

Abstract
Rationale:
Mitochondrial mutations are associated with a wide spectrum of clinical abnormalities. More than half of these mutations are distributed in the 22 mitochondrial tRNA genes, including tRNALeu(UUR). In particular, the A3243G mutation in the tRNALeu(UUR) gene causes mitochondrial encephalomyopathy.
Patient concerns:
A 12-year-old boy was admitted to Shaoxing People's Hospital because there is a reduction in the volume of speech, dysphonia, unable to write, recognize words, and unable to wear clothes, accompanied by unstable walking after treatment of unexplained fever and somnolence.
Diagnoses:
The proband underwent a thorough examination in our hospital and was diagnosed as mitochondrial encephalomyopathy. The proband carried the pathogenic heteroplasmic mutation A3243G mutation in mitochondrial 12S rRNA gene. Although his parents did not carry the mutation.
Interventions:
Intravenous acyclovir, ceftriaxone, and dexamethasone were used for the patient's antiviral, antimicrobial, and anti-inflammatory therapy, respectively. Intravenous mannitol was gradually tapered for reducing intracranial pressure with furosemide for inducing diuresis. Intravenous arginine could help to treat alkalosis and supple some essential amino acids. Oral oxiracetam capsules, vitamin B1, and coenzyme Q10 were used for providing nutrition and improving energy. His medications were 30 mg vitamin B1, 0.1 g vitamin C, and mecobalamin 750 μg daily after discharge from our hospital.
Outcomes:
The patient was able to walk and talk slowly with improved writing skills and no stroke-like episodes. The neurological examination was negative and muscle tension was identified as grade V.
Lessons:
Mitochondrial encephalomyopathy has different phenotypes, in addition to traditional examinations, it is important for clinicians to be familiar with genetic testing methodology as well as applications of these tests in clinic to get an accurate diagnosis.
Keywords: A3243G, mitochondrial DNA, mitochondrial encephalomyopathy, tRNALeu(UUR)
1. Introduction
Mitochondrial mutations are associated with a wide spectrum of clinical abnormalities, including neuromuscular disorders, heart failure, diabetes, and hearing loss.[1–4] More than half of these mutations are distributed in the 22 mitochondrial tRNA genes, including tRNALeu(UUR).[5] In particular, the A3243G mutation in the tRNALeu(UUR) gene causes mitochondrial encephalomyopathy.[6]
In the present study, we report a boy diagnosed with mitochondrial encephalomyopathy carried the mitochondrial A3243G heterplastic mutation. We present detailed clinical data and further delineate the phenotype associated with this disease.
2. Case presentation
The proband was a 12-year-old boy on first visiting to our hospital. The proband was the first and the only child of his healthy, nonconsanguineous parents of Han Chinese descentand was born at 38 weeks gestation by spontaneous delivery. The proband had no postnatal adaptation, and the Apgar score was unknown. He was admitted to the hospital because there is a reduction in the volume of speech, dysphonia, unable to write, recognize words, and unable to wear clothes, accompanied by unstable walking after the treatment of unexplained fever and somnolence. The highest temperature was 37.8°C, accompanied by vomiting 4 to 5 times on the same day, with insufficient spirits.
The study was conducted in accordance with the Declaration of Helsinki and was approved by the local ethics committee of our hospital (no. 2018021). Informed written consent was obtained from all participants prior to their enrollment in this study.
The proband underwent a thorough examination in our hospital, including electroencephalogram, echocardiogram, electrocardiogram, brain magnetic resonance imaging (MRI), magnetic resonance spectroscopy (MRS), physical examination, laboratory examination, and mitochondrial gene detection.
Genomic DNA from the family was extracted from peripheral whole blood samples using the genomic DNA extraction kit (Qiagen) according to the manufacturer's instructions. The entire mitochondrial genomes of the family were PCR amplified in 24 overlapping fragments by using sets of the light strand and the heavy strand oligonucleotide primers, as described elsewhere.[7] Each fragment was purified and subsequently Sanger sequencings were conducted by TSINGKE Biological Technology(Beijing, China). The resultant sequence data were compared with the revised consensus Cambridge sequence (GenBank accession no. NC-012920).
His electroencephalogram showed clear increase of 4–5c/s theta waves, 2–3c/s delta waves, with a small amount of sharp waves. His brain MRI revealed abnormal signal, brain tissue swelling, and midline structure was shifted slightly to the right (Fig. 1A). A large lactate peak and decreased N-acetylaspartate were found in this region on proton MRS (Fig. 1B). Normal structure and ejection fraction were observed via echocardiography.
Figure 1.
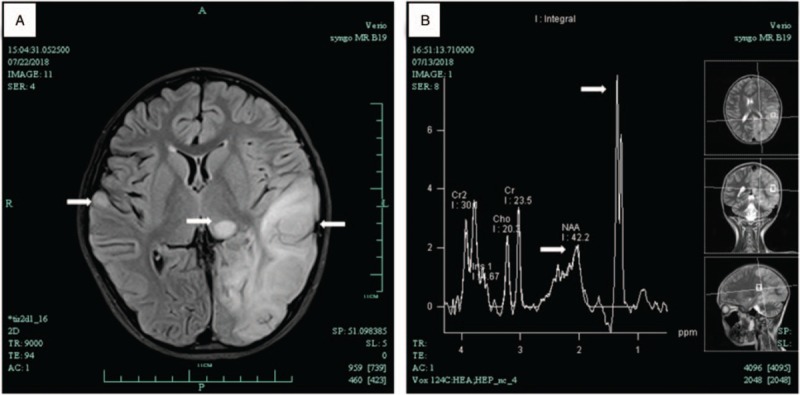
Brain MRI of patient showed the left temporal, occipital and parietal lobe, cortex and subcortical of right temporal lobe, and left thalamus had abnormal signals (A) MRS showed NAA peak in the lesion area decreased significantly, and raised lactate peak in the lesion at 1.3 ppm (B). MRI = magnetic resonance imaging, MRS = magnetic resonance spectroscopy, NAA = N-acetyl aspartate.
Serum lactate dehydrogenase, creatine kinase, α-hydroxybutyrate dehydrogenase were significantly higher than normal, whereas creatinine, sodium, and chloride were significantly lower than normal. In the cerebrospinal fluid examination, the leucocyte count was zero, red blood cell count was 210×106 L, chloride was slightly lower, the protein and sugar quantifications were in normal control range.
Mitochondrial sequencing revealed that the proband carried the pathogenic heteroplasmic mutation A3243G mutation in mitochondrial 12S rRNA gene, while his parents did not carry the mutation (Fig. 2).
Figure 2.
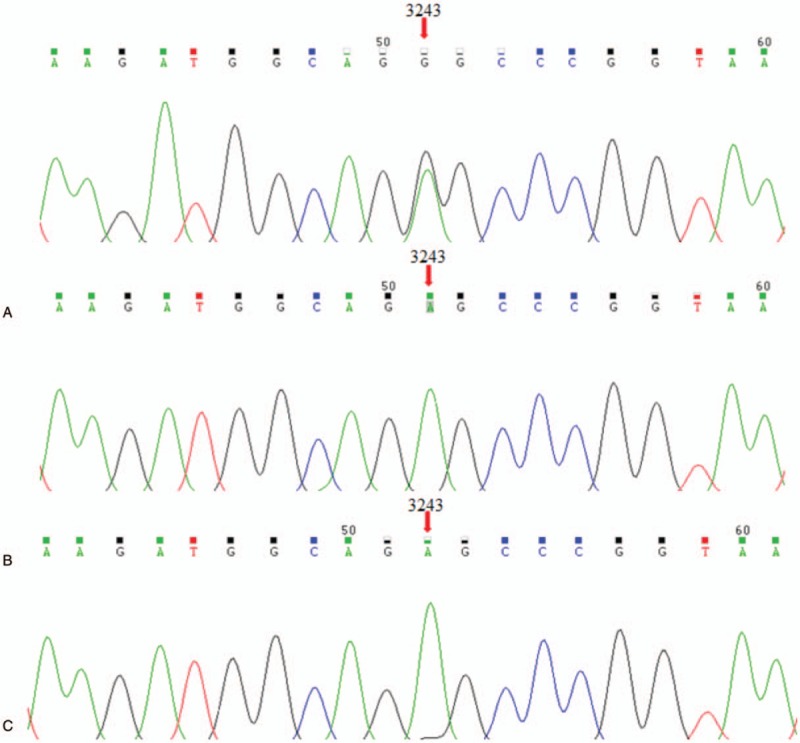
Identification of the mitochondrial A3243G mutation using polymerase chain reaction and Sanger sequencing. The proband carried the pathogenic heteroplasmic mutation A3243G mutation in mitochondrial 12S rRNA gene (A). While his father and mother did not carry the mutation (B and C, respectively).
The patient was treated according to the diagnosis of mitochondrial encephalomyopathy. Intravenous acyclovir, ceftriaxone and dexamethasone were used for antiviral, antimicrobial, and anti-inflammatory therapy, respectively. Intravenous mannitol was gradually tapered for reducing intracranial pressure with furosemide for inducing diuresis. Intravenous arginine could help to treat alkalosis and supple some essential amino acids. Oral oxiracetam capsules, vitamin B1, and coenzyme Q10 were used for providing nutrition and improving energy. His medications were 30 mg vitamin B1, 0.1gm vitamin C and mecobalamin 750 μg daily after discharge from our hospital.
The patient was able to walk and talk slowly with improved writing skills and no stroke-like episodes. The neurological examination was negative and muscle tension was identified as grade V.
3. Discussion
Mitochondrial encephalomyopathy was a heterogeneous group of disorders affecting primarily the central nervous system (CNS) and skeletal muscle. Conventional examination including MRI, MRS, electroencephalogram, and laboratory examination were regularly used for auxiliary diagnosis, the patient was considered to have mitochondrial encephalomyopathy. Even though the pathology of mitichondrial diseases is still underlying, several studies had suggested common causes of mitochondrial encephalomyopathy are dysfunction caused by mutations in tRNALeu(UUR) gene.[8]
In our study, mitochondrial A3243G mutation was detected only in the samples of the proband, but not in his parents. It may be related to the low mutation rate and the limitation of detection sensitivity. However, it suggests that A3243G may have a sporadic mutation during oocytes formation or fertilization. Further study of heteroplasmic rate of the A3243G mutation should be investigated including skin, urinary sediment, hair, saliva, and nail samples. Mitochondrial A3243G mutation in mitochondrial DNA is the most common pathogenic point mutation, causing a variety of phenotypes, including mitochondrial encephalomyopathy.[9,10] Interesting, the mitochondrial mutation A3243T was also reported.[11]
The mitochondrial A3243G mutation causes a defect in mitochondrial translation leading to impaired oxidative phosphorylation (OXPHOS), with deficient activity of respiratory chain complexes I and/or IV, results in modifications of tRNAs structure leading to deficiency in enzymes of electron transport chain and reduced mitochondria's ability to generate ATPs.[12,13]
Although muscle biopsy was not performed, MRI and MRS findings, elevation of lactate and genetic testing results suggested the diagnosis of mitochondrial encephalomyopathy in this case. Identification of the mitochondrial encephalomyopathy pathogenic gene will help to elucidate the pathogenesis of mitochondrial encephalomyopathy, not only to perform prenatal diagnosis, but also to determine the severity and provide a better treatment. For mitochondrial encephalomyopathy suspicious patients, perfect genetic identification can appropriately reduce the frequency of follow-up and, more importantly, reduce the psychological stress caused by the disease itself.
In summary, since mitochondrial encephalomyopathy has different phenotypes, in addition to traditional examinations, it is important for clinicians to be familiar with genetic testing methodology as well as applications of these tests in clinic to get an accurate diagnosis.
Author contributions
Conceptualization: Qi Liu.
Funding acquisition: Qi Liu.
Investigation: Xiao-Qun Liu.
Methodology: Qi Liu.
Project administration: Shao-Qing Shen.
Resources: Xiao-Qun Liu.
Validation: Qi Liu.
Writing – original draft: Guo-Can Yang.
Writing – review & editing: Qi Liu.
Footnotes
Abbreviations: ATP = adenosine triphosphate, CNS = central nervous system, MRI = magnetic resonance imaging, MRS = magnetic resonance spectroscopy, OXPHOS = oxidative phosphorylation.
XQL and SQS have contributed equally to this work.
Written informed consent was obtained from the patients for publication of this case report and any accompanying images. This study has been approved by the local ethics committee of our hospital.
This work is supported by grants from Shaoxing Bureau of Science and Technology (no. 2017B70023).
The authors have no conflicts of interest to disclose.
References
- [1].Tanahashi C, Nakayama A, Yoshida M, et al. MELAS with the mitochondrial DNA 3243 point mutation: a neuropathological study. Acta Neuropathol 2000;99:31–8. [DOI] [PubMed] [Google Scholar]
- [2].Jia Z, Wang X, Qin Y, et al. Coronary heart disease is associated with a mutation in mitochondrial tRNA. Hum Mol Genet 2013;22:4064–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kadowaki T, Kadowaki H, Mori Y, et al. A subtype of diabetes mellitus associated with a mutation of mitochondrial DNA. N Engl J Med 1994;330:962–8. [DOI] [PubMed] [Google Scholar]
- [4].von Ameln S, Wang G, Boulouiz R, et al. A mutation in PNPT1, encoding mitochondrial-RNA-import protein PNPase, causes hereditary hearing loss. Am J Hum Genet 2012;91:919–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Scaglia F, Wong LJ. Human mitochondrial transfer RNAs: role of pathogenic mutation in disease. Muscle Nerve 2008;37:150–71. [DOI] [PubMed] [Google Scholar]
- [6].Kovalenko SA, Tanaka M, Yoneda M, et al. Accumulation of somatic nucleotide substitutions in mitochondrial DNA associated with the 3243 A-to-G tRNA(leu)(UUR) mutation in encephalomyopathy and cardiomyopathy. Biochem Biophys Res Commun 1996;222:201–7. [DOI] [PubMed] [Google Scholar]
- [7].Xu Y, Wang L, He J, et al. Prevalence and control of diabetes in Chinese adults. JAMA 2013;310:948–59. [DOI] [PubMed] [Google Scholar]
- [8].Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990;348:651–3. [DOI] [PubMed] [Google Scholar]
- [9].Bargagli M, Primiano G, Primiano A, et al. Recurrent kidney stones in a family with a mitochondrial disorder due to the m.3243A>G mutation. Urolithiasis 2018;doi:10.1007/s00240-018-1087-1. [DOI] [PubMed] [Google Scholar]
- [10].Niedermayr K, Polzl G, Scholl-Burgi S, et al. Mitochondrial DNA mutation “m.3243A>G”-Heterogeneous clinical picture for cardiologists (“m. 3243A>G”: A phenotypic chameleon). Congenit Heart Dis 2018;13:671–7. [DOI] [PubMed] [Google Scholar]
- [11].Ikeda T, Osaka H, Shimbo H, et al. Mitochondrial DNA 3243A>T mutation in a patient with MELAS syndrome. Hum Genome Var 2018;5:25Accessed May 10, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rahman S, Poulton J, Marchington D, et al. Decrease of 3243 A-->G mtDNA mutation from blood in MELAS syndrome: a longitudinal study. Am J Hum Genet 2001;68:238–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ou YH, Chen AW, Fan JY, et al. Aminoglycoside-associated nonsyndromic deafness and speech disorder in mitochondrial A1555G mutation in a family: a case report. Medicine (Baltimore) 2018;97:e12878. [DOI] [PMC free article] [PubMed] [Google Scholar]