

Abstract
Introduction.
Lafora disease (LD) is autosomal recessive progressive myoclonus epilepsy with late childhood-to teenage-onset caused by loss-of-function mutations in either EPM2A or EPM2B genes encoding laforin or malin, respectively.
Development.
The main symptoms of LD, which worsen progressively, are: myoclonus, occipital seizures, generalized tonic-clonic seizures, cognitive decline, neuropsychiatric syptoms and ataxia with a fatal outcome. Pathologically, LD is characterized by the presence of polyglucosans deposits (named Lafora bodies, LBs), in the brain, liver, muscle and sweat glands. Diagnosis of LD is made through clinical, electrophysiological, histological and genetic findings. Currently, there is no treatment to cure or prevent the development of the disease. Traditionally, antiepileptic drugs are used for the management of myoclonus and seizures. However, patients become drug-resistant after the initial stage.
Conclusions.
LD is a rare disease that has serious consequences for patients and their caregivers despite its low prevalence. Therefore, continuing research in order to clarify the underlying mechanisms and hopefully developing new palliative and curative treatments for the disease is necessary.
Keywords: Lafora disease, EPM2A, EPM2B, laforin, malin, progressive myoclonus epilepsy
Abstract
Introducción.
La enfermedad de Lafora (LD) es una forma de epilepsia mioclónica progresiva de herencia autosómica recesiva, de inicio en la infancia tardía o en la adolescencia, y producida por mutaciones de pérdida de función en los genes EPM2A o EPM2B, los cuales codifican para las proteínas laforina y malina, respectivamente.
Desarrollo.
Los principales síntomas de la enfermedad, que empeoran progresivamente, son: mioclonías, crisis occipitales, crisis tónico-clónicas generalizadas, deterioro cognitivo, síntomas neuropsiquiátricos, y ataxia. El curso es progresivo y fatal. Patológicamente, la LD se caracteriza por la presencia de depósitos de poliglucosanos llamados cuerpos de Lafora (LBs, del inglés Lafora bodies) en el cerebro, hígado, músculo, y glándulas sudoríparas. El diagnóstico de LD se realiza mediante hallazgos clínicos, electrofisiológicos, histológicos y genéticos. Actualmente, no existe un tratamiento que erradique o prevenga el desarrollo de la enfermedad. Tradicionalmente, se utilizan fármacos antiepilépticos para el manejo de las mioclonías y las convulsiones, aunque aparecen resistencias a los mismos.
Conclusiones.
La LD es una enfermedad rara que, pese a su baja prevalencia, supone graves consecuencias para los pacientes y sus cuidadores. Así pues, resulta necesario continuar la investigación para clarificar los mecanismos subyacentes y desarrollar nuevos tratamientos paliativos y curativos de la enfermedad.
Keywords: Enfermedad de Lafora, epilepsia mioclónica progresiva, EPM2A, EPM2B, laforina, malina
Introducción
La enfermedad de Lafora (LD, OMIM#254780 ORPHA501) es una forma de epilepsia mioclónica progresiva de herencia autosómica recesiva, causada por mutaciones en los genes que codifican para la laforina (EPM2A) o para la malina (EPM2B o NHLRC1). Fue descrita por primera vez en 1911 por el neuropsiquiatra español Gonzalo Rodríguez Lafora, discípulo de Santiago Ramón y Cajal y Luis Simarro, y se refirió a ella como “epilepsia mioclónica con demencia de inicio adolescente y con depósitos de amiloide” [1]. Es particularmente frecuente en algunos países mediterráneos, en Oriente Medio, Norte de África y sur de India, así como en regiones con una tasa alta de consanguineidad [2].
La sintomatología de la LD tiene su inicio entre la infancia tardía y la adolescencia en personas con un desarrollo neurológico previo normotípico. Puede aparecer en un rango de edad que va desde los 5 hasta los 20 años, siendo más frecuente entre los 14 y los 16 años [2]. Se caracteriza por la aparición de mioclonías y crisis epilépticas (desde tónico-clónicas generalizadas hasta atónicas o ausencias), degeneración progresiva del sistema nervioso y deterioro del funcionamiento cognitivo. El pronóstico es progresivo y fatal. La enfermedad presenta una evolución de 5 a 10 años, en los que aumenta progresivamente la frecuencia y variedad de los ataques, así como la dificultad para controlarlos mediante fármacos, hasta llegar al estatus epiléptico y el fallecimiento del paciente [3]. Con frecuencia también aparecen alucinaciones visuales o ceguera transitoria (no siempre asociadas a las crisis epilépticas occipitales), síntomas neuropsiquiátricos (apatía, depresión, cambios en el comportamiento…), disartria, mutismo o alteraciones respiratorias [4,5].
La LD está causada por mutaciones de pérdida de función en los genes EPM2A [6,7] o EMP2B (NHLRC1) [8], los cuales codifican para las proteínas laforina y malina respectivamente. La primera de ellas es una fosfatasa con especificidad dual, mientras que en el segundo caso se trata de una E3 ubiquitina ligasa.
La patología distintiva de esta enfermedad es la acumulación intracelular de los denominados cuerpos de Lafora, unos agregados que contienen poliglucosanos (moléculas de glucógeno mal ramificadas) así como proteínas ubiquitinadas. Estas inclusiones se acumulan en diferentes órganos del cuerpo, y típicamente se han descrito en el cerebro, el hígado, el músculo cardíaco y esquelético y las glándulas sudoríparas [1,9,10].
Actualmente, no existe un tratamiento que erradique o prevenga el desarrollo de la enfermedad. Tradicionalmente, se utilizan fármacos antiepilépticos para el manejo de las mioclonías y las convulsiones, aunque pronto aparecen resistencias a los mismos [2]. Por otro lado, las principales líneas de investigación actuales están dedicadas a clarificar los mecanismos subyacentes a la enfermedad, así como a la búsqueda de un tratamiento eficaz. Para ello, y dado que se trata de una enfermedad con unos índices de prevalencia muy bajos, se han de utilizar modelos animales. Estos modelos consisten en alterar en roedores el gen Epm2a [11] (ratones Epm2a−/−) o Epm2b [12–14] (ratones Epm2b−/−), y manifiestan alteraciones neurológicas y comportamentales similares a las descritas en pacientes con LD, incluyendo la neurodegeneración y el desarrollo de cuerpos de Lafora en diferentes órganos. Cabe destacar que también se ha descrito la LD en perros, los cuales presentan una alteración en el gen EPM2B y manifiestan un fenotipo similar a la forma humana [15].
Se estima que la LD aparece con una prevalencia de 4 casos por cada millón de habitantes en el mundo [2]. Se han descrito alrededor de 250 pacientes y/o familias con esta enfermedad, de los cuales el 42% está causado por mutaciones en el gen EPM2A, mientras que el resto está causado por mutaciones en el gen EPM2B. Sin embargo, la proporción de casos de LD con mutaciones EPM2A/EPM2B varía según la población o zona geográfica [16], siendo ciertas mutaciones específicas de grupos étnicos y/o localizaciones geográficas concretos (Figura 1).
Figura 1.
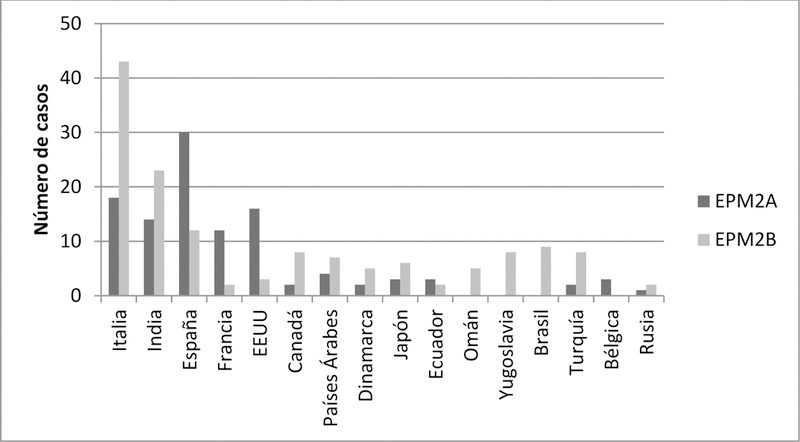
Número de casos de pacientes con mutaciones en los genes EPM2A y EPM2B según país/etnicidad conocidos hasta 2016. Sólo figuran los países/etnicidades que cuentan con más de un caso. Obtenido de Turnbull et al., 2016. Epileptic Dis. 18:S38-S62 [2].
El objetivo del presente trabajo es revisar y unificar los distintos hallazgos aparecidos en la literatura científica en relación a la LD. Dado que se trata de una enfermedad rara y actualmente no se conoce ningún tratamiento eficaz ni tan siquiera para paliar los síntomas de manera sostenida, resulta fundamental unificar los principales conocimientos que se han documentado hasta la fecha.
Características clínicas y evolución de la enfermedad
Los primeros síntomas de la enfermedad suelen ser: crisis epilépticas occipitales parciales o generalizadas, que pueden ser fotoconvulsivas, y suelen asociarse con ceguera transitoria o alucinaciones visuales elementales o complejas; episodios de caída por ausencias o atonía, y mioclonías, tanto en reposo como por sensibilidad a ciertos estímulos. Al inicio de los síntomas, pueden no observarse anormalidades en EEG o que aparezca un fondo ligeramente ralentizado de actividad. Posteriormente, aparecen picos sobre las regiones occipitales, así como descargas generalizadas punta-onda que aumentan mediante estimulación fótica. Sin embargo, la MRI no muestra irregularidad alguna [17]. Después, y de forma progresiva e insidiosa, aparecen y van aumentando en frecuencia las crisis tónico-clónicas generalizadas, y aparece la mioclonía de forma casi continua, sobre todo de las extremidades superiores e inferiores. Esta mioclonía es la primera causa para la dependencia de la silla de ruedas [17].
Desde las fases tempranas, comienza un deterioro rápidamente progresivo del funcionamiento cognitivo y neurológico. Los primeros síntomas se suelen reflejar en la caída del rendimiento académico, pero poco después el deterioro comienza a afectar de forma devastadora al resto de funciones, produciéndose disartria y afasia, apraxia, ataxia cerebelosa, disminución del tono muscular, espasticidad y ceguera cortical. En cuanto al funcionamiento cognitivo, los estudios coinciden en la observación de dificultades en atención y funciones ejecutivas [17–21] lenguaje [4,22,23], memoria [4,23] y agnosia visual [20]. Además del deterioro neurológico y cognitivo, es frecuente que aparezcan de manera temprana diferentes síntomas psiquiátricos como pueden ser alteraciones conductuales, depresión, apatía, confusión o sintomatología psicótica.
Los pacientes que sufren la LD acaban finalmente postrados en la cama y en un estado de dependencia absoluta. En esta última fase, puede observarse atrofia cerebral y cerebelar. Sin embargo, otros órganos no están afectados [17]. Aproximadamente a los 10 años del inicio de los síntomas, se produce la muerte, normalmente en status epilepticus, y debido a neumonía por aspiración, o complicaciones relacionadas con la degeneración del sistema nervioso [17].
Clínicamente, la enfermedad es bastante homogénea en cuanto al inicio en la pubertad o adolescencia y la aparición del deterioro cognitivo poco después del mismo. Sin embargo, el curso y la severidad de los síntomas son muy heterogéneos, y la edad exacta de inicio (dentro del rango observado de 5 a 20 años) es muy variable [24]. Esta variabilidad se ha observado incluso en pacientes pertenecientes a una misma familia y con mutaciones genéticas idénticas, lo que nos puede estar indicando que hay otros modificadores genéticos y/o ambientales que pueden influir en el espectro de la caracterización clínica de la LD [24].
Bases genéticas y moleculares de la enfermedad de Lafora. Modelos animales.
Genética
La herencia de la LD se produce siguiendo un patrón autosómico recesivo. Tal y como se ha mencionado anteriormente, se han descrito dos genes responsables de esta enfermedad, ambos localizados en el cromosoma 6: el gen EPM2A (locus 6q24) y el gen EPM2B (locus 6p22). Se ha propuesto un tercer gen, el PRDM8 (locus 4q2121), responsable de una forma de LD de inicio temprano [25]. Sin embargo, esta forma de la enfermedad presenta diferencias significativas respecto de la LD típica.
El gen EPM2A se identificó primeramente como responsable de la LD en 1998 [6,7], y codifica para la laforina, una fosfatasa con especificidad dual implicada en la defosforilación del glucógeno y carbohidratos complejos, entre otros procesos [26,27]. El gen EPM2B o NHLRC1, descrito como causante de la LD en 2003 [8,28], codifica la proteína malina, una E3 ubiquitina ligasa implicada en la poliubiquitinación de proteínas relacionadas con el metabolismo del glucógeno y con procesos de autofagia, así como en la degradación de la propia laforina [29].
Se han encontrado casi 100 mutaciones diferentes en ambos genes en 200 familias independientes con LD. Casi la mitad de estas mutaciones eran de sentido erróneo (missense), y una cuarta parte eran deleciones [16]. Sin embargo, la enfermedad causada por mutaciones diferentes en dichos genes presenta manifestaciones clínicas similares.
Tradicionalmente, se asoció la LD causada por mutaciones en el gen EPM2B con una evolución más lenta de la enfermedad que cuando se debe a mutaciones en el gen EPM2A (30). Sin embargo, posteriormente se encontró que las mutaciones en el gen EPM2B no tienen por qué estar asociadas necesariamente con un curso lento de la enfermedad, ya que algunos pacientes con este genotipo tienen sintomatología extremadamente severa [31]. Por tanto, la heterogeneidad clínica de la LD no puede asociarse a la presencia de la mutación causante de la enfermedad en uno u otro gen.
Los cuerpos de Lafora
Histopatológicamente, la LD se caracteriza, en primer lugar, por la acumulación intracelular de cuerpos de Lafora y, en segundo lugar, por la neurodegeneración [32]. Esto ocurre de igual manera tanto si la mutación se encuentra en el gen EPM2A como si se encuentra en el gen EPM2B [12,26,33].
Los cuerpos de Lafora son depósitos de poliglucosanos, esto es, polisacáridos fibrilares de una forma ramificada anormal de glucógeno. Estas estructuras, a diferencia del glucógeno normal, contienen menos ramificaciones glucosídicas, tienen mayores niveles de fosfato (están hiperfosforilados), y son insolubles. Los poliglucosanos son más similares en composición y estructura a la amilopectina de las plantas o el almidón, e incluso a los cuerpos amiláceos del cerebro, que al propio glucógeno [32]. Los cuerpos de Lafora tienen un tamaño entre 3 y 40 micras, contienen un núcleo denso, y se tiñen fuertemente con la tinción de ácido Peryódico Schiff (PAS) [34].
Tradicionalmente, los cuerpos de Lafora se han localizado en las neuronas del cerebro, los hepatocitos de zona periportal, la retina, los miocitos del músculo esquelético y cardíaco, y las células mioepiteliales de glándulas sudoríparas apocrinas y ecrinas [35]. Sin embargo, sólo en el sistema nervioso central suelen causar sintomatología [2].
A nivel neuronal, los cuerpos de Lafora se localizan en el soma o en las dendritas, pero nunca en los axones. Esta diferenciación es importante en tanto que los cuerpos de Lafora son similares morfológicamente a los poliglucosanos que aparecen en otras enfermedades neurológicas con una sintomatología clínica muy diferente, como la deficiencia de la enzima ramificadora del glucógeno (APBD, del inglés enfermedad de acúmulo de poliglucosanos del adulto). En esta última, las inclusiones se localizan en los axones de las neuronas. Se ha sugerido que la localización de los cuerpos de Lafora en las dendritas puede relacionarse con el inicio, la progresión y la intratabilidad de la enfermedad, ya que estas tienen un papel esencial en la excitabilidad neuronal [2]. En un modelo de ratón de la enfermedad sin laforina (Epm2a−/−), se vio que la mayoría de cuerpos de Lafora se encuentran específicamente ubicados en neuronas gabaérgicas de la corteza cerebral [36]. Además, también observaron que en ratones jóvenes que aún no presentaban cuerpos de Lafora, había un menor número de neuronas gabaérgicas, y estas presentaban un deterioro en la señalización de p75NTR y neurotrofinas [36].
Dado que la acumulación de los cuerpos de Lafora parece ser fundamental en la LD, es esencial entender cómo la deficiencia de laforina o malina causa la acumulación de esta forma insoluble de glucógeno.
El glucógeno cerebral en los mamíferos se encuentra en unas concentraciones mucho menores que en otros tejidos. Éste se localiza mayoritariamente en los astrocitos, por lo que, en condiciones normales, las neuronas no lo acumulan [37]. Recientemente, se han encontrado evidencias de que el glucógeno cerebral está implicado no sólo en suministrar energía en casos de emergencia, sino también en otras funciones cerebrales superiores, como el aprendizaje y la memoria [38–40]. A pesar de que las neuronas no almacenan glucógeno en condiciones normales, sí poseen la “maquinaria” necesaria para sintetizar glucógeno, pues expresan la isoforma muscular de la glucógeno sintasa (MGS), aunque ésta se mantiene inactiva [37]. La laforina y la malina se unen formando un complejo que estimula la degradación de la MGS y la PTG a través del sistema ubiquitina-proteasoma, manteniendo así inactivo el mecanismo de síntesis de glucógeno neuronal [37]. De esta manera, laforina y malina inhiben la formación de cuerpos de Lafora y, así, previenen el desarrollo de la LD.
En la enfermedad de Lafora, además, se da un proceso de neurodegeneración progresiva de todas las regiones del sistema nervioso central, aunque en diferentes grados. Además de la alteración en el metabolismo del glucógeno, se cree que la LD conlleva alteraciones en los procesos de autofagia, y que podrían ser estos la base de la neurodegeneración observada en la LD [41].
Una pregunta clave es si los LB son la causa o la consecuencia de la LD, cuya respuesta ha sido fuente de controversia entre los investigadores. Sin embargo, la evidencia se inclina hacia la primera opción. Algunos estudios han encontrado que la reducción de la síntesis de glucógeno previene la formación de cuerpos de Lafora, así como la neurodegeneración y la susceptibilidad a convulsiones epilépticas, por lo que evita el desarrollo de la LD [42–44]. Esta reducción de síntesis de glucógeno se llevó a cabo tanto por reducción parcial de la síntesis de glucógeno mediante la eliminación genética de PTG en ratones Epm2a−/− [42] y Epm2b−/− [44], como por la eliminación total o parcial de glucógeno cerebral mediante la interrupción del gen Gys1 tanto en ratones Epm2a−/− como en ratones Epm2b−/− [41,43]. Por tanto, la regulación a la baja de la síntesis de glucógeno podría ser un tratamiento potencial para la LD. En humanos no se puede eliminar completamente la GS, ya que causaría otro tipo de patologías, pero podría considerarse como una posibilidad terapéutica la reducción parcial de la GS [45].
Laforina y malina
Tras la identificación del gen de la laforina en 1998, la investigación se centró en descubrir cuál era el sustrato de la laforina y cómo la ausencia de esta proteína se relaciona con la LD. Se ha propuesto que la laforina funciona como regulador de la GS [46], así como glucano fosfatasa [27], aunque recientemente se ha descrito que la actividad fosfatasa de la laforina no parece ser un requerimiento para prevenir la enfermedad [47,48]. De hecho, la hiperfosforilación del glucógeno no se relaciona causalmente con la formación de cuerpos de Lafora [47,48]. La lista de proteínas que interactúan con la laforina es cada vez mayor, e incluye los siguientes: malina, subunidades reguladoras de la proteína fosfatasa 1 (PP1), las unidades α y β de la proteína quinasa activada por AMP (AMPK), proteína tau, HIRIP5 y EPM2AIP1 [49].
Existe una fuerte evidencia de la interacción entre la laforina y la malina mediante la formación de un complejo que parece esencial en la prevención de la LD [37,46,50,51]. Sin embargo, la mayoría de los estudios se han realizado in vitro, por lo que resulta difícil replicar biológicamente (in vivo) los hallazgos encontrados.
La malina es una E3 ubiquitina ligasa con capacidad de auto-ubiquitinarse in vitro, así como de ubiquitinar la laforina promoviendo su degradación [29]. Además, la malina parece ser capaz de ubiquitinar la GS [37], la enzima ramificante (AGL) [52] y la PTG [50,51]. La función de la poliubiquitinación por parte del complejo laforina-malina relacionaría los procesos de autofagia con el metabolismo del glucógeno.
Nuevas perspectivas. Más allá del metabolismo del glucógeno
En la última década, se han propuesto nuevas perspectivas dirigidas a clarificar los mecanismos de la LD, en las que el foco de atención es diferente del de la alteración en el metabolismo del glucógeno. Por ejemplo, se ha propuesto que los niveles altos de neuronatina en diferentes regiones cerebrales, así como la señalización aberrante de Ca2+ inducida por esta proteína, o el estrés oxidativo y el estrés del retículo endoplasmático, podrían componer el núcleo patogénico de la LD [53,54]. En relación a estos hallazgos, se ha propuesto recientemente que podría ser la disfunción mitocondrial un factor subyacente a algunos aspectos neuropatológicos de la LD, siendo la laforina y la malina reguladores de la homeostasis mitocondrial [55]. Por otro lado, se ha visto que la homeostasis del transportador del glutamato GLT-1, denominado EAAT2 en humanos y responsable de la recaptación del 90% del glutamato en el sistema nervioso central, se ve comprometida en ratones Epm2a−/− y Epm2b−/−, observándose niveles disminuidos en la membrana plasmática de los astrocitos. Esto podría asociarse a la hiperexcitabilidad y la muerte neuronal que se produce en la LD [56]. Asimismo, se ha visto un incremento del número de astrocitos reactivos y microglía en ratones modelo de la enfermedad de ambos genes, así como un incremento de la expresión de genes que codifican citoquinas y mediadores de la respuesta inflamatoria [57].
Diagnóstico
El diagnóstico de la LD se basa en hallazgos clínicos (crisis epilépticas parciales –occipitales- o generalizadas, mioclonías, agnosia visual, deterioro cognitivo y neurológico, y síntomas neuropsiquiátricos), electrofisiológicos (cambios en el EEG), histológicos (acumulación de cuerpos de Lafora PAS positivos) y genéticos [2]. Debido a las similitudes en las etapas iniciales de la enfermedad, puede que los pacientes al inicio sean diagnosticados con epilepsia mioclónica juvenil (JME, por sus siglas en inglés) o con epilepsia idiopática generalizada (IGE, por sus siglas en inglés). La presencia de una actividad de fondo formada por ritmo lento en EEG, las convulsiones resistentes a los fármacos, las crisis occipitales y el deterioro cognitivo y neurológico en el paciente debe ser un indicio de sospecha para investigar si podría tratarse de LD.
Tradicionalmente se ha utilizado la biopsia de piel de las glándulas sudoríparas como procedimiento principal para realizar el diagnóstico de la enfermedad, pero la interpretación de la muestra conlleva gran riesgo tanto de falsos-negativos, especialmente en pacientes con síntomas en etapas tempranas [58], como de falsos-positivos por la dificultad de distinguir los cuerpos de Lafora de otros polisacáridos normales PAS positivos de las glándulas apocrinas [59].
Por ello se propone la realización de un análisis genético con el fin de confirmar LD. En el caso de que no se confirmase el diagnóstico en un paciente que presenta las características de la LD después de analizar los exones de los genes EPM2A y EPM2B, se debe considerar un análisis genómico en el que se incluya las secuenciaciones completas del genoma [45]. Turnbull et al. [2] proponen realizar el diagnóstico en base a tres niveles diferentes de evidencia, que se recogen representados en el esquema de la Figura 2.
Figura 2.
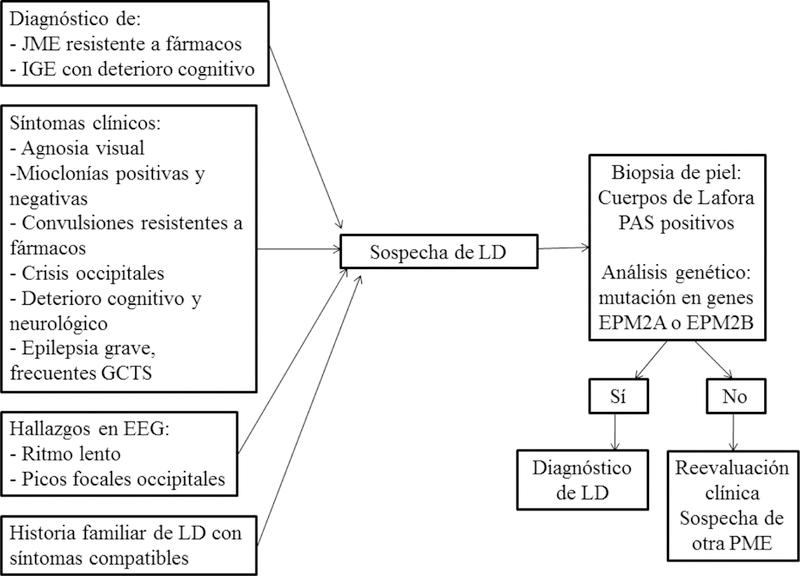
Proceso diagnóstico de LD. Cuando algunas de las condiciones que se muestran en la parte de la izquierda del diagrama sean presentadas por un paciente entre 5 y 20 años de edad, debe sospecharse la posibilidad de que se trate de la enfermedad de Lafora. En ese caso, para confirmar el diagnóstico, debe realizarse una biopsia de piel y, sobre todo, un análisis genético, dadas las limitaciones de la interpretación de los hallazgos histológicos en relación a la presencia o no de cuerpos de Lafora (riesgo de falsos negativos y falsos positivos). En el caso de que el análisis genético sea negativo, habrá que realizar una reevaluación clínica y descartar otros tipos de epilepsia mioclónica progresiva. Si el análisis genético es positivo, se confirma el diagnóstico de LD.
Por otro lado, debe realizarse el diagnóstico diferencial con otras epilepsias mioclónicas progresivas (Tabla I), así como con IGE, JME o epilepsia fotosensible resistente a la farmacoterapia y con empeoramiento progresivo [2].
Tabla I.
Características distintivas y diagnóstico de las PME de tipo hereditario más comunes
| Epilepsia mioclónica progresiva |
Tipo de herencia |
Edad de inicio |
Signos y síntomas indicativos |
Diagnóstico |
|---|---|---|---|---|
| Enfermedad de Unverricht-Lundborg (EPM1) | ARa | 6–15 | Progresión lenta Deterioro cerebeloso leve tardío Demencia ausente o tardía |
Análisis genético: mutación en EPM1 (CSTB) |
| Enfermedad de Lafora (EPM2) | AR | 5–20 | Síntomas visuales | Biopsia de piel: cuerpos de Lafora Análisis genético: mutación en EPM2A o EPM2B (NHLRC1) |
| Epilepsia mioclónica con fibras rojas rasgadas (MERRF) | Maternal | Cualquier edad | Acidosis láctica | Lactato y piruvato en plasma Biopsia muscular: fibras rojas rasgadas Análisis genético: mutación en MT-TK |
| Ceiroidolipofuscinosis neuronales (la forma juvenil es conocida como enfermedad de Batten) | AR o ADb | Variable | Degeneración macular y deterioro visual (excepto en la forma adulta) | Biopsia de piel: depósitos granulares osmiofílicos (GROD) Análisis enzimático: PPT1, TPP1, CTSD Análisis genético: mutación en CLN1/PPT1, CLN2/TPP1, CLN3, CLN4/DNAJC5, CLN5, CLN6, CLN7/MFSD8, CLN8, CLN10/CTSD, CLN11/GRN, CLN12/ATP13A2, CLN13/CTSF, CLN14/KCTD7 |
| Sialidosis | AR | 8–15 | Deterioro cerebeloso gradual | Orina: Sialo-oligosacáridos Análisis enzimático: Neuroaminidasa Análisis genético: mutación en NEU1 |
AR: autosómica recesiva
AD: autosómica dominante
Manejo y tratamiento multidisciplinar en la enfermedad de Lafora
En la actualidad, no se dispone de un tratamiento específico para la LD, por lo que será, en todo caso, paliativo. Sin embargo, se recomienda un abordaje multidisciplinario con el objeto de brindar mayores posibilidades de mejoría sintomática.
Intervención farmacológica
-
-
Fármacos antiepilépticos. La terapia combinada con valproato (el más comúnmente utilizado), fenobarbital, benzodiacepinas, piracetam, levetiracetam y zonisamida son útiles para el tratamiento sintomático [17]. Con el fin de evitar el empeoramiento de las mioclonías, debe evitarse el uso de vigabatrina, carbamazepina, fenitoina, gabapentina, pregabalina, tiagabina, y probablemente lamotrigina, ya que producen un efecto paradójicamente agravante de los síntomas [17]. Sin embargo, con el paso del tiempo los síntomas se vuelven resistentes. Además, este tipo de tratamiento no tiene ningún efecto en la progresión de los síntomas cognitivos y comportamentales [60].
-
-
Perampanel. Se trata de un antagonista no competitivo y selectivo de los receptores ionotrópicos de glutamato de tipo AMPA en las neuronas postsinápticas. Dos estudios de caso muestran efectos beneficiosos en la frecuencia de las convulsiones, mioclonías y crisis tónico-clónicas generalizadas, así como en la disfunción cognitiva y neurológica tras iniciar un tratamiento con perampanel [21,61].
-
-
Estimulación del nervio vago. Se han descrito dos casos de pacientes en los que se ha producido una mejora de los síntomas mediante el implante de un estimulador del nervio vago [62,63]. El mecanismo por el cual esta técnica funciona no está claro, pero debe implicar alteraciones en la modulación de neurotransmisores de la irritabilidad cortical [62].
-
-
Ácido 4-fenilbutírico (4-PBA, por sus siglas en inglés) y metformina. En ratones Epm2b−/− se ha observado una disminución del número de cuerpos de Lafora, una reducción de la neurodegeneración y de la gliosis, y una mejora en el comportamiento motor y la memoria [64], tras el tratamiento con estos fármacos. Además, también se ha descrito una disminución de la susceptibilidad a las convulsiones, produciéndose una reducción tanto en el número como en la duración de las mismas, así como un efecto positivo en la hipersensibilidad al agente epileptogénico pentilenetetrazol (PTZ) [65].
-
-
Selenato sódico. En ratones, se ha visto que mejora algunos déficits, así como la degeneración neuronal y la gliosis [66].
Intervención no farmacológica
En la LD, el apoyo psicosocial es, al menos, tan importante como el tratamiento médico [2]. En este sentido, las organizaciones y asociaciones de pacientes y familiares adquieren un gran protagonismo. Se recomienda comunicar el diagnóstico a los familiares de los pacientes cuando éste sea definitivo, así como llevar a cabo una intervención psicoeducativa mediante la cual se suministre información a los afectados sobre los principales aspectos de la enfermedad de forma individualizada y adecuada al nivel sociocultural de los mismos. Dado que la enfermedad se produce por herencia bilateral de una co-ocurrencia muy poco común de genes anormales, cabe esperar que aparezcan sentimientos no sólo de pérdida y tristeza, sino también de culpa, resentimiento y preocupación por hermanos menores asintomáticos del paciente y/o futuros hermanos que aún no hayan nacido. Cabe señalar que no hay ningún tratamiento recomendado en casos de LD pre-sintomáticos para prevenir o retrasar la aparición y progresión de los síntomas [2].
El tratamiento psicológico es recomendable, sobre todo, en las primeras etapas de la enfermedad [2], aunque puede ser necesario realizar un acompañamiento durante todo el proceso de la misma. Es aconsejable mantener los contactos sociales y la escolaridad durante el mayor tiempo posible mediante adaptaciones curriculares sucesivas, así como realizar medidas adaptativas estructurales en el hogar (evitar escaleras, cercanía al cuarto de baño, accesibilidad para la silla de ruedas, etc.). Por otro lado, el tratamiento fisioterapéutico está indicado para mantener una buena condición muscular y preservar la movilidad durante el mayor tiempo posible. En caso de institucionalización, ésta debe ser especializada, y debe considerarse la posibilidad de mantener al paciente con otros pacientes de epilepsia de edad parecida y desarrollar actividades sociales y educativas.
Conclusiones
La presente revisión ha pretendido aunar los principales hallazgos de la LD, desde los aspectos clínicos y diagnósticos hasta los aspectos terapéuticos, pasando por las bases genéticas y moleculares de la enfermedad. La LD es una enfermedad rara que no puede considerarse un problema general de salud pública, ya que los costes están limitados por el reducido número de personas afectadas. Sin embargo, resulta evidente que tiene graves consecuencias para los pacientes y sus cuidadores. Así pues, y dado que actualmente los tratamientos conocidos son meramente sintomáticos, resulta necesario continuar las líneas de investigación centradas, tanto en descifrar los mecanismos subyacentes a la enfermedad, como en desarrollar nuevas estrategias terapéuticas dirigidas a paliar los síntomas y erradicar y/o prevenir la aparición de la enfermedad.
Ayudas o fuentes de financiación total o parcial
Este trabajo no ha sido financiado por ninguna entidad.
Footnotes
Conflictos de interés
No existen conflictos de interés
Bibliografía
- 1.Lafora G, Glueck B. Contribution to the histopathology of the myoclonic epilepsies. Bull Gov Hosp Insa. 1911;6:1–14. [Google Scholar]
- 2.Turnbull J, Tiberia E, Striano P, Genton P, Carpenter S, Ackerley CA, et al. Lafora disease. Vol. 18, Epileptic Disorders. 2016. p. S38–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Heycop Ten Ham M, De Jager H. Progressive myoclonus epilepsy with Lafora bodies. Epilepsia. 1963;4(1–4):95–115. [DOI] [PubMed] [Google Scholar]
- 4.El Tahry R, de Tourtchaninoff M, Vrielynck P, Van Rijckevorsel K. Lafora disease: psychiatric manifestations, cognitive decline, and visual hallucinations. Vol. 115, Acta Neurologica Belgica. 2015. p. 471–4. [DOI] [PubMed] [Google Scholar]
- 5.Casciato S, Gambardella S, Mascia A, Quarato PP, D’Aniello A, Ackurina Y, et al. Severe and rapidly-progressive Lafora disease associated with NHLRC1 mutation: a case report. Int J Neurosci. 2017;127(12):1150–3. [DOI] [PubMed] [Google Scholar]
- 6.Minassian B a, Lee JR, Herbrick J a, Huizenga J, Soder S, Mungall a J, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20(october):171–4. [DOI] [PubMed] [Google Scholar]
- 7.Serratosa JM, Gómez-Garre P, Gallardo ME, Anta B, Beltrán-Valero De Bernabé D, Lindhout D, et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum Mol Genet. 1999;8(2):345–52. [DOI] [PubMed] [Google Scholar]
- 8.Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35(2):125–7. [DOI] [PubMed] [Google Scholar]
- 9.Harriman DGF, Millar JHD, Stevenson AC. Progressive familial myoclonic epilepsy in three families: Its clinical features and pathological basis: An appendix on the genetic aspects. Brain. 1955;78(3):325–49. [DOI] [PubMed] [Google Scholar]
- 10.Berkovic SF, Andermann F, Carpenter S, Wolfe LS. Progressive Myoclonus Epilepsies: Specific Causes and Diagnosis. N Engl J Med. 1986;315(5):296–305. [DOI] [PubMed] [Google Scholar]
- 11.Ganesh S, Delgado-Escueta AV, Sakamoto T, Avila MR, Machado-Salas J, Hoshii Y, et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet. 2002;11(11):1251–62. [DOI] [PubMed] [Google Scholar]
- 12.DePaoli-Roach AA, Tagliabracci VS, Segvich DM, Meyer CM, Irimia JM, Roach PJ. Genetic depletion of the malin e3 ubiquitin ligase in mice leads to Lafora bodies and the accumulation of insoluble laforin. J Biol Chem. 2010;285(33):25372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valles-Ortega J, Duran J, Garcia-Rocha M, Bosch C, Saez I, Pujadas L, et al. Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol Med. 2011;3(11):667–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Criado O, Aguado C, Gayarre J, Duran-Trio L, Garcia-Cabrero AM, Vernia S, et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum Mol Genet. 2012;21(7):1521–33. [DOI] [PubMed] [Google Scholar]
- 15.Márquez M, Pérez L, Serafín A, Teijeira S, Navarro C, Pumarola M. Characterisation of Lafora-like bodies and other polyglucosan bodies in two aged dogs with neurological disease. Vet J. 2010;183(2):222–5. [DOI] [PubMed] [Google Scholar]
- 16.Singh S, Ganesh S. Lafora progressive myoclonus epilepsy: a Meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Vol. 30, Human Mutation. 2009. p. 715–23. [DOI] [PubMed] [Google Scholar]
- 17.Striano P, Zara F, Turnbull J, Girard J-M, Ackerley CA, Cervasio M, et al. Typical progression of myoclonic epilepsy of the Lafora type: a case report. Nat Clin Pract Neurol. 2008;4(2):106–11. [DOI] [PubMed] [Google Scholar]
- 18.Baykan B, Striano P, Gianotti S, Bebek N, Gennaro E, Gurses C, et al. Late-onset and slow-progressing Lafora disease in four siblings with EPM2B mutation. Epilepsia. 2005;46(10):1695–7. [DOI] [PubMed] [Google Scholar]
- 19.Pichiecchio A, Veggiotti P, Cardinali S, Longaretti F, Poloni GU, Uggetti C. Lafora disease: Spectroscopy study correlated with neuropsychological findings. Eur J Paediatr Neurol. 2008;12(4):342–7. [DOI] [PubMed] [Google Scholar]
- 20.Jennesson M, Milh M, Villeneuve N, Guedj E, Marie PY, Vignal JP, et al. Posterior glucose hypometabolism in Lafora disease: Early and late FDG-PET assessment. Epilepsia. 2010;51(4):708–11. [DOI] [PubMed] [Google Scholar]
- 21.Schorlemmer K, Bauer S, Belke M, Hermsen A, Klein KM, Reif PS, et al. Sustained seizure remission on perampanel in progressive myoclonic epilepsy (Lafora disease). Vol. 1, Epilepsy and Behavior Case Reports. 2013. p. 118–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genton P, Borg M, Vigliano P, Pellissier J-F, Roger J. Semi-late onset and rapidly progressive case of Lafora’s disease with predominant cognitive symptoms. Eur Neurol. 1989;29(6):333–8. [DOI] [PubMed] [Google Scholar]
- 23.Lanoiselée H-M, Genton P, Lesca G, Brault F, De Toffol B. Are c.436G>A mutations less severe forms of Lafora disease? A case reporte. Clin Genet. 2014;89(1):104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomez-Abad C, Afawi Z, Korczyn AD, Misk A, Shalev SA, Spiegel R, et al. Founder effect with variable age at onset in Arab families with Lafora disease and EPM2A mutation. Epilepsia. 2007;48(5):1011–4. [DOI] [PubMed] [Google Scholar]
- 25.Turnbull J, Girard JM, Lohi H, Chan EM, Wang P, Tiberia E, et al. Early-onset Lafora body disease. Brain. 2012;135(9):2684–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tagliabracci VS, Turnbull J, Wang W, Girard J-M, Zhao X, Skurat AV, et al. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci U S A. 2007;104(49):19262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Worby CA, Gentry MS, Dixon JE. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem. 2006;281(41):30412–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan EM, Bulman DE, Paterson AD, Turnbull J, Andermann E, Andermann F, et al. Genetic mapping of a new Lafora progressive myoclonus epilepsy locus (EPM2B) on 6p22. J Med Genet. 2003;40(9):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gentry MS, Worby CA, Dixon JE. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci U S A. 2005;102(24):8501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomez-Abad C, Gomez-Garre P, Gutierrez-Delicado E, Saygi S, Michelucci R, Tassinari CA, et al. Lafora disease due to EPM2B mutations: a clinical and genetic study. Neurology. 2005;64(6):982–6. [DOI] [PubMed] [Google Scholar]
- 31.Brackmann FA, Kiefer A, Agaimy A, Gencik M, Trollmann R. Rapidly progressive phenotype of lafora disease associated with a novel NHLRC1 mutation. Pediatr Neurol. 2011;44(6):475–7. [DOI] [PubMed] [Google Scholar]
- 32.Roach PJ. Glycogen phosphorylation and Lafora disease. Vol. 46, Molecular Aspects of Medicine. 2015. p. 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turnbull J, Wang P, Girard JM, Ruggieri A, Wang TJ, Draginov AG, et al. Glycogen hyperphosphorylation underlies Lafora body formation. Ann Neurol. 2010;68(6):925–33. [DOI] [PubMed] [Google Scholar]
- 34.Carpenter S, Karpati G. Sweat gland duct cells in Lafora disease: diagnosis by skin biopsy. Neurology. 1981;31(12):1564–8. [DOI] [PubMed] [Google Scholar]
- 35.Shirozu M, Hashimoto M, Tomimatsu M, Nakazawa Y, Anraku S, Nagata M. Lafora disease diagnosed by skin biopsy. Kurume Med J. 1985;32(4):311–3. [DOI] [PubMed] [Google Scholar]
- 36.Ortolano S, Vieitez I, Agis-Balboa RC, Spuch C. Loss of GABAergic cortical neurons underlies the neuropathology of Lafora disease. Mol Brain. 2014;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vilchez D, Ros S, Cifuentes D, Pujadas L, Vallès J, García-Fojeda B, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10(11):1407–13. [DOI] [PubMed] [Google Scholar]
- 38.Duran J, Saez I, Gruart A, Guinovart JJ, Delgado-García JM. Impairment in Long-Term Memory Formation and Learning-Dependent Synaptic Plasticity in Mice Lacking Glycogen Synthase in the Brain. J Cereb Blood Flow Metab. 2013;33(4):550–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Obel LF, Müller MS, Walls AB, Sickmann HM, Bak LK, Waagepetersen HS, et al. Brain glycogen - new perspectives on its metabolic function and regulation at the subcellular level. Frontiers in Neuroenergetics. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, et al. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144(5):810–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duran J, Gruart A, García-Rocha M, Delgado-García JM, Guinovart JJ. Glycogen accumulation underlies neurodegeneration and autophagy impairment in lafora disease. Hum Mol Genet. 2014;23(12):3147–56. [DOI] [PubMed] [Google Scholar]
- 42.Turnbull J, DePaoli-Roach AA, Zhao X, Cortez MA, Pencea N, Tiberia E, et al. PTG depletion removes lafora bodies and rescues the fatal epilepsy of lafora disease. PLoS Genet. 2011;7(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pederson BA, Turnbull J, Epp JR, Weaver SA, Zhao X, Pencea N, et al. Inhibiting glycogen synthesis prevents lafora disease in a mouse model. Ann Neurol. 2013;74(2):297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turnbull J, Epp JR, Goldsmith D, Zhao X, Pencea N, Wang P, et al. PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann Neurol. 2014;75(3):442–6. [DOI] [PubMed] [Google Scholar]
- 45.Kecmanović M, Keckarević-Marković M, Keckarević D, Stevanović G, Jović N, Romac S. Genetics of lafora progressive myoclonic epilepsy: Current perspectives. Vol. 9, Application of Clinical Genetics. 2016. p. 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lohi H, Ianzano L, Zhao X-C, Chan EM, Turnbull J, Scherer SW, et al. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum Mol Genet. 2005;14(18):2727–36. [DOI] [PubMed] [Google Scholar]
- 47.Gayarre J, Duran-Trío L, Criado Garcia O, Aguado C, Juana-López L, Crespo I, et al. The phosphatase activity of laforin is dispensable to rescue Epm2a −/− mice from lafora disease. Brain. 2014;137(3):806–18. [DOI] [PubMed] [Google Scholar]
- 48.Nitschke F, Sullivan MA, Wang P, Zhao X, Chown E, Perri A, et al. Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in Lafora disease. Mol Med. 2017;9(7):906–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gentry MS, Romá-Mateo C, Sanz P. Laforin, a protein with many faces: Glucan phosphatase, adapter protein, et al.ii. FEBS J. 2013;280(2):525–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Worby CA, Gentry MS, Dixon JE. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J Biol Chem. 2008;283(7):4069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Solaz-Fuster MC, Gimeno-Alcañiz JV, Ros S, Fernandez-Sanchez ME, Garcia-Fojeda B, Criado Garcia O, et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum Mol Genet. 2008;17(5):667–78. [DOI] [PubMed] [Google Scholar]
- 52.Cheng A, Zhang M, Gentry MS, Worby CA, Dixon JE, Saltiel AR. A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori’s disease. Genes Dev. 2007;21(19):2399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma J, Mukherjee D, Rao SNR, Iyengar S, Shankar SK, Satishchandra P, et al. Neuronatin-mediated aberrant calcium signaling and endoplasmic reticulum stress underlie neuropathology in Lafora disease. J Biol Chem. 2013;288(13):9482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Romá-Mateo C, Aguado C, García-Giménez JL, Ibáñez-Cabellos JS, Seco-Cervera M, Pallardó FV., et al. Increased Oxidative Stress and Impaired Antioxidant Response in Lafora Disease. Mol Neurobiol. 2015;51(3):932–46. [DOI] [PubMed] [Google Scholar]
- 55.Upadhyay M, Agarwal S, Bhadauriya P, Ganesh S. Loss of laforin or malin results in increased Drp1 level and concomitant mitochondrial fragmentation in Lafora disease mouse models. Neurobiol Dis. 2017;100:39–51. [DOI] [PubMed] [Google Scholar]
- 56.Muñoz-Ballester C, Berthier A, Viana R, Sanz P. Homeostasis of the astrocytic glutamate transporter GLT-1 is altered in mouse models of Lafora disease. Biochim Biophys Acta - Mol Basis Dis. 2016;1862(6):1074–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.López-González I, Viana R, Sanz P, Ferrer I. Inflammation in Lafora Disease: Evolution with Disease Progression in Laforin and Malin Knock-out Mouse Models. Mol Neurobiol. 2017;54(5):3119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lesca G, Boutry-Kryza N, De Toffol B, Milh M, Steschenko D, Lemesle-Martin M, et al. Novel mutations in EPM2A and NHLRC1 widen the spectrum of Lafora disease. Epilepsia. 2010;51(9):1691–8. [DOI] [PubMed] [Google Scholar]
- 59.Andrade DM, Ackerley C a, Minett TSC, Teive H a G, Bohlega S, Scherer SW, et al. Skin biopsy in Lafora disease: genotype-phenotype correlations and diagnostic pitfalls. Neurology. 2003;61(11):1611–4. [DOI] [PubMed] [Google Scholar]
- 60.Andrade DM, Turnbull J, Minassian BA. Lafora disease, seizures and sugars. Vol. 26, Acta Myologica. 2007. p. 83–6. [PMC free article] [PubMed] [Google Scholar]
- 61.Dirani M, Nasreddine W, Abdulla F, Beydoun A. Seizure control and improvement of neurological dysfunction in Lafora disease with perampanel. Epilepsy Behav Case Reports. 2014;2:164–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mikati MA, Tabbara F. Managing Lafora body disease with vagal nerve stimulation. Epileptic Disord. 2017;19(1):82–6. [DOI] [PubMed] [Google Scholar]
- 63.Hajnsek S, Petelin Gadze Z, Borovecki F, Nankovic S, Mrak G, Gotovac K, et al. Vagus nerve stimulation in Lafora body disease. Epilepsy Behav Case Reports [Internet]. 2013;1:150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berthier A, Payá M, García-Cabrero AM, Ballester MI, Heredia M, Serratosa JM, et al. Pharmacological Interventions to Ameliorate Neuropathological Symptoms in a Mouse Model of Lafora Disease. Mol Neurobiol. 2016;53(2):1296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sánchez-Elexpuru G, Serratosa JM, Sanz P, Sánchez MP. 4-Phenylbutyric acid and metformin decrease sensitivity to pentylenetetrazol-induced seizures in a malin knockout model of Lafora disease. Neuroreport. 2017;28(5):268–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sánchez-Elexpuru G, Serratosa JM, Sánchez MP. Sodium selenate treatment improves symptoms and seizure susceptibility in a malin-deficient mouse model of Lafora disease. Epilepsia. 2017;58(3):467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiménez Caballero PE. Epilepsia mioclónica progresiva: Descripción de un caso de enfermedad de Lafora con autopsia. Vol. 28, Neurologia. 2013. p. 584–93.22703635 [Google Scholar]
- 68.Kälviäinen R Progressive myoclonus epilepsies. Semin Neurol. 2015;35(3):293–9. [DOI] [PubMed] [Google Scholar]