

Abstract
ON1 is a novel genotype of human respiratory syncytial virus (HRSV) subtype A, in children with acute respiratory tract infections (ARTIs). However, there is not much data on the prevalence and clinical and molecular characterization in China.
Our study is based on the children who had respiratory infections positive for RSV-A admitted by Gansu Provincial Maternity and Child-care Hospital in Lanzhou (northwestern China) during the last 7 epidemic seasons from 2010 to 2017.
In our study, different strains of the novel RSV-A genotype ON1, first identified in Canada in December 2010, were first detected in Gansu Provincial Maternity and Child-care Hospital in August 2012 and then followed by an abrupt expansion in the number of ON1 variants in the beginning of 2014 and eventually replaced all other RSV-A strains from 2015 to 2017. ON1 is characterized by a 72-nt duplication in the C-terminal region of the highly variable attachment glycoprotein (G), predicted to lengthen the polypeptide with 24 amino acids, including a 23-aa duplication, which likely changes antigenicity. New N-glycosylation sites occurred within the 23-aa duplication and 24-aa insertion of the ON1 viruses in our study. Notably, RSV infections occurred later, but peaked sooner from the 2014/2015 to 2016/2017 epidemic seasons, compared with the previous 4 seasons.
Our study concluded that genotype ON1 has caused larger outbreaks and became the predominate genotype for HRSV subgroup A in Lanzhou from 2013 to 2017, and became the sole genotype of RSV-A in 2015/2016 and 2016/2017. Our data indicate that northwest of China and the world will eventually be dominated by the ON1 RSV-A genotype, including the possibility for vaccine development. Based on trends seen in RSV-B BA genotype, which predominated for decades, there is a possibility to develop a vaccine for children in the next 10 years.
Keywords: Lanzhou, ON1 genotype, respiratory syncytial virus A
1. Introduction
First identified in 1956, human respiratory syncytial virus (HRSV) is a member of the Pneumovirus subfamily of the family Paramyxoviridae. HRSV is a major cause of lower respiratory tract infections (LRTIs) among infants and young children worldwide and is also a major cause of morbidity in children under 1 year old.[1] Moreover, people who had an RSV infection during childhood remain susceptible to RSV upper respiratory tract reinfection throughout life.[2] However, up till now, no licensed RSV vaccines have been developed.
RSV is a nonsegmented virus in the Paramyxoviridae family of Pneumovirus with a single-stranded, negative-sense RNA genome of 15222-nucleotide length, coding for 11 proteins.[3] Among the 11 proteins encoded by the RSV genome, the attachment glycoprotein (G) is the most variable protein and has been shown to be able to accumulate amino acid changes over time. In this transmembrane glycoprotein, the first 37 amino acids of N-terminal are cytoplasmic tail, and the amino acids from 38 to 66 belong to transmembrane domain, and the ones from 67 to the end of C-terminal are extracellular region, which further divides into the first hypervariable region (HVR1, amino acids from 67 to 163) and the secondary hypervariable region (HVR2, the 187th amino acids to the end of C-terminal).[4] Most molecular epidemiological studies have analyzed the G gene's hypervariable C-terminal domain, and more importantly, some studies[5] also reveal that the C-terminal part of the G gene reflects the evolutionary patterns and clades of the RSV genome. Based on reactions with specific antibodies against the glycoproteins G and the distinction of RSV genome, RSV has been divided into subtypes A and B. RSV-A and RSV-B can be further classified into various genomes, for instance, RSV-A has fallen into GA1-GA7, SAA1, NA1-NA4, CB-A, and the recent ON1; and RSV-B includes GB1-GB13, SAB1-SAB4, URU1-URU2, and BA.[6] It has been found in the studies that multiple genotypes can co-circulate during successive epidemic seasons. The fact that antigenic diversity and variability in the virus, especially in the second hypervariable region of the G protein, are more possible to lead to the increasing cases of RSV reinfection and the difficulty in developing a vaccine.[7–10]
So far, researchers from all over the world have detected 2 novel RSV genotypes (BA and ON1) with large duplications of amino acids in the attachment G glycoprotein. In 1999, the BA genotype with a 60-nt insertion in G (generating a 20-aa insertion in the C-terminal region) in an RSV-B strain was detected in Buenos Aires, Argentina.[11] The BA variant subsequently spread slowly and sequentially throughout the world,[12] becoming the predominant group B genotype, and in some regions replaced all previous circulating RSV-B genotypes with a divergent BA lineage.[13–15] More recently, the RSV-A genotype ON1, which had a larger insertion than BA, was first identified in specimens collected at the end of 2010 in Ontario, Canada. In the ensuing years, the ON1 genotype has been found in Malaysia,[16] South Korea,[17] Italy, Japan, and South Africa,[18] which indicates that it spread rapidly and diversified globally. However, in China, there are no detailed reports about the population of the ON1 genotype, particularly in northwestern China. However, it is not known whether this new genotype could be more pathogenic, which is the information that would be of high interest to researchers worldwide. Research on the ON1 genotype has developed gradually. Investigation on the spread of ON1 is important to understand to which extent genetic variability can modify the epidemic behavior of RSV at the population level.
In China, there is no ongoing national surveillance published about circulating RSV, and only one study has investigated the genetic diversity of RSV-A in different seasons, till 2014,[19] but till 2009 in northwestern China.[20] In this study, we have analyzed sequence data of the G gene of RSV-A strains circulating during 2010 to 2017 (7 RSV epidemics seasons) in Lanzhou. Unexpectedly, in August 2012, we detected ON1, the new RSV-A variant of the GA2 genotype identified in Ontario (Canada) in 2010,[11] which demonstrated its rapid spread in the following year. Therefore, we performed an in-depth analysis of the RSV-A genotype ON1 epidemiology in Lanzhou, northwestern China. Analysis of case distribution and clinical patient data showed differences between infections with the ON1 genotype and those with previously circulating RSV-A strains. In our study, the change of ON1 was observed in a large time range from 2010 (ON1 emerged abroad) to 2017 (recently), to collect data for the development of an RSV vaccine in China and internationally. Which is of public health interest is whether increased fitness is associated with increased severity and immune evasion (with potential vaccine modality implications).
2. Materials and methods
2.1. Study location, population, and samples
From December 2010 to June 2017, 2175 nasopharyngeal aspirates (NPAs) were collected from outpatients and inpatients children aged 1 month to <5 years with ARTI in the Gansu Maternal and Child Health Care Hospital (Lanzhou, China). Demographic and clinical data were taken from the medical files. Informed consent was sought from the children's parents for participation in the study and it was approved by the ethics committee of the hospital. A 3-point (0–2, age adjusted) clinical severity score based on use of supplemental oxygen, duration of hospital, and admission to an intensive care unit (ICU) was determined on admission.[11,17] All NPA specimens were collected and transported immediately to the laboratory at the National Institute for Viral Disease Control and Prevention, China CDC, and stored at −80°C until further analysis.
2.2. RNA extraction and cDNA synthesis
Total RNA was extracted with the QIAamp extraction kit (QIAGEN, Valencia, CA) from a starting clinical specimen volume of 200 μL and final elution volume of 80 μL. RNA was stored at −80°C until use. For viral detection, RNA was reverse transcribed into cDNA by using random hexamer primers with Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA).
2.3. RSV screening and subgroup identification
For RSV screening, reverse transcription polymerase chain reaction (RT-PCR) was performed with primers RSV-NF and RSV-NR to amplify a 279 bp fragment of the RSV N gene.[21] Subgroup-specific products were amplified with the subgroup A-specific 5′-primer RSV-GPA, and subgroup B-specific 5′-primer RSV-GPB and 3’ primer RSV-GF.[21] Briefly, 2.5 μL of cDNA was added to a 22.5 μL PCR master mixture containing 12.5 μL Premix, 0.5 μL GPA primer (GPB primer), 0.5 μL RSV-GR primer and 9 μL ddH2O. The thermocycling protocol was 95°C for 5 minutes, followed by 40 cycles of 98°C for 10 seconds, 50°C for 10 seconds (for RSV-A) or 55°C (for RSV-B) for 10 seconds and 72°C for 1 minute, with a final extension for 7 minutes at 72°C and hold at 4°C. The amplified products of 550 bp were resolved by electrophoresis on a 1.5% agarose gel. The gel was stained with nucleic acid stain (SBS Genetech, Shanghai, China) and visualized under UV light.
2.4. Nucleotide sequence analysis
All positive PCR products were sequenced by the Beijing Tianyi-Huiyuan Gene Company, purified and sequenced in the forward and the reverse directions. The nucleotide and amino acid sequences of the group A were edited using Bioedit v7.1.3 and aligned with reference sequences available from the GenBank database using CLUSTAL W (version 1.81). Phylogenetic trees were constructed by the neighbor-joining method in MEGA software 7.0.[22]
The statistical significance of the tree topology was tested by bootstrapping (1000 replicates). The nucleotide sequences of a fragment which contain the C-terminal second hypervariable region of the G gene encoding the G protein (452-nt, corresponding to codon positions 144–298) from RSV-A isolates in our study were determined in our study and compared with those of reference strains representing different RSV-A genotypes deposited in GenBank. The viruses detected were designated by city/group/year of isolation/isolate number.
3. Statistical analysis
We explored associations among demographic, clinical, or outcome variables and RSV genotypes for all RSV-positive cases. Significant differences in rates between various groups were tested with a χ2, Student t test, and a Mann–Whitney U test. The index of severity was tested by logistic regression. A P value of <0.05 was considered significant. Analyses were performed with SPSS 17.0 software.
4. Results
4.1. RSV-positive patients
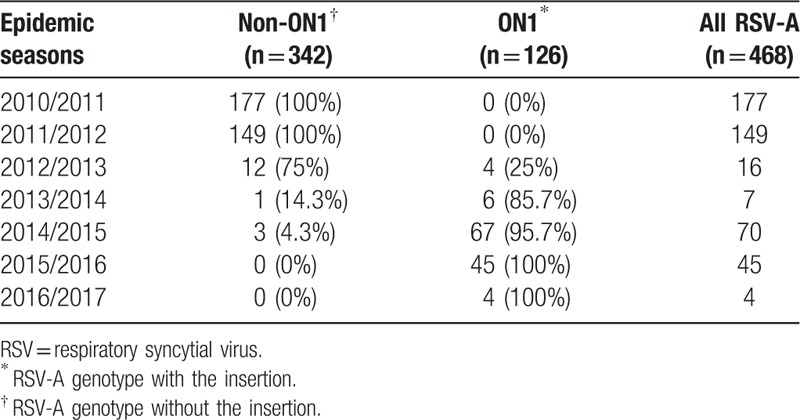
From December 2010 to June 2017 (7 RSV epidemics), 2430 samples were collected from eligible children (average age: 5.1 months; median age: 2.95 months; range: 1–36 months). For the samples, 2175 (89.5%) were tested for RSV, 584 (26.9%) were RSV-positive and 449 (78.2%) were RSV-A-positive. The G gene was successfully sequenced in 574 (98.3%) samples. All patients were hospitalized and clinical serviced for respiratory conditions. As summarized in Table 1, 177 cases were from 2010/2011, 149 from 2011/2012, 16 from 2012/2013, 7 from 2013/2014, 70 from 2014/2015, 45 from 2015/2016, and 4 cases from 2016/2017. Among the 449 RSV-A positive samples, all were successfully sequenced and categorized based on the presence of a 72-nt insert in the G gene (Table 1).
Table 1.
Sequence numbers of respiratory syncytial virus-A-positive strains in relation to the presence of a 72-nt insert, Lanzhou, northwest China, December 2010 to June 2017 (n = 468).
4.2. RSV cases distribution by season
Analysis by month of presentation highlighted a different case distribution in the last 7 seasons in Lanzhou. In 2010/2011 to 2013/2014, the earliest RSV-associated infections occurred in December and peaked in February and ended in May. However, in 2014/2015 to 2016/2017, the cases started in October, peaked during December to January, and ended in April. The positive numbers of RSV in different seasons are shown in Figure 1.
Figure 1.
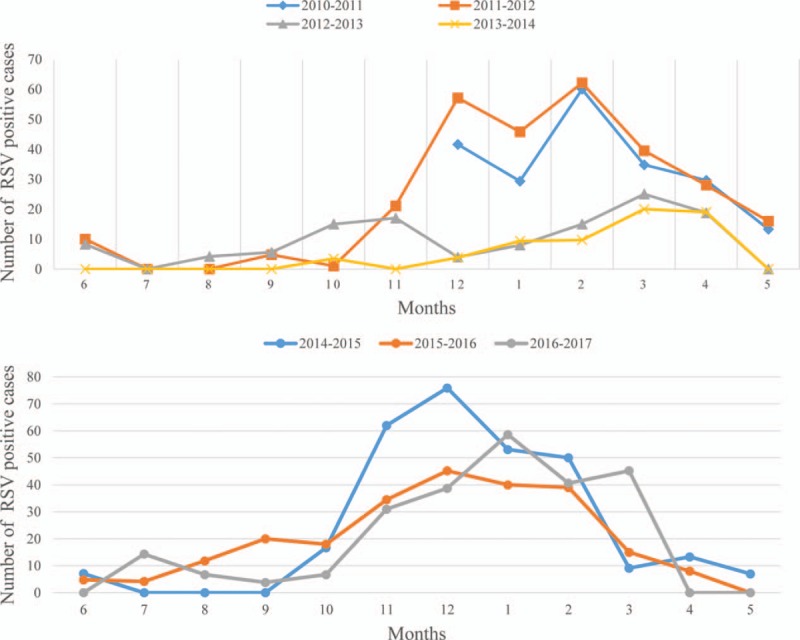
Respiratory syncytial virus cases, distribution of children, Lanzhou, northwestern China, December 2010 to May 2017 (n = 2175).
4.3. ON1 Circulation
We found that 2 RSV-A genotypes were circulating in Lanzhou during 2010 to 2017, ON1 (n = 136, 30.3%) and NA1 (n = 313, 69.7%). The temporal prevalence of RSV-A, and genotypes ON1 and non-ON1, is shown in Table 1. ON1 was first detected in Lanzhou in the 2012/2013 season (August 2012). The prevalence of ON1 in Lanzhou was 0% in the 2010/2011 and 2011/2012, 25% in 2012/2013 when ON1 was first detected in Lanzhou, 85.7% in 2013/2014 and 95.7% in 2014/2015 epidemics, respectively, and finally 100% in the recent 2015/2016 and 2016/2017 epidemics, that is, we observed rapid replacement of the previously circulating dominant non-ON1 genotypes by ON1, and 2 years after its first being detected abroad in 2010/2011 (November 2010) and prevalence in 2012/2013 in Canada. In addition, RSV-A predominated in 4 nonconsecutive RSV epidemics in 2010/2011, 2011/2012, 2014/2015, and 2015/2016.
4.4. Sequence analysis
According to the evolutionary tree, in Lanzhou and between 2010 and 2017, there are 2 epidemic RSV-A genomes in Lanzhou, that is, NA1 and ON1 genotypes. Among the 449 RSV-A isolates, 44 different G genomes exist, and combining reference sequences, genetic evolutionary tree was drawn. The bacteria strains NA1 and ON1 were both derived from GA2 genome, and more specifically, ON1 came from NA1 genotype. ON1 is characterized by a 72-nt insertion in G, resulting in 24 extra amino acids of which 23 are duplications of 261 to 283.
Between G genome of NA1 genotype in Lanzhou and the reference NA1 genotype, the homology of the nucleotide level is 93.5% to 97.6% and the amino acid level is 88.9% to 99.1%. However, these 2 ratios dropped respectively to 74.6% and 73.8%, showing they are different (Fig. 2) despite the fact that ON1 genotype stem from NA1 genotype. Among the new RSV-A ON1 genotype, there are 3 unique substitutions of amino acid, and they are E232G, T253K, and P314L. Compared with the reference strain, there are 7 amino acids substitutions among G protein of ON1 genotype in Lanzhou as E232G, K209 M, L208P, E204G/K, I243T, L248F, and T249I. Six N-glycosylation sites, 2 ON1 genotypes (codons 237 and 318), and 4 NA1 genotypes (237, 251, 273, and 294) were identified in RSV-G gene sequences in Lanzhou. Notably in NA1 genotype, N- glycosylation sites 237 and 273 were mutually excluded. Only 1 of the 2 occurred and with a high ratio. But in ON1 genotype, the 273-glycosylation site completely disappeared and the 237 site was a stable glycosylation site. Notably for 5 ON1 strains in the 2016/2017 seasons, among two ones, a new N-glycosylation site 275 was found in 261 to 283 duplication, which was never shown before. In Figure 3, the gray areas represent N-glycosylation sites.
Figure 2.
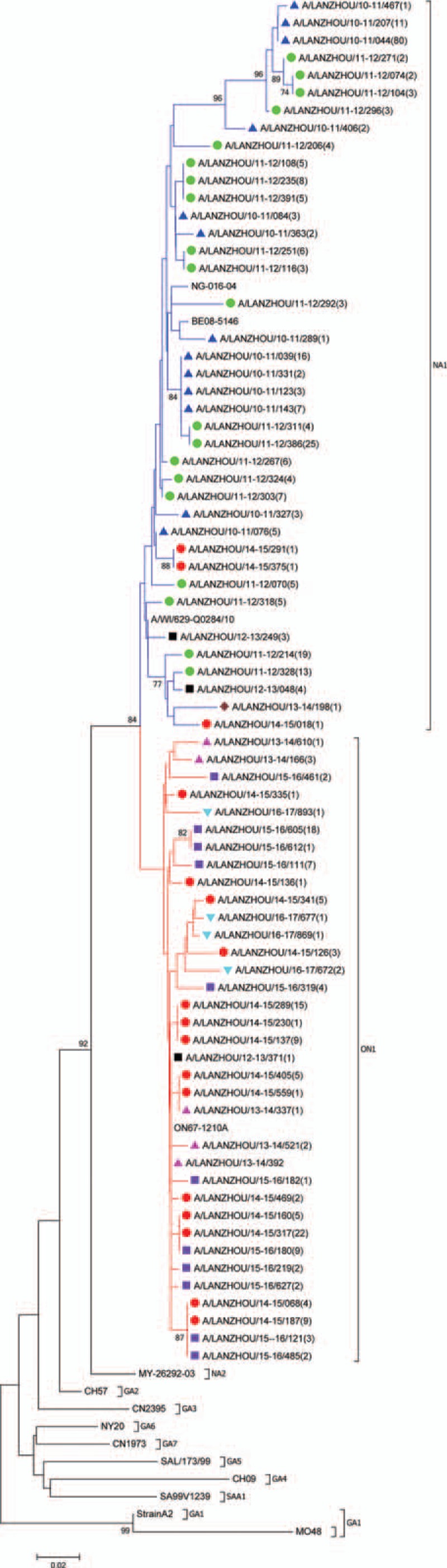
Phylogenetic tree based on the second hypervariable region of the G protein gene, Lanzhou December 2010 to June 20137 (n = 75 unique Lanzhou sequences). Multiple sequences alignment and phylogenetic trees were constructed using Clustal W and neighbor-joining algorithm running within MEGA 6 software. The scale bar shows the proportions of nucleotide substitutions per site. Numbers at nodes are bootstrap values for 1000 iterations; only bootstrap values of >70% are shown. Numbers in round brackets indicate the total number of strains with an identical sequence. Reference strains representing known genotypes are indicated in bold. Branches are color-coded according to the deduced amino acid sequence, identifying subtrees and genotypes—red: sequences with insertion clustering with the novel ON1 genotype; blue: sequences of NA1 genotype. Symbol color indicates epidemic season—blue: 2010/2011; green: 2011/2012; black: 2012/2013;brown: 2013/2014; red: 2014/2015; purple 2015/2016; turquoise blue: 2016/2017.
Figure 3.
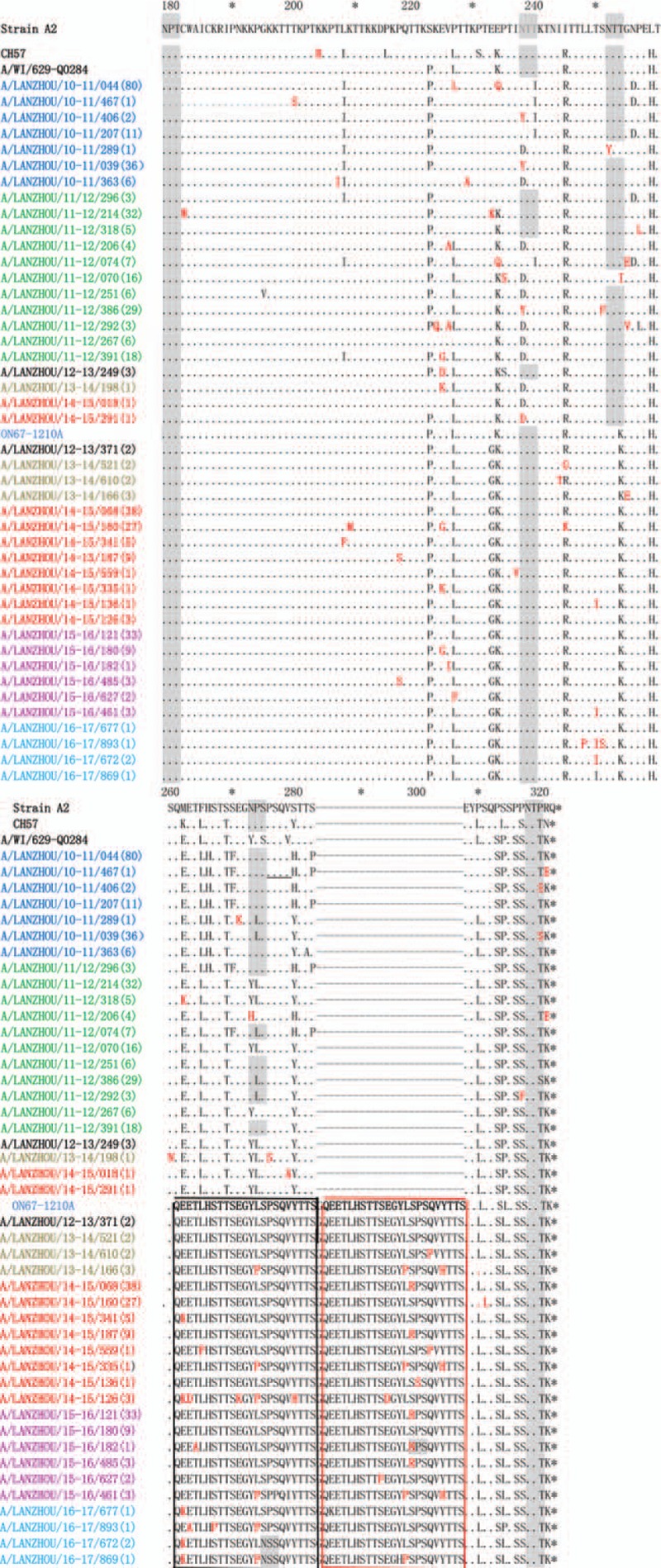
Alignment of deduced amino acid sequence of the G protein of RSV-A strains isolated in Lanzhou during the 2010 to 2017 epidemic seasons. Alignments are shown relative to the sequences of prototype strain A2, genotype GA2 strain (CH57), NA1 strain (A/WI/629-Q0284), and ON1 strain (ON67-1210A). The AAs shown correspond to positions 179 to 298 of the second hypervariable region of RSV-A strain A2 G protein. Numbers in parentheses indicate the total number of identical strains. Dots indicate nucleotides identical to the strain A2; the sequence of the ON1 prototype is shown only for clarity. Dashes indicate the gap corresponding to the nucleotide insertions; asterisks indicate stop codons. Black and red boxes indicate the duplicated region (homologous portion and insertion); mutations are shown in red and bold. Predicted N-glycosylation sites are shaded in gray. Color of sequence names indicates the epidemic season—blue: 2010/2011; green: 2011/2012; black: 2012/2013; brown: 2013/2014; red: 2014/2015; purple: 2015/2016; turquoise blue: 2016/2017.
Compared with NA1 and GA2 genotypes, there are some significant amino-acid substitutions, parts of which have been found in Lanzhou and some not. Amino acid positions 215 and 230 are highly variable compared with reference strain GA2. However, this variation either in NA1 or ON1 genotypes was never shown, and the amino-acid substitutions L215P and S230P were conservatively replaced. What have been confirmed in our study is that there are 3 amino acid substitutions, that is, ON1 (232G, 253K, 290L) and NA1 (232E, 253T, 290P) viruses. Importantly, G gene in ON1 genotypes lacks 2 N-glycosylation sites due to the mutations of 2 amino acids T253K and N273Y, which has also been found in our study. Notably, compared with the ON1 prototype, because of the mutations of 2 amino acids P274S and S299N, new N-glycosylation sites occurred within the 23aa duplication and 24aa insertion of the ON1 viruses in our study, which had not been previously described.
4.5. Patient data and clinical diagnosis
In our study, we analyzed demographic and clinical data from 468 RSV-A cases and found differences between infections with ON1 versus non-ON1 strains. ON1 patients were more likely to be younger males receiving inpatient treatment (P = .044), similar to data record in an Italian report for 2011 to 2013. Infections were more severe than those caused by non-ON1 strains. ON1 infection is more commonly diagnosed with bronchiolitis and wheezing is a more common symptoms. However, for the ON1 patients, the PICU occupancy rate is not high (not statistically different from non-ON1) (P = .4), which is different from Italy and German Studies. In addition, ON1 patients spend more days in the hospital (P = .002).
5. Discussion
HRSV is one of the most common pathogens causing severe ARTIs, accounting for 15% to 40% of pneumonia/bronchiolitis cases in children. RSV has been detected for 61 years, but a vaccine has not yet been developed due to constant changes in RSV antigens. The biggest change reported is a 20-aa insertion in the G protein in RSV-B BA strains detected in Buenos Aires in 1999,[8] until the detection of the RSV-A ON1 genotype. Emerging RSV variants that possess a selective advantage in terms of genetic diversity can spread to neighboring areas, gradually replacing dominant genotypes over several years.[11]
We documented circulation of 2 genotypes of NA1 (98.6% of RSV-A isolates detected from 2010 to 2013) and ON1 (97.1% of RSV-A isolates detected from 20132017) in Lanzhou. In addition, ON1 is a novel genotype, characterized by a 72-nt duplication in the C-terminal region of the G gene, first reported in Canada. All RSV-A G-gene variants described in this report are derived from the GA2 genotype and are genetically close to the recently characterized NA1 genotype, which are the most recent strains circulating worldwide.[11,16] Our findings document the presence of the novel ON1 genotype in Northwestern China in the 2012/2013 epidemic season (August 2012) and its rapid spread in 2013/2014, which is 2 years later than its prevalence in some of the foreign countries (e.g., Italy). We provide a detailed analysis on the spread and the associated demographic, clinical, and evolutionary characteristics of the novel RSV-A genotype ON1 in Lanzhou.
In addition to the epidemiological impact, significant genetic variation in circulating strains may involve different pathogenicity and virulence. A study from Kilif showed a change in the alternation of RSV subgroup dominance patterns because ON1 was introduced into this community. Previously, RSV-A predominated in up to 2 consecutive epidemics in Kilifi, whereas it predominated over RSV-B in 3 consecutive epidemics from 2012/2013 to 2014/2015. However, we did not find this phenomenon in our study. The first ON1 strain was found in Lanzhou in 2012 and in following years, RSV-A and B subtypes are still prevalent every 2 years. A study in Italy reported that the beginning and peak times of RSV were earlier than before because of the emergence of the ON1 genotype, and the phenomenon was also been found in our research.
The ON1 genotype was first detected in Ontario in the winter of 2010/2011 (November 2010).[11] To the best of our knowledge, the earliest ON1 sequence from a non-Canadian isolate was detected in Malaysia in November 2011.[16] In the ensuing years, the ON1 genotype has been found in South Africa,[18] Italy, Germany,[23] and South Korea,[17] indicating its rapid global dissemination during the 2012/2013 epidemic season. In China, the ON1 genotype was first reported in Beijing in 2013 (detected in November 2012),[24] and then in 2014, cities such as Chongqing, Shanghai, Shenzhen, Guangzhou, and other places also reported appearance of the novel genotype.[25,26] It can be inferred that Lanzhou, reported in our report, may be the earliest region of the emerging ON1 genotype in China (detected in August 2012 and spreading quickly in 2014/2015). Therefore, we can draw the conclusion that ON1 appeared and erupted 2 years later than abroad. These data demonstrate that ON1 was introduced into China from abroad, and in China's northwest, ON1 is spreading fast and completely replacing the previously genotypes, which may be the current situation of ON1 in China and worldwide.
The prevalence of ON1 varies from place to place. Among some countries such as China,[27] Germany,[28] and Ontario of Canada where ON1 was first detected, its prevalence remains <20%[29]. However, in Italy,[30] the United States, and South Korea,[31] the prevalence of ON1 was reported between 20% and 70%. There is also study claiming that ON1 was the only RSVA genotype in Argentina, in 2014.[32] These data show the different prevalence of ON1 in different places and the increasing rate over time. In our study, it can be seen ON1 has strong transmission dynamics: in Lanzhou, the prevalence of ON1 reached 100% in 2 successive years. In the past, GA2 replaced GA5 as dominant genotype for 7 years, and for 5 years with BA genotype to replace RSV-B non-BA genotype, but it only took <2 years for ON1 to completely replace RSV-A. Therefore, it is concluded that the strong transmission of BA genotype would be likely to be repeated by ON1 and even stronger, and ON1 would become the sole epidemic genotype of RSV-A in the next 10 years or more.
High homology among wild-type NA1 and ON1 genotypes in Lanzhou and isolates from GenBank reveals the global distribution of these 2 genotypes. Site-specific evolutionary analysis of the C-terminal hypervariable region of the G protein among Ontario NA1 isolates revealed strong evolutionary selection pressure, resulting in 19 positively selected sites compared with the NA1 reference genotype. For ON1 genotype, the substitutions at amino acids 274 and 290 resulted in loss of group- and strain-specific epitopes.[33,34] The variability and apparent evolution seen at the positively selected site 274 in the ON1 cluster of sequences is particularly interesting. The aa 237 mutation, presenting in 56% of the Ontario NA1 isolates, suggested the gain of a potential N-glycosylation site. These reverted mutations, particularly in the epitope regions, may decrease the antigen avidity to the current circulating strain-specific antibodies. The longer attachment protein of the 72-nt duplication in ON1 viruses seems to offer more opportunities for variable changes and thus greater diversity and increased fitness over previous group A genotypes. Notably, new N-glycosylation sites were discovered in our study, 2 in 23aa in 2015/2016 epidemic season and 1 in 24-aa duplication area in 2016/2017. Overall, a variety of genetic changes could be responsible for the spread of ON1 strains, conferring low cross-protection by preexisting antibodies against RSV-A strains previously circulating in Italy: the 24aa insertion, the loss of a further potential glycosylation site due to the T253K substitution, and even other amino acid changes.
In our study, ON1-infected cases have the following characteristics: predominantly male young and more severe clinical symptoms such as more wheezing. However, studies in other countries report different results. According to Duvvuri's study,[29] the infected subjects are mostly women, which is different from our conclusion. In Cyprus, it is reported that the clinical symptoms of ON1 was milder compared with other RSV infections.[31] In Vitenam,[35] the upper respiratory infection caused by RSV-ON1 was closely related to clinically severe manifestations such as wheezing, tachypnea, and difficulty in breathing, according to Yoshihara's reports. There is report in Germany that it is more common for ON1 infectors to admit into PICU than cases caused by other RSV-A strains. But researchers suggest that it is possibly related to the epidemic peak resulted from the sudden diffusion of ON1 due to genetic differences. Moreover, it is also reported that there are no differences among clinical symptoms from different RSV-A genotypes. But it is clear that differences in results may be resulted from differences in analyses, study designs, inadequate sample sizes, differences between viruses in different locations, host/environmental differences, or even different viral characteristics all could lead to different results. In our study, the clinical symptoms of ON1 infection in children are more severe; the infection-lasting time is longer but PICU admission rate is lower and the prognosis was good. But there is another possibility that ON1 is more easily combined with other pathogens, which lead to severe symptoms, rather than ON1. However, in our study, there are no evidences of mixed infection. Therefore, a more comprehensive and standardized research is needed for further investigation of clinical features of ON1.
The possibility exists that ON1 and other similar new RSV variants (e.g., the BA genotype) gain dominance by evading host immunity. The appearance of ON1 makes RSV-A “reset to zero,” which provides a new possibility for further development of an RSV vaccine. ON1 may be similar to the BA genotype and be the predominant strain of RSV-A for decades. Therefore, it is reasonable to assume the appearance of ON1 could lead to evasion of future vaccine-induced protection, lessening the herd immunity potential of vaccination, yet to provide a new possibility for manufacture of an RSV vaccine. Because of the emergence of ON1, RSV-A “reset to zero” and human immunity set to zero, the vaccine development can also been seen as zero to some extent. In conclusion, the emergence of ON1 is a challenge and an opportunity for the development of an RSV vaccine, and has important public health significance. Continued surveillance for cases and collection of detailed standardized clinical data are warranted.
Acknowledgments
We thank our colleagues of the Laboratory of Clinical Virology for helpful comments and critical reading of the manuscript.
Author contributions
XL, D-HL, and R-FZ conceived and designed the experiments. XL, D-HL, DC, LG, HY, YX, G-RL, Y-SS, Y-JW, W-KW, Z-PX, H-CG, and Z-JD performed the experiments and analyzed the data.
Conceptualization: Xuan Liang, Rong-Fang Zhang.
Data curation: Dong-Hai Liu, De Chen, Li Guo, Hui Yang, Yong-Sheng Shi, Yong-Jun Wang, Wei-Kai Wang, Zhi-Ping Xie, Han-Chun Gao, Zhao-Jun Duan, Rong-Fang Zhang.
Formal analysis: Dong-Hai Liu, Rong-Fang Zhang.
Funding acquisition: Rong-Fang Zhang.
Investigation: Xuan Liang, Dong-Hai Liu, De Chen, Hui Yang.
Resources: Yong-Jun Wang, Wei-Kai Wang.
Software: Xuan Liang, De Chen, Li Guo, Yong-Sheng Shi.
Supervision: Zhi-Ping Xie, Han-Chun Gao, Zhao-Jun Duan, Rong-Fang Zhang.
Validation: Xuan Liang, Rong-Fang Zhang.
Visualization: Xuan Liang.
Writing – original draft: Xuan Liang, Rong-Fang Zhang.
Writing – review and editing: Xuan Liang, Dong-Hai Liu, De Chen, Li Guo, Hui Yang, Yong-Sheng Shi, Yong-Jun Wang, Wei-Kai Wang, Zhi-Ping Xie, Han-Chun Gao, Zhao-Jun Duan, Rong-Fang Zhang.
Footnotes
Abbreviations: ARTIs = acute respirator tract infections, HRSV = human respiratory syncytial virus, HVR1 = first hypervariable region, HVR2 = secondary hypervariable region, ICU = intensive care unit, LRTIs = lower respiratory tract infections, NPAs = nasopharyngeal aspirates, RT-PCR = reverse transcription polymerase chain reaction.
XL and D-HL equally contributed to this work.
XL, D-HL, and R-FZ wrote the manuscript. All authors read and approved the final manuscript.
This study was supported by the National Natural Science Foundation of China (Grant No. 81260001).
The authors have no conflicts of interest to disclose.
References
- [1].Anderson LJ, Parker RA, Strikas RL. Association between respiratory syncytial virus outbreaks and lower respiratory tract deaths of infants and young children. J Infect Dis 1990;161:640–6. [DOI] [PubMed] [Google Scholar]
- [2].Agoti CN, Mwihuri AG, Sande CJ, et al. Genetic relatedness of infecting and reinfecting respiratory syncytial virus strains identified in a birth cohort from rural Kenya. J Infect Dis 2012;206:1532–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cane PA, Pringle CR. Evolution of subgroup A respiratory syncytial virus: evidence for progressive accumulation of amino acid changes in the attachment protein. J Virol 1995;69:2918–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cane PA. Molecular epidemiology of respiratory syncytial virus. Rev Med Virol 2001;11:103–16. [DOI] [PubMed] [Google Scholar]
- [5].Rebuffo-Scheer C, Bose M, He J, et al. Whole genome sequencing and evolutionary analysis of human respiratory syncytial virus A and B from Milwaukee, WI 1998–2010. PLoS One 2011;6:e25468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mufson MA, Orvell C, Rafnar B, et al. Two distinct subtypes of human respiratory syncytial virus. J Gen Virol 1985;66(Pt. 10):2111–24. [DOI] [PubMed] [Google Scholar]
- [7].Shobugawa Y, Saito R, Sano Y, et al. Emerging genotypes of human respiratory syncytial virus subgroup A among patients in Japan. J Clin Microbiol 2009;47:2475–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Trento A, Galiano M, Videla C, et al. Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J Gen Virol 2003;84(Pt. 11):3115–20. [DOI] [PubMed] [Google Scholar]
- [9].Venter M, Madhi SA, Tiemessen CT, et al. Genetic diversity and molecular epidemiology of respiratory syncytial virus over four consecutive seasons in South Africa: identification of new subgroup A and B genotypes. J Gen Virol 2001;82(Pt. 9):2117–24. [DOI] [PubMed] [Google Scholar]
- [10].Katzov-Eckert H, Botosso VF, Neto EA, et al. Phylodynamics and dispersal of HRSV entails its permanence in the general population in between yearly outbreaks in children. PLoS One 2012;7:e41953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Eshaghi A, Duvvuri VR, Lai R, et al. Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS One 2012;7:e32807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Trento A, Casas I, Calderon A, et al. Ten years of global evolution of the human respiratory syncytial virus BA genotype with a 60-nucleotide duplication in the G protein gene. J Virol 2010;84:7500–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Houspie L, Lemey P, Keyaerts E, et al. Circulation of HRSV in Belgium: from multiple genotype circulation to prolonged circulation of predominant genotypes. PLoS One 2013;8:e60416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].van Niekerk S, Venter M. Replacement of previously circulating respiratory syncytial virus subtype B strains with the BA genotype in South Africa. J Virol 2011;85:8789–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zlateva KT, Vijgen L, Dekeersmaeker N, et al. Subgroup prevalence and genotype circulation patterns of human respiratory syncytial virus in Belgium during ten successive epidemic seasons. J Clin Microbiol 2007;45:3022–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Khor CS, Sam IC, Hooi PS, et al. Displacement of predominant respiratory syncytial virus genotypes in Malaysia between 1989 and 2011. Infect Genet Evol 2013;14:357–60. [DOI] [PubMed] [Google Scholar]
- [17].Lee WJ, Kim YJ, Kim DW, et al. Complete genome sequence of human respiratory syncytial virus genotype A with a 72-nucleotide duplication in the attachment protein G gene. J Virol 2012;86:13810–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Valley-Omar Z, Muloiwa R, Hu NC, et al. Novel respiratory syncytial virus subtype ON1 among children, Cape Town, South Africa, 2012. Emerg Infect Dis 2013;19:668–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hu P, Zheng T, Chen J, et al. Alternate circulation and genetic variation of human respiratory syncytial virus genotypes in Chengdu, West China, 2009-2014. J Med Virol 2017;89:32–40. [DOI] [PubMed] [Google Scholar]
- [20].Zhang RF, Jin Y, Xie ZP, et al. Human respiratory syncytial virus in children with acute respiratory tract infections in China. J Clin Microbiol 2010;48:4193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Peret TC, Hall CB, Schnabel KC, et al. Circulation patterns of genetically distinct group A and B strains of human respiratory syncytial virus in a community. J Gen Virol 1998;79(Pt. 9):2221–9. [DOI] [PubMed] [Google Scholar]
- [22].Kumar S, Tamura K, Nei M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 2004;5:150–63. [DOI] [PubMed] [Google Scholar]
- [23].Prifert C, Streng A, Krempl CD, et al. Novel respiratory syncytial virus a genotype, Germany, 2011-2012. Emerg Infect Dis 2013;19:1029–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cui G, Qian Y, Zhu R, et al. Emerging human respiratory syncytial virus genotype ON1 found in infants with pneumonia in Beijing. China Emerg Microbes Infect 2013;2:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu J, Mu Y, Dong W, et al. Genetic variation of human respiratory syncytial virus among children with fever and respiratory symptoms in Shanghai, China, from 2009 to 2012. Infect Genet Evol 2014;27:131–6. [DOI] [PubMed] [Google Scholar]
- [26].2014;Ren L, Xia Q, Xiao Q, et al. The genetic variability of glycoproteins among respiratory syncytial virus subtype A in China between 2009 and 2013JT Infect Genet Evol. 27:339–47. [DOI] [PubMed] [Google Scholar]
- [27].Yu X, Kou Y, Xia D, et al. Human respiratory syncytial virus in children with lower respiratory tract infections or influenza-like illness and its co-infection characteristics with viruses and atypical bacteria in Hangzhou, China. J Clin Virol 2015;69:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pretorius MA, van Niekerk S, Tempia S, et al. Replacement and positive evolution of subtype A and B respiratory syncytial virus G-protein genotypes from 1997-2012 in South Africa. J InfectDis 2013;208Suppl. 3:S227–37. [DOI] [PubMed] [Google Scholar]
- [29].Duvvuri VR, Granados A, Rosenfeld P, et al. Genetic diversity and evolutionary insights of respiratory syncytial virus A ON1 genotype: global and local transmission dynamics. Sci Rep 2015;5:14268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pierangeli A, Trotta D, Scagnolari C, et al. Rapid spread of the novel respiratory syncytial virus A ON1 genotype, central Italy, 2011 to 2013. Euro Surveill 2014;19: [DOI] [PubMed] [Google Scholar]
- [31].Panayiotou C, Richter J, Koliou M, et al. Epidemiology of respiratory syncytial virus in children in Cyprus during three consecutive winter seasons (2010-2013): age distribution, seasonality and association between prevalent genotypes and disease severity. Epidemiol Infect 2014;142:2406–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Viegas M, Goya S, Mistchenko AS. Sixteen years of evolution of human respiratory syncytial virus subgroup A in Buenos Aires, Argentina: GA2 the prevalent genotype through the years. Infect Genet Evol 2016;43:213–21. [DOI] [PubMed] [Google Scholar]
- [33].Lazar I, Canaan A, Weibel C, et al. Novel mutations in the respiratory syncytial virus G gene identified in viral isolates from a girl with severe combined immune deficiency treated with intravenous immune globulin. J Clin Virol 2006;37:168–73. [DOI] [PubMed] [Google Scholar]
- [34].Martinez I, Dopazo J, Melero JA. Antigenic structure of the human respiratory syncytial virus G glycoprotein and relevance of hypermutation events for the generation of antigenic variants. J Gen Virol 1997;78(Pt 10):2419–29. [DOI] [PubMed] [Google Scholar]
- [35].Yoshihara K, Le MN, Okamoto M, et al. Association of RSV-A ON1 genotype with increased pediatric acute lower respiratory tract infection in Vietnam. Sci Rep 2016;6:27856. [DOI] [PMC free article] [PubMed] [Google Scholar]