

Abstract
The lung epithelium is derived from the endodermal germ layer, which undergoes a complex series of endoderm–mesoderm-mediated signaling events to generate the final arborized network of conducting airways (bronchi, bronchioles) and gas-exchanging units (alveoli). These stages include endoderm induction, anterior–posterior and dorsal–ventral patterning, lung specification, lung budding, branching morphogenesis, and, finally, maturation. Here we describe a protocol that recapitulates several of these milestones in order to differentiate human pluripotent stem cells (hPSCs) into ventral–anterior foregut spheroids and further into two distinct types of organoids: human lung organoids and bud tip progenitor organoids. The resulting human lung organoids possess cell types and structures that resemble the bronchi/bronchioles of the developing human airway surrounded by lung mesenchyme and cells expressing alveolar-cell markers. The bud tip progenitor organoids possess a population of highly proliferative multipotent cells with in vitro multilineage differentiation potential and in vivo engraftment potential. Human lung organoids can be generated from hPSCs in 50–85 d, and bud tip progenitor organoids can be generated in 22 d. The two hPSC-derived models presented here have been benchmarked with human fetal tissue and found to be representative of human fetal-like tissue. The bud tip progenitor organoids are thus ideal for exploring epithelial fate decisions, while the human lung organoids can be used to model epithelial–mesenchymal cross-talk during human lung development. In addition to their applications in developmental biology, human lung organoids and bud tip progenitor organoids may be implemented in regenerative medicine, tissue engineering, and pharmaceutical safety and efficacy testing.
Introduction
Development of the protocol
During development, the endodermal germ layer gives rise to the lining of the gut tube, which is patterned along the anterior–posterior axis of the embryo into distinct morphological and molecular domains1. The lung is specified in the ventral–anterior foregut endoderm region of the gut tube, and development begins as two primordial lung buds emerge from this region. The lung buds possess a population of multipotent epithelial progenitors at the tips (‘bud tip progenitors’) and are surrounded by mesenchyme. As the lung grows, the epithelium undergoes repeated rounds of bifurcation in a process known as branching morphogenesis, in order to establish the arborized architecture of the adult lung. During the branching process, bud tip progenitors are maintained, continuously proliferate, and give rise to all lung epithelial cell types. Early in development, branching establishes the network of tubes that will conduct air (bronchi, bronchioles). Later during development, when the branching program is completed, bud tip progenitors that remain at the end of the airways give rise to alveolar epithelial cells2,3, as confirmed by lineage tracing experiments in mice4.
Several studies have demonstrated that the recapitulation of key stages of embryonic development through a series of steps in vitro, known as directed differentiation, is an effective method to generate cell and tissue lineages of interest from hPSCs. Directed differentiation has been used to generate 3D mid- and hindgut spheroids that give rise to small and large human intestinal organoids5–8, as well as to generate 2D monolayers of ventral foregut cells and lung epithelial cell types9–15. Here we describe protocols based on published work for the generation of 3D ventral–anterior foregut spheroids from hPSCs15,16 and the differentiation of foregut spheroids into two distinct types of lung organoids: human lung organoids15 and bud tip progenitor organoids17 (Fig. 1). Human lung organoids are generated if foregut spheroids are cultured with high levels of FGF10 and 1% FBS, and possess airway-like epithelium surrounded by a diffuse network of mesenchymal cells and epithelial cells that express alveolar-cell-type markers. The transcriptional profile of these organoids is highly similar to that of fetal lung. The presence of mesenchyme and organized airway structures is a strength of this system, making it ideal for studies of mesenchymal epithelial cross-talk during fetal lung development. Bud tip progenitor organoids are generated when foregut spheroids are cultured in a serum-free environment with FGF7, CHIR-99021, and all-trans retinoic acid (ATRA). After 22 d in culture, bud tip progenitor organoids contain a highly enriched and proliferative population of SOX2+- SOX9+ID2+NKX2.1+ cells that are transcriptionally similar to human fetal bud tip progenitors. These cells can be expanded in culture for more than 16 weeks. Given that bud tip progenitors are a precursor cell to all epithelial cell types during development, bud tip progenitor organoids are uniquely suited for studying mechanisms involved in epithelial cell fate decisions in the developing human lung.
Fig. 1 |. Schematic of protocol and timeline.

hPSCs in a monolayer are directed to endoderm and then anterior foregut endoderm over the course of 9–10 d. Foregut spheroids self-aggregate and lift away from the monolayer to float in the media. These spheroids are then cultured in a 3D Matrigel droplet, where they can be directed to become airway-like human lung organoids or bud tip progenitor organoids.
Overview of the experimental design
The generation of ventral–anterior foregut spheroids takes ~10 d. Subsequent differentiation into human lung organoids takes 50–85 d, and differentiation into bud tip progenitor organoids takes ~22 d (Fig. 1). The protocol begins with hPSCs grown in a monolayer, and differentiation is directed through several stages, including definitive endoderm (4 d) and ventral–anterior foregut spheroids (5–6 d). Cells begin as 2D monolayers of undifferentiated hPSCs, and during the specification into ventral–anterior foregut tissue, cells self-assemble into small clusters called spheroids and form free-floating structures that detach from the cell monolayer. Foregut spheroids are then encapsulated in a 3D extracellular matrix or synthetic hydrogel18 and are overlaid with specific growth factors and small molecules to generate either human lung organoids (50–85 d)15 or bud tip progenitor organoids (22 d)17.
Foregut spheroids are differentiated in serum-free media. If subsequently embedded in a droplet of Matrigel and grown in high levels of FGF10 and 1% (vol/vol) FBS, they give rise to human lung organoids, which form airway-like structures and cell types surrounded by mesenchymal populations. Further, some cells that express AECI and AECII markers are present in the organoids. These organoids contain optimal airway-like tissue at 50–85 d of culture, but airway-like tissue has been observed in organoids cultured for more than 100 d.
In contrast, foregut spheroids embedded in Matrigel and grown in serum-free medium containing FGF7, CHIR-99021 and ATRA give rise to bud tip progenitor organoids, which are composed of SOX9+SOX2+ bud-tip-progenitor-like cells and have a transcriptome similar to that of bud tip progenitors found in native human fetal lung during branching morphogenesis. If left unperturbed in Matrigel droplets for several weeks, spheroids treated with FGF7, CHIR-99021 and ATRA will form patterned branch-like structures with interior regions that express proximal airway markers, and distal bud tip regions. Bud tip progenitors are highly proliferative and can be easily needle-passaged for expansion/enrichment of progenitor populations. Bud tip progenitor organoids can be serially passaged every 2 weeks and maintain a relatively uniform multipotent population of progenitor cells in vitro. Manipulation of the in vitro growth factor milieu can promote the differentiation of bud tip progenitor cells into differentiated lung epithelial cells that express both airway/bronchiolar cell markers and alveolar cell markers17.
Experimental design
A schematic of the protocol is shown in Fig. 1. The first 9 d of the protocol are the same for both types of organoids described here: hPSCs are first directed to definitive endoderm with activin A, then directed to anterior foregut endoderm via inhibition of transforming growth factor β (TGF-β) and bone morphogenic protein (BMP) signaling by SB-431542 and NOGGIN, respectively, in addition to simultaneous activation of the WNT (CHIR-99021), Hedgehog (SAG) and fibroblast growth factor 4 (FGF4) pathways. Foregut spheroids will self-assemble and can be transferred into a droplet of Matrigel. After plating in Matrigel, differentiation into different lung lineages is controlled by the growth factor signaling milieu (Fig. 1). Treatment of foregut spheroids with serum-free media containing FGF7, CHIR-99021 and ATRA will result in bud tip progenitor organoids that possess bud tip progenitors after 14 d in culture. This population can be easily expanded and maintained by needle passaging, or, if left unpassaged, will give rise to lung epithelial structures with proximal–distal patterning and a population of bud tip progenitors at budded tips after ~40 d in culture. Conversely, treatment of foregut spheroids with 1% FBS and a high concentration of FGF10 for ~50 d will result in human lung organoids that contain fetal airway-like structures with surrounding lung mesenchyme and alveolar-progenitor-like cells. Specific mRNA and protein markers identified from human lung development are used to determine whether cells have differentiated as expected.
Application of the method
The generation of self-aggregating foregut spheroids from hPSCs is the first step in this protocol for either type of organoid (Fig. 1). Depending on the signaling milieu applied to foregut spheroids, the researcher can grow foregut spheroids that will give rise either to human lung organoids or to bud tip progenitor organoids. Human lung organoids and bud tip progenitor organoids have been compared to both human adult lung and fetal lung, and possess cells that are highly similar to the developing human lung. Therefore, both systems are ideal for studying developmental biology and tissue engineering.
Human lung organoids
These contain airway-like structures surrounded by lung mesenchymal cells and cells that stain positive for AECI and AECII markers. Bulk RNA-seq analysis shows that these organoids are more similar to native human fetal lung than to adult lung15. Because of the presence of mesenchyme, airway structures and immature alveolar-like epithelial cells, human lung organoids are ideal for studying mesenchymal–epithelial interactions during human lung development and for modeling human fetal lung malformations using patient-specific cell lines or mutations. For example, our ongoing work with these organoids is focused on epithelial mesenchymal cross-talk during pulmonary fibrosis, and on congenital malformations19 related to Wnt signaling (data not shown). Human lung organoids can be microinjected20,21 with drugs or bacteria for investigation of their effects in the context of development or disease (Fig. 2a), transplanted onto a bioengineered scaffold to form mature airway structures and cell types (Fig. 2b–d), or seeded onto a decellularized lung scaffold to generate mature airway cell types (Fig. 2e). These organoids are ideal for use after ~50–85 d in culture but are long-lived and stable in vitro, so experiments that last several weeks can easily be carried out. Owing to these unique features, the human lung organoid model is ideal for studies of epithelial–mesenchymal interactions during lung development, including maturation of alveolar cell types, and may be ideal for modeling infections common to the immature lung, such as respiratory syncytial virus.
Fig. 2 |. Applications of the protocol.
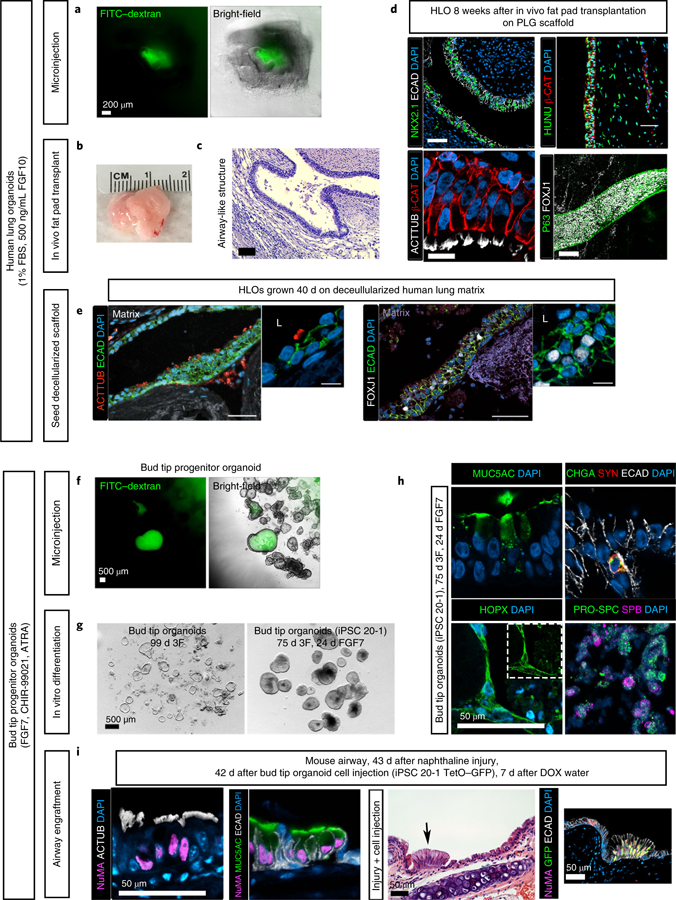
a–e, Potential applications for human lung organoids. a, Organoid lumens can be microinjected, as done here with 1 µL of 4-kDa FITC–dextran suspended in PBS at a concentration of 2 mg/mL (green)20. Images show organoids 1 h after injection. Scale bar, 200 µm (applies to both images). b, Human lung organoids can be seeded on a bioengineered poly(lactic-co-glycolic acid) (PLG) scaffold and transplanted into the mouse fat pad (full methods can be found in ref. 16). This structure can grow to more than 1 cm in diameter in ~8 weeks. c, Transplanted human lung organoids contain airway-like structures, as visualized here by H&E staining after 8 weeks16. Scale bar, 200 µm. d, After 8 weeks of transplantation, human lung organoids (HLOs) contain NKX2.1+ airway epithelial structures that stain positive for human nuclear antigen (HUNU) and exhibit multiciliated cells that stain positive for acetylated tubulin (ACTTUB; a video showing the resulting functional beating ciliated cells is available from ref. 16). Further, these structures contain epithelial tubes with P63+ basal-like cells lining the basolateral surface of the tube and FOXJ1+ ciliated cells lining the interior luminal side of the pseudostratified epithelium. Scale bars, 50 µm. e, Roughly 50 human lung organoids were seeded onto a decellularized human lung matrix punch in a 96-well plate. Human lung organoid seeded cells gave rise to epithelial structures expressing multiciliated-cell markers acetylated tubulin and FOXJ1 after 40 d of culture. Scale bars, 100 µm. f–i, Applications for bud tip progenitor organoids. f, Bud tip progenitor organoids can be microinjected with fluids, as done here with 1 µL of 4-kDa FITC–dextran suspended in PBS at a concentration of 2 mg/ml (green)20. Five organoids were injected. Images show organoids 1 h after injection. Scale bar, 500 µm. g, Bud tip progenitor cells in bud tip progenitor organoids are capable of undergoing multilineage differentiation into airway and alveolar-like cells, and this system can be used to interrogate mechanisms of lineage decisions in vitro. Bud tip progenitor organoids treated for 24 d with FGF7 alone generated cells that expressed the AECI marker HOPX and AECII markers pro-surfactant protein C (PRO-SPC) and surfactant protein B (SFTPB). Alevolar-like cells made up roughly 50% of these organoids. Airway-like cells were also generated, including cells that stained positive for goblet cell marker MUC5AC and neuroendocrine markers chromagranin A (CHGA) and synaptophysin (SYN). Scale bar, 50 µm. i, Bud tip progenitor organoids can be expanded in culture and injected directly into the injured mouse airway, where they can engraft. In this experiment, bud tip progenitor organoids were generated from the iPSC 20–1 tet-O–GFP cell line to provide doxycycline (DOX)-inducible GFP expression. Organoids were grown, expanded and dissociated to single cells, and roughly 500,000 bud tip progenitor organoid cells were injected into the mouse airway 24 h after airway injury with naphthaline. Mice were allowed to recover for 6 weeks. During the fifth week after cell injection, mice were given DOX-treated water; surviving human cells with access to the host blood stream could then begin expressing GFP upon exposure to DOX17. Engrafted cells expressed the human nuclear marker NuMA, as well as markers of differentiation including multiciliated-cell markers (acetylated tubulin) and the goblet cell marker MUC5AC. These cells integrated seamlessly with the host epithelium. Further, cells were able to receive signals from the host bloodstream, as bud tip progenitor cells derived from a tet-O–GFP iPSC line were able to turn on GFP expression when the host mouse was given DOX in drinking water. Scale bars, 50 µm. Detailed information about antibodies used for staining can be found in Table 3. b–d, Reproduced with permission from ref. 16, eLife Sciences Publications (licensed under CC BY-NC-ND 4.0, https://creativecommons.org/licenses/by/4.0/). e, Reproduced with permission from ref. 15, eLife Sciences Publications (licensed under CC BY-NC-ND 4.0, https://creativecommons.org/licenses/by/4.0/). g–i, Reproduced with permission from ref. 17, Elsevier (licensed under CC BY-NC-ND 4.0, https://creativecommons.org/licenses/by/4.0/).
Bud tip progenitor organoids
These can be generated from hPSCs in 22 d (ref. 17). By 22 d, organoids contain a highly enriched population of epithelial bud tip progenitor cells that are similar to bud tip progenitors residing at the tips of branching buds in native human fetal lung in terms of their transcriptional profile, protein expression and functional characterization. These cells are ideally suited for research on the mechanisms of epithelial cell fate decisions in the developing lung, as bud tip progenitors are obligate precursors to all lung epithelial cell types. Our work has shown that bud tip progenitor organoids have multilineage potential in vitro, where they can give rise to airway-like and alveolar-like cells (Fig. 2g,h), and ongoing studies in our laboratory have used this model system to study lineage decisions, as bud tip progenitors give rise to multiple epithelial cell lineages (data not shown). We speculate that this model may also be useful as a predictive preclinical model for many human lung diseases22,23, or for testing pharmaceutical safety during pregnancy24. Because they are highly expandable, bud tip progenitor organoids can be expanded to accommodate large screens to test pharmaceuticals for potential effects on proliferation, apoptosis, metabolic function and changes in lineage decisions. Large, easily identifiable lumens within these organoids allow drugs or pathogens to be delivered by microinjection20,21 or in the surrounding medium (Fig. 2f). One unique feature of cells derived from bud tip progenitor organoids is their ability to successfully engraft, proliferate and repopulate differentiated airway cells in the injured mouse airway17. Thus, the high expandability and multilineage potential of these organoids might make them suitable for cell-based regenerative medicine applications, such as reseeding damaged airway epithelium in vivo (Fig. 2i).
Human lung organoids and bud tip progenitor organoids engraft in mice
Both organoids can engraft into immunocompromised mice. Human lung organoids are able to engraft into ectopic vascular beds (i.e., epididymal fat pad; Fig. 2b–d), and bud tip progenitor organoids have been shown to engraft into the injured mouse airway (Fig. 2i), suggesting potential in regenerative medicine. However, a deeper understanding of these cells is required before they could be used in a clinical setting.
Comparison to other methods
The protocol described here outlines methods to generate 3D foregut spheroids, which can be further cultured in a 3D environment such as Matrigel to generate (i) human lung organoids containing lung epithelium and mesenchyme or (ii) a highly expandable population of bud tip progenitor organoids containing SOX9+SOX2+ bud tip progenitors that are similar to bud tip progenitors in the native developing fetal lung. Other methods to direct the differentiation of hPSCs to endoderm25, foregut13, and lung lineages on a monolayer11,12,14 and in 3D culture have been described15,17,26–31. Each of the different 2D and 3D systems described has unique features, and each has strengths and limitations that should be considered to ensure that the model system being used matches the scientific question at hand.
The first reported studies of directed differentiation of hPSCs into a lung lineage used monolayer cultures11,12,32–34 in which hPSCs were directed first to definitive endoderm25, then to anterior foregut endoderm13, and finally to an NKX2.1+ lung lineage.
More recent reports, including our own, have focused on the generation of 3D lung tissue in attempts to recapitulate more complex elements of a native organ15,17,26–31. The organoid models described here and in other reports are summarized in Table 1. In a recent study, Chen et al.28 directed hPSC differentiation on a monolayer to early NKX2.1+ lung progenitors, which formed clumps of cells that could be dissociated from the monolayer and placed in a droplet of Matrigel with FGF7, FGF10, CHIR-99021 and ATRA, giving rise to 3D structures termed lung bud organoids (LBOs). A subset of LBOs cultured in Matrigel quickly formed branched structures that could be manipulated by plating density. Epithelial cells expressed markers of goblet cells and AECII cells, although many other cell types (P63+ cells, SCGB3A2+ secretory and multiciliated cells) were not detected. Although mesenchymal cells were present in these cultures, the proportion declined to <2% of total cells after prolonged culture in Matrigel. Transplantation of LBOs into mouse kidney capsule yielded NKX2.1+ epithelial tubes that formed branch-like structures surrounded by lung mesenchyme. Tips of these branch-like structures did not express SOX2 but were positive for the AECII markers SFTPB and SFTPC, which suggests that these structures were patterned into alveolar-like structures. The stalks of these tubules contained cells that expressed multiciliated and mucus-producing cell markers. Mucus was found within lumen, suggesting functionality of mucus-producing cells. Of note, when LBOs grown in vitro in Matrigel were treated with respiratory syncytial virus, they exhibited epithelial shedding, characteristic of the physiological lung response28. This LBO model is thus well suited for experiments exploring the effect of infections or diseases that affect the mucus-producing cells or AECIIs of the lung epithelium, such as influenza. Further, LBOs could be valuable tools for further exploration of the mechanisms regulating epithelial branching events.
Table 1 |.
Comparison and contrast of methods to derive lung organoid model systems
| Bud tip progenitor organoid (routine passaging) | Patterned lung organoid (intact structure, no passage) | Human lung organoid | Lung bud organoid | Proximal airway organoids | Alveolar spheres | Alveolar organoids | Airway epithelial spheroids | ||
|---|---|---|---|---|---|---|---|---|---|
| ref. 17 | ref. 17 | refs. 15,16 | ref. 28 | ref. 27 | ref. 26 | refs. 29,35 | ref. 30 | ||
| In vitro | Time required for generation from hPSCs | ~3 weeks | ~6 weeks | ~8 weeks | ~10 weeks | ~3 weeks | ~4 weeks | ~4 weeks | ~4 weeks |
| Cell-sorting step required | No | No | No | No | Yes | Yes | Yes | Yes | |
| Mesenchyme present | No | No | Yes | Yes; minimal | No | No | No | No | |
| Airway epithelial structures | No | Yes | Yes | Yes | Yes | No | No | Yes | |
| Airway-like cells | No | Yes: SCGB1A1+ Club-like cells and MUC5AC/B+ goblet-like cells |
Yes: FOXJ1+ ACTTUB+ multiciliated-like cells, SCGB1A1+ Club-like cells, P63+ basal-like cells |
Yes: MUC5AC+MUC1+ goblet-like cells |
Yes: FOXJ1+ACTTUB+ multiciliated-like cells, SCGB3A2 Club-like cells, P63+KRT5+ basal-like cells, MUC5AC+ goblet-like cells |
No | No | Yes: FOXJ1+ACTTUB+ multiciliated cells, SCGB3A2 + Club-like cells, KRT5+ basal-like cells, MUC5AC+ goblet-like cells |
|
| Functional airwaycells | No | Yes: mucus secreted into organoid lumens | Not tested | Yes: mucus secreted into organoid lumens | Yes: mucus secreted into organoid lumens, beating multiciliated cells, and forskolin-induced swelling | No | No | Yes: mucus secreted into organoid lumen, beatingmulticiliated cells | |
| Alveolar-like cells | No | Yes: AECI and AECII-like cells (FGF7 alone in media) |
Yes: AECI and AECII-like cells |
Yes: AECII-like cells |
Yes: AECII-like cells |
Yes: AECII-like SFTPC+ cells |
Yes: AECI and AECII-like cells |
No | |
| Functional alveolarcells | No | Not tested | Not tested | Yes: cells uptake tagged SFTPB | Not tested | Yes: functional surfactant production | Not tested | No | |
| Epithelial budded/branch-like structures | No | Yes | No | Yes | No | No | No | No | |
| Functional (self-renewing, multilineage potential) bud tip progenitors | Yes | Yes | No | Not tested | Not tested | No | No | No | |
| Expandable organoids | Yes | No | No | No | No | Yes | Yes | No | |
| In vivo maturation | Time required for maturation in vivo | 6 weeks | Not tested | 8 weeks | 6–18 weeks | Not tested | Not tested | Not tested | Not tested |
| Transplantation into mouse fat pad/kidney capsule | Not tested | Not tested | Yes; maturation of adult airway structures and airway cell types including beating cilia and mucus secretion | Yes; maturation of airway structures and cell types including beating cilia and mucus secretion; maturation of alveolar type I and II cells | Not tested | Not tested | Not tested | Not tested | |
| Transplantation into injured mouse airway | Yes; successful engraftment and differentiation | Not tested | Not tested | Not tested | Not tested | Not tested | Not tested | Not tested | |
| Best suited for studies exploring: | • Epithelial lineage fate decisions • Fetal lung development • High-throughput drug/toxicology testing • Pathogen infection |
• Epithelial morphogenesis • Fetal lung development • Pathogen infection • Mucous cell metaplasia |
• Mesenchymal–epithelial interactions during development and disease (e.g., pulmonary fibrosis) • Pathogen infection |
• Epithelial morphogenesis • Pathogen infection • Mucous cell metaplasia |
• Adult diseases involving mucociliary function (CF, asthma) • Pathogen infection |
• Genetic alveolar disease • High-throughput drug/toxicology testing • Pathogen infection |
• Genetic alveolar disease • High-throughput drug/toxicology testing • Pathogen infection |
• Adult diseases involving mucociliary function (CF, asthma) • Pathogen infection |
|
| Not well suited for studies exploring: | • Adult epithelial disease modeling (e.g., CF, asthma) • Mesenchymal–epithelial interactions |
• Some adult disease modeling (e.g. CF, asthma) • Mesenchymal–epithelial interactions |
• Adult diseases involving mucociliary function (CF, asthma) • High-throughput drug/toxicology testing |
• Adult epithelial disease modeling (e.g. CF, asthma) • Mesenchymal–epithelial interactions |
• Mesenchymal–epithelial interactions • Alveolar diseases • Fetal lung development |
• Mesenchymal–epithelialinteractions • Airway diseases • Fetal lung development |
Mesenchymal–epithelial interactions; airway diseases | • Mesenchymal–epithelial interactions • Alveolar diseases • Fetal lung development |
Following directed differentiation to foregut endoderm/lung lineages, several reports have used cell-sorting techniques to isolate early-lung-lineage progenitors for subsequent 3D culture26,27,29–31. Gotoh et al.29 identified carboxypeptidase M (CPM) as a marker of ventral foregut endoderm. 3D culture of sorted CPM+ cells in coculture with human fetal fibroblasts gave rise to organoids with cells expressing alveolar epithelial cell markers and lamellar bodies. This technique was later built upon by Konishi et al.30, who reported that CPM+ organoids could be cultured in a commercially available medium specialized for air–liquid interface culture and supplemented with DAPT. After 42–56 d in culture, the CPM+ cells gave rise to airway-like organoids with functional multiciliated and mucus-producing cells. Given the presence of functional multiciliated and mucus-producing cells, this model is ideal for interrogating diseases affecting the airway, such as ciliopathies, cystic fibrosis or the process of goblet cell hyperplasia after infection. More recently, Yamamoto et al.35 demonstrated that CPM+ cells can be differentiated into AECII-like organoids via coculture with fetal fibroblasts, or in defined medium.
Hawkins et al.31 described the generation of an NKX2.1-reporter hPSC line (RUES2), which allowed isolation of NKX2.1+ cells. NKX2.1+ cells plated in 3D formed epithelial-only organoid structures. Transcriptomic profiling of these cells identified cell-surface markers (CD47hiCD26lo) that could allow for the prospective isolation of NKX2.1+ cells without the use of reporter cell lines. This method was built upon by Jacob et al.26 and McCauley et al.27, who showed that modulation of growth factor signaling in these cultures leads to the generation of distal (alveolar-like) or proximal (airway-like) organoids that are epithelial only, respectively. Proximal airway organoids have functional multiciliated and mucus-producing cells and have been shown to be able to model some hallmarks of cystic fibrosis when CF-iPSC cell lines are used. These systems are ideal for studying airway diseases that affect functional epithelial cells, such as cystic fibrosis and ciliopathies, and are also highly expandable, which makes them potential candidates for large drug-screening platforms.
In addition to hPSC-derived lung organoid models, it should be noted that many more in vitro human lung models have been generated from primary human adult or fetal lung tissue. These systems have been thoroughly reviewed elsewhere36–38 and are ideal for interrogating lung diseases, as samples can be patient specific; they also may be critical for personalized medicine and screening for therapeutic responsiveness.
Several of the models described above have excelled at generating functional and differentiated epithelial cell types that may be uniquely suited to the interrogation of disease phenotypes. In contrast, the protocols described here generate fetal-like lung tissue that is ideal for studies of human lung development. One additional difference between other methods and those described here is that self-aggregating 3D foregut spheroids form spontaneously and do not require cell dissociation, cell sorting or other specialized techniques. Foregut spheroids can be cultured in a Matrigel droplet and further differentiated into two distinct 3D lung organoid models (Fig. 1).
Limitations of the protocol
The HLO model (Fig. 1a) generates fetal-like airway epithelium and surrounding mesenchyme, as well as a population of cells that express alveolar cell markers. However, as with all other 3D organoid models described here, transplantation into a mouse is required for the generation of fully mature structures and cell types. The largest limitation to using this model to study disease and development is that HLOs take ~60 d to form and are not highly expandable, which limits the amount of material available for study. For both protocols, the use of Matrigel or other animal-derived products such as serum (used during endoderm differentiation) remains a barrier to translation. However, serum-free alternatives have been developed for other organoid models28,39, and therefore the protocol should be adaptable to the use of defined serum (dFBS), serum-free protocols, or fully synthetic hydrogels, which will aid in progress toward translation18,40. For example, human intestinal organoid protocols5 initially developed for use with serum now have commercially available serum-free alternatives (e.g., STEMdiff intestinal organoid kit, Stemcell Technologies), which suggests that these systems are fairly flexible and can be modified depending on the specific needs of the end user. In some cases, such as those where maintenance of a mesenchymal population is desirable (e.g., human lung organoids15), serum may be required, as the growth of lung organoids plus mesenchyme in serum-free medium was shown to lead to mesenchymal loss over time28.
The bud tip progenitor organoid model (Fig. 1b) generates a highly enriched population of lung bud tip progenitors from hPSCs in 22 d in a serum-free environment, but this population is composed largely of undifferentiated progenitor cells. Thus, this system is not ideal for studying differentiated cell function (e.g., airway function or alveolar function). Understanding the mechanisms by which bud tip progenitor cells differentiate into a specific functional epithelial cell type remains a large gap in knowledge in the field, which the bud tip progenitor organoid model will help to close. Moreover, our initial attempts to differentiate bud tip progenitors used a stochastic differentiation strategy in which cells were provided with a supportive growth environment containing only FGF7 and were allowed to spontaneously differentiate. In this context, bud tip progenitors gave rise to immature alveolar and airway cell types. However, we noted that these cells exhibited a bias toward mucus-producing cells in vitro; this result is not well understood. Much further progress will be required before truly controlled directed differentiation is achieved and becomes a viable option for regenerative medicine.
Expertise needed to implement the protocol
Any student or postdoctoral fellow with hPSC culture experience can use this protocol. No core facilities are required. All equipment is standard to most cell culture facilities, and reagents can be purchased from standard scientific vendors.
Materials
Biological materials
hESCs or hiPSCs. This protocol can be performed with any hESC or human iPSC line. We have successfully used hESC lines H1, H9, UM63–1 and UM77–1 (NIH registry nos. 0277, 0062, 0043 and 0278, respectively). H9 and H1 cell lines were obtained from the WiCell Research Institute, and UM used iPSC line 20–1 (ref. 5). In our experience, genetically modified reporter lines derived from parental cell lines perform as well as their unmodified counterparts (e.g., H9 mCherry and iPSC 20–1 TetO-GFP). ! CAUTION Research using hPSCs must be conducted in accordance with federal, state, local and institutional ethical guidelines and regulations. ! CAUTION Cell lines should be regularly checked for mycoplasma contamination and tested via cell line authentication methods (e.g., short tandem repeat profiling) to ensure proper cell line identity.
Reagents
Cell growth media and supplements
1-thioglycerol (Sigma-Aldrich, cat. no. M6145)
Activin A (R&D Systems, cat. no. 338-AC)
Advanced DMEM (Thermo Fisher Scientific, cat. no. 12491015)
ATRA (Stemgent, cat. no. 04–0021, CAS number 302-79-4)
B27 supplement (Thermo Fisher Scientific, cat. no. 17504044)
BSA (Sigma-Aldrich, cat. no. A9647)
CHIR-99021 (Stemcell Technologies, cat. no. 72054)
DMEM/F12 (no glutamine (DMEM/F12); Thermo Fisher Scientific, cat. no. 21331020 or 21331–020)
FBS (Thermo Fisher Scientific, cat. no. 16000044)
FGF10 (recombinant human fibroblast growth factor 10; R&D Systems, cat. no. 345-FG, or made in-house as previously described17)
FGF4 (recombinant human fibroblast growth factor 4; R&D Systems, cat. no. 7460-F4, or made in-house as previously described17)
FGF7 (recombinant human fibroblast growth factor 7; R&D Systems, cat. no. 251-KG/CF)
GlutaMAX (100×; Thermo Fisher Scientific, cat. no. 35050061)
HEPES buffer (1 M; Thermo Fisher Scientific, cat. no. 15630080)
HyClone-defined FBS (dFBS; Thermo Fisher Scientific, cat. no. 10437010)
L-ascorbic acid (Sigma-Aldrich, cat. no. A4544, CAS number 50-81-7)
mTeSR™1 (Stemcell Technologies, cat. no. 85850)
N-2 supplement (Thermo Fisher Scientific, cat. no. 17502048)
NOGGIN (R&D Systems, cat. no. 6057)
Penicillin–streptomycin (100×; Thermo Fisher Scientific, cat. no. 15140122)
RPMI 1640 medium (Thermo Fisher Scientific, cat. no. 11875119)
SB431542 (Stemgent, cat. no. 04–0010)
Smoothened agonist (SAG; Enzo Life Sciences, cat. no. ALX-270-426-M001)
Matrices, enzymes and other reagents
Dispase (Thermo Fisher Scientific, cat. no. 17105041)
Dimethyl sulfoxide (DMSO; sterile-filtered; Sigma-Aldrich, cat. no. D2650, CAS number 67-68-5)
Matrigel basement membrane matrix growth factor reduced (Corning, cat. no. 354230). Separate the volume of Matrigel into aliquots that will yield 100 µg/mL when diluted in 12 mL of liquid. A proteinconcentration is provided with the product specification sheet. ▲ CRITICAL Matrigel is used for coating plates to maintain hPSCs.
Matrigel basement membrane matrix (Corning, cat. no. 354234) ▲ CRITICAL This reagent is used for culturing organoids in droplets. ▲ CRITICAL Lot-to-lot variation occurs. Ensure that the Matrigel has a protein concentration greater than 8.0 mg/mL. If the protein concentration is higher, dilute to 8.0 mg/mL with DMEM/F12. If the protein concentration is too low, organoids will sink to the bottom of the droplet and adhere to the plastic culture dish, which will cause them to lose their 3D structure and is likely to change the fate of the cells.
Parafilm M wrapping film (Thermo Fisher Scientific, cat. no. S37440)
Sterile phosphate-buffered saline (PBS; pH 7.4; Thermo Fisher Scientific, cat. no. 10010049)
Sterile dH2O (Thermo Fisher Scientific, cat. no. 15230170)
Equipment
Tissue culture hood
Cell culture incubator
Dissecting microscope
Centrifuge
Pipet-Aid (or similar; e.g., Fisher Scientific, cat. no. 1368106)
Cell scraper (VWR, cat. no. 76036–004)
Steriflip (Millipore, cat. no. EW-29969–24)
Six-well plates (tissue-culture treated; Thermo Fisher Scientific, cat. no. 140675)
24-well plates (tissue-culture treated; Thermo Fisher Scientific, cat. no. 142475)
10-mL serological pipettes (Thermo Fisher Scientific, cat. no. 07200574)
1.5-mL tubes (Thermo Fisher Scientific, cat. no. 02682000)
15-mL conical centrifuge tubes (Falcon; Thermo Fisher Scientific, cat. no. 1495953A)
50-mL conical centrifuge tubes (Falcon; Thermo Fisher Scientific, cat. no. 1443222)
27-gauge needles (BD PrecisionGlide, single-use needles; Thermo Fisher Scientific, cat. no. 1482113B)
1-mL syringes (BD disposable syringes with Luer-Lok Tips; Thermo Fisher Scientific, cat. no. 1482330)
Reagent setup
Human pluripotent stem cell culture
Culture of hPSCs has been described elsewhere, such as in the Harvard Stem Cell Institute StemBook Protocols (http://www.stembook.org/protocols/pluripotent-cells). Detailed instructions are provided here to ensure the successful completion of this protocol. More technical information regarding the maintenance of hPSCs with mTeSR™1 can be found at the Stemcell Technologies website (https://www.stemcell.com/guide-to-passaging-human-pluripotent-stem-cells-using-mtesr1.html).
Aliquoting Matrigel for plate coating to maintain hPSCs
Thaw matrix growth-factor-reduced (BM GF-reduced Matrigel) Matrigel at 4 °C overnight. Separate BM GF-reduced Matrigel into aliquots as indicated on the manufacturer product sheet (lot dependent). The product specification sheet will provide a protein concentration for each lot of Matrigel. Separate the volume of Matrigel into aliquots that will lead to a 100 µg/mL hESC-qualified GF-reduced Matrigel when diluted into 12 mL of cold DMEM F/12. 12 mL of 100 µg/mL Matrigel is enough to coat one 24-well plate or two 6-well plates. Aliquots can be stored at −80 for up to 6 months.
Coating plates with basement membrane growth-factor-reduced Matrigel ● Timing 15–30 min to set up; 1 h to overnight incubation
Six-well tissue culture plates are coated with a thin layer of Matrigel to improve adhesion of stem cells. For routine maintenance of hPSCs, six-well plates are used. For differentiation, 24-well plates are used, and the number of wells can be scaled according to the needs of an individual experiment. The following procedure provides instructions for the preparation of two 6-well plates or one 24-well plate. Plates can be prepared 1 h before passaging of cells and left to coat at room temperature (20–25 °C) in a sterile hood, or plates can be coated up to 1 week before cells are split and stored at 4 °C with the plate wrapped in Parafilm to prevent evaporation. When you are ready to coat plates, remove one aliquot of basement membrane growth-factor-reduced Matrigel from −80 °C and leave it on ice until completely thawed. This aliquot corresponds to a volume of Matrigel that will yield a concentration of 100 µg/mL of protein when diluted into 12 mL of liquid. Each lot of hESC-qualified Matrigel comes with a protein concentration supplied by the manufacturer. Gently pipette the thawed basement membrane growth-factor-reduced Matrigel into 12 mL of cold DMEM/F12. Use immediately or store at 4 °C for up to 1 week. Do not agitate Matrigel while it is thawing, to avoid premature polymerization. Add 1 mL of diluted Matrigel to each well of a 6-well plate or 0.5 mL of thawed Matrigel to each well of a 24-well plate. Gently swirl or tap the plate to ensure that the entire bottom of each well is covered. Let plates sit for 1 h at room temperature (20–25 °C) before use, or wrap the edges of the plate with Parafilm and store at 4 °C for up to 1 week.
Preparing Matrigel before making Matrigel droplets to culture organoids
Basement membrane Matrigel should be stored at −80 °C and thawed overnight at 4 °C before use. Dilute basement membrane Matrigel with DMEM/F12 to a concentration of 8.2 mg/mL of protein.▲ CRITICAL When working with Matrigel, keep it on ice at all times and work quickly. Matrix will begin to solidify as it warms up.
Dispase
Prepare a 5 mg/mL solution of dispase powder with DMEM/F12. Make 2-mL aliquots in 15-mL conical tubes and store at −20 °C for up to 1 year. When it is time to use the solution, add 8 mL of DMEM/F12 to a 2-mL aliquot of dispase for a working concentration of 1 mg/mL. Store at 4° for up to 1 month.
Growth factors and small molecules
Dilute, separate into aliquots, and store according to the manufacturer’s instructions. Once thawed, growth factors and small molecules can be kept at 4 °C for up to 2 weeks.
Ascorbic acid
To make a 50 mg/mL solution, dissolve 500 mg of l-ascorbic acid into 10 mL of tissue-culture-grade sterile water in a 50-mL conical tube. In a tissue culture hood, filter this solution using a 0.22-µm Steriflip filter. Make aliquots of 50 µL and store at −20 °C for up to 1 year. Use a fresh aliquot each time; do not store a thawed aliquot for reuse.
Definitive endoderm differentiation medium for use on day 2
Dilute HyClone FBS in RPMI basal medium to 0.2% (vol/vol) concentration for day-2 endoderm differentiation. To make day-2 endoderm medium for all wells of a single 24-well plate, add 24 µL of HyClone FBS to 12 mL of RPMI medium. Details are also included in Table 2.
Table 2 |.
Media components
| Media | Definitive endoderm, day 1 | Definitive endoderm, day 2 | Definitive endoderm, day 3 | Definitive endoderm, day 4 | Anterior foregut endoderm, days 5–9 | Human lung organoid maintenance | Bud tip progenitor organoid medium |
|---|---|---|---|---|---|---|---|
| Basal media | RPMI 1640 | RPMI 1640 0.2% (vol/vol) HyClone FBS | RPMI 1640 2% (vol/vol) HyClone FBS | RPMI 1640 2% (vol/vol) HyClone FBS | (Foregut basal) Advanced DMEM/F12, 1× N-2, 1× B27, 10 mM HEPES buffer, 1× L-glutamine (2 mM), 1× penicillin–streptomycin (5,000 U/mL) | (Foregut basal) Advanced DMEM/F12, 1× N-2, 1× B27, 10 mM HEPES buffer, 1× L-glutamine (2 mM), 1× penicillin–streptomycin (5,000 U/mL) | (Bud tip basal) DMEM F12, 1× N-2, 1× B27, 1× L-glutamine (200 mM), 1× penicillin–streptomycin (5,000 U/mL), 0.05% (vol/vol) BSA |
| Add on day of use | Activin A, 100 ng/mL | Activin A, 100 ng/mL | Activin A, 100 ng/mL | Activin A, 100 ng/mL | 10 µM SB431542, 200 ng/mL Noggin, 1 µM SAG, 500 ng/mL FGF4, 2 µM CHIR-99021 | 500 ng/mL FGF10, 1% (vol/vol) FBS | 0.4 µM monothio-glycerol, 50 µg/mL ascorbic acid, 10 ng/mL FGF7, 50 nM ATRA, 3 µM CHIR-99021 |
Definitive endoderm differentiation medium for use on days 3 and 4
Dilute HyClone FBS in RPMI basal medium to a 2% concentration for days 3 and 4 of endoderm differentiation. To make basal medium for 2 d for a single 24-well plate, add 480 µL of HyClone FBS to 24 mL of RPMI medium. Store at 4 °C overnight between uses. Details are also included in Table 2.
Foregut differentiation medium
Make foregut basal medium by adding 1× N-2, 1× B27, 10 mM HEPES, 1× GlutaMAX and 1× pen–strep to advanced DMEM/F12 in a sterile hood. Store at 4 °C for up to 2 months. On the day of use, add 10 µM SB431542, 200 ng/mL Noggin, 1 µM SAG, 500 ng/mL FGF4 and 2 µM CHIR99021. Once growth factors and small molecules have been added, use on the same day; do not store. Details are also included in Table 2.
Human lung organoid medium
On the day of use, add 1% (vol/vol) FBS and 500 ng/mL FGF10 to foregut differentiation medium. 50 mL is enough to feed a 24-well plate for roughly 2 weeks. Store at 4 °C for up to 2 weeks. Details are also included in Table 2.
Bud tip progenitor organoid basal medium
First, prepare BSA by taking a 500-mL bottle of DMEM/F12 and transferring 10 mL of DMEM/F12 from the 500-mL bottle into a 50-mL conical tube. Weigh out 0.25 g of BSA and add to the DMEM/ F12 aliquot. Warm the aliquot in a water bath for 15 min with occasional gentle agitation to dissolve the BSA. In a tissue culture hood, filter the dissolved solution through a Steriflip filter. Add the BSA solution back to the DMEM/F12 bottle in a sterile hood.
Add 1× N-2, 1× B27, 1× glutamine and 1× pen–strep to the DMEM/F12 + BSA in a sterile hood to complete the bud tip progenitor organoid basal medium. Store at 4 °C for up to 2 months. Details are also included in Table 2.
Bud tip progenitor organoid complete medium for the generation and maintenance of bud tip progenitor organoids
When ready to generate or maintain bud tip progenitor organoids, add the appropriate supplements to the bud tip progenitor organoid basal medium. To make a 50-mL volume of bud tip progenitor organoid complete medium, place 50 mL of bud tip progenitor organoid basal medium in a 50-mL conical tube and add 50 µg/mL ascorbic acid and 0.4 µM monothioglycerol. This is the complete medium.
Then add 3 µM CHIR99021, 10 ng/mL FGF7 and 50 nM ATRA. 50 mL of medium is enough to feed a single 24-well plate for ~2 weeks. Store at 4 °C for up to 2 weeks.
Procedure
Splitting of human pluripotent stem cell culture ● Timing 30–60 min
▲ CRITICAL Proper maintenance of hPSCs is essential for successful completion of this protocol. Seeding density and colony size must be closely monitored to ensure that no premature differentiation occurs. See Figs. 3 and 4 for visual guides. Note that we do not use feeder layers to maintain hPSCs.
Fig. 3 |. Human pluripotent stem cell splitting and directed differentiation.
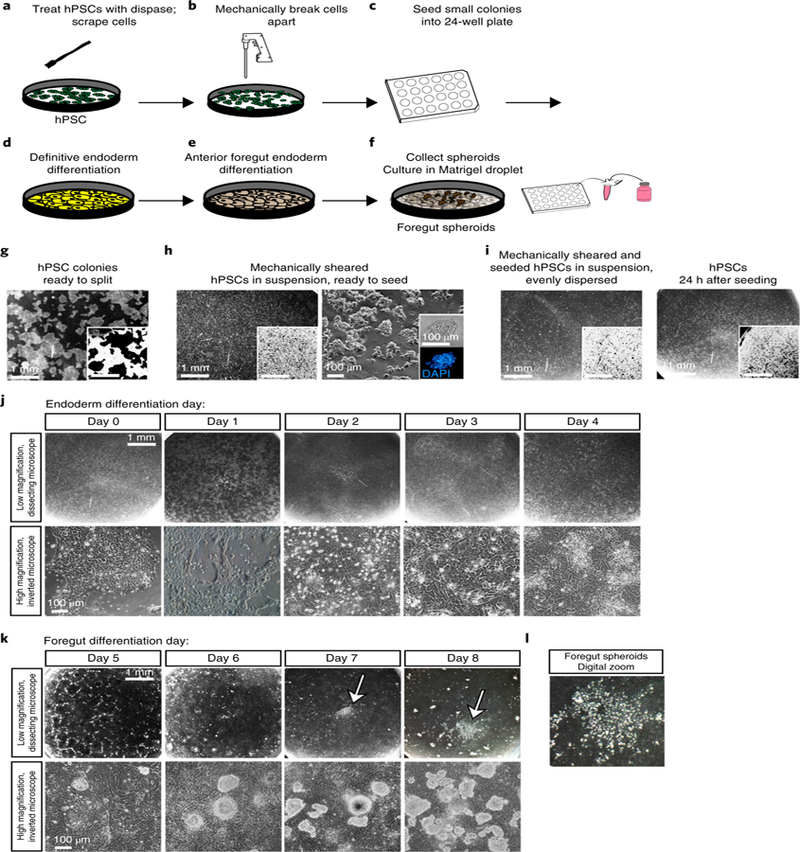
a–f, Schematic of the timeline and experimental procedures. g–i, Brightfield images of hPSCs during growth and splitting procedures. Insets are inverted images taken at the same magnification to help visualize cell density and colony shape. Scale bars, 1 mm or 100 µm as noted. a, First, treat hPSCs with dispase. g, Bright-field image indicates proper confluency and healthy colony shape. These cells are ready to split. b, Wash cells, scrape them up with a cell scraper, and use a serological pipette to mechanically break apart colonies. h, Bright-field image shows the proper size of broken-up colonies in suspension. These cells are ready to be seeded into a 24-well plate. c, Seed cells into a 24-well plate. i, Bright-field image showing floating colonies. These colonies are evenly dispersed throughout the well and can be placed in the incubator overnight. 24 h after seeding, colonies should cover roughly 50% of the bottom of the well and should be evenly dispersed. d, Begin endoderm differentiation protocol. j–l, Bright-field images showing typical morphology in the cell monolayer throughout endoderm differentiation at low magnification through a dissecting scope (j, top row; scale bar, 1 mm) or at higher magnification under an inverted microscope (j, bottom row; scale bar, 100 µm). e, Begin anterior foregut differentiation protocol. k, Bright-field images showing typical morphology in the cell monolayer throughout anterior foregut differentiation. Foregut spheroids begin to form around day 3–4 of anterior foregut differentiation (k, top row, white arrows). Shown at low magnification through a dissecting scope (k, top row; scale bar, 1 mm) and at higher magnification under an inverted microscope (k, bottom row; scale bar, 100 µm). l, A digitally zoomed image shows what foregut spheroids look like on the culture plate on day 8 of the differentiation protocol. f, Collect floating spheroids, mix them with Matrigel, and put them into droplets on a fresh 24-well plate.
Fig. 4 |. Common errors and troubleshooting.
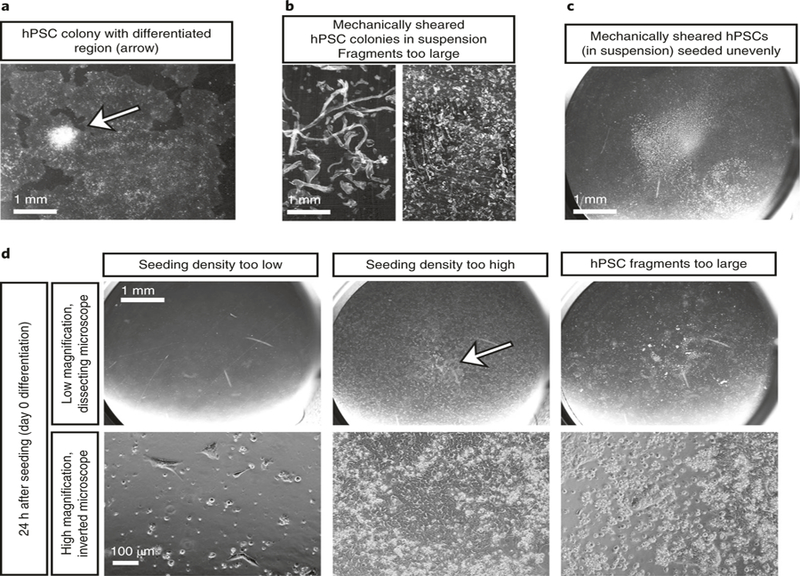
a, Before splitting hPSCs, check that there are no obvious differentiated colonies on the plate. Scrape away any abnormal colonies using a sterile pipette tip before splitting hPSCs into 24-well plates. The arrow indicates an area of hPSC differentiation visible as a raised white structure that should be scraped away before splitting. Scale bar, 1 mm. b, When breaking up hPSC colonies for splitting, ensure that the colonies are broken up to a small enough size. These images show colonies that are still too large. Continue breaking up colonies before proceeding with splitting cells. Scale bar, 1 mm. c, After hPSC colonies have been placed in a 24-well dish, shake the plate vigorously under a dissecting microscope to ensure that the cells are evenly dispersed. In this image the majority of cells are still clustered in the center of the well. This will cause an uneven growth pattern, and the differentiation will not yield spheroids. Shake cells before proceeding with differentiation. Scale bar, 1 mm. d, Bright-field images at low (top row; scale bar, 1 mm) and high (bottom row; scale bar, 100 µm) magnification showing culture conditions on day 0 of differentiation that are likely to fail to form spheroids. If the seeding density is too low or too high or if colony sizes are too big because the hPSC colonies were not sufficiently sheared after splitting, the differentiation protocol will not be successful. The arrow in the high-density seeding column indicates cells that have become confluent in the center of the well, which will lead to uneven differentiation and likely failure of the protocol to yield foregut spheroids.
▲ CRITICAL This procedure was developed and optimized for hPSCs maintained in six-well tissue culture dishes coated with 100 µg/mL basement membrane growth-factor-reduced Matrigel and fed daily with mTeSR™1. For routine maintenance, cells are kept in a tissue-culture-grade incubator with 95% air and 5% CO2 and are maintained in a sterile environment.
▲ CRITICAL This section describes how to establish cell cultures before beginning the differentiation protocol. It describes splitting 4 wells of a 6-well plate into all the wells of one 24-well plate.
Alternatively, one well of a six-well plate can be split into six wells of a six-well plate to maintain hPSCs.
-
1
Undifferentiated hPSC colonies that are ~75–85% confluent will appear rounded with well-defined smooth edges (Fig. 3a,g). Colonies with jagged edges, many bright white raised spots or ‘donut holes’ have probably begun to differentiate, and should not be used for this protocol (Fig. 4a). If there are only a few differentiated areas, scrape differentiated colonies off with a pipette tip under a dissecting microscope before splitting (Fig. 4a, arrow).
-
2
Warm dispase, mTeSR™1 and DMEM/F12 in a 37 °C water bath for ~10 min.
-
3
Add 1 mL of 1 mg/mL dispase solution to each well that will be split. Return to incubator for 5–10 min. Observe colonies under a microscope, and proceed when the edges of the colonies begin to lift off the tissue culture dish.
-
4
Carefully aspirate dispase. Colonies will still be attached to the dish.
-
5
Gently wash cells three times with prewarmed DMEM/F12.
-
6
Add 3.5 mL of mTeSR™1 medium to each well that will be split. This includes an extra 0.5 mL of medium per well to accommodate for the volume that may be lost during trituration and transfer to new plates.
-
7
Use a cell scraper to gently scrape all cells off the bottom of the dish.
-
8
Using a 10-mL serological pipette and automatic dispenser, triturate colonies into small clumps by doing the following: pull all cells up into the pipette, place the pipette firmly on the bottom of the dish and pipette down to shear the cell colonies apart. Check under a dissecting scope under low magnification (images in Fig. 2b shown at 0.7× magnification) to assess the size of the sheared colonies. Repeat this process until colonies are small enough that they do not immediately sink to the bottom of the dish. Figure 4b shows sheared colonies in suspension under a dissecting microscope that are too large. Aim to shear colonies until their size is similar to what is shown in Fig. 3h. Cell clumps should be small enough to remain floating in suspension, but should not be triturated all the way to single cells.
▲ CRITICAL STEP If sheared cell colonies are too large during splitting, proper differentiation will be difficult to achieve during this protocol (Fig. 4d).
-
9
Aspirate the diluted Matrigel media that was used to coat each well of the 24-well dish. If splitting into a six-well dish, add 1 mL of fresh, prewarmed mTeSR™1 plus 0.5 mL of triturated hPSC clumps. If splitting into a 24-well dish for differentiation, add 0.5 mL of triturated hPSCs to each well.
-
10
Under the dissecting microscope, aggressively shake the plate in a side-to-side motion to ensure that hPSCs are evenly distributed throughout the entire well, as seen in Fig. 2c. One can also achieve this by tapping the side of the 24-well plate several times.
▲ CRITICAL STEP If all of the hPSCs congregate to the center of the well (as shown in Fig. 4c),
Directed differentiation into endoderm and anterior foregut spheroids ● Timing 9–12 d: 4 d definitive endoderm differentiation, 4–5 d anterior foregut endoderm differentiation
-
11
Allow passaged cells to become 45–75% confluent before starting the protocol. This may take 1–2 d after passaging.
▲ CRITICAL STEP If confluency is too low, insufficient cells will remain after endoderm differentiation. If confluency is too high, differentiation in the center of the colonies may be incomplete and result in reduced spheroid formation. Figure 4d shows images of improper seeding density and of colonies that are too large. Proper confluency of cells in a 24-well plate ready to proceed to endoderm differentiation is shown in Fig. 3i.
?TROUBLESHOOTING
-
12Feed cells for 4 d with definitive endoderm differentiation medium (each day, change the medium by removing the existing medium and replacing it with 0.5 mL of medium per well of the 24-well plate). The appropriate media to use each day are listed below and in Table 2. Also check that cell morphology is as expected by comparing with the representative images shown in Fig. 3j.
Day Medium 1 RPMI + 0% (vol/vol) FBS + 100 ng/mL activin A 2 RPMI + 0.2% (vol/vol) FBS + 100 ng/mL activin A 3 RPMI + 2% (vol/vol) FBS + 100 ng/mL activin A 4 RPMI + 2% (vol/vol) FBS + 100 ng/mL activin A -
13
On day 5, replace medium with anterior foregut endoderm differentiation medium (0.5 mL of foregut basal media + 10 µM SB431542 + 200 ng/mL Noggin + 1 µM SAG + 500 ng/mL FGF4 + 2 µM CHIR99021, as listed in Table 2). Replace medium on days 6, 7, 8 and 9. On days 5–9, change media daily. Also check that cell morphology is as expected by comparing it with the representative images shown in Fig. 3k.
-
14
Observe that detached, floating spheroids begin to form around days 8 and 9 of culture (representative images are shown in Fig. 3k,l).
Collection of floating anterior foregut spheroids ● Timing 15–60 min
▲ CRITICAL Ensure sterile technique and a sterile environment throughout the process. This procedure is routinely performed in a laminar flow hood equipped with a stereomicroscope, or in a semi-sterile hood (e.g., Labconco 3970321) equipped with a stereomicroscope.
▲ CRITICAL A video presentation of the generation of Matrigel droplets for intestinal organoid culture has been published and can be used as a helpful reference for setting up lung organoid cultures41 (found at the 4:15 mark of the video https://www.jove.com/video/53359/organoids-as-model-for-infectious-diseases-culture-human-murine).
-
15
On day 8 or 9, place Matrigel on ice in the sterile hood.
-
16
16 Cut the tip off of a sterile p200 pipette filter tip (~5 mm) using sterile scissors or a scalpel and place an empty 1.5-mL snap-cap tube and a new (uncoated) 24-well plate in a hood with a dissecting microscope.
▲ CRITICAL STEP Some 24-well tissue culture dishes will allow Matrigel to form 3D droplets, whereas Matrigel disperses across the surface of other dishes. Nunc cell-culture treated multidishes allow droplets to form; other brands should be tested before transfer of Matrigel plus spheroids.
-
17
Place the 24-well culture plate that contains the foregut spheroids under a dissecting microscope. Observe one well under the stereomicroscope to identify free-floating spheroids. Without disturbing the monolayer, gently remove the free-floating spheroids by pipetting with a cut p200 pipette tip and transfer them into an empty 1.5-mL snap-cap tube. Spheroids from multiple wells can be pooled together in a single tube. We typically pool one entire 24-well plate into one 1.5-mL tube. Allow 5–10 min for the spheroids to settle to the bottom of the tube. Although spheroids should settle by gravity, they can be gently spun down with a microcentrifuge on the lowest setting for ~10 s if necessary.
-
18
Plate spheroids in basement membrane Matrigel (8.0 mg/mL protein concentration). To do this, gently remove as much media as possible from the tube under the dissecting microscope using a fresh p200 pipette. Once media has been removed, cut the tip off a fresh p200 tip. Draw up ~200 µL of Matrigel. Place Matrigel in the tube with the spheroids and gently pipette up and down several times to mix the spheroids in the Matrigel. Work quickly so the Matrigel does not congeal. Pipette with care to avoid making bubbles. Draw up all the Matrigel and spheroids, and pipette ~25 µL of the Matrigel–spheroid mixture into the center of a single well of the 24-well plate. 200 µL of starting mixture will be sufficient to make eight individual wells with a Matrigel droplet containing spheroids.
▲ CRITICAL STEP We routinely draw up the entire contents of wells containing Matrigel and spheroids into a single cut p200 tip and estimate 25 µL of droplets, but it is also acceptable to pipette individual 25-µL droplets if greater accuracy is desired.
▲ CRITICAL STEP Keep the Matrigel on ice throughout the protocol to ensure that it does not polymerize inside the bottle.
-
19
Place the plate in the incubator for ~10 min until Matrigel droplets have solidified. Once droplets have solidified, add 0.5 mL of desired media to generate either human lung organoids or epithelial bud tip progenitor organoids (see the next step for details).
Generation of human lung organoids or bud tip progenitor organoids
-
20Generate either human lung organoids (option A), which contain airway-like structures surrounded by lung mesenchyme and a population of cells with alveolar markers, or bud tip progenitor organoids (options B and C). Use option B if doing needle passaging. Use option C if using bulk passaging.
- Generation and maintenance of human lung organoids ● Timing Organoids with airway-like structures and surrounding mesenchyme will take ~50 d to grow and are optimal for use on days 50–85, as this is when the organoids have the maximum amount of airway-like structures. They need to be re-embedded in fresh Matrigel every 2–3 weeks.
- Add 1% (vol/vol) FBS and 500 ng/mL of FGF10 to foregut basal medium. Add 500 µL of media to each well of spheroids, ensuring that the media completely covers the Matrigel droplets. Change media every 3–5 d. See Table 2 for a summary of the media components.
- Re-embedding of organoids. Every 2–3 weeks, or sooner if organoids appear to be accumulating cellular debris within lumens or sinking to the bottom of the Matrigel droplet, cut the tip off a P1000 pipette tip and gently scrape the bottom of the well to dislodge the Matrigel droplet(s) containing organoids.
- Pick up the entire droplet and surrounding media with the pipette tip and move to a Petri dish. Under a dissecting scope in a sterile environment, use a scalpel and 27-gauge needle to gently cut and remove the old Matrigel away from the organoids. Be cautious not to cut the tissue. Once finished, move the now-cleaned organoids to a 1.5-mL snap-cap tube and remove any media.
- Cut the tip off a p200 pipette tip and transfer 200 µL of fresh Matrigel (kept on ice) to the tube with the organoids; mix and draw up all Matrigel into the p200 tip. Serially pipette ~50 µL of the Matrigel–organoid mixture into single wells in a fresh 24-well plate. Aim to put one to three organoids in each droplet.
- Place plate in the incubator for ~10 min, or until Matrigel droplets have solidified. Once droplets have solidified, add 500 µL of media.
- Maintain organoids for the desired time period, repeating Step 20A(ii–v) to re-embed organoids every 2–3 weeks. Once foregut spheroids are placed in Matrigel, it takes roughly 50 d to obtain pseudostratified epithelial structures that contain basal cells, mucus-producing cells and ciliated cells, as well as surrounding lung mesenchymal cell types and primitive alveolar cell types. The organoids can be maintained for more than 100 d; however, we find that the epithelium is gradually lost over time, and utilization between 60 and 85 d is optimal.
- Generation and maintenance of bud tip progenitor organoids using serial needle passaging ● Timing Epithelium-only cysts will form after ~2 weeks and will contain a nearly homogeneous population of bud tip progenitor cells. If culture is not homogeneous, organoids with clear epithelial cystic structures can be isolated by hand at this time point (2 weeks). This population of epithelial organoids can be maintained and expanded by serial needle passaging for over 120 d in culture.
- On the day of use, add 50 µg/mL l-ascorbic acid, 0.04 µL/mL monothioglycerol, 10 ng/mL FGF7, 50 nM ATRA and 3 µM CHIR-99021 to basal medium composed of DMEM/F12 + N-2 supplement + B27 supplement + 1× l-glutamine + 1× pen–strep to make bud tip progenitor organoid complete medium (as also detailed in Table 2). Feed foregut spheroids with 500 µL of bud tip progenitor organoid complete medium.
- Replace media every 3–5 d.
- Needle passage. After 2–3 weeks, or sooner if organoids appear to be accumulating cellular debris within lumens or sinking to the bottom of the Matrigel droplet, use a P1000 pipette tip to dislodge Matrigel droplets containing organoids. Place the dislodged droplets together into a 1.5-mL snap-cap tube.
- Using a 1-mL syringe and a 27-gauge needle, draw the Matrigel and cells into the syringe and forcefully expel the contents back into the tube. Repeat two to three times.
- Spin down the contents of the tube in a benchtop minicentrifuge (e.g., Thermo Fisher Scientific mySPIN 6 Mini Centrifuge, cat. no. 75004061) at full speed (6,300g) for ~3–5 s. It is also acceptable to spin cells down at 300g for 3 min at 4 °C. Epithelial fragments will settle to the bottom, and Matrigel will remain suspended in the media.
- Under a dissecting scope in a sterile hood, use the needle and syringe to remove the media and Matrigel from the tube, leaving behind the cell pellet.
- Add ~200 µL of fresh ice-cold Matrigel to the tube using a p200 pipette with the tip cut off. Mix the cells and Matrigel, avoiding bubbles. Draw up all 200 µL of the Matrigel mixture and serially pipette ~25 µL of the mixture into a single well of a 24-well plate.
- Place the plate in the incubator for ~10 min or until Matrigel droplets have solidified. Add 750 µL of bud tip progenitor organoid complete medium.
- Repeat the needle passage (Step 20B(iii–viii)) every 2–3 weeks, or at any time to select for the bud tip progenitor population.
-
Generation and maintenance of bud tip progenitor organoids using bulk passaging ● Timing Epithelium-only cysts will form after ~2 weeks and will contain a nearly homogeneous population of bud tip progenitor cells. To generate patterned lung organoids with discrete bud tip and airway-like regions, keep their structure intact during passaging. Bud tip progenitor organoids that are not needle passaged will develop budded epithelial structures and elements of proximal–distal patterning, as well as functional mucus-producing cells on the interior of the structure, after roughly 6 weeks in culture.▲ CRITICAL Bud tip progenitor organoids that are not needle passaged will secrete mucus into the lumen of the organoid, causing a dense appearance and leading to increased cell death; therefore, these organoids must be cleaned out regularly.
- On the day of use, add 50 µg/mL l-ascorbic acid, 0.04 µL/mL monothioglycerol, 10 ng/mL FGF7, 50 nM ATRA and 3 µM CHIR-99021 to basal medium composed of DMEM/F12 + N-2 supplement + B27 supplement + 1× l-glutamine + 1× pen–strep to make bud tip progenitor organoid complete medium (as also detailed in Table 2). Feed foregut spheroids with 500 µL of bud tip progenitor organoid complete medium.
- Replace media every 3–5 d.
- Passage of bud tip progenitor organoids without a needle. After 2–3 weeks, or sooner if organoids appear to be accumulating cellular debris within lumens or sinking to the bottom of the Matrigel droplet, scrape Matrigel droplets containing bud tip progenitor organoids using a cut P1000 pipette and transfer both the media within the well and the Matrigel droplet to a Petri dish.
- Use a scalpel to cut away old Matrigel without disturbing the tissue structure, and reembed in fresh Matrigel droplets as described in Step 20A(ii–v) above. These organoids contain bud tip progenitor cells in budded structures at the periphery of the organoid. Interior regions will predominantly show SOX2+ airway-like structures with differentiated mucus-producing cells. They can be needle passaged as described in Step 20B(iii–viii) at any time to select for the bud tip progenitor population.
- Clearing mucus from non-needle-passaged organoids. Mucus or cellular debris will often accumulate within the lumens of organoids and will appear as an optically dense, dark and granulated substance within the lumen after ~2 weeks in culture without passage. To clear this debris, get a sterile scalpel, a clean 27-gauge needle, a 1-mL syringe and a fresh aliquot of prewarmed DMEM/F12. Transfer organoids plus Matrigel to a Petri dish with prewarmed DMEM/F12 and transfer to a sterile hood with a stereomicroscope.
- Under the stereomicroscope, manually cut organoids in half with the sterile scalpel. Using the 1-mL syringe with the 27-gauge needle, draw up clean prewarmed DMEM/F12 into the syringe. Gently expel the media through the needle to wash the mucus away from the organoids.
● Timing See Fig. 1 for an overview of the timeline of the protocol. Splitting hPSCs to establish the cell cultures required takes 0.5–1 h (Steps 1–10). The culture reaches appropriate confluency after 1–2 d. The generation of foregut spheroids from hPSCs takes 9 d (Steps 11–14). It takes roughly 1 h to collect foregut spheroids and culture them in a Matrigel droplet (Steps 15–20). From there, human lung organoids with airway-like structures and multiple immature epithelial and mesenchymal cell types can be generated after roughly 50 d in culture (41 d from foregut spheroid stage; Step 20A(i–v)), whereas bud tip progenitor organoids can be generated within 22 d (14 d from foregut spheroids stage; Step 20B(i,ii)) and can be maintained as proliferating progenitors (Step 20B (iii–ix)) or as budded patterned lung organoids with proximal–distal patterning (Step 20C(i–vi)).
Troubleshooting
The most common problem we have identified is a failure of foregut spheroids to form during the directed differentiation of hPSCs (Steps 11–14). Common causes of failure to form spheroids are as follows: (i) the initial seeding density of hPSCs is too dense; (ii) hPSC colonies are allowed to grow too large in the 24-well plate; (iii) growth factors were stored at 4 °C for too long and lost potency; and (iv) hPSC lines were not maintained well (they contained spiky, unhealthy colonies) or differentiation was started at too high a passage number. Generally, hPSCs with a passage number < 90 should be used. We have also noticed that high-passage (>70 passages) hPSCs have reduced spheroid-forming ability. If spheroids fail to form, the experiment must be attempted again from the beginning.
To ensure that the seeding density and colony size of the hPSC starting material are correct, we recommend comparing results to those shown in Figs. 3h–l and 4b–d. Preliminary and unpublished experiments in our lab have suggested that stem cells can be broken into colonies as small as single cells and plated at a density of 125,000 cells per single well of a 24-well plate to generate foregut spheroids; this could be attempted if mechanical trituration is not working well. Our experience is that small colonies differentiate far more robustly than large colonies. Review Figs. 3 and 4 as a guide for appropriate colony sizes.
We also recommend that you record how long growth factors have been stored at 4 °C. When in doubt, throw them out.
Once spheroids have been plated in Matrigel, another common problem is that the spheroids and resulting organoids appear to drop to the bottom of the Matrigel droplet and adhere to the plastic culture dish (Steps 20A(i) and 20B(i)). If this happens, it is crucial to replate the organoids in a fresh Matrigel droplet as soon as possible; any organoids that have adhered to the plate and have a ‘flat’ morphology should be discarded. Possible reasons for organoids ‘bottoming out’ are that (i) it has been longer than 2–3 weeks since the last passage, (ii) the Matrigel has been at 4 °C for too long and the protein concentration has decreased owing to adhesion to the container, or (iii) the Matrigel droplets were too large and do not have a high dome shape that allows ample room for 3D growth.
Timing
Coating of culture plates: 1.25 h or overnight
Steps 1–10, splitting of hPSC culture: 30–60 min
Steps 11–14, directed differentiation into endoderm and anterior foregut spheroids: 9–12 d: 4 d definitive endoderm differentiation, 4–5 d anterior foregut endoderm differentiation
Steps 15–19, collection of floating anterior foregut spheroids: 15–60 min
Step 20A, generation of human lung organoids: ~50 d
Step 20B, generation of bud tip progenitor organoids by serial needle passaging: ~14–120 d (14 d for initial growth, 120+ d for maintenance and serial passaging)
Step 20C, generation of budded, patterned lung organoids by bulk passaging: ~6 weeks
Anticipated results
Foregut spheroids should self-aggregate, detach from the monolayer and float freely in the media by the fourth day of foregut differentiation (Fig. 3k–l). The number of spheroids formed in each well can vary. Generally, a single well will produce roughly 100–400 spheroids by day 5 of foregut differentiation. They appear as small irregular cell clumps (Fig. 5a) that express NKX2.1 and SOX2 (Fig. 5b,c).
Fig. 5 |. Expected outcomes of the protocol.

a, Bright-field image of foregut spheroids on the day of collection, cultured in a Matrigel droplet. Scale bar, 200 µm. b,c, The overwhelming majority of foregut spheroid cells are (b) NKX2.1+ and (c) SOX2+. Scale bars, 50 µm. d–j, Expected outcomes of the human lung organoid protocol. d, Bright-field images showing expected growth patterns for human lung organoids. Scale bars, 500 µm. e, Human lung organoids have persisting epithelial structures and surrounding mesenchyme, as shown by H&E staining at day 100 of culture. Scale bar, 500 µm. f, Epithelial structures contain P63+ basal-like cells and an organized airway-like epithelium. Scale bar, 50 µm. g, Surrounding mesenchyme is positive for smooth muscle marker smooth muscle actin (SMA), and epithelial structures contain P63+ cells on the basolateral surface, with FOXJ1+ cells on the luminal side of the epithelium. P63+ cells constituted, on average, roughly 40% of all cells within organoids. Though FOXJ1+ cells composed ~5% of the total cells in the HLOs, mature ACTUB structures were not seen at day 65 and were rare at 85 d (data not shown). Scale bar, 50 µm. h–j, Scale bars, 50 µm. h, Cells surrounding day-65 HLO ECAD+ epithelial structures were positive for multiple mesenchymal-cell-type markers, including PDGFRα and vimentin (VIM). i, Rare cells within HLOs express SOX9 and markers of the AECII marker surfactant protein C (SFTPC). These cells make up roughly 5% of all cells within an organoid. j, Rare cells within HLOs express SOX9 and the AECI marker HOPX. These cells make up roughly 5% of all cells within an organoid. k–m, Expected outcomes of the bud tip progenitor organoid protocol if organoids are maintained by regular needle passaging. k, Epithelial structures form and can be isolated by day 12 in culture. Scale bar, 500 µm. l, By day 14 of culture, over 95% of epithelial cells are NKX2.1+SOX2+. Scale bars, 50 µm. m, Needle-passaged bud tip progenitor organoids have a simple epithelial structure composed of SOX9+SOX2+ cells similar to that observed in the native human lung bud tip progenitors before week 16 of gestation. Scale bars, 50 µm. n–r, Expected outcomes of the bud tip progenitor organoid protocol if organoids are not maintained by needle passage and are instead allowed to grow intact into complex epithelial structures. n, Expected growth patterns over time of bud tip progenitor organoids. Epithelial structures predictably begin to fold at ~3 weeks, and form branch-like structures at 5–6 weeks. Scale bar, 200 µm; all images taken at the same magnification. o, Bright-field images over 5 d in culture, showing bifurcation of an epithelial bud structure. Bifurcated bud tips are marked with an asterisk. Scale bar, 200 µm; all images taken at the same magnification. p, Cartoon representation of the interior and bud tip regions of non-needle-passaged bud tip progenitor organoids. q, In the interior regions of these structures, roughly 5% of cells exhibit SCGB1A1 staining, and roughly 2% of cells stain positive for goblet cell marker MUC5AC. Scale bar, 50 µm. r, The bud tip regions of these structures maintain the expression of bud tip progenitor markers SOX9 and pro-surfactant protein C (SFTPC). Scale bars, 50 µm. Detailed information about antibodies used for staining can be found in Table 3. g–j, Reproduced with permission from ref. 15, eLife Sciences Publications (licensed under CC BY-NC-ND 4.0, https://creativecommons.org/licenses/by/4.0/). l–r, reproduced with permission from ref. 17, Elsevier (licensed under CC BY-NC-ND 4.0, https://creativecommons.org/licenses/by/4.0/).
Human lung organoids
Spheroids that are treated with FGF10 and 1% (vol/vol) FBS will give rise to human lung organoids. Spheroids grown in these conditions will generate clear epithelial structures by day 12 in culture. As the organoids grow, mesenchymal populations will be visible surrounding epithelial structures (Fig. 5d). Epithelial structures will persist until ~100 d in culture, as visualized by H&E staining (Fig. 5e). By 65 d in culture, human lung organoids will have pseudostratified epithelial structures with P63+ basal stem-cell-like cells lining the basal surface (Fig. 5f), as well as a population of cells with positive staining for the ciliated cell marker FOXJ1 (~5% of all cells; Fig. 5g) and alveolar cell markers SFTPC and HOPX (~5% of all cells; Fig. 5i,j)15,16. Cells surrounding the epithelial structures stain positive for multiple mesenchymal cell markers, including smooth muscle actin (Fig. 5g), PDGFR-α and vimentin (Fig. 5h).
Bud tip progenitor organoids
For spheroids that are cultured in serum-free medium with FGF7, CHIR99021 and ATRA, by day 12 of culture epithelial structures will form that are positive for SOX2 and NKX2.1 (Fig. 5k,l). To maintain a highly enriched population of bud tip progenitors, one can needle passage the organoids. This can be done starting at 2 weeks of culture, or at any time thereafter. Needle-passaged organoids form round cysts (Fig. 5m), and over 88% of these cells are SOX2+SOX9+ (Fig. 5m)17 and share similar molecular profiles with bud tip progenitors in the human fetal lung before week 16 of gestation17. If cultures are kept intact and are not broken up, they will form organoids with clear budded structures by day 45 (Fig. 5n). Epithelial bud tip structures will undergo clear bifurcation events (Fig. 5o). These ‘patterned lung organoids’ exhibit bud-tip-like and airway-like regions (Fig. 5p). Interior regions of the organoid contain SOX2+ airway-like cells and mucus-producing cells. Roughly 5% of cells in these organoids stain positive for Club cell marker SCGB1A1, and roughly 2% of cells stain positive for goblet cell marker MUC5AC (Fig. 5q). Bud tip regions of these organoids will maintain SOX9+, pro–surfactant protein C (ProSFTPC)+ cells, indicating maintenance of bud tip progenitors (Fig. 5r). Mucus will build up within these organoids, causing the interior to become dense and optically opaque. Mucus can be cleared during passaging to a fresh Matrigel droplet.
Supplementary Material
Table 3 |.
Antibody information
| Primary antibody | Source | Cat. no. | Dilution (sections) | Clone |
|---|---|---|---|---|
| Chicken anti-GFP | Abcam | Ab13970 | 1:500 | Polyclonal |
| *Biotin–goat anti-TP63 | R&D systems | BAF1916 | 1:500 | |
| *Biotin–mouse anti-MUC5AC | Abcam | ab79082 | 1:500 | Monoclonal |
| Goat anti-CC10 (SCGB1A1) | Santa Cruz Biotechnology | sc-9770 | 1:200 | C-20 |
| Goat anti-chromogranin A (CHGA) | Santa Cruz Biotechnology | sc-1488 | 1:100 | C-20 |
| Goat anti-SOX2 | Santa Cruz Biotechnology | Sc-17320 | 1:200 | Polyclonal |
| Goat anti-vimentin (VIM) | Santa Cruz Biotechnology | sc-7558 | 1:100 | S-20 |
| Mouse anti-acetylated tubulin (ACTUB) | Sigma-Aldrich | T7451 | 1:1,000 | 6–11B-1 |
| anti-Mouse α-smooth muscle actin (SMA)*Cy3-conjugated | Sigma | C6198 | 1:400 | Monoclonal |
| Mouse anti-E-cadherin (ECAD) | BD Transduction Laboratories | 610181 | 1:500 | 36/E-cadherin |
| Mouse anti-FOXJ1 | eBioscience | 14-9965-82 | 1:500 | 2A5 |
| Mouse anti-human nuclear antigen (HuNu) | Abcam | ab191181 | 1:250 | Monoclonal |
| Mouse anti-surfactant protein B (SFTPB) | Seven Hills Bioreagents | Wmab-1B9 | 1:250 | Monoclonal |
| Rabbit anti-HOPX | Santa Cruz Biotechnology | Sc-30216 | 1:250 | Polyclonal |
| Rabbit anti-NKX2.1 | Abcam | ab76013 | 1:200 | EP1584Y |
| Rabbit anti-nuclear mitotic aparatus—human (NuMa) | Thermo Scientific | PA5–22285 1:500 | Polyclonal | |
| Rabbit anti-PDGFRα | Santa Cruz Biotechnology | sc-338 | 1:100 | C-20 |
| Rabbit anti-pro-surfactant protein C (pro-SFTPC) | Seven Hills Bioreagents | Wrab-9337 1:500 | Polyclonal | |
| Rabbit anti-SOX9 | Millipore | AB5535 | 1:500 | Polyclonal |
| Rabbit anti-synaptophysin | Abcam | AB32127 | 1:500 | Monoclonal |
| Secondary antibody | Source | Cat. no. | Dilution |
|---|---|---|---|
| Donkey anti-goat 488 | Jackson ImmunoResearch | 705-545-147 | 1:500 |
| Donkey anti-goat 647 | Jackson ImmunoResearch | 705-605-147 | 1:500 |
| Donkey anti-goat Cy3 | Jackson ImmunoResearch | 705-165-147 | 1:500 |
| Donkey anti-mouse 488 | Jackson ImmunoResearch | 715-545-150 | 1:500 |
| Donkey anti-mouse 647 | Jackson ImmunoResearch | 415-605-350 | 1:500 |
| Donkey anti-mouse Cy3 | Jackson ImmunoResearch | 715-165-150 | 1:500 |
| Donkey anti-rabbit 488 | Jackson ImmunoResearch | 711-545-152 | 1:500 |
| Donkey anti-rabbit 647 | Jackson ImmunoResearch | 711-605-152 | 1:500 |
| Donkey anti-rabbit Cy3 | Jackson ImmunoResearch | 711-165-102 | 1:500 |
| Donkey anti-goat 488 | Jackson ImmunoResearch | 705-545-147 | 1:500 |
| Donkey anti-goat 647 | Jackson ImmunoResearch | 705-605-147 | 1:500 |
| Donkey anti-goat Cy3 | Jackson ImmunoResearch | 705-165-147 | 1:500 |
| Donkey anti-mouse 488 | Jackson ImmunoResearch | 715-545-150 | 1:500 |
| Donkey anti-mouse 647 | Jackson ImmunoResearch | 415-605-350 | 1:500 |
| Donkey anti-mouse Cy3 | Jackson ImmunoResearch | 715-165-150 | 1:500 |
| Donkey anti-rabbit 488 | Jackson ImmunoResearch | 711-545-152 | 1:500 |
| Donkey anti-rabbit 647 | Jackson ImmunoResearch | 711-605-152 | 1:500 |
| Donkey anti-rabbit Cy3 | Jackson ImmunoResearch | 711-165-102 | 1:500 |
| Streptavidin 488 | Jackson ImmunoResearch | 016-540-084 | 1:500 |
Acknowledgements
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01HL119215 to J.R.S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests
J.R.S., B.R.D. and A.J.M. hold patents pertaining to the lung organoid technologies described. The other authors declare no competing interests.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Some of the data presented in the current study were generated for and published in previous reports. Original data used for figures in this paper are available at the following links: Fig. 2b–d (Dye et al.16), https://doi.org/10.7554/eLife.19732; Figs. 2e and 5g–j (Dye et al.15), https://doi.org/10.7554/eLife.05098; Figs. 2g–i and 5l–r (Miller et al.17), https://doi.org/10.1016/j.stemcr.2017.11.012.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41596-018-0104-8.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zorn AM & Wells JM Vertebrate endoderm development and organ formation. Annu. Rev. Cell Dev. Biol 25, 221–251 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang DR et al. Lung epithelial branching program antagonizes alveolar differentiation. Proc. Natl. Acad. Sci. USA 110, 18042–18051 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rawlins EL Lung epithelial progenitor cells: lessons from development. Proc. Am. Thorac. Soc 5, 675–681 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rawlins EL, Clark CP, Xue Y & Hogan BLM The Id2+ distal tip lung epithelium contains individual multipotent embryonic progenitor cells. Development 136, 3741–3745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spence JR et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCracken KW, Howell JC, Wells JM & Spence JR Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc 6, 1920–1928 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Múnera JO et al. Differentiation of human pluripotent stem cells into colonic organoids via transient activation of BMP signaling. Cell Stem Cell 21, 51–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai Y-H et al. In vitro patterning of pluripotent stem cell-derived intestine recapitulates in vivo human development. Development 144, 1045–1055 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hannan NRF, Sampaziotis F, Segeritz C-P, Hanley NA & Vallier L Generation of distal airway epithelium from multipotent human foregut stem cells. Stem Cells Dev 24, 1680–1690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hannan NRF et al. Generation of multipotent foregut stem cells from human pluripotent stem cells. Stem Cell Rep 1, 293–306 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Longmire TA et al. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Stem Cell 10, 398–411 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang SXL et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat. Biotechnol 32, 84–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green MD et al. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat. Biotechnol 29, 267–272 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang SXL et al. The in vitro generation of lung and airway progenitor cells from human pluripotent stem cells. Nat. Protoc 10, 413–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dye BR et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife 4, e05098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dye BR et al. A bioengineered niche promotes in vivo engraftment and maturation of pluripotent stem cell derived human lung organoids. Elife 5, e19732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller AJ et al. In vitro induction and in vivo engraftment of lung bud tip progenitor cells derived from human pluripotent stem cells. Stem Cell Rep 10, 101–119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cruz-Acuña R et al. Synthetic hydrogels for human intestinal organoid generation and colonic wound repair. Nat. Cell Biol 19, 1326–1335 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szenker-Ravi E et al. RSPO2 inhibition of RNF43 and ZNRF3 governs limb development independently of LGR4/5/6. Nature 557, 564–569 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Hill DR et al. Bacterial colonization stimulates a complex physiological response in the immature human intestinal epithelium. Elife 6, e29132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill DR, Huang S, Tsai Y-H, Spence JR & Young VB Real-time measurement of epithelial barrier permeability in human intestinal organoids. J. Vis. Exp 10.3791/56960 (2017). [DOI] [PMC free article] [PubMed]
- 22.Perrin S Preclinical research: make mouse studies work. Nature 507, 423–425 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Ruggeri BA, Camp F & Miknyoczki S Animal models of disease: pre-clinical animal models of cancer and their applications and utility in drug discovery. Biochem. Pharmacol 87, 150–161 (2014). [DOI] [PubMed] [Google Scholar]
- 24.van der Laan JW, Chapin RE, Haenen B, Jacobs AC & Piersma A Testing strategies for embryo-fetal toxicity of human pharmaceuticals. Animal models vs. in vitro approaches: a workshop report. Regul. Toxicol. Pharmacol 63, 115–123 (2012). [DOI] [PubMed] [Google Scholar]
- 25.D’Amour KA et al. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol 23, 1534–1541 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Jacob A et al. Differentiation of human pluripotent stem cells into functional lung alveolar epithelial cells. Cell Stem Cell 21, 472–488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCauley KB et al. Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of Wnt signaling. Cell Stem Cell 20, 844–857 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y-W et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol 19, 542–549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gotoh S et al. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell Rep 3, 394–403 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konishi S et al. al. Directed induction of functional multi-ciliated cells in proximal airway epithelial spheroids from human pluripotent stem cells. Stem Cell Rep 6, 18–25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawkins F et al. Prospective isolation of NKX2-1-expressing human lung progenitors derived from pluripotent stem cells. J. Clin. Invest 127, 2277–2294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D, Haviland DL, Burns AR, Zsigmond E & Wetsel RA A pure population of lung alveolar epithelial type II cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 104, 4449–4454 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Firth AL et al. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 111, E1723–E1730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Haute L, De Block G, Liebaers I, Sermon K & De Rycke M Generation of lung epithelial-like tissue from human embryonic stem cells. Respir. Res 10, 105 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto Y et al. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat. Methods 14, 1097–1106 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Nikolić MZ & Rawlins EL Lung organoids and their use to study cell-cell interaction. Curr. Pathobiol. Rep 5, 223–231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller AJ & Spence JR In vitro models to study human lung development, disease and homeostasis. Physiology 32, 246–260 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clevers H Modeling development and disease with organoids. Cell 165, 1586–1597 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Hannan NRF, Segeritz C-P, Touboul T & Vallier L Production of hepatocyte-like cells from human pluripotent stem cells. Nat. Protoc 8, 430–437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gjorevski N et al. Designer matrices for intestinal stem cell and organoid culture. Nature 539, 560–564 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Bartfeld S & Clevers H Organoids as model for infectious diseases: culture of human and murine stomach organoids and microinjection of Helicobacter pylori. J. Vis. Exp 2015, e53359 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.