

Abstract
Rationale:
IgG4-related disease (IgG4-RD) is a multiorgan disease of unestablished prevalence that is characterized histopathologically by a dense lymphoplasmacytic infiltrate enriched with IgG4-expressing plasma cells and associated with storiform fibrosis. Tubulointerstitial nephritis (TIN) is the most common renal manifestation of IgG4-RD, but membranous nephropathy (MN) has also been described and often occurs in the context of concurrent TIN. Patients with IgG4-related MN have been characteristically negative for autoantibodies to the phospholipase A2 receptor (PLA2R).
Patient concerns:
A 45-year-old man presented with abdominal pain and lower extremity edema.
Diagnosis:
Histopathological evaluation of pancreas and liver biopsies established a diagnosis of IgG4-RD. Renal biopsy confirmed a diagnosis of PLA2R-associated MN without evidence of concurrent TIN.
Interventions:
The patient was treated with rituximab, a short course of low-dose, oral cyclophosphamide, and a rapid glucocorticoid taper.
Outcomes:
The patient achieved remission of MN after 8 months of therapy and maintained remission of IgG4-RD.
Lessons:
PLA2R-associated MN may be a rare manifestation of IgG4-RD. Systematic evaluation of larger cohorts of IgG4-RD patients for the presence of PLA2R autoantibodies and the investigation of PLA2R-associated MN cohorts for evidence of IgG4-RD would facilitate the understanding of the nature of the relationship between these observations.
Keywords: IgG4-related disease, membranous nephropathy, nephrotic syndrome, PLA2R antibody, proteinuria
1. Introduction
IgG4-related disease (IgG4-RD) is a systemic disease characterized by tissue infiltration of lymphocytes and plasma cells enriched for IgG4-expression and fibrosis arranged in a “storiform” pattern.[1] IG4-RD can involve almost any organ including the pancreas, salivary glands, liver, and kidneys, and often affects multiple organs simultaneously.[2] Renal involvement in IgG4-RD typically consists of tubulointerstitial nephritis (TIN) and, less commonly, membranous nephropathy (MN).[3–5] Autoantibodies to the phospholipase A2 receptor (PLA2R), which are present in the majority of patients with primary MN, are notably absent in reported cases of IgG4-related MN. In a case series of patients with IgG4-related MN, all 8 cases with available immunostaining were found to be negative for antibodies against PLA2R.[5]
We report a case of resistant PLA2R-associated MN occurring in a patient with typical extrarenal manifestations IgG4-RD. The potential relationship between IgG4-RD and PLA2R-associated MN is discussed. The patient provided informed consent for publication of this report. Institutional board review approval was not necessary for this case report.
2. Case report
In August 2016, a 45-year-old white Caucasian man presented to a local emergency department with several months of progressive abdominal pain. Computed tomography (CT) of the abdomen and pelvis demonstrated a pancreatic mass, diffuse pancreatic edema, and abdominal lymphadenopathy. Endoscopic biopsy of the pancreatic mass was negative for malignancy but revealed a lymphoplasmacytic infiltrate with abundant IgG4+ plasma cells (not quantified precisely), associated with mild tissue eosinophilia. Serum total IgG and IgG4 levels were 551 mg/dL (normal 767–1590) and 28.9 mg/dL (normal 2.4–121), respectively. He was given a presumptive diagnosis of type 1 autoimmune pancreatitis. Prednisone 40 mg daily was initiated and tapered to discontinuation over 2 months.
Repeat CT imaging in October 2016 demonstrated resolution of the pancreatic mass. However, a surveillance CT scan in December 2016 showed recurrence of the pancreatic mass in addition to new mass lesions within the liver. Biopsy of a hepatic lesion showed a marked lymphoplasmacytic infiltrate admixed with eosinophils, accompanied by storiform fibrosis (Fig. 1A). An immunohistochemical stain for IgG4 showed abundant IgG4+ plasma cells within the infiltrate (up to 60 per high-power field) (Fig. 1B). Prednisone was restarted at 40 mg daily for relapsing IgG4-RD.
Figure 1.
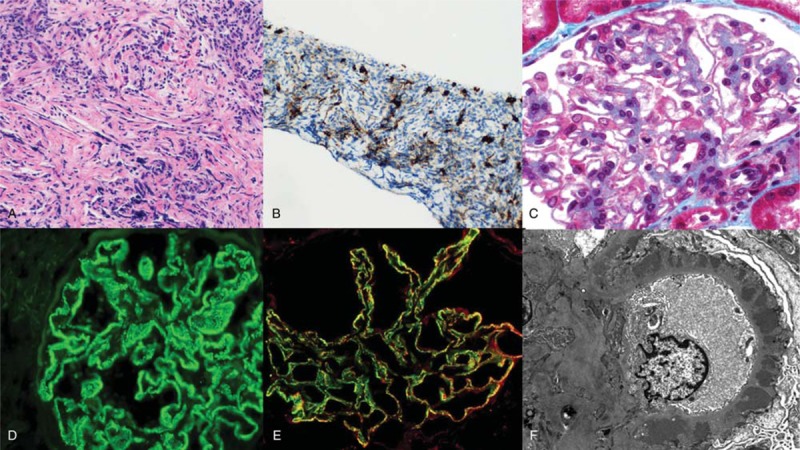
Pathology of IgG4-related disease (IgG4-RD) and membranous nephropathy (MN). A, Liver biopsy showing marked lymphoplasmacytic infiltrate with admixed eosinophils and associated storiform fibrosis. B, An immunohistochemical stain for IgG4 shows abundant IgG4-positive plasma cells within the infiltrate in the liver. C, Kidney biopsy shows diffuse thickening of glomerular capillary walls with fuchsinophilic deposits on trichrome stain. D, Immunofluorescence on frozen tissue shows diffuse granular staining of the glomerular basement membrane (GBM) for IgG, diagnostic of MN. E, Immunofluorescence on frozen tissue shows extensive colocalization of IgG4 (green) and phospholipase A2 receptor (PLA2R; red) within the deposits in the GBM, indicative of PLA2R-associated MN. F, Electron microscopy shows numerous subepithelial electron dense deposits associated with diffuse podocyte foot process effacement, and no mesangial or subendothelial deposits.
In January of 2017, the patient was evaluated for 2 months of progressive lower extremity edema and was found to have the nephrotic syndrome with a spot urine albumin:creatinine ratio of 7 g/g and a serum albumin of 2.1 g/dL. Renal function was normal (serum creatinine 0.9 mg/dL). A workup that included serologies for hepatitis B and C infections, antinuclear antibodies, and complement components 3 and 4 was unrevealing. The nephrotic syndrome was presumed to be a manifestation of IgG4-RD. He was started on quinapril 10 mg/day and maintained on prednisone 40 mg daily. In March 2017, two doses of rituximab (1000 mg per dose) were administered and prednisone was tapered to discontinuation. In April 2017, repeat abdominal CT imaging revealed resolution of the pancreatic and liver masses. However, high-grade proteinuria persisted, with 9 g of protein on a 24-hour urine collection.
In July 2017, the patient was referred to a nephrologist due to persistent nephrotic syndrome and a kidney biopsy was performed. Light microscopy showed histopathologic features characteristic of MN (diffuse thickening of glomerular capillary walls with fuchsinophilic deposits on trichrome stain, Fig. 1C) without evidence of an interstitial infiltrate. Immunofluorescence staining on frozen tissue showed diffuse granular staining of the glomerular basement membrane (GBM) for IgG, diagnostic of MN (Fig. 1D). No tubular basement membrane deposits were observed. Immunofluorescence staining also showed extensive colocalization of IgG4 and PLA2R within the deposits in the GBM (Fig. 1E). Electron microscopy showed numerous subepithelial electron dense deposits associated with diffuse podocyte foot process effacement, but no mesangial or subendothelial deposits (Fig. 1E). Testing for serum antibodies to PLA2R was positive by indirect immunofluorescence on PLA2R-expressing cultured cells and by enzyme-linked immunosorbent assay with a titer of 51 RU/mL (reference range: negative <14 RU/mL).
In September 2017, the patient was referred to our center, where repeat laboratory testing revealed a spot urine protein:creatinine ratio (UPCR) of 10 g/g, a serum albumin of 1.9 g/dL, persistence of PLA2R autoantibodies, and B cell reconstitution. He was initiated on combination therapy with rituximab dosed every 4 months with the aim of continuous B cell depletion, an 8-week course of low-dose, oral cyclophosphamide, and an accelerated prednisone taper, as has been previously described.[6] By May 2018, he had achieved remission with a UPCR of 1.34 g/g and a serum albumin of 3.8 g/dL. Trends of serum albumin, UPCR, and serum PLA2R autoantibodies following initiation of combination therapy are shown in Figure 2. The patient's renal function has remained normal and relapse of IgG4-RD has not occurred.
Figure 2.
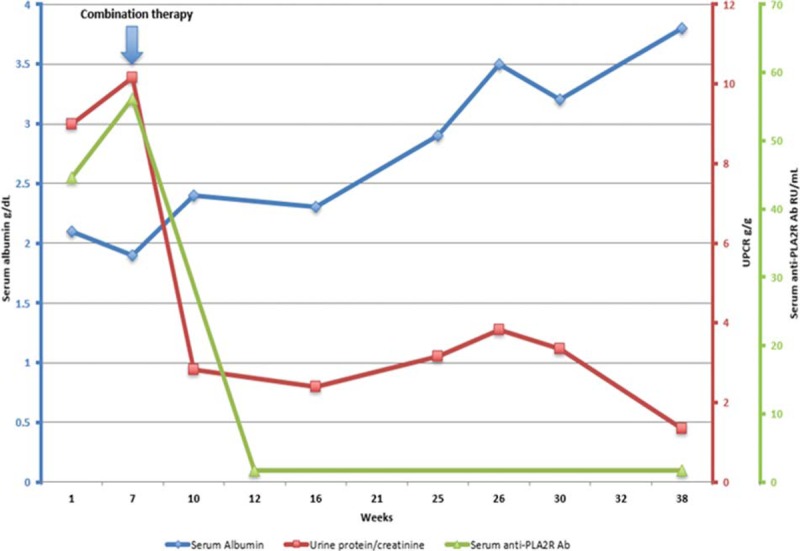
Response to therapy. Shown are trends of serum albumin (blue), spot urine protein:creatinine ratio (red), and serum anti-phospholipase A2 receptor (PLA2R) antibodies (green). Arrow denotes initiation of combination therapy with rituximab, cyclophosphamide, and prednisone. Ab = antibody, UCPR = spot urine protein:creatinine ratio.
3. Discussion
IgG4-RD is a systemic fibroinflammatory disease that typically presents as tumefactive lesions that can involve essentially any organ system, including the pancreas, biliary system, lung, salivary glands, thyroid, pachymeninges, and kidneys.[7] An elevated serum IgG4 concentration is present in the majority of cases, but a normal level does not exclude the diagnosis.[8,9] Two hallmark histopathologic features predominate regardless of the involved organ: a lymphoplasmacytic infiltrate enriched in IgG4+ plasma cells and storiform fibrosis.[1] The kidney is unique among the organs affected by IgG4-RD in that 2 distinct histologic patterns of involvement have been described.
TIN, the most common renal manifestation of IgG4-RD, generally occurs in the setting of concomitant extrarenal manifestations of the disease, although patients with apparently isolated renal disease have been described.[4] Clues to the presence of IgG4-related TIN are a progressive decline in renal function, hypocomplementemia, and the presence of characteristic hypodense nodular lesions on contrast-enhanced CT.[3,4] The pathological findings in IgG4-related TIN are typical of those of other organs affected by the disease, including a lymphoplasmacytic infiltrate with an increased number of IgG4+ plasma cells (generally >30–40 per high-power field) and fibrosis.[3]
MN, a rare manifestation of IgG4-RD, was present in approximately 7% of patients in the largest biopsy series of IgG4-related TIN.[3,4] IgG4-related MN can occur with or without TIN. As is true with primary MN, the autoreactive antibodies found in the GBM by immunostaining are predominantly of the IgG4 subclass.[10] The predominance of IgG4 antibodies in both primary MN and IgG4-related MN stands in contrast to other causes of secondary MN, such as lupus and malignancy, in which the other IgG subclasses typically predominate.[11,12] The finding of subendothelial and mesangial deposits in addition to subepithelial deposits is also suggestive of a secondary etiology. In the largest reported series of IgG4-related MN, 3 of 9 patients had either subendothelial or mesangial deposits or both.[5] The variability in pathologic findings raises the possibility of heterogenous autoantigen specificities leading to the development of MN in the context of IgG4-RD.
Because approximately 75% to 80% of cases historically termed “primary MN” are now known to be caused by autoantibodies to PLA2R, this entity is now termed more appropriately as “PLA2R-antibody associated MN.”[10] In the largest series of IgG4-related MN, none of the 8 cases tested showed positive staining for anti-PLA2R antibodies.[5] Moreover, in a report of 28 patients with IgG4-RD involving various organs, no patients had circulating autoantibodies to PLA2R.[13] More recently, autoantibodies to another transmembrane protein expressed on podocytes, namely thrombospondin type-1 domain-containing 7A (THSD7A), have been implicated in a minority of cases of primary MN and in a subset of malignancy-associated cases.[14,15] The testing of serum from patients with IgG4-related MN for autoantibodies to THSD7A is an important study that needs to be undertaken.
Treatment with glucocorticoids led to a swift response of the patient's autoimmune pancreatitis and hepatic lesions but these features recurred following discontinuation of treatment, a common finding in IgG4-RD. In contrast, overt nephrotic syndrome persisted after prolonged treatment with glucocorticoids and 8 months following the initial course of rituximab. The differential timing of treatment response of MN compared with the typical cellular inflammatory lesions of IgG4-RD is not unexpected. Indeed, immune complex mediated injury in MN is characteristically slow to resolve, and remission often does not ensue for many months following a course of successful therapy.[16] Thus, the difference in treatment response does not necessarily imply that IgG4-RD and MN were unrelated disease processes. The patient's resistant MN was successfully treated with continued B cell depletion, 8 weeks of low-dose cyclophosphamide, and a rapid prednisone taper.[6] The resolution of PLA2R autoantibodies preceded clinical remission, as has been described before.[17]
The patient described had typical features of IgG4-RD involving the pancreas and liver, whereas simultaneously exhibiting the characteristic features of PLA2R-associated MN. The immunoglobulin deposits were IgG4 predominant and exclusively subepithelial in location. They also colocalized with staining for PLA2R on immunofluorescence. To our knowledge, only 1 previous case of PLA2R-associated MN occurring in a patient with IgG4-RD has been described. In that case, nephrotic syndrome occurred 4 years after a diagnosis of autoimmune pancreatitis that had become clinically quiescent.[18] In our case, the MN developed in the setting of active IgG4-RD raising the question of a shared pathogenesis. However, it is also possible this represents a coincidental occurrence of 2 distinct disease processes.
4. Conclusions
PLA2R autoantibodies have been characteristically absent in cases of IgG4-related MN. Here we have described a case of a 45-year-old man with IgG4-RD and persistent nephrotic syndrome with kidney biopsy findings diagnostic of PLA2R-associated MN. A better understanding of the pathogenesis of IgG4-RD and the systematic evaluation of additional cases of MN occurring in the context of IgG4-RD may further elucidate the relationship, if any, of IgG4-RD and PLA2R-associated MN.
Author contributions
Conceptualization: Saif A. Muhsin, Frank B. Cortazar.
Data curation: Ricard Masia.
Writing – original draft: Saif A. Muhsin, Frank B. Cortazar.
Writing – review and editing: Saif A. Muhsin, Ricard Masia, Rex N. Smith, Zachary S. Wallace, Cory A. Perugino, John H. Stone, John L. Niles, Frank B. Cortazar.
Footnotes
Abbreviations: CT = computed tomography, GBM = glomerular basement membrane, IgG4-RD = IgG4-related disease, MN = membranous nephropathy, PLA2R = phospholipase A2 receptor, THSD7A = thrombospondin type-1 domain-containing 7A, TIN = tubulointerstitial nephritis, UPCR = urine protein: creatinine ratio.
Consent for publication: Informed written consent was obtained from the patient for publication of this case report and accompanying images.
The authors have no conflicts of interest to disclose.
References
- [1].Deshpande V, Zen Y, Chan JKC, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012;25:1181–92. [DOI] [PubMed] [Google Scholar]
- [2].Stone J, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012;366:539–51. [DOI] [PubMed] [Google Scholar]
- [3].Saeki T, Nishi S, Imai N, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int 2010;78:1016–23. [DOI] [PubMed] [Google Scholar]
- [4].Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol 2011;22:1343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Alexander MP, Larsen CP, Gibson IW, et al. Membranous glomerulonephritis is a manifestation of IgG4-related disease. Kidney Int 2013;83:455–62. [DOI] [PubMed] [Google Scholar]
- [6].Cortazar FB, Leaf DE, Owens CT, et al. Combination therapy with rituximab, low-dose cyclophosphamide, and prednisone for idiopathic membranous nephropathy: a case series. BMC Nephrol 2017;18:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kamisawa T, Zen Y, Pillai S, et al. IgG4-related disease. Lancet 2015;385:1460–71. [DOI] [PubMed] [Google Scholar]
- [8].Carruthers MN, Khosroshahi A, Augustin T, et al. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis 2015;74:14–8. [DOI] [PubMed] [Google Scholar]
- [9].Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-related disease: Baseline clinical and laboratory features in 125 patients with biopsy-proven disease. Arthritis rheumatol 2016;67:2466–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Beck LH, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 2010;361:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kuroki A, Shibata T, Honda H, et al. Glomerular and serum IgG subclasses in diffuse proliferative lupus nephritis, membranous lupus nephritis, and idiopathic membranous nephropathy. Intern Med 2002;41:936–42. [DOI] [PubMed] [Google Scholar]
- [12].Lönnbro-Widgren J, Ebefors K, Mölne J, et al. Glomerular IgG subclasses in idiopathic and malignancy-associated membranous nephropathy. Clin Kidney J 2015;8:433–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Khosroshahi A, Ayalon R, Beck LH, et al. IgG4-related disease is not associated with antibody to the phospholipase A2 receptor. Int J Rheumatol 2012;2012:139409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tomas NM, Beck LH, Meyer-Schwesinger C, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med 2015;371:2277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hoxha E, Beck LH, Wiech T, et al. An indirect immunofluorescence method facilitates detection of thrombospondin type 1 domain–containing 7A–specific antibodies in membranous nephropathy. J Am Soc Nephrol 2017;28:520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ponticelli C, Altieri P, Scolari F, et al. A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol 1998;9:444–50. [DOI] [PubMed] [Google Scholar]
- [17].Beck LH, Fervenza FC, Beck DM, et al. Rituximab-induced depletion of anti-PLA2R autoantibodies predicts response in membranous nephropathy. J Am Soc Nephrol 2011;22:1543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Acevedo Ribó M, Ahijado Hormigos FJ, Díaz F, et al. IgG4-related disease and idiopathic membranous nephropathy: “The clothes do not make the man.”. Clin Nephrol 2016;86:345–8. [DOI] [PubMed] [Google Scholar]