

Abstract
Zinc (Zn) is an essential trace metal required for all forms of life, but is toxic at high concentrations. While the toxic effects of high levels of Zn are well documented, the mechanism of cell death appears to vary based on the study and concentration of Zn. Zn has been proposed as an anti-cancer treatment against non-small cell lung cancer (NSCLC). The goal of this analysis was to determine the effects of Zn on metabolism and cell death in A549 cells. Here, high throughput multi-omics analysis identified the molecular effects of Zn intoxication on the proteome, metabolome, and transcriptome of A549 human NSCLC cells after 5 min to 24 h of Zn exposure. Multi-omics analysis combined with additional experimental evidence suggests Zn intoxication induces ferroptosis, an iron and lipid peroxidation-dependent programmed cell death, demonstrating the utility of multi-omics analysis to identify cellular response to intoxicants.
Graphical Abstract

INTRODUCTION
Zinc (Zn) is a d-block transition metal that is essential for all forms of life. The human proteome is estimated to be comprised of 10% Zn-binding proteins, where Zn serves catalytic and structural roles 1, 2. However, at high concentrations, Zn is toxic to cells, likely due to mismetallation 3, 4. Acute exposure to Zn by inhalation causes severe Zn toxicity. Inhalation of zinc oxide (ZnO) particles, such as from welding fumes, can cause metal fume fever, which is a flu-like disease that typically resolves in 1–3 weeks 3. In contrast, exposure to smoke bombs in military training or combat can cause zinc chloride (ZnCl2) smoke inhalation injury (ZCSII), which is uncommon, and leads to progressive diffuse lung injury that is often fatal 3, 5. Excess dietary Zn also leads to gut oxidative stress, disruption of the microbiota that increases susceptibility to infection, and iron and copper deficiency associated with decreased hemoglobin and neurologic disease 6–11. It has been proposed that Zn toxicity can be harnessed as an anti-tumor therapeutic, as ZnO nanoparticles offer a drug delivery platform and preferentially kill tumor cells in vitro 12, 13.
Zn toxicity leads to loss of cellular viability, but reports of the mechanism vary based on Zn concentration and cell type studied. High levels of Zn (e.g. > 500 μM) have been shown to inhibit apoptosis 3, 4, 14–16. Specifically, Zn blocks respiration and induces reactive oxygen species (ROS) generation that leads to caspase-independent apoptotic-like death in neuronal cells 17, and Zn has been shown to inhibit caspase-3 directly in lymphoblast cells 18. Conversely, moderately high levels of Zn (200 μM) induce mitochondrial damage in C6 rat brain glioma cells 16. Additional characterization of the cell death induced by Zn is warranted, particularly in light of novel cell death pathways more recently described including oxidative glutamate toxicity and ferroptosis 19. Understanding how Zn induces cell death will allow development of treatments for acute Zn intoxication induced by metal fume fever or ZCSII, as well as development of Zn intoxication for therapeutic purposes such as in the treatment of cancer.
Here, the comprehensive effects of Zn toxicity on the proteome, metabolome, and transcriptome of A549 human lung epithelial cells were measured. The goal of this research was to understand how lung cells respond to Zn intoxication and identify the mechanism of cell death. We previously showed that the response of HL-60 human promyeloblasts to Zn is primarily mediated by Nrf2 20. However, 10–30% of human lung cancers have KEAP1/Nrf2 mutations that lead to constitutive Nrf2 activation, or Nrf2 addiction, which is associated with worse clinical outcome 21. The prevalence of Nrf2 addiction suggests that the non-small cell lung cancer (NSCLC) response to Zn might be different than that of HL-60 cells. A549 cells have a KEAP1 mutation and aberrant promoter methylation that prevents inhibition of Nrf2, thus leading to constitutively active Nrf2 22. Zn has been proposed as a pro-apoptotic treatment for NSCLC cells alone or in combination with docetaxel 23, suggesting that understanding the effects of Zn on A549 NSCLC cells may also improve assessment of the potential for Zn as a lung cancer therapeutic.
EXPERIMENTAL
Cell culture
A549 cells were obtained from ATCC (Manassas, VA) and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Waltham, MA) with 10% v/v heat-inactivated fetal bovine serum (Atlanta Biologicals) and 1% penicillin/streptomycin at 37˚C with 5% CO2 atmosphere. Cells were plated at the following densities: 5,000 cells/well in 384-well plates; 10,000 cells/well in 96-well plates; 60,000 cells/well in 24-well plates; 296,000 cells/well in 6-well plates; and 1.72X106 cells/plate in 10-cm dishes. Cells were plated 24 h prior to treating with ZnCl2 or ddH2O vehicle control, as indicated.
Assessment of cellular viability and death
To measure viability following Zn intoxication, A549 cells were plated to 96-well plates 23.25 h prior to intoxication. ZnCl2 was added at the indicated concentrations (0–1000 μM) and the cells were incubated for the indicated periods (1–24 h). Viability was assessed by measuring ATP using CellTiter-Glo (Promega, Madison, WI) diluted 6-fold in PBS and then by manufacturer’s instructions, or cellular reducing environment by alamarBlue (ThermoFisher, Waltham, MA) added 1 h prior to measurement by excitation/emission 555/560 nm at the time point indicated. Necrosis was assessed by CytoTox-Glo (Promega), apoptosis was measured by ApoOne (Promega) and fluorescence was measured with excitation/emission 485/528 nm. All luminescence and fluorescence was assessed in a BioTek plate reader (Winooski, VT). For CellTiter-Glo, CytoTox-Glo, and ApoOne, luminescence or fluorescence were monitored every 10 min for 60 min, and typically the 30-min reading was used for analysis.
For treatment with NAD+, desferoxamine (DFO), and α-tocopherol (vitamin E), 24 h prior to intoxication A549 cells were plated to 384-well plates. At 30 min prior to intoxication, 100 μM NAD+, 100 μM DFO, 100 μM vitamin E, or appropriate vehicle control were added. ZnCl2 was then added to the indicated concentrations (0–1000 μM) and incubated for 24 h at 37˚C with 5% CO2 atmosphere before measuring viability with CellTiter-Glo. All assays were carried out in at least biological triplicate.
RNA Sequencing
For RNA sequencing sample preparation, A549 cells were plated in 6-well plates in triplicate. RNA was isolated from biological triplicate untreated and ZnCl2-treated samples using the RNeasy Mini Kit (Qiagen, Venlo, Netherlands). RNA was sequenced and analyzed by the Genomics Services Lab at HudsonAlpha (Huntsville, AL). RNA-seq was performed using poly(A) selection on an Illumina HiSeq v4 sequencing platform. Reads were paired-end with a read length of 50 bp and 20 million reads per sample.
Proteomics and metabolomics
For mass spectrometry sample preparation, A549 cells were plated in 6-well plates in biological triplicate and incubated post-Zn intoxication for the indicated times. Cells were then immediately placed on ice, supernatant was removed, and the adherent A549 cells were washed and prepared as previously described 20, with the exception that separate samples were used for proteomics and metabolomics. Stable isotope labeling by amino acids in cell culture (SILAC), and phospho-enriched proteomics cells and samples were prepared as previously described 24, and analyzed with the modifications previously described 20.
Computational analysis, data mining, visualization, and statistics
All instrument output files (transcriptomics, metabolomics, and proteomics) were uploaded to a central in-house database and arranged into an exportable file with fold change represented as treated/control and significance determinations as previously described 20. Graphs were generated using GraphPad Prism 6 (La Jolla, CA) and additional statistical tests were as follows: cell death assays, one-way ANOVA with Dunnett’s multiple comparisons to untreated control; addition of NAD+, DFO, or vitamin E, two-way ANOVA with Sidak’s multiple comparisons; lipid peroxidation, two-way ANOVA with Tukey’s multiple comparisons. For graphs, significance is indicated as follows: * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. Venn diagrams were generated with nVenn 25. All figures were generated in Canvas X16. Gene ontology (GO) annotation was determined based on UniProt ID using the QuickGo tool for GO slim annotations 26, 27.
Lipid peroxidation
A549 cells were plated in 96-well plates one day prior to Zn intoxication. Cells were washed thrice with HBSS, then incubated with 30 μL of the Image-iT lipid peroxidation kit (Thermo Fisher, Waltham, MA) diluted at 10 μM HBSS. NAD+, desferoxamine, α-tocopherol (dissolved in 70% ethanol) or vehicle control were added to 100 μM from a 1 mM stock. After 1 h incubation, ZnCl2 or ddH2O vehicle controls were added to each well to the final concentrations indicated. Cumene hydroperoxide (100 μM) was added as a positive control. Fluorescence was monitored using a Biotek Cytation 5 (Winooski, VT) with excitation/emission 574/595 nm to measure reduced and 488/515 nm to measure oxidized reagent over 19 h.
Inductively coupled mass spectrometry
A549 cells were plated in 6-well plates one day prior to Zn intoxication. At 24 h post Zn intoxication, cells were removed from the plate by trypsin digestion to a metal-free 15-mL conical tube, washed twice in PBS, and resuspended in 110 μL PBS. A 10-μL sample was removed for cell counting by hemocytometer and trypan exclusion. The remaining 100 μL cells were digested in 0.5 mL Optima grade nitric acid (Fisher) overnight at 60°C and then diluted to 5 mL with ddH2O. Optima grade nitric acid is strongly corrosive and toxic if inhaled; it should be used in a well-ventilated area with appropriate personal protective equipment. Elemental quantification on acid-digested liquid samples was performed using an Agilent 7700 inductively coupled plasma mass spectrometer (Agilent, Santa Clara, CA). The following settings were fixed for the analysis Cell Entrance = −40 V, Cell Exit = −60 V, Plate Bias = −60 V, OctP Bias = −18 V, and collision cell Helium Flow = 4.5 mL/min. Optimal voltages for Extract 2, Omega Bias, Omega Lens, OctP RF, and Deflect were determined empirically before each sample set was analyzed. Element calibration curves were generated using ARISTAR ICP Standard Mix (VWR, Radnor, PA). Samples were introduced by peristaltic pump with 0.5 mm internal diameter tubing through a MicroMist borosilicate glass nebulizer (Agilent). Samples were initially up taken at 0.5 rps for 30 s followed by 30 s at 0.1 rps to stabilize the signal. Samples were analyzed in Spectrum mode at 0.1 rps collecting three points across each peak and performing three replicates of 100 sweeps for each element analyzed. Data were acquired and analyzed using the Agilent Mass Hunter Workstation Software version A.01.02.
Zinquin fluorescence
To assess bioavailable Zn, A549 cells were plated to a black-walled 96-well plate and intoxicated with Zn. After 23.5 h, cells were washed twice with PBS, and 100 μL PBS containing 50 μM Zinquin ethyl ester (Enzo Life Sciences, Farmingdale, NY) and 5 μM SytoOrange 82 (ThermoFisher). After 30 min incubation, fluorescence was assessed using a Biotek Cytation 5 with excitation/emission 368/490 for Zinquin and 531/593 for SytoOrange. To normalize Zinquin fluorescence, Zinquin fluorescence was divided by SytoOrange fluorescence following background subtraction for both.
RESULTS
A549 cells lose viability in response to Zn, but not by necrosis or apoptosis
To determine how A549 cells respond to Zn intoxication, A549 cell cultures were treated with a ZnCl2 two-fold dilution series from 0–1000 μM. Viability was assessed at 1 h, 6 h, and 24 h post intoxication using ATP concentrations (Fig. 1A) and the reducing environment of the cell (Fig. 1B) as proxies. These results showed that A549 cells treated with ≥ 31.2 μM Zn have an initial drop in ATP compared to untreated. By 6 h, this decrease in ATP is largely recovered; we hypothesized that at early time points the recovery of ATP levels was due to induction of Zn-chelating proteins such as metallothioneins that protect the electron transport chain from Zn intoxication (see below). When measuring viability by the reduction of resazurin dye by the cellular reducing environment, there is no loss of viability at 1 h post Zn intoxication (Fig. 1B). Viability is significantly reduced with ≥ 500 μM Zn intoxication at 6 h, but the cells maintain >60% dye reduction capability (Fig. 1B). At 24 h, A549 cells treated with 250 μM Zn have significantly lower ATP and viability, but maintain 81% ATP and 45% dye reduction capability (Fig. 1A-B, S1A-B). In contrast, A549 cells treated with ≥ 500 μM Zn have significantly lower ATP and viability (<25%) and the majority are dead at 24 h (Fig. 1A-B, S1C). Together, these results suggest that Zn intoxication of A549 cells induces a decrease in ATP within 1 h that is recovered by 6 h, and by 24 h post intoxication, A549 cells cannot survive Zn intoxication in excess of 250 μM.
Figure 1. A multi-omics approach to identify molecular changes during zinc intoxication of A549 cells.
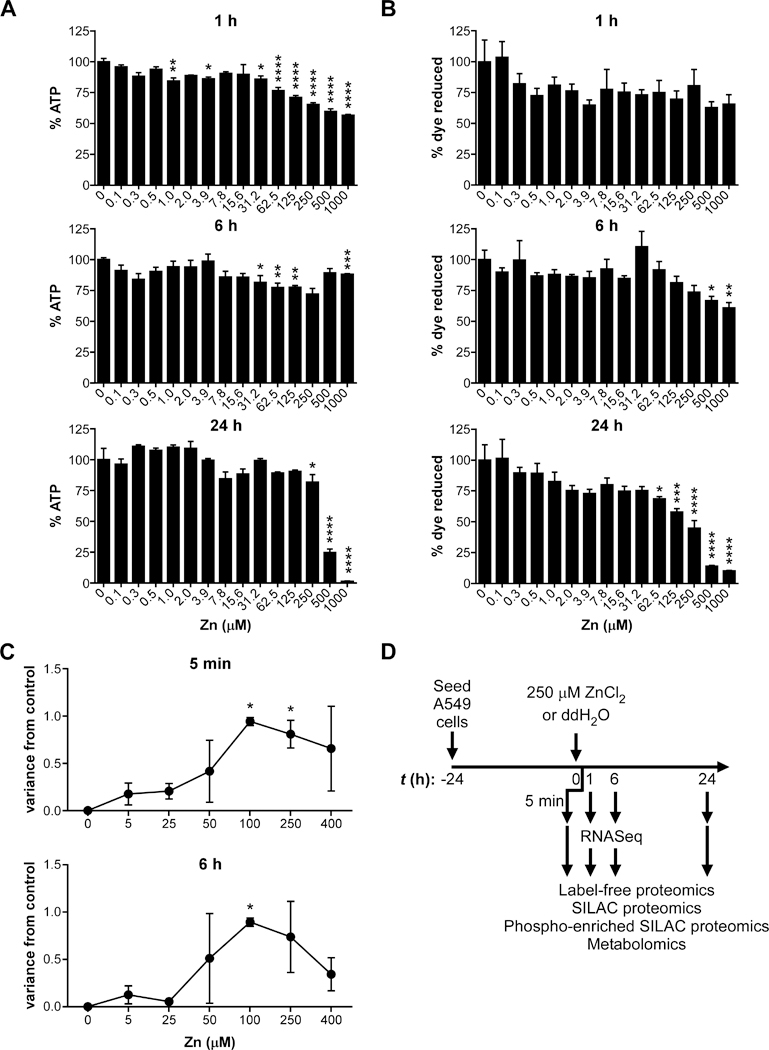
(A) Application of Zn leads decreased ATP in A549 cells at doses above 250 μM by 24 h (% ATP of 0 μM Zn). (B) Application of Zn leads decreased viability in A549 cells at doses above 62.5 μM by 24 h (% dye reduction of alamarBlue resazurin with 0 μM Zn). (C) Molecular variance analysis shows that 100–250 μM Zn induce the most molecular changes by molecular variance score, a metric that projects the high dimensional variation between mass spectra of treated and control samples into one dimension using principal component analysis. (D) Schematic for multi-omics data collection. Twenty-four h after plating, A549 cells were intoxicated with 250 μM Zn or vehicle control (ddH2O). At 5 min, 1 h, 6 h, and 24 h post-intoxication, cells were harvested for proteomics and metabolomics; at 1 h and 6 h post-intoxication, cells were also harvested for RNA-Seq.
To determine whether the decrease in viability is due to apoptosis or necrosis, caspase-3/7 cleavage and protease release were assayed. Neither apoptosis nor necrosis were detected at any time points measured over 24 h (Fig. S1D-H). Therefore, a high-throughput multi-omics analysis was designed to identify the mechanism by which A549 cells respond to Zn intoxication over time based on our previously described platform 20, 24. First, a molecular variance analysis was used to identify the Zn concentration (0–400 μM) that induced the highest number of molecular changes at 5 min and 6 h. Briefly, this molecular variance analysis uses matrix-assisted laser desorption ionization (MALDI) mass spectrometry to rapidly profile proteomic changes without identifications 24. Principle component analysis of the changes determines a 1-dimensional molecular variance score that represents the overall molecular distance between the untreated control and the intoxicated samples. The results of the molecular variance analysis revealed that the greatest molecular changes were induced with 100–250 μM ZnCl2 (Fig. 1C). In order to capture the A549 cell molecular response and mechanism of death by Zn intoxication while retaining sufficient viability over 24 h, 250 μM Zn was chosen for temporal dynamics studies using multi-omics analysis (Fig. 1D).
Multi-omics analysis determines that the response to Zn intoxication varies with time
In order to capture early and late responses, molecular changes in response to intoxication with 250 μM ZnCl2 were measured at 5 min, 1 h, 6 h, and 24 h post Zn intoxication by mass spectrometry-based label-free proteomics, SILAC proteomics, phospho-enriched proteomics, and untargeted metabolomics; transcriptomic changes were measured by RNA-sequencing at 1 and 6 h post Zn intoxication to corroborate and extend mass spectrometry findings (Fig. S1). Across these four time points and five modalities, 77,001 analytes or transcripts were detected, and 13.4% were significantly changed in Zn intoxicated cells compared to control vehicle-treated cells. To compare molecular changes over time, quasi-proportional Venn diagrams of the proteomics, phospho-proteomics, metabolomics, and transcriptomics datasets were generated with the nVenn online tool 25; label-free and SILAC proteomics were combined for this comparison because both measure protein abundance. For each modality, there is substantial overlap in the detected analytes at each time point (Fig. 2A-C, S2A). By contrast, there is little overlap in the significantly changed analytes over time (Fig. 2D-F, S2B). This comparison suggests that the A549 response to Zn intoxication varies with time, consistent with the changes in ATP/viability with time observed in Fig. 1A. Overlap in detected metabolites over time was less substantial than proteomic or transcriptomic changes, which could be due to the fact that the metabolomics comparison was identity-independent or suggest that a smaller set of key metabolites are detected at all time points. Comparison of the percent significantly changed analytes compared to vehicle-treated control at each time point shows that Zn intoxication induces significant changes in 18–19% of proteins and 27–28% of detected metabolites at 5 min, 1 h, and 6 h post intoxication. By contrast, at 24 h post Zn intoxication, only 12% of proteins and 16% of metabolites are significantly changed, suggesting that A549 cells reach a new homeostatic state in this condition that reduce changes to the overall proteome and metabolome, likely by induction of Zn chelating proteins such as metallothioneins.
Figure 2. Multi-omics analysis reveals time-dependent response of A549 cells to zinc intoxication.
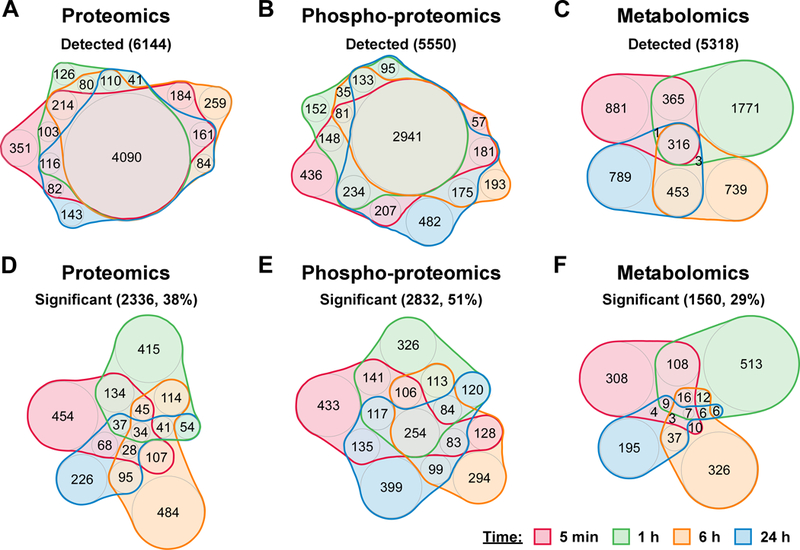
(A-C) Detected proteins, phospho-proteins, or metabolites are displayed across time. (D-F) Significantly changed proteins, phospho-proteins, or metabolites are displayed across time.
Analysis of top proteomic changes shows induction of Zn-binding proteins after 6 h.
The finding that metabolome changes were lessened by 24 h post intoxication with 250 μM Zn led to the hypothesis that A549 cells induced proteomic changes in Zn chelating proteins that minimized the effect of Zn intoxication on overall metabolism. To identify these changes, the top 10 significantly increased peptides or proteins at each time point were analyzed by Gene Ontology (GO) annotation. This analysis is represented in Fig. 3A, where the size of each circle represents the number of proteins for that GO annotation that were among the top 10 significantly changed at that time point. These data demonstrate that the early (5 min-1 h) most significantly upregulated proteins are involved in “cell differentiation.” The fold change for peptides and proteins among the top 10 significantly upregulated species that mapped to the “cell differentiation” GO annotation are shown over time (Fig. 3B), and by 24 h “cell differentiation” peptides or proteins are unchanged in Zn intoxicated compared to the control samples. In contrast, at later time points the top 10 most significantly induced proteins are annotated as “response to stress” (Fig. 3C). By analyzing “response to stress” protein fold changes over time, the late (24 h) response to Zn intoxication is dominated by upregulation of metallothioneins MT1F (+451 fold), MT1X (+9–54 fold), and MT2A (+30 fold). Metallothioneins are cysteine-rich metal binding proteins induced in response to stress and metal intoxication 28, 29. Metallothioneins have been shown to be induced following heavy metal intoxication of A549 cells 30, 31. These data are consistent with a model where at low levels of Zn intoxication, i.e. <250 μM Zn, A549 cells can effectively limit Zn toxicity by 24 h through metallothioneins.
Figure 3. Biological process analysis of top significantly increased proteins in zinc intoxicated A549 cells by QuickGo.
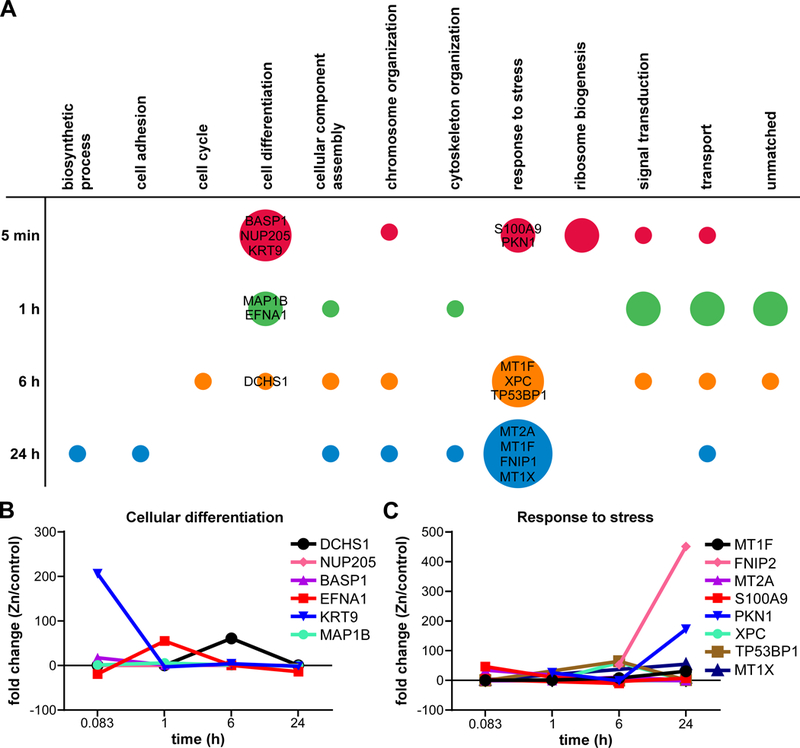
(A) Gene ontology analysis identifies the biological processes of the top 10 significantly increased proteins at each time point. The color represents each time point, while the size of circle represents the number of top 10 significantly increased proteins for each biological process. (B) Fold change over time of the top significantly increased proteins in the “cellular differentiation” biological process. (C) Fold change over time of top significantly increased proteins in the “response to stress” biological process.
Metabolomics analysis reveals changes in NAD+ biosynthesis
Because A549 cells intoxicated with 250 μM Zn had a slight but significant reduction in viability at 24 h (Fig. 1A), metabolome and proteome differences were interrogated to identify the mechanism of reduced viability. To predict metabolic network activity, the metabolomics hydrophilic interaction liquid chromatography (HILIC) and reversed-phase liquid chromatography (RPLC) datasets were combined and analyzed by mummichog 1.0.5 32. Mummichog predicts predicting pathway activity directly from untargeted metabolomics liquid chromatography/mass spectrometry feature tables without requiring metabolite identification. Vitamin B3 (nicotinate and nicotinamide) metabolism was a predicted top pathway at both 6 h and 24 h post Zn intoxication (p-value 0.007 and 0.006, respectively) and was the only significantly affected pathway at 24 h by this analysis. Nicotinate (NA) and nicotinamide (NAM) are the vitamin B3 precursors to NAD+ and NADP+ that are critically important for cellular redox chemistry. Specifically, mummichog analysis identified changes in N-methyl-nicotinamide (mNAM; +1.2 to 1.3X at 6 h, and +1.4X at 24 h), nicotinate-D-ribonucleoside (NAR; +20.6X at 24 h), and NAM (−1.7X at 6 h) (Fig. 4A). NAM is available in the DMEM medium used to culture the A549 cells and is the dominant NAD+ precursor in mammals 33. NAM is thought to be actively imported by mammalian cells, but its transporter is unknown 34–36.
Figure 4. Zinc intoxication disrupts NAD+ metabolism but NAD+ deficiency is not the cause of loss of viability.
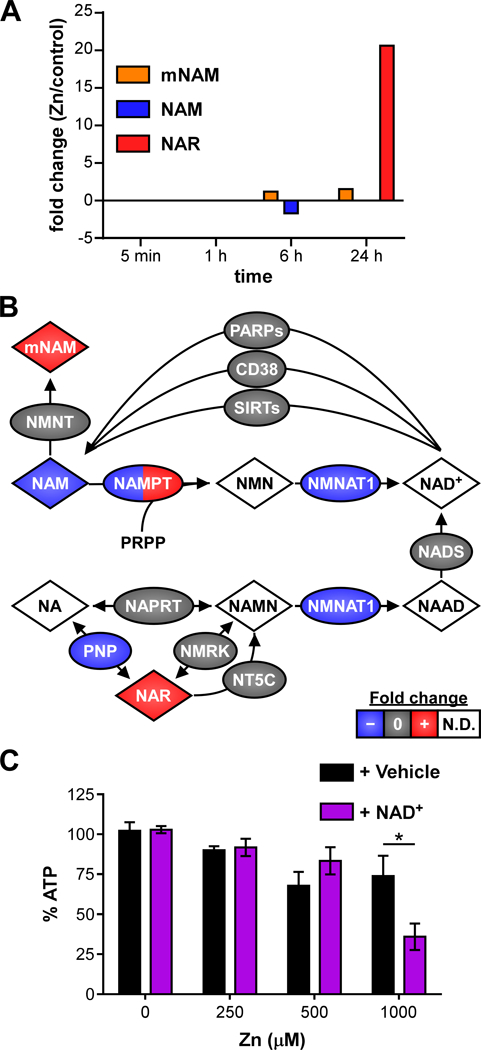
(A) Untargeted metabolomics identifies changes in NAD+ metabolites. (B) Schematic of NAD+ metabolic network is painted with results from multi-omics analysis. Species painted with two colors indicates time-dependent changes in fold-change. (C) Addition of NAD+ (100 μM) exacerbates reduced viability at 1000 μM Zn at 24 h. Abbreviations: mNAM: N-methyl-nicotinamide; NAM: nicotinamide; NMN: nicotinamide mononucleotide; NA: nicotinic acid; NAMN: nicotinic acid mononucleotide; NAAD: nicotinic acid adenine dinucleotide; N.D.: not detected.
The NAD+ metabolic network was manually reconstructed and the network nodes were painted based on the multi-omics dataset (Fig. 4B). This analysis revealed that many NAD+ salvage pathway enzymes were downregulated in the proteomics and RNA-Seq datasets. Nicotinamide phosphoribosyltransferase (NAMPT) is the major regulator of the human intracellular NAD+ pool and converts NAM to nicotinamide mononucleotide (NMN) for NAD+ salvage 37. Analysis of the multi-omics datasets determined that in response to Zn intoxication, NAMPT protein levels decrease by −2.8 fold at 6 h. However, NAMPT transcript
levels are increased +2.1 fold in Zn intoxicated cells at 6 h and by 24 h, NAMPT protein has increased +1.5 fold, consistent with the earlier transcriptional upregulation. Together these results suggest that the cells sense and respond to lower NAMPT levels. Nicotinamide mononucleotide adenylyltransferase (NMNAT) enzymes convert NMN to NAD+ and nicotinic acid mononucleotide (NAMN) to nicotinic acid adenine dinucleotide (NAAD). Analysis of the RNA-Seq datasets showed that NMNAT2 was detected and unchanged at 1 h and 6 h. NMNAT1 was not detected in most datasets, but was 3.8-fold lower in Zn intoxicated cells at 5 min, which could explain buildup of upstream precursors such as mNAM and NAR. NAR is converted to NAMN by nicotinamide riboside kinase 1 or 2 (NMRK), which were unchanged by RNA-Seq. Members of the 5’, 3’-Nucleotidase, Cytosolic (NT5C) family, which can also convert NAR to NAMN, were detected and unchanged at all time points. Alternatively, NAR can be converted to NA by purine nucleoside phosphorylase (PNP), which was 3.1-fold lower in Zn intoxicated cells at 1 h. Together these data suggested Zn intoxication reduces expression of the NAD+ salvage enzymes at early time points.
Therefore, the proteomics data were interrogated to determine whether there were lower levels of enzymes that convert NAD+ to NAM that might lead to decreased NAM levels. Overall, there were not differences in the NAD+-utilizing enzymes. The poly ADP-ribose polymerase (PARP) family post-translationally modifies protein with the ADP-ribose moiety from NAD+. In the proteomics dataset, PARP1 was detected and unchanged at every time point, and PARP14 was mildly downregulated 1.5-fold as measured by RNA-Seq. Cluster of differentiation 38 (CD38) synthesizes cyclic ADP-ribose from NAD+; CD38 was detected at every time point but unchanged. Finally, the Sir2-like family “sirtuin” protein deacylases utilize NAD+ and release NAM; SIRT1–6 proteins were detected and unchanged at various time points, and SIRT6 was upregulated only +1.4-fold at 6 h as measured by RNA-Seq. Overall, there were few and very minor differences in protein or expression levels of the NAD+-utilizing enzymes, suggesting accumulation of precursors was not due to increased levels of NAD+ breakdown.
If Zn intoxication led to increased upstream precursors and decreased NAD+, decreased NAD+ could be responsible for loss of viability with Zn intoxication. Therefore, the ability of NAD+ to rescue viability of A549 cells intoxicated with Zn was tested. At 24 h, addition of 100 μM NAD+ did not improve viability of Zn intoxicated cells, and at 1000 μM Zn NAD+ exacerbated loss of viability (Fig. 4C). Together, these data demonstrate that Zn intoxication disrupts NAD+ biosynthesis, but that NAD+ does not rescue loss of viability caused by Zn intoxication.
Zn intoxication induces ferroptosis
Mummichog analysis also identified changes in glutathione and oxidized glutathione. At 6 h post Zn intoxication, analytes identified as oxidized glutathione (GSSG) were decreased 1.4 to 1.9-fold and glutathione (GSH) was decreased 1.1 to 3.5-fold, together suggesting that Zn intoxication led to glutathione depletion at 6 h (Fig. 5A). At 24 h, Mummichog analysis did not identify any changes in glutathione metabolites, suggesting glutathione homeostasis is restored by 24 h post intoxication with 250 μM Zn, consistent with overall maintenance of viability (Fig. 1A). Glutathione depletion is associated with two forms of non-apoptotic cell death: oxidative glutamate toxicity and ferroptosis 19. Two key differences between oxidative glutamate toxicity and ferroptosis are that oxidative glutamate toxicity is associated with mitochondrial ROS, while ferroptosis is iron-dependent and caused by lipid peroxidation. We previously reported that Zn induced low level mitochondrial ROS in A549 cells 20. However, inhibition of mitochondrial ROS with MitoTEMPO did not protect viability during Zn intoxication (Fig. S3), suggesting oxidative glutamate toxicity is not the mechanism of death caused by Zn intoxication.
Figure 5. Zinc intoxication leads to ferroptosis in A549 cells.
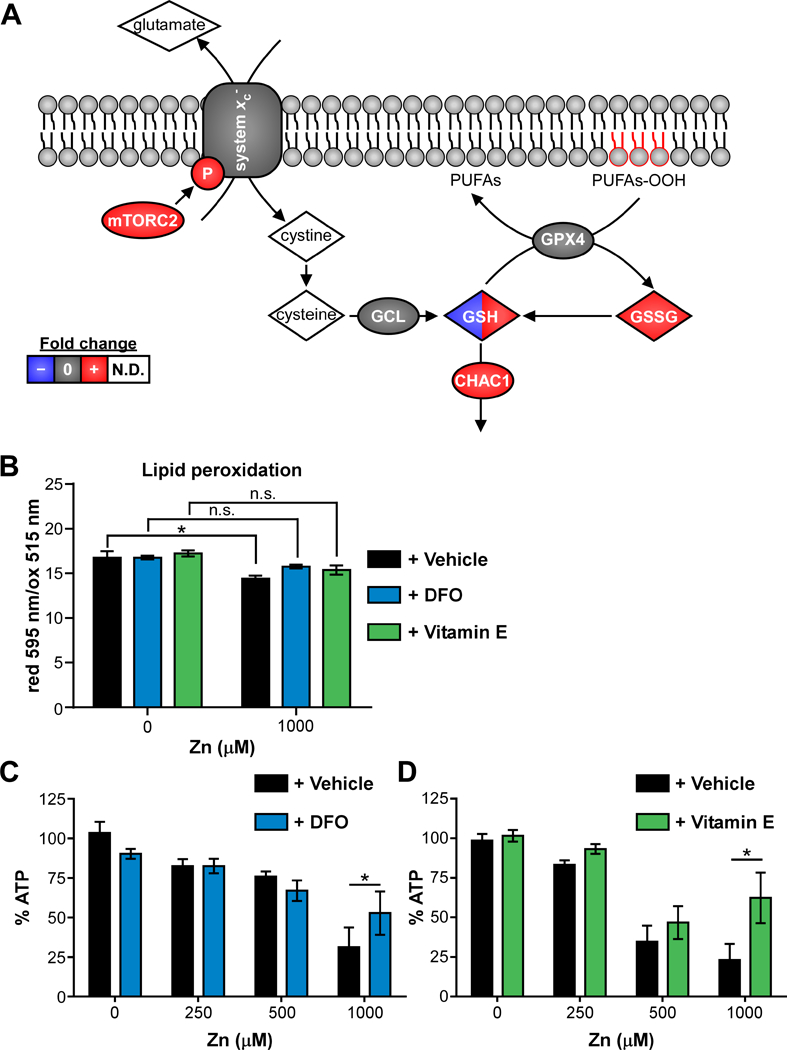
(A) Schematic of ferroptosis network is painted with results from multi-omics analysis. Species painted with two colors indicates time-dependent changes in fold-change. (B) Lipid peroxidation is significantly increased (lower reduced/oxidized) in A549 cells treated with 500 μM Zn, but not in the presence of desferoxamine (DFO, 100 μM) or α-tocopherol (Vitamin E, 100 μM). (C-D) Addition of DFO or vitamin E (100 μM) limits loss of viability at 24 h when A549 cells are intoxicated with 1000 μM Zn (% of 0 μM). Abbreviations: GSH: reduced glutathione; GSSG: oxidized glutathione; N.D.: not detected; PUFAs: polyunsaturated fatty acids; PUFAs-OOH: polyunsaturated fatty acid peroxides.
Therefore, the ferroptosis cell death network was reconstructed, which determined that multiple components are increased following Zn intoxication (Fig. 5A). Ferroptosis is a regulated form of non-apoptotic cell death that is iron-dependent and was first described in 2012 38. System xc-is a cystine/glutamate antiporter that is encoded by SLC7A11. Inhibition of system xc-can initiate either oxidative glutamate toxicity or ferroptosis 19. In the multi-omics datasets, system xc-abundance was not changed, but phosphorylation of system xc-Ser26, which inhibits its activity, was increased +1.6-fold in the Zn intoxicated cells. Mammalian target of rapamycin complex 2 (mTORC2), a complex of mTOR and Rapamycin-insensitive companion of mammalian target of rapamycin (RICTOR), is capable of phosphorylating SLC7A11 of system xc-at this site 39. RICTOR was +1.7–3X more abundant in Zn intoxicated cells at 5 min, consistent with increased phosphorylation of system xc-and decreased system xc-activity. Gamma-glutamyl cysteine ligase (GCL) and glutathione peroxidase 4 (GPX4) were detected at every time point but not changed, consistent with previous reports that Zn does not affect GPX function 23. In addition to decreases in functional system xc-and glutathione discussed above, the ferroptosis marker CHAC1 was significantly upregulated by +1.6-fold at 1 h and +5.8-fold at 6 h by RNA-Seq 40. Because lipid peroxidation is a key executioner of ferroptosis-mediated death, the metabolomics datasets were searched for evidence of polyunsaturated fatty acid lipid peroxides (PUFAs-OOH). Candidate lipid peroxides were not detected in the metabolomics dataset, likely because lipid peroxides do not ionize well in positive ion mode. Therefore, lipid peroxidation was measured using the image-iT lipid peroxidation kit, with which the ratio of reduced (595 nm) over oxidized (515 nm) BODIPY® 581/591 C11 fluorescence decreases with lipid peroxidation. The induction of lipid peroxidation by Zn was measured at 12 h post intoxication, the time point at which lipid peroxidation induced by the cumene hydroperoxide positive control was strongest (not shown). At 12 h, 500 μM Zn induced a significant reduction in the ratio of reduced/oxidized BODIPY® 581/591 C11, indicating Zn induces lipid peroxidation (Fig. 5B).
Ferroptosis can be rescued by cell permeable iron chelators (e.g. desferoxamine (DFO)) or lipophilic antioxidants (e.g. vitamin E/α-tocopherol) 19, 41, 42. Both DFO and vitamin E prevented lipid peroxidation measured by BODIPY® 581/591 C11 (Fig. 5B), indicating that lipid peroxidation was iron dependent. Finally, both DFO and vitamin E limited Zn-induced loss of viability at 24 h (Fig. 5C-D). DFO binds iron preferentially but can also bind Zn at approximately 5-fold lower affinity 43; therefore, we assessed whether DFO chelation of Zn could contribute to rescue of viability in two ways. First, the effect of DFO on total iron and Zn levels in the cells was measured by ICP-MS (Fig. S4A-B). These data show that at 500 μM Zn, there is a trend toward decreased total iron in the cells pre-treated with DFO (Fig. S4A). In contrast, there is no significant difference in total cellular Zn levels based on DFO treatment at 0, 250 or 500 μM Zn (Fig. S4B), and there may be a slight trend toward increased total cellular Zn in the presence of DFO (Fig. S4B). Next, the effect of DFO on bioavailable Zn was measured using the cell permeant dye Zinquin, which increases fluorescence in a Zn-specific manner in the presence of physiological concentrations of Ca2+ and Mg2+. Zinquin fluorescence was normalized to fluorescence by the nucleic acid dye SytoOrange 82, to control for decreased adherence of Zn-intoxicated cells in PBS washing. These results showed that Zinquin fluorescence increased upon Zn intoxication, and was further increased in the presence of DFO (Fig. S4C). The results of ICP-MS and Zinquin fluorescence suggest that DFO may increase total and bioavailable Zn during Zn intoxication and therefore that Zn chelation by DFO is likely not the mechanism by which DFO protects from Zn-induced loss of viability. Together, these data show that Zn induces lipid peroxidation and cell death in an iron-dependent mechanism consistent with ferroptosis. In summary, a comprehensive multi-omics analysis of Zn intoxication in A549 cells led to the discovery that Zn intoxication induces ferroptosis.
DISCUSSION
A high throughput multi-omic strategy was used to identify the cellular response and mechanism of cell death when A549 cells were intoxicated with Zn. A549 cells effectively manage low level Zn intoxication, up to approximately 250 μM Zn. This low-level Zn intoxication is likely mitigated by metallothionein proteins that buffer toxic metals in the cytoplasm (Fig. 3C). However, at Zn levels above 250 μM Zn, A549 cells suffer loss of viability by an apoptosis-and necrosis-independent mechanism. Multi-omics data were analyzed for potential mechanisms of cell death, and complementary experimentation suggests that A549 cells induce ferroptosis in response to high levels of Zn.
Previous work showed that Zn can selectively kill cancer cells and potentiate apoptosis-inducing cancer drugs, such as docetaxel, suggesting Zn might be an effective adjuvant in the treatment of cancer 12, 13, 23. However, other work on A549 cells, BEAS-2B lung cells, and HL-60 cells reported that Zn induced an inflammatory response including IL-8 production 4, 20, 44. Consistent with this finding, the gene encoding IL-8, CXCL8, was induced +3X at 1 h and +22X at 6 h in the present study. IL-8 has been shown to increase angiogenesis of NSCLC and therefore promote tumor growth 45, 46. Therefore, stimulation of IL-8 production may limit therapeutic potential of Zn intoxication for cancer.
In addition, the multi-omics data reported here suggest that NAD+ biosynthesis is disrupted in Zn-intoxicated A549 cells. This is consistent with data from neuronal cells, where Zn intoxication leads to decreased NAD+ levels and subsequent inhibition of glycolysis and energy generation 47. However, the addition of NAD+ to A549 cells exacerbated loss of viability at high Zn, suggesting that low NAD+ was not the cause of cell death in A549 cells.
Consistent with ferroptotic death, previous work in NSCLC cell lines showed that Zn intoxication led to decreased GSH and increased GSSG levels due to reduced glutathione reductase activity 48. Zn intoxication of Hep-2 cells was also reported to decrease ATP and increase peroxides, and loss of viability by a caspase 3-independent mechanism 49. Similarly, Zn intoxication of neuronal cells led to cell death with features of both apoptosis and necrosis that was inhibited by a vitamin E analog 50. Together these results indicate that the response to Zn intoxication in A549 lung cells is similar to findings previously reported in other cells and that Zn may also induce ferroptosis in other cell types.
The rescue of cell viability by addition of the iron-chelating molecule DFO at high Zn levels suggests that iron plays an intermediary role in Zn-induced loss of viability. Previous research suggests that Zn intoxication displaces iron atoms in respiratory chain proteins, leading to increased oxidative stress 32. Therefore, Zn intoxication displacing iron may lead to increased free intracellular iron, which is essential for the execution of ferroptosis. The initial signal responsible for Zn-induced ferroptosis remains unknown. Based on multi-omics data analysis, Zn intoxication may activate mTORC2 or other kinases capable of inhibiting system xc-function by phosphorylation. Alternatively, Zn may inhibit system xc-, glutamate biosynthesis, or GPX4 directly. Future work should clarify the initiating events in Zn-induced ferroptosis.
Together, the results of this study demonstrate the value of multi-omics analyses to identify the mechanism of cell death of potential therapeutic agents. This analysis identified a novel link between Zn intoxication and ferroptosis, a newly described mechanism of programmed cell death. While the finding that Zn induces ferroptosis, a non-inflammatory cell death, would suggest Zn may serve as a valuable anti-tumor therapeutic, the additional finding that Zn induces IL-8 expression from unbiased multi-omics datasets suggests that the therapeutic potential may be limited. Therefore, unbiased multi-omics datasets can be harnessed to identify both the primary mechanism of action (i.e. cell death mechanism), and secondary and potentially harmful effects of intoxicants.
CONCLUSIONS
Together, the data included here suggest that Zn intoxication of A549 lung epithelial cells induces lipid peroxidation and cell death that can be limited by incubation with the iron chelator desferoxamine or the lipophilic antioxidant vitamin E, consistent with ferroptosis. Consideration of other reports suggests Zn intoxication may induce ferroptosis in other cell types, including liver cells and neuronal cells. More broadly, the results demonstrate that an unbiased approach such as the multi-omics time course analysis used here can identify mechanisms of cell death in response to intoxicants.
Supplementary Material
ACKNOWLEDGMENTS
We thank John P. Wikswo for his contributions to development of the high-throughput multi-omics platform. We thank James C. Pino, Michael Ripperger, and Jay Holman for their contributions of the custom-built biocomputational tools utilized for data analysis. We thank members of the Skaar laboratory for critical reading of the manuscript. We also acknowledge the Proteomics Core of the Mass Spectrometry Research Center at Vanderbilt University for their preparation and analysis of the phosphoproteomics samples, and the Center for Innovative Technology for the untargeted metabolomics. Research was sponsored by an American Asthma Foundation grant and NIH R01 AI101171 to E.P.S. and the U.S. Army Research Office and the Defense Advanced Research Projects Agency and was accomplished under Cooperative Agreement no. W911 NF-14–2-0022. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the Army Research Office, DARPA, or the U.S. Government. The U.S. Government is authorized to reproduce and distribute reprints for Government purposes notwithstanding any copyright notation herein.
Footnotes
CONFLICTS OF INTEREST
The authors declare no financial or other conflicts of interest.
REFERENCES
- 1.Andreini C, Banci L, Bertini I and Rosato A, Counting the zinc-proteins encoded in the human genome, J Proteome Res, 2006, 5, 196–201. [DOI] [PubMed] [Google Scholar]
- 2.Eide DJ, Zinc transporters and the cellular trafficking of zinc, Biochim Biophys Acta, 2006, 1763, 711–722. [DOI] [PubMed] [Google Scholar]
- 3.Wu W, Bromberg PA and Samet JM, Zinc ions as effectors of environmental oxidative lung injury, Free Radic Biol Med, 2013, 65, 57–69. [DOI] [PubMed] [Google Scholar]
- 4.Plum LM, Rink L and Haase H, The essential toxin: impact of zinc on human health, Int J Environ Res Public Health, 2010, 7, 1342–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu HH, Tzao C, Chang WC, Wu CP, Tung HJ, Chen CY and Perng WC, Zinc chloride (smoke bomb) inhalation lung injury: clinical presentations, high-resolution CT findings, and pulmonary function test results, Chest, 2005, 127, 2064–2071. [DOI] [PubMed] [Google Scholar]
- 6.Podany A, Rauchut J, Wu T, Kawasawa YI, Wright J, Lamendella R, Soybel DI and Kelleher SL, Excess dietary zinc intake in neonatal mice causes oxidative stress and alters intestinal host-microbe interactions, Mol Nutr Food Res, 2018, DOI: 10.1002/mnfr.201800947, e1800947. [DOI] [PubMed] [Google Scholar]
- 7.Zackular JP, Moore JL, Jordan AT, Juttukonda LJ, Noto MJ, Nicholson MR, Crews JD, Semler MW, Zhang Y, Ware LB, Washington MK, Chazin WJ, Caprioli RM and Skaar EP, Dietary zinc alters the microbiota and decreases resistance to Clostridium difficile infection, Nat Med, 2016, 22, 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox DH and Harris DL, Effect of excess dietary zinc on iron and copper in the rat, J Nutr, 1960, 70, 514–520. [DOI] [PubMed] [Google Scholar]
- 9.Cox DH and Hale OM, Liver iron depletion without copper loss in swine fed excess zinc, J Nutr, 1962, 77, 225–228. [DOI] [PubMed] [Google Scholar]
- 10.O’Neil-Cutting MA, Bomford A and Munro HN, Effect of excess dietary zinc on tissue storage of iron in rats, J Nutr, 1981, 111, 1969–1979. [DOI] [PubMed] [Google Scholar]
- 11.Nations SP, Boyer PJ, Love LA, Burritt MF, Butz JA, Wolfe GI, Hynan LS, Reisch J and Trivedi JR, Denture cream: an unusual source of excess zinc, leading to hypocupremia and neurologic disease, Neurology, 2008, 71, 639–643. [DOI] [PubMed] [Google Scholar]
- 12.Hanley C, Layne J, Punnoose A, Reddy KM, Coombs I, Coombs A, Feris K and Wingett D, Preferential killing of cancer cells and activated human T cells using ZnO nanoparticles, Nanotechnology, 2008, 19, 295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasmussen JW, Martinez E, Louka P and Wingett DG, Zinc oxide nanoparticles for selective destruction of tumor cells and potential for drug delivery applications, Expert Opin Drug Deliv, 2010, 7, 1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fraker PJ and Telford WG, A reappraisal of the role of zinc in life and death decisions of cells, Proc Soc Exp Biol Med, 1997, 215, 229–236. [DOI] [PubMed] [Google Scholar]
- 15.Truong-Tran AQ, Carter J, Ruffin RE and Zalewski PD, The role of zinc in caspase activation and apoptotic cell death, Biometals, 2001, 14, 315–330. [DOI] [PubMed] [Google Scholar]
- 16.Watjen W, Haase H, Biagioli M and Beyersmann D, Induction of apoptosis in mammalian cells by cadmium and zinc, Environ Health Perspect, 2002, 110 Suppl 5, 865–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA and Lipton SA, Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels, Neuron, 2004, 41, 351–365. [DOI] [PubMed] [Google Scholar]
- 18.Perry DK, Smyth MJ, Stennicke HR, Salvesen GS, Duriez P, Poirier GG and Hannun YA, Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis, J Biol Chem, 1997, 272, 18530–18533. [DOI] [PubMed] [Google Scholar]
- 19.Cao JY and Dixon SJ, Mechanisms of ferroptosis, Cell Mol Life Sci, 2016, 73, 2195–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gutierrez DB, Gant-Branum RL, Romer CE, Farrow MA, Allen JL, Dahal N, Nei YW, Codreanu SG, Jordan AT, Palmer LD, Sherrod SD, McLean JA, Skaar EP, Norris JL and Caprioli RM, An integrated, high-throughput strategy for multiomic systems level analysis, J Proteome Res, 2018, 17, 3396–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitamura H and Motohashi H, NRF2 addiction in cancer cells, Cancer Sci, 2018, 109, 900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taguchi K and Yamamoto M, The KEAP1-NRF2 system in cancer, Front Oncol, 2017, 7, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kocdor H, Ates H, Aydin S, Cehreli R, Soyarat F, Kemanli P, Harmanci D, Cengiz H and Kocdor MA, Zinc supplementation induces apoptosis and enhances antitumor efficacy of docetaxel in non-small-cell lung cancer, Drug Des Devel Ther, 2015, 9, 3899–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Norris JL, Farrow MA, Gutierrez DB, Palmer LD, Muszynski N, Sherrod SD, Pino JC, Allen JL, Spraggins JM, Lubbock AL, Jordan A, Burns W, Poland JC, Romer C, Manier ML, Nei YW, Prentice BM, Rose KL, Hill S, Van de Plas R, Tsui T, Braman NM, Keller MR, Rutherford SA, Lobdell N, Lopez CF, Lacy DB, McLean JA, Wikswo JP, Skaar EP and Caprioli RM, Integrated, high-throughput, multiomics platform enables data-driven construction of cellular responses and reveals global drug mechanisms of action, J Proteome Res, 2017, 16, 1364–1375. [DOI] [PubMed] [Google Scholar]
- 25.Perez-Silva JG, Araujo-Voces M and Quesada V, nVenn: generalized, quasi-proportional Venn and Euler diagrams, Bioinformatics, 2018, 34, 2322–2324. [DOI] [PubMed] [Google Scholar]
- 26.Huntley RP, Sawford T, Martin MJ and O’Donovan C, Understanding how and why the Gene Ontology and its annotations evolve: the GO within UniProt, Gigascience, 2014, 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Binns D, Dimmer E, Huntley R, Barrell D, O’Donovan C and Apweiler R, QuickGO: a web-based tool for Gene Ontology searching, Bioinformatics, 2009, 25, 3045–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kimura T and Kambe T, The functions of metallothionein and ZIP and ZnT transporters: an overview and perspective, Int J Mol Sci, 2016, 17, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krezel A and Maret W, The functions of metamorphic metallothioneins in zinc and copper metabolism, Int J Mol Sci, 2017, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang YJ, Clapper JA and Enger MD, Enhanced cadmium cytotoxicity in A549 cells with reduced glutathione levels is due to neither enhanced cadmium accumulation nor reduced metallothionein synthesis, Cell Biol Toxicol, 1989, 5, 249–259. [DOI] [PubMed] [Google Scholar]
- 31.Foldbjerg R, Irving ES, Hayashi Y, Sutherland DS, Thorsen K, Autrup H and Beer C, Global gene expression profiling of human lung epithelial cells after exposure to nanosilver, Toxicol Sci, 2012, 130, 145–157. [DOI] [PubMed] [Google Scholar]
- 32.Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, Jones DP and Pulendran B, Predicting network activity from high throughput metabolomics, PLoS Comput Biol, 2013, 9, e1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collins PB and Chaykin S, The management of nicotinamide and nicotinic acid in the mouse, J Biol Chem, 1972, 247, 778–783. [PubMed] [Google Scholar]
- 34.Olsson A, Olofsson T and Pero RW, Specific binding and uptake of extracellular nicotinamide in human leukemic K-562 cells, Biochem Pharmacol, 1993, 45, 1191–1200. [DOI] [PubMed] [Google Scholar]
- 35.Reyes AM, Bustamante F, Rivas CI, Ortega M, Donnet C, Rossi JP, Fischbarg J and Vera JC, Nicotinamide is not a substrate of the facilitative hexose transporter GLUT1, Biochemistry, 2002, 41, 8075–8081. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki E, Okuda H, Nishida K, Fujimoto S and Nagasawa K, Protective effect of nicotinamide against poly(ADP-ribose) polymerase-1-mediated astrocyte death depends on its transporter-mediated uptake, Life Sci, 2010, 86, 676–682. [DOI] [PubMed] [Google Scholar]
- 37.Garten A, Schuster S, Penke M, Gorski T, de Giorgis T and Kiess W, Physiological and pathophysiological roles of NAMPT and NAD metabolism, Nat Rev Endocrinol, 2015, 11, 535–546. [DOI] [PubMed] [Google Scholar]
- 38.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd and Stockwell BR, Ferroptosis: an iron-dependent form of nonapoptotic cell death, Cell, 2012, 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu Y, Albuquerque CP, Braas D, Zhang W, Villa GR, Bi J, Ikegami S, Masui K, Gini B, Yang H, Gahman TC, Shiau AK, Cloughesy TF, Christofk HR, Zhou H, Guan KL and Mischel PS, mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT, Mol Cell, 2017, 67, 128–138 e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS and Stockwell BR, Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis, Elife, 2014, 3, e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dixon SJ and Stockwell BR, The role of iron and reactive oxygen species in cell death, Nat Chem Biol, 2014, 10, 9–17. [DOI] [PubMed] [Google Scholar]
- 42.Hao S, Liang B, Huang Q, Dong S, Wu Z, He W and Shi M, Metabolic networks in ferroptosis, Oncol Lett, 2018, 15, 5405–5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hider RC, Bittel D and Andrews GK, Competition between iron(III)-selective chelators and zinc-finger domains for zinc(II), Biochem Pharmacol, 1999, 57, 1031–1035. [DOI] [PubMed] [Google Scholar]
- 44.Saptarshi SR, Feltis BN, Wright PF and Lopata AL, Investigating the immunomodulatory nature of zinc oxide nanoparticles at sub-cytotoxic levels in vitro and after intranasal instillation in vivo, J Nanobiotechnology, 2015, 13, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith DR, Polverini PJ, Kunkel SL, Orringer MB, Whyte RI, Burdick MD, Wilke CA and Strieter RM, Inhibition of interleukin 8 attenuates angiogenesis in bronchogenic carcinoma, J Exp Med, 1994, 179, 1409–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arenberg DA, Kunkel SL, Polverini PJ, Glass M, Burdick MD and Strieter RM, Inhibition of interleukin-8 reduces tumorigenesis of human non-small cell lung cancer in SCID mice, J Clin Invest, 1996, 97, 2792–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheline CT, Behrens MM and Choi DW, Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis, J Neurosci, 2000, 20, 3139–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walther UI, Wilhelm B, Walther S, Muckter H and Fichtl B, Zinc toxicity in various lung cell lines is mediated by glutathione and GSSG reductase activity, Biol Trace Elem Res, 2000, 78, 163–177. [DOI] [PubMed] [Google Scholar]
- 49.Rudolf E, Depletion of ATP and oxidative stress underlie zinc-induced cell injury, Acta Medica (Hradec Kralove), 2007, 50, 43–49. [PubMed] [Google Scholar]
- 50.Kim YH, Kim EY, Gwag BJ, Sohn S and Koh JY, Zinc-induced cortical neuronal death with features of apoptosis and necrosis: mediation by free radicals, Neuroscience, 1999, 89, 175–182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.