11). Since PDE1 breaks down both cAMP and cGMP, we then sought to test the role of PKA or PKG activation in the acceleration of GFPu degradation by PDE1 inhibition. Both PKG inhibitor (KT5823) and PKA inhibitor (H89) significantly attenuated the reduction of GFPu by IC86430 treatment (Fig. 6, D to G), indicating that both PKA and PKG activation mediate PDE1 inhibition–induced proteasomal function enhancement in cardiomyocytes.
Fig. 6. Accelerated degradation of GFPu by PDE1 inhibition in cardiomyocytes is PKA and PKG dependent.
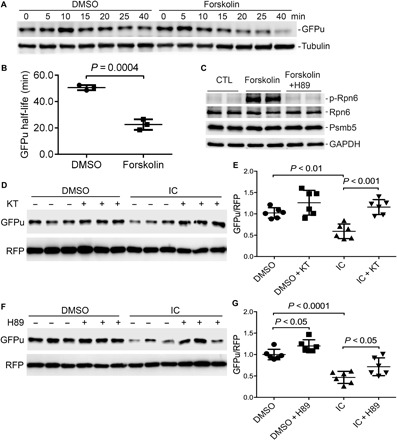
(A and B) CHX chase assay for changes in GFPu protein stability induced by increasing cAMP in cardiomyocytes. NRVMs were infected with Ad-GFPu in serum-free medium for 2 hours; the medium was then switched to 10% FBS medium for the remainder of the experiment. Forty-eight hours later, the treatment with forskolin (10 μM), a potent adenylyl cyclase activator, or with the vehicle control (DMSO) was initiated, which was 2 hours before blocking protein synthesis with CHX (100 mg/ml). Cell lysates harvested at the indicated time point after CHX was subject to SDS-PAGE and Western blot analysis for GFPu and β-tubulin. Example Western blot images (A) and the estimated GFPu protein half-lives (B) are shown. Two-tailed unpaired t test with Welch’s correction. (C) Western blot analyses for p-Rpn6 and other indicated proteins in NRVMs subject to the indicated treatment for 6 hours. The PKA inhibitor H89 (5 μM) was added 5 min before forskolin (10 μM). (D to G) Western blot analyses for GFPu and RFP coexpressed in cardiomyocytes subject to the indicated treatments. Cultured NRVMs were coinfected with Ad-GFPu and Ad-RFP in serum-free medium for 6 hours. The cells were then cultured in DMEM containing 2% serum for 24 hours before treatment indicated below. The treatment with a PKG inhibitor KT5823 (KT; 1 μM) or volume-corrected vehicle (DMSO) was started 3 hours before IC86430 treatment (30 μM). The cells were harvested 9 hours later (D and E). For the PKA experiment, NRVMs were pretreated with IC86430 (30 μM) or with volume-corrected vehicle DMSO for 3 hours before H89 (5 μM), or volume-corrected vehicle DMSO was administered. The cells were harvested 6 hours later (F and G). Representative Western blot analyses (D and F) for GFPu with RFP probed as the loading control and a summary of the pooled densitometry (E and G) are shown. Two-way ANOVA followed by Tukey’s multiple comparisons test; n = 6 repeats per manipulation.
DISCUSSION
Prior to the present study, Miller et al. (17) demonstrated that PDE1A was up-regulated in hypertrophied mouse hearts and isolated cardiomyocytes exposed to hypertrophic stimuli such as ISO or angiotensin II (AngII). Both pharmacological inhibition of PDE1 with IC86340 and gene silencing of PDE1A attenuated cardiac hypertrophy, an effect associated with activation of the cGMP/PKG axis (17). PDE1C was reported to play a critical role in pressure overload–induced pathological cardiac remodeling and dysfunction in a PKA-dependent manner (18). In addition, PDE1 inhibition with IC86340, as shown by Zhang et al. (25), protected against doxorubicin-induced cardiac toxicity and dysfunction in vivo. Recently, emerging evidence showed that patients in the end-state HF display increased protein levels of PDE1A (26) and increased mRNA levels of PDE1C (18, 26). In the present study, we used both in vitro and in vivo approaches to investigate the therapeutic effects and underlying mechanism of PDE1 inhibition on HFpEF induced by proteotoxicity and have gained the following novel findings: (i) PDE1A expression at both mRNA and protein levels is up-regulated in mouse hearts with advanced cardiac proteinopathy induced by CryABR120G, a human misfolded protein; (ii) CryABR120G mice develop HFpEF at 4 months of age, and chronic PDE1 inhibition initiated at this stage effectively blocks the progression of HFpEF and delays mouse premature death in this model; and (iii) PDE1 inhibition, likely acting on the cAMP/PKA axis and the cGMP/PKG pathway, promotes proteasomal degradation of misfolded proteins, thereby protecting cardiomyocytes from proteotoxicity. Together, we demonstrate here that duo-activation of PKA and PKG by PDE1 inhibition facilitates proteasomal degradation of misfolded proteins and thereby protects against cardiac diastolic malfunction caused by IPTS.
Potential mechanisms underlying the protection of PDE1 inhibition against proteinopathy
The mechanisms underlying the beneficial effects of PDE1 inhibition on cardiac function are likely quite complex, given the potentially multipronged involvement of PDE1 (a dual PDE) in cardiac regulation. As elaborated below, findings of this study strongly support the notion that improving cardiac PQC via enhancing proteasomal degradation of misfolded proteins represents an important molecular mechanism underlying the therapeutic effect of PDE1 inhibition against HFpEF due to the αB-crystallin mutation.
First, our data compellingly support that PDE1 inhibition–induced UPS enhancement protects the heart from proteotoxic stress. The degradation of individual misfolded proteins is primarily performed by the UPS. When UPS proteolytic function is impaired or becomes inadequate, misfolded proteins undergo aberrant protein aggregation with the formation of oligomers in the initial step. The intermediate oligomers are believed to be toxic to the cell (3). In the present study, we showed that the NP-40–insoluble but SS fraction of CryABR120G (i.e., the oligomers formed by misfolded CryAB) was significantly decreased by IC86430 treatment (Fig. 4, A and B). In addition, PDE1 inhibition also reduced the oligomeric CryABR120G in cultured NRVMs (Fig. 5, I and J). The reduction of CryABR120G oligomers by PDE1 inhibition in cardiomyocytes is proteasome dependent because the reduction was prevented by proteasome inhibition (Fig. 5, K and L). Thus, the improvement of cardiac function by IC86430 treatment in proteinopathic mice is at least in part attributable to the enhanced clearance of misfolded CryABR120G by the UPS. Accompanied with the reduction of misfolded proteins, the myocardial protein level of p-Rpn6 was markedly up-regulated in IC86430-treated CryABR120G tg mice (Fig. 4, C and D). The 26S proteasome consists of a 20S proteolytic core particle (the 20S) and a 19S regulatory particle (the 19S) attached at one or both ends of the 20S. The 19S is composed of at least 17 subunits, including regulatory particle non–adenosine triphosphatases (ATPases) 1 to 12 and regulatory particle triple-A+ ATPases 1 to 6 (5). Recent studies revealed that phosphorylation of Rpn6 at Ser14 by PKA increased proteasome activity and thereby enhanced proteasomal degradation of misfolded proteins in the tested cells, which are noncardiac cells (12); here, we were able to confirm that PKA activation increases p-Rpn6 and facilitates degradation of a UPS surrogate substrate (GFPu) in cardiomyocytes as well (Fig. 6, A to C). Furthermore, in cultured NRVMs, we found that 26S proteasome peptidase activities were substantially increased (Fig. 5, A to C), and the degradation of GFPu was remarkably accelerated (Fig. 5, D to H) by PDE1 inhibition. The accelerated GFPu degradation by IC86430 was discernibly attenuated by inhibition of either PKA or PKG (Fig. 6, D to G), indicating that improving UPS functioning in cardiomyocytes by PDE1 inhibition depends on the activation of both PKA and PKG, which is also consistent with PDE1 being a duo-substrate PDE. Our previous study has shown that PKG activation enhances proteasome peptidase activity and promotes degradation of misfolded proteins (11). Hence, it is very likely that PDE1 inhibition enhances both cAMP/PKA– and cGMP/PKG–dependent activation of the proteasome, thereby promoting proteasomal degradation of misfolded proteins in the heart.
Second, PFI is a major pathogenic factor for the model we used, and correction of PFI is known to be cardioprotective. Enhancing cardiac proteasome function by cardiomyocyte-restricted overexpression of proteasome activator 28α remarkably reduces aberrant protein aggregation, slows down the progression of proteinopathy, delays mouse premature death in this CryABR120G-based proteinopathy mouse model, and protects against acute myocardial I/R injury, demonstrating that PFI is a major pathogenic factor in heart disease with IPTS (7). In agreement with findings of the present study, increasing cGMP and thereby activating PKG by PDE5 inhibition with sildenafil also has been demonstrated to increase myocardial proteasome activities, decrease myocardial CryABR120G protein levels, and slow down the disease progression in CryABR120G mice (11), the first in vivo demonstration that proteasome function can be pharmacologically enhanced to treat disease. The activation of PKA by PDE4 inhibition has been shown to protect against a mouse model of Alzheimer’s disease (27). No reported study has tested directly the effect of PKA activation on cardiac proteotoxicity, but pharmacologically induced PKA activation was shown to increase myocardial proteasome assembly and proteasome activities (28). The present study also demonstrates that increasing cAMP via acute PDE4 inhibition is sufficient to increase PKA-mediated p-Rpn6 and proteasome peptidase activities in proteinopathic hearts (Fig. 4, E to G). Therefore, there is compelling evidence that, through PKA- and PKG-mediated proteasomal activation, PDE1 inhibition with IC86430 facilitates degradation of misfolded proteins in cardiomyocytes and thereby reduces cardiac proteotoxicity and protects the heart against IPTS. Although the cardiac proteasome impairment hypothesis had not been proposed until years after neural scientists hypothesized a pathogenic role for UPS impairment in neurodegeneration, cardiac experimentalists were able to take the lead to establish both genetically and pharmacologically that impaired proteasome function plays a major role in cardiac pathogenesis (7, 10, 11, 29, 30), even before colleagues of neuroscience did so for neural disorders (27). Pathological studies have revealed that accumulation of ubiquitinated proteins and preamyloid oligomers and reduction of proteasome activities are prevalent in explanted failing human hearts (6, 31, 32), further supporting a major pathogenic role for proteasome impairment in a large subset of HF in humans (3). Therefore, findings of the present study provide a new compelling rationale for PDE1 inhibition to treat HF.
Loss of titin distensibility increases cardiomyocyte stiffness, which can, in turn, contribute to increased stiffness of human failing myocardium. CryAB can bind titin and thereby protect cardiomyocytes from titin aggregation–induced stiffening (33). A recent report has further shown that in vitro administration of CryAB reduced the stiffness of cardiomyocytes from patients with HFpEF due to aortic stenosis probably through relief of titin aggregation and thereby improving titin distensibility (34). Therefore, a potential mechanism underlying the observed therapeutic efficacy of PDE1 inhibition could be the reduction of cardiomyocyte stiffness due to improved titin elasticity. There are two possible scenarios for the mutant CryAB to reduce titin distensibility: First, similar to wild-type CryAB, the mutant and aggregation-prone CryAB may also bind to titin, but, unlike wild-type CryAB, the mutant CryAB may promote titin aggregation, thereby reducing titin distensibility. Second, the mutant CryAB dominant negatively inhibits wild-type CryAB and prevents the latter from protecting titin. In either scenario, enhanced proteasomal removal of the mutated CryAB proteins would conceivably attenuate the direct or indirect toxic effects of this misfolded protein on titin elasticity, thereby improving cardiomyocyte stiffness.
Potential contributions of proteasome enhancement to most reported pharmacological benefits of PDE1 inhibition
One of the beneficial effects of PDE1 inhibition, as demonstrated by Miller et al. (17), is protection against pathological cardiac hypertrophy. They found that PDE1 inhibition by IC86340 and PDE1A down-regulation by PDE1A-specific small hairpin RNA attenuated phenylephrine-induced hypertrophic responses in isolated rat cardiomyocytes in a PKG-dependent manner, highly suggesting a PDE1A-cGMP/PKG axis in regulating cardiac hypertrophy in cardiomyocytes. However, the underlying mechanism by which PDE1A-mediated cGMP/PKG regulates cardiac hypertrophy remains elusive. More recently, Knight et al. (18) reported that global PDE1C knockout significantly attenuated transverse aortic constriction–induced cardiac remodeling and dysfunction, characterized by decreases in chamber dilation, myocardial hypertrophy, and interstitial fibrosis in mice. In line with the reported antihypertrophic effect of PDE1 inhibition (16, 17), we also observed a statistically significant decrease of LVPWd wall thickness at the end of the 4-week IC86430 treatment in CryABR120G mice (table S2). Pathological cardiac hypertrophy is a maladaptive response to increased stress and requires increased protein synthesis, which inevitably increases production of misfolded proteins and proteotoxic stress (3). This is because (i) it has been reported that approximately one-third of the newly synthesized polypeptides never became mature proteins and instead were cotranslationally degraded by the UPS (35) and (ii) a hypertrophied cardiomyocyte demands for a greater PQC capacity to maintain the quality of the mature proteins. Hence, for quality control purposes, the demand for UPS-mediated proteolysis is increased in response to cardiac hypotrophy (3). UPS inadequacy and PFI have been observed in pressure overload cardiomyopathy, and PFI activates the calcineurin-nuclear factor of activated T-cells (NFAT) pathway (3, 29, 36, 37), an important signaling pathway to pathological hypertrophy. PQC improvement by removal of misfolded proteins and protein aggregates has been shown to attenuate cardiac hypertrophy (7, 22). Proteasome enhancement was also shown to protect against pressure overload–induced right ventricular failure (29). Therefore, it is conceivable that PQC improvement resulting from PKA- and PKG-mediated proteasome enhancement contributes to the antihypertrophic effect of PDE1 inhibition.
Reduction of cardiomyocyte death is another reported benefit from PDE1 inhibition. PDE1C depletion or PDE1 inhibition using IC86340 abolished AngII- or ISO-induced cell death in isolated cardiomyocytes. The anti-apoptotic effects of PDE1C deficiency or inhibition were mediated by the cAMP/PKA axis (18). Zhang et al. (25) further demonstrated that PDE1C colocalized with adenosine A2 receptors (A2Rs) in cardiomyocyte membrane and T-tubules and that the anti-apoptotic effect of PDE1 inhibition is dependent on cAMP-generating A2Rs in cardiomyocytes, suggesting that PDE1C selectively regulates A2R/cAMP signaling in cardiomyocytes. Since impaired PQC resulting from PFI is sufficient to cause cardiomyocytes to die, proteasome enhancement may also contribute to the prosurvival effect of PDE1 inhibition. Overexpression of a polyglutamine oligomer or CryABR120G results in cardiomyocyte death, and conversely, accelerating the degradation of misfolded proteins improves cardiomyocyte survival (11, 38). Proteasome inhibition by N-carbobenzyloxy-l-leucyl-l-leucyl-l-leucinal (MG-132) alone is sufficient to induce cell death in cultured NRVMs (3). Beyond PQC, proteasomal degradation also positively regulates cell signaling pathways that support cardiomyocyte survival. Akt-mediated prosurvival signaling was impaired in a mouse genetic model of cardiomyocyte-restricted proteasomal inhibition during myocardial I/R injury, likely due to impairment of proteasome-mediated degradation of phosphatase and tensin homolog (PTEN), rendering cardiomyocytes more vulnerable to apoptosis in response to I/R (30).
In addition to acting on cardiomyocytes, PDE1 inhibition and PDE1A down-regulation attenuated AngII- or transforming growth factor–β (TGF-β)–induced cardiac myofibroblast activation and extracellular matrix synthesis in cultures (16). The antifibrotic effect is dependent on the suppression of both cAMP/Epac1/Rap1 signaling and cGMP/PKG signaling (16). PDE1C, which is only expressed in cardiomyocytes and undetectable in cardiac fibroblasts, also regulates TGF-β–induced fibroblast activation, likely through a paracrine-dependent mechanism (18). Hence, both in vitro and in vivo evidence compellingly support an antifibrotic effect for PDE1 inhibition. It is well known that myocardial fibrosis can be induced by loss of cardiomyocytes, known as replacement fibrosis, which is more often seen in HFrEF (2); HFpEF on the other hand is often associated with reactive fibrosis, where a paracrine mechanism may be more relevant (1, 2). Proteasome enhancement also could potentially participate in PDE1 inhibition’s antifibrotic effect, probably because proteasome enhancement may curtail TGF-β signaling; both TGF-β receptors and intracellular signaling mediators Smad2/3 are degraded by the UPS, and conversely, proteasome inhibition was shown to promote the TGF-β signaling (39).
PDE1 inhibition as a potential strategy to treat HFpEF
Human patients with HFpEF often have both cardiac (e.g., defective ventricular filling, concentric hypertrophy, reduced SV and CO, and preserved EF) and noncardiac manifestations (e.g., obesity, insulin resistance, and type 2 diabetes), and an integrative molecular basis for these manifestations remains murky (1, 2). Despite the prevalence of HFpEF in humans, there is an apparent shortage of animal models that can completely mimic human HFpEF conditions, hindering the pathogenic and therapeutic exploration of HFpEF. Previous characterization of the CryABR120G tg mouse line used here revealed that these mice display no discernible abnormal cardiac phenotype at 1 month of age, develop concentric cardiac hypertrophy and diastolic malfunction at 3 months, and die of HF between 6 and 7 months (19). In the present study, IC86430 treatment of CryABR120G mice was initiated at the disease stage when the animal displays all cardiac characteristics of moderate HFpEF as reflected by decreases in LVEDV, SV, CO, and LV endodiastolic internal diameter (LVIDd), increased LVPWd, and normal EF (fig. S2 and table S1). During the entire treatment session, as well as the follow-up echocardiographic surveillance, the EF in CryABR120G mice remained comparable to that in NTG mice (Fig. 3C), whereas the cardiac morphometric and functional changes characteristic of HFpEF became more pronounced in the TG-Vehicle group, indicating that the CryABR120G model is primarily an HFpEF model, at least during the treatment period. Human cardiac proteinopathies, including those caused by the CryABR120G mutation, often present in the clinic as restrictive cardiomyopathy (4), a cousin, if not an etiology, of HFpEF. We have demonstrated that a defined period of PDE1 inhibition with IC86430 can effectively diminish HFpEF-associated cardiac morphological and functional deficits, including normalizing LVEDV and SV (Fig. 3, A and G) and thereby improving CO (Fig. 3I), resulting in a significant delay in the premature death of diseased mice (Fig. 3K). This is an extremely exciting discovery as HFpEF lacks an established medical treatment.
Recently, Hashimoto et al. (26) reported that acute PDE1 inhibition using a compound (ITI-214) different from what we used here exerted cardioprotective effect on HF dogs induced by tachypacing. They observed that acute PDE1 inhibition exerts positive inotropic, lusitropic, chronotropic, and arterial vasodilatory effects in mammalian hearts expressing primarily PDE1C and that those effects are likely derived from modulation of cAMP; however, they were unable to detect the inotropic effects, although a mild systemic vasodilation and increase in heart rate, in normal mice where the dominant isoform is PDE1A. We did not test the acute effect of IC86430 treatment on cardiac mechanical performance in normal mice, but we observed cardiac proteasome functional enhancement by a single dose of IC86430 in GFPdgn mice (Fig. 2), suggesting that IC86430 and ITI-214 may differ in their targeting preference of PED1 isoforms. By contrast, we report here that chronic PDE1 inhibition with IC86430 does not discernibly alter heart rate and aortic pressure in anesthetized proteinopathic mice (figs. S4 and S5) but effectively improves cardiac diastolic function and animal survival in a well-tested mouse model of cardiac proteotoxicity likely through enhanced proteasomal degradation of misfolded proteins in a cAMP- and cGMP-dependent manner. This seeming discrepancy may arise from multiple aspects of difference between the two studies. First of all, different PDE1 inhibitors were used; different PDE1 inhibitors might have preference in not only PDE1 isoforms but also subcellular compartments as implicated by inhibitors of other PDE families (40). It is known that PDE1A and PDE1C regulate cardiac structural remodeling and function likely with distinct mechanistic actions (40). Our pan-PDE1 inhibitor (IC86430) likely inhibits both PDE1A and PDE1C, while ITI-214 is claimed as PDE1C selective; therefore, it is expected that they yield different therapeutic effects. Second, the duration of treatment differs between the studies; mice were treated continuously for 4 weeks in our study, whereas only acute effects were determined by Hashimoto et al. (26). It should be pointed out that both our in vivo and in vitro experiments reveal that PDE1 inhibition by IC86430 can acutely enhance proteasome proteolytic function in murine cardiomyocytes as well, suggesting that difference in targeting preference of PDE1 isoforms between the two PDE1 inhibitors is likely the primary cause.
Notably, the IC86430 treatment in our survival study was initiated at an overt disease stage when aberrant protein aggregates, cardiac hypertrophy, and diastolic malfunction are discernible. The Kaplan-Meier survival results are notable, indicating that the drug’s overall therapeutic efficacy is very encouraging regardless of its underlying mechanism. A couple of clinically translatable therapeutic approaches, including exercise and doxycycline treatment (22, 23), have been shown to improve the survival of the proteinopathy mouse model used in the present study, and their survival benefit seemed to be more impressive than PDE1 inhibition by IC86430 treatment. In both previous reports, the intervention started earlier (at 1 month of age for the exercise experiment and 16 weeks of age for doxycycline treatment), and both treatments lasted for the entire remaining lifetime of the mice treated; the IC86430 treatment in the present study, however, lasted for only 4 weeks. Thus, the observed life span elongation effects are not comparable. It remains to be tested but very likely that the survival improvement by IC86430 would be much greater should the treatment be started earlier or last longer.
Solo activation of either the cAMP/PKA axis or the cGMP/PKG (e.g., PDE5 inhibition) pathway has so far failed to show positive effects in clinic trials treating HFpEF for reasons remaining elusive (1, 2); however, we have demonstrated in the present study that, likely through duo-activation of the cAMP/PKA and the cGMP/PKG pathways, chronic PDE1 inhibition improves cardiac PQC, effectively blocks the progression of HFpEF, and improves survival in an animal model of cardiac proteinopathy. Together, our data and other existing reports provide compelling evidence that PDE1 inhibition shall be explored as a new therapeutic strategy for HFpEF and heart disease with IPTS.
MATERIALS AND METHODS
Animal models
The National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals was followed throughout the study. Animal protocols used in this study were approved by the University of South Dakota Institutional Animal Care and Use Committee. Animals were given ad libitum access to food and water and were housed in controlled rooms with optimum temperature (22° to 24°C) with 12-hour light/12-hour dark cycle. The creation and characterization of the tg mouse model expressing GFPdgn were reported before (21). GFPdgn is a slightly shorter version of GFPu. GFPu is an enhanced GFP that is modified by carboxyl fusion of degron CL1. Both GFPu and GFPdgn are surrogates for misfolded proteins and well-proven UPS substrates (21). The tg mice with cardiomyocyte-restricted overexpression of CryABR120G or CryABWT were described previously (19). CryABR120G, CryABWT, and GFPdgn tg mice were maintained in the FVB/N inbred background. Tg mice were identified by PCR analysis of genomic DNA isolated from toe or tail clips.
Mouse PDE1 or PDE4 inhibitor treatment
A PDE1-selective inhibitor IC86430 was provided by Eli Lilly and Company (Indianapolis, IN, USA). Age- and sex-matched GFPdgn mice were treated with IC86430 (3 mg/kg, ip) or equivalent amount of vehicle control (DMSO). At 6 hours after the injection, ventricular myocardium was collected for total RNA isolation and protein extraction for further analyses. Treatment to cohorts of age- and sex-matched line 134 CryABR120G tg mice with IC86430 (3 mg/kg per day) or vehicle control was initiated at exactly 4 months of age and lasted for 4 weeks using subcutaneous mini-osmotic pumps (Alzet 2004 model). Dose determination was based on previous reports (16, 17).
To test the effect of acute PDE4 inhibition, a separate cohort of line 134 CryABR120G tg mice at 4 months of age were treated with a selective PDE4 inhibitor, piclamilast (catalog number 4525; Tocris a Bio-Techne brand, Minneapolis, MN). Piclamilast (0.03, 0.1, 0.3, or 1.0 mg/kg body weight) or equivalent amount of vehicle control (0.1% DMSO in normal saline) was administered via intraperitoneal injections (twice daily at a 12-hour interval for 3 days). Animals were sacrificed at 6 hours after the sixth injection, and ventricular tissues were collected and snap-frozen for subsequent analyses.
Echocardiography
Transthoracic echocardiography was performed on mice using the VisualSonics Vevo 2100 system and a 30-MHz probe to assess LV morphometric changes, as previously described (11).
LV catheterization and hemodynamic measurements
Aortic pressure and LV pressure-volume loop assessments were performed, as previously described (30). Briefly, at the end of the 4-week treatment period, a cohort of six mice per group was used for in vivo hemodynamic measurements. Mice were anesthetized via inhaling isoflurane (3 to 4%), intubated through the mouth, and mechanically ventilated. Via the right common carotid artery, a high-fidelity 1.4-F Millar Mikro-Tip pressure-volume transducer catheter (model SPR-839, Millar Instruments, TX) was inserted into the transverse aorta for measuring aortic blood pressure before advancing to the LV chamber for recording the LV pressure-volume loops. After stabilization for 15 min, LV pressure and volume loops were recorded using a PowerLab data acquisition system (AD Instruments, Colorado Springs, CO) and analyzed with associated software. Both saline calibration and cuvette calibration were applied before data exportation.
NRVM culture and adenoviral infection
NRVMs were isolated from the ventricles of 2-day-old Sprague-Dawley rats using the Cellutron Neomyocytes isolation system (Cellutron Life Technology, Baltimore, MD), plated on 6-cm plates at a density of 2.0 × 106 cells in 10% fetal bovine serum, and cultured as described previously (11). The plated cells were then infected with adenoviruses harboring the expression cassette for GFPu (Ad-GFPu), RFP (Ad-RFP), or HA-tagged CryABR120G (Ad-HA-CryABR120G) (11).
Extraction of the NP-40–soluble and NP-40–insoluble fractions
NRVMs were infected with Ad-HA-CryABR120G in serum-free Dulbecco’s minimum essential medium (DMEM) for 6 hours. The cells were then cultured in DMEM containing 2% serum for 24 hours before protein extraction. Cells were washed in cold phosphate buffered saline at pH 7.4, harvested into cell lysis buffer [50 mM tris (pH 8.8), 100 mM NaCl, 5 mM MgCl2, 0.5% NP-40, and 2 mM dithiothreitol (DTT)] containing 1× complete protease and phosphatase cocktail (T-2496, A.G. Scientific Inc.), and incubated on ice for 30 min. Cell lysates were then centrifuged at 17,000g for 15 min at 4°C.The supernatant was collected as the NP-40–soluble fraction. The pellet containing NP-40–insoluble fractions (NI) was resuspended in cell pellet buffer [20 mM tris (pH 8.0), 15 mM MgCl2, 2 mM DTT, 1× complete protease, and phosphatase cocktail], followed by 30-min incubation on ice. The NI fractions were further solubilized in 3× SDS boiling buffer [6% SDS, 20 mM tris (pH 8.0), and 150 mM DTT] and boiled for 5 min. The NI fractions were then subject to SDS–polyacrylamide gel electrophoresis (PAGE) and Western blot analysis for the misfolded species of CryAB.
Western blot analysis
Total proteins were extracted from ventricular myocardium or cultured NRVMs with 1× sampling buffer (41 mM tris-HCl, 1.2% SDS, and 8% glycerol). Protein concentration was determined with bicinchoninic acid (BCA) reagents (Pierce Biotechnology, Rockford, IL). Equal amounts of proteins were loaded to each lane of the SDS–polyacrylamide gel and fractionated via electrophoresis, transferred to a polyvinylidene difluoride (PVDF) membrane using a Trans-Blot apparatus (Bio-Rad, Hercules, CA) and immunodetected with anti-PDE1A (1:800; SC-50480, Santa Cruz Biotechnology), anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1000; G8795, Sigma-Aldrich), anti-HA (1:1000; 3724, Cell Signaling), anti-GFP (1:1000; SC-9996, Santa Cruz Biotechnology), anti-tubulin (1:1000; E7-s, DSHB), anti-CryAB (1:1000; ADI-SPA-222, Enzo Life Sciences), anti-Psmb5 [1:1000; custom-made (30)], or anti–p-Rpn6 [1:5000; custom-made (13)] as the primary antibodies and appropriate horse radish peroxidase–conjugated secondary antibodies. The bound secondary antibodies were detected using the enhanced chemiluminescence detection reagents (GE Healthcare, Piscataway, NJ). Blots were imaged and quantified using the Quantity One or Image Lab software (Bio-Rad). For some of the blots, the total protein content derived from the stain-free protein imaging technology was used as in-lane loading control (8).
CHX chase assay
To determine the protein half-life of GFPu or HA-tagged CryABR120G, NRVMs were cultured in DMEM in the presence of CHX (50 μM; Sigma-Aldrich), which was used to block further protein synthesis. Cells were collected at various time points after administration of CHX, and total proteins were extracted for Western blot analysis for GFPu or HA-tagged CryABR120G. This was done as we previously reported (11).
RNA isolation and RT-PCR
Total RNA was extracted from ventricular myocardium using the TRI Reagent (Molecular Research Center Inc., Cincinnati, OH) as described previously (11). The concentration of RNA was determined using Agilent RNA 6000 Nano assay (Agilent technologies Inc., Germany) following the manufacturer’s instruction. For RT reaction, 1 μg of RNA was used as a template to generate complementary DNA using the SuperScript III First-Strand Synthesis Kit (Invitrogen), and the RT was performed by following the manufacturer’s instructions. For the duplex PCR of a gene of interest and GAPDH, 2 μl of solution from the RT reaction and specific primers toward the target gene and GAPDH were used. The mRNA levels of the gene of interest were quantified by PCR at the minimum number of cycles (15 cycles) capable of detecting the PCR products within the linear amplification range. Mouse PDE1A, GFPdgn, and GAPDH were measured using the following primer pairs: PDE1A, 5′-CTAAAGATGAACTGGAGGGATCTTCG GAAC-3′ (forward) and 5′-TGGAGAAAATGGAAGCCCTAATTCAGC-3′ (reverse); GFPdgn, 5′-TCTATATCATGGCCGACAAGCAGA-3′ (forward) and 5′-ACTGGGTGCTCAGGTAGTGGTTGT-3′ (reverse); and GAPDH, 5′-ATGACATCAAGAAGGTGGTG-3′ (forward) and 5′-CATACCAGGAAATGAGCTTG-3′ (reverse).
Native gel electrophoresis and in-gel proteasome peptidase activity assays
In-gel proteasome peptidase activity assays were performed as described previously (13), with minor modification. Cultured NRVMs were lysed on ice in cytosolic extraction buffer [50 mM tris-HCl (pH 7.5), 250 mM sucrose, 5 mM MgCl2, 0.5 mM EDTA, 1 mM DTT, and 1 mM adenosine 5′-triphosphate (ATP)]. The cell lysates or myocardium homogenates were plunged through a 29-gauge needle eight times with an insulin syringe before centrifugation at 4°C for 30 min (15,000g). Protein concentration was determined with BCA reagents, and samples were diluted with 4× native gel loading buffer [200 mM tris-HCl (pH 6.8), 60% (v/v) glycerol, and 0.05% (w/v) bromophenol blue]. Each well of a 4.0% native polyacrylamide gel was loaded with 40 μg of sample, and the gel electrophoresis was performed at a constant current of 15 mA for 8 hours. After electrophoresis, the gel was incubated in developing buffer [50 mM tris-HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, and 1 mM ATP] containing 50 μM Suc-LLVY-AMC at 37°C for 30 min and visualized in an ultraviolet trans-illuminator with a wavelength of 365 nm. Immediately after the in-gel peptidase activity assay, the proteins from the native gel were transferred to a PVDF membrane at a constant current of 250 mA for 90 min and immunodetected with anti-Psmb5 antibodies.
Fluorescence confocal microscopy
Confocal microscopy was performed as described (11). Ventricular myocardium from GFPdgn tg mice was fixed with 3.8% paraformaldehyde and processed for obtaining cryosections. The myocardial sections (6 μm) were stained with Alexa Fluor 568–conjugated phalloidin (Invitrogen) to reveal F-actin and identify cardiomyocytes. GFPdgn direct fluorescence (green) and the stained F-actin (red) were visualized and imaged using a Leica TCS SP8 confocal microscope (Leica Microsystems Inc., Buffalo Grove, IL).
Proteasome peptidase activity assays in test tubes
Proteasome peptidase activity assays were performed as described previously (11). Briefly, ventricular myocardial tissues were homogenized in the crude protein extraction buffer [50 mM tris-HCl (pH 7.5), 250 mM sucrose, 5 mM MgCl2, 0.5 mM EDTA, 1 mM DTT, and 0.025% digitonin] on ice. The homogenates were centrifuged at 6800g at 4°C for 10 min, and then, the supernatant was collected and measured for protein concentrations using the BCA reagent. The protein samples were then used for 26S proteasome chymotrypsin-like activity assays in 96-well microplates (Greiner Bio-One, Germany). Each assay reaction used 10 μg of myocardial proteins and took place in 200 μl of proteasome assay buffers [50 mM tris-HCl (pH 7.5), 40 mM KCl, 5 mM MgCl2, 1 mM DTT, bovine serum albumin (0.5 μg/ml), and 0.2 mM ATP] containing the synthetic fluorogenic peptidase substrate Suc-LLVY-AMC (18 μM; Calbiochem, San Diego, CA). The assays were carried out in the presence or absence of the proteasome inhibitor BZM (0.28 μM; LC Laboratories, Woburn, MA). The fluorescence intensity was read every 18 min for a duration of 180 min using the Victor X3 Microplate Reader (Perkin Elmer, Shelton, CT) with an excitation wavelength of 380 nm and an emission wavelength of 460 nm. The proteasome inhibitor-suppressible peptidase activity was attributed to the proteasome. The slope of the log phase of the reaction curve was used as in the indicator of proteasome chymotrypsin activity.
Statistical analysis
Data were statistically analyzed using GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA). Western blot and PCR densitometry data are presented as scatter dot plots with means ± SD superimposed. Differences between the two groups were evaluated using two-tailed unpaired Student’s t test. When the difference among three or more groups was evaluated, one-way analysis of variance (ANOVA) or, when appropriate, two-way ANOVA followed by Tukey’s multiple comparisons test or Dunnett’s multiple comparisons test was used. Serial echocardiography data were examined using two-way repeated measures ANOVA followed by Bonferroni’s test to assess interaction of time and drug intervention. For a dataset with unequal variances, two-tailed unpaired t test with Welch’s correction was also used and is indicated in figure legends.
Supplementary Material
Acknowledgments
We thank A. Jahn and M. Lewno for excellent assistance in tg mouse colonies maintenance and genotype determination. We thank the Physiology Core of the Division of Basic Biomedical Sciences for assistance with echocardiography. Funding: X.W. received funding to support this research through NIH grants (HL072166, HL085629, and HL131667). H.Z. was supported by an American Heart Association Predoctoral Fellowship (16PRE27790059). Author contributions: X.W. and H.Z. designed the research studies. H.Z., B.P., N.P., and P.W. conducted experiments. H.Z., B.P., N.P., and P.W. acquired data, X.W., H.Z., and A.L.G. analyzed data. M.D.R. and A.L.G. provided reagents, H.Z., X.W., A.L.G., M.D.R., and B.P. wrote or revised the manuscript. Competing interests: M.D.R. is employed by Eli Lilly and Company (Indianapolis, IN, USA). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The tg mouse models and recombinant adenoviral vectors can be provided by the corresponding author (X.W.), pending scientific review and a completed material transfer agreement. Requests for the tg mouse models and recombinant adenoviral vectors should be submitted to University of South Dakota Technology Transfer Officer (tto@usd.edu). Additional data related to this paper maybe requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaaw5870/DC1
Fig. S1. Verification of the PDE1A antibody.
Fig. S2. Changes in LV echocardiographic parameters in CryABR120G tg mice at 4 months of age are consistent with HFpEF.
Fig. S3. Representative images of M mode echocardiograms taken from sex- and age-matched NTG and CryABR120G tg mice at the completion of 4 weeks of treatment with the PDE1 inhibitor IC86430 (TG-LSN) or vehicle (TG-Vehicle).
Fig. S4. Hemodynamic analysis from sex- and age-matched NTG and CryABR120G tg mice at the end of 4 weeks of treatment with the PDE1 inhibitor IC86430 (TG-IC) or vehicle (TG-Vehicle).
Fig. S5. Assessments of aortic pressure at the end of 4 weeks of treatment with the PDE1 inhibitor IC86430 (TG-IC) or vehicle (TG-Vehicle).
Fig. S6. Western blot analysis for CryAB levels in the SS fraction of NTG and R120G mouse hearts.
Table S1. Echocardiographic measurements at 4 months of age immediately before IC86430 treatment.
Table S2. Echocardiographic measurements at the end of 4 weeks of treatment.
REFERENCES AND NOTES
- 1.Gladden J. D., Chaanine A. H., Redfield M. M., Heart failure with preserved ejection fraction. Annu. Rev. Med. 69, 65–79 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Shah S. J., Kitzman D. W., Borlaug B. A., van Heerebeek L., Zile M. R., Kass D. A., Paulus W. J., Phenotype-specific treatment of heart failure with preserved ejection fraction: A multiorgan roadmap. Circulation 134, 73–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X., Robbins J., Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell. Cardiol. 71, 16–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arbustini E., Morbini P., Grasso M., Fasani R., Verga L., Bellini O., Dal Bello B., Campana C., Piccolo G., Febo O., Opasich C., Gavazzi A., Ferrans V. J., Restrictive cardiomyopathy, atrioventricular block, and mild to subclinical myopathy in patients with desmin-immunoreactive material deposits. J. Am. Coll. Cardiol. 31, 645–653 (1998). [DOI] [PubMed] [Google Scholar]
- 5.VerPlank J. J. S., Goldberg A. L., Regulating protein breakdown through proteasome phosphorylation. Biochem. J. 474, 3355–3371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Day S. M., The ubiquitin proteasome system in human cardiomyopathies and heart failure. Am. J. Physiol. Heart Circ. Physiol. 304, H1283–H1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J., Horak K. M., Su H., Sanbe A., Robbins J., Wang X., Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J. Clin. Invest. 121, 3689–3700 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu C., Tian Y., Xu H., Pan B., Terpstra E. M., Wu P., Wang H., Li F., Liu J., Wang X., Inadequate ubiquitination-proteasome coupling contributes to myocardial ischemia-reperfusion injury. J. Clin. Invest. 128, 5294–5306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Divald A., Kivity S., Wang P., Hochhauser E., Roberts B., Teichberg S., Gomes A. V., Powell S. R., Myocardial ischemic preconditioning preserves postischemic function of the 26S proteasome through diminished oxidative damage to 19S regulatory particle subunits. Circ. Res. 106, 1829–1838 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Li J., Ma W., Yue G., Tang Y., Kim I.-m., Weintraub N. L., Wang X., Su H., Cardiac proteasome functional insufficiency plays a pathogenic role in diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 102, 53–60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ranek M. J., Terpstra E. J. M., Li J., Kass D. A., Wang X., Protein kinase G positively regulates proteasome-mediated degradation of misfolded proteins. Circulation 128, 365–376 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lokireddy S., Kukushkin N. V., Goldberg A. L., cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Natl. Acad. Sci. U.S.A. 112, E7176–E7185 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.VerPlank J. J. S., Lokireddy S., Zhao J., Goldberg A. L., 26S Proteasomes are rapidly activated by diverse hormones and physiological states that raise cAMP and cause Rpn6 phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 116, 4228–4237 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knight W., Yan C., Therapeutic potential of PDE modulation in treating heart disease. Future Med. Chem. 5, 1607–1620 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vandeput F., Wolda S. L., Krall J., Hambleton R., Uher L., McCaw K. N., Radwanski P. B., Florio V., Movsesian M. A., Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. J. Biol. Chem. 282, 32749–32757 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Miller C. L., Cai Y., Oikawa M., Thomas T., Dostmann W. R., Zaccolo M., Fujiwara K., Yan C., Cyclic nucleotide phosphodiesterase 1A: A key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res. Cardiol. 106, 1023–1039 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller C. L., Oikawa M., Cai Y., Wojtovich A. P., Nagel D. J., Xu X., Xu H., Florio V., Rybalkin S. D., Beavo J. A., Chen Y.-F., Li J.-D., Blaxall B. C., Abe J.-i., Yan C., Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ. Res. 105, 956–964 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knight W. E., Chen S., Zhang Y., Oikawa M., Wu M., Zhou Q., Miller C. L., Cai Y., Mickelsen D. M., Moravec C., Small E. M., Abe J., Yan C., PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc. Natl. Acad. Sci. U.S.A. 113, E7116–E7125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X., Osinska H., Klevitsky R., Gerdes A. M., Nieman M., Lorenz J., Hewett T., Robbins J., Expression of R120G–αB-crystallin causes aberrant desmin and αB-crystallin aggregation and cardiomyopathy in mice. Circ. Res. 89, 84–91 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Zong C., Gomes A. V., Drews O., Li X., Young G. W., Berhane B., Qiao X., French S. W., Bardag-Gorce F., Ping P., Regulation of murine cardiac 20S proteasomes: Role of associating partners. Circ. Res. 99, 372–380 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Kumarapeli A. R. K., Horak K. M., Glasford J. W., Li J., Chen Q., Liu J., Zheng H., Wang X., A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. FASEB J. 19, 2051–2053 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Zheng H., Tang M., Zheng Q., Kumarapeli A. R. K., Horak K. M., Tian Z., Wang X., Doxycycline attenuates protein aggregation in cardiomyocytes and improves survival of a mouse model of cardiac proteinopathy. J. Am. Coll. Cardiol. 56, 1418–1426 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhuiyan M. S., Pattison J. S., Osinska H., James J., Gulick J., McLendon P. M., Hill J. A., Sadoshima J., Robbins J., Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest. 123, 5284–5297 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo L., Prall W., Yang X., Assays for the degradation of misfolded proteins in cells. J. Vis. Exp., e54266 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y., Knight W., Chen S., Mohan A., Yan C., Multiprotein complex with TRPC (transient receptor potential-canonical) channel, PDE1C (phosphodiesterase 1C), and A2R (adenosine A2 receptor) plays a critical role in regulating cardiomyocyte cAMP and survival. Circulation 138, 1988–2002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hashimoto T., Kim G. E., Tunin R. S., Adesiyun T., Hsu S., Nakagawa R., Zhu G., O'Brien J. J., Hendrick J. P., Davis R. E., Yao W., Beard D., Hoxie H. R., Wennogle L. P., Lee D. I., Kass D. A., Acute enhancement of cardiac function by phosphodiesterase type 1 inhibition. Circulation 138, 1974–1987 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myeku N., Clelland C. L., Emrani S., Kukushkin N. V., Yu W. H., Goldberg A. L., Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 22, 46–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asai M., Tsukamoto O., Minamino T., Asanuma H., Fujita M., Asano Y., Takahama H., Sasaki H., Higo S., Asakura M., Takashima S., Hori M., Kitakaze M., PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J. Mol. Cell. Cardiol. 46, 452–462 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Rajagopalan V., Zhao M., Reddy S., Fajardo G., Wang X., Dewey S., Gomes A. V., Bernstein D., Altered ubiquitin-proteasome signaling in right ventricular hypertrophy and failure. Am. J. Physiol. Heart Circ. Physiol. 305, H551–H562 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian Z., Zheng H., Li J., Li Y., Su H., Wang X., Genetically induced moderate inhibition of the proteasome in cardiomyocytes exacerbates myocardial ischemia-reperfusion injury in mice. Circ. Res. 111, 532–542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanbe A., Osinska H., Saffitz J. E., Glabe C. G., Kayed R., Maloyan A., Robbins J., Desmin-related cardiomyopathy in transgenic mice: A cardiac amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 101, 10132–10136 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rainer P. P., Dong P., Sorge M., Fert-Bober J., Holewinski R. J., Wang Y., Foss C. A., An S. S., Baracca A., Solaini G., Glabe C. G., Pomper M. G., Van Eyk J. E., Tomaselli G. F., Paolocci N., Agnetti G., Desmin phosphorylation triggers preamyloid oligomers formation and myocyte dysfunction in acquired heart failure. Circ. Res. 122, e75–e83 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kötter S., Unger A., Hamdani N., Lang P., Vorgerd M., Nagel-Steger L., Linke W. A., Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J. Cell Biol. 204, 187–202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franssen C., Kole J., Musters R., Hamdani N., Paulus W. J., α-B crystallin reverses high diastolic stiffness of failing human cardiomyocytes. Circ. Heart Fail. 10, e003626 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Schubert U., Antón L. C., Gibbs J., Norbury C. C., Yewdell J. W., Bennink J. R., Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404, 770–774 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Ranek M. J., Zheng H., Huang W., Kumarapeli A. R., Li J., Liu J., Wang X., Genetically induced moderate inhibition of 20S proteasomes in cardiomyocytes facilitates heart failure in mice during systolic overload. J. Mol. Cell. Cardiol. 85, 273–281 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang M., Li J., Huang W., Su H., Liang Q., Tian Z., Horak K. M., Molkentin J. D., Wang X., Proteasome functional insufficiency activates the calcineurin-NFAT pathway in cardiomyocytes and promotes maladaptive remodelling of stressed mouse hearts. Cardiovasc. Res. 88, 424–433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pattison J. S., Sanbe A., Maloyan A., Osinska H., Klevitsky R., Robbins J., Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation 117, 2743–2751 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao S., Alarcón C., Sapkota G., Rahman S., Chen P.-Y., Goerner N., Macias M. J., Erdjument-Bromage H., Tempst P., Massagué J., Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-β signaling. Mol. Cell 36, 457–468 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen S., Knight W. E., Yan C., Roles of PDE1 in pathological cardiac remodeling and dysfunction. J. Cardiovasc. Dev. Dis. 5, E22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaaw5870/DC1
Fig. S1. Verification of the PDE1A antibody.
Fig. S2. Changes in LV echocardiographic parameters in CryABR120G tg mice at 4 months of age are consistent with HFpEF.
Fig. S3. Representative images of M mode echocardiograms taken from sex- and age-matched NTG and CryABR120G tg mice at the completion of 4 weeks of treatment with the PDE1 inhibitor IC86430 (TG-LSN) or vehicle (TG-Vehicle).
Fig. S4. Hemodynamic analysis from sex- and age-matched NTG and CryABR120G tg mice at the end of 4 weeks of treatment with the PDE1 inhibitor IC86430 (TG-IC) or vehicle (TG-Vehicle).
Fig. S5. Assessments of aortic pressure at the end of 4 weeks of treatment with the PDE1 inhibitor IC86430 (TG-IC) or vehicle (TG-Vehicle).
Fig. S6. Western blot analysis for CryAB levels in the SS fraction of NTG and R120G mouse hearts.
Table S1. Echocardiographic measurements at 4 months of age immediately before IC86430 treatment.
Table S2. Echocardiographic measurements at the end of 4 weeks of treatment.