

Methylmercaptopropionate (MMPA) is an environmentally significant molecule produced by degradation of the abundant marine metabolite dimethylsulfoniopropionate, which plays a significant role in the biogeochemical cycles of both carbon and sulfur, with ramifications for ecosystem productivity and climate homeostasis. Detailed knowledge of the mechanisms for MMPA production and consumption is key to understanding steady-state levels of this compound in the biosphere. Unfortunately, the biochemistry required for MMPA catabolism under anoxic conditions is poorly characterized. The data reported here validate the suggestion that the MtpA protein catalyzes the first step in the methanogenic catabolism of MMPA. However, the enzyme does not catalyze a proposed second step required to produce the key intermediate, methyl coenzyme M. Therefore, the additional enzymes required for methanogenic MMPA catabolism await discovery.
KEYWORDS: Methanosarcina, methylmercaptopropionate, methylsulfide, methyltransferase
ABSTRACT
Methanogenesis from methylated substrates is initiated by substrate-specific methyltransferases that generate the central metabolic intermediate methyl-coenzyme M. This reaction involves a methyl-corrinoid protein intermediate and one or two cognate methyltransferases. Based on genetic data, the Methanosarcina acetivorans MtpC (corrinoid protein) and MtpA (methyltransferase) proteins were suggested to catalyze the methylmercaptopropionate (MMPA):coenzyme M (CoM) methyl transfer reaction without a second methyltransferase. To test this, MtpA was purified after overexpression in its native host and characterized biochemically. MtpA catalyzes a robust methyl transfer reaction using free methylcob(III)alamin as the donor and mercaptopropionate (MPA) as the acceptor, with kcat of 0.315 s−1 and apparent Km for MPA of 12 μM. CoM did not serve as a methyl acceptor; thus, a second unidentified methyltransferase is required to catalyze the full MMPA:CoM methyl transfer reaction. The physiologically relevant methylation of cob(I)alamin with MMPA, which is thermodynamically unfavorable, was also demonstrated, but only at high substrate concentrations. Methylation of cob(I)alamin with methanol, dimethylsulfide, dimethylamine, and methyl-CoM was not observed, even at high substrate concentrations. Although the corrinoid protein MtpC was poorly expressed alone, a stable MtpA/MtpC complex was obtained when both proteins were coexpressed. Biochemical characterization of this complex was not feasible, because the corrinoid cofactor of this complex was in the inactive Co(II) state and was not reactivated by incubation with strong reductants. The MtsF protein, composed of both corrinoid and methyltransferase domains, copurifies with the MtpA/MtpC, suggesting that it may be involved in MMPA metabolism.
IMPORTANCE Methylmercaptopropionate (MMPA) is an environmentally significant molecule produced by degradation of the abundant marine metabolite dimethylsulfoniopropionate, which plays a significant role in the biogeochemical cycles of both carbon and sulfur, with ramifications for ecosystem productivity and climate homeostasis. Detailed knowledge of the mechanisms for MMPA production and consumption is key to understanding steady-state levels of this compound in the biosphere. Unfortunately, the biochemistry required for MMPA catabolism under anoxic conditions is poorly characterized. The data reported here validate the suggestion that the MtpA protein catalyzes the first step in the methanogenic catabolism of MMPA. However, the enzyme does not catalyze a proposed second step required to produce the key intermediate, methyl coenzyme M. Therefore, the additional enzymes required for methanogenic MMPA catabolism await discovery.
INTRODUCTION
Methanosarcina species comprise a versatile group of methane-producing archaea capable of growth on a variety of methylated one-carbon (C1) compounds, including methanol, methylamines, and methylsulfides. The catabolism of these C1 substrates is initiated by the transfer of the methyl moiety to coenzyme M ([CoM] mercaptoethanesulfonate), generating methyl-CoM. This central metabolic intermediate is then disproportionated to produce carbon dioxide and methane or reduced directly to methane using hydrogen as an electron donor (1, 2). Multiple mechanistically distinct types of C1-dependent CoM-methylating enzyme complexes are known in methanogenic archaea.
The activation of methanol or methylamines is mediated by a 3-component system, which produces methyl-CoM via two distinct methyl transfer reactions designated methyltransferase I (MT1) and methyltransferase II (MT2). MT1 enzymes are heterodimeric complexes that transfer the methyl moiety from the substrate to the cob(I)alamin cofactor of their corrinoid-binding subunit, generating a protein-bound methyl-cob(III)alamin intermediate. A distinct MT2 protein then methylates CoM using this protein-bound intermediate as a methyl donor (3–7). The corrinoid-binding subunits of MT1 enzymes comprise a homologous family of proteins related to B12-dependent methionine synthases, whereas the substrate-specific methyltransferase subunits are unrelated to each other (8). In contrast, MT2 methyltransferases comprise a family of homologous proteins with relatively broad substrate specificity (8, 9).
Methanosarcina barkeri is known to initiate catabolism of dimethylsulfide (DMS) or methylmercaptopropionate (MMPA) via a 2-component system in which a bifunctional protein homologous to MT2 enzymes, designated MtsA, catalyzes both half-reactions via a ping-pong mechanism (10, 11). In the “MT1” reaction, the corrinoid-binding protein MtsB is methylated by MtsA using a methylsulfide donor. In the “MT2” reaction, MtsA methylates CoM using methyl-MtsB as the C1 donor. The chemical similarity of the substrate (DMS or MMPA) and product (methyl-CoM), both of which contain thioether linkages, provides a satisfying explanation for the ability of the MtsA to catalyze both half-reactions (11). This is in stark contrast to the activation of methylamines or methanol by 3-component systems, in which the MT1 and MT2 half-reactions are chemically distinct.
A third type of activation system is found in Methanosarcina acetivorans, where genes encoding a family of proteins (MtsD, MtsF, and MtsH) comprised of a corrinoid-binding domain fused to an MT2 domain is required for use of methanethiol (MeSH) and DMS (9, 12). Based on the precedent of the M. barkeri MtsA/MtsB system and the chemical similarity of the methyl-thiol substrates, it has been suggested that these proteins catalyze a methylthiol:CoM methyltransferase reaction analogous to the 2-component reaction catalyzed by MtsA/MtsB (9, 12). However, in this case, a single polypeptide carries both the methyltransferase and corrinoid-binding domains. Although purified MtsF (also known as CmtA), is capable of methyl transfer from DMS to the corrinoid cofactor, in vitro assays suggest that methyl-tetrahydromethanopterin is a superior substrate. This observation led to the idea that MstF acts to bypass the membrane-bound ion motive force-dependent methyl-tetrahydromethanopterin:CoM methyltransferase encoded by the mtrA-H operon (13). Significantly, the genetic and biochemical observations are not mutually exclusive, and it has been suggested that this family of proteins may serve to channel C1 units from methylsulfides into both the oxidative and reductive branches (i.e., tetrahydromethanopterin and CoM, respectively) of the methylotrophic pathway for methanogenesis (9).
Recently, we showed that growth of M. acetivorans using MMPA requires two genes, mtpC and mtpA, which encode members of the MT1 corrinoid-binding subunit and MT2 methyltransferase families, respectively (9). Based on these genetic data, we predicted that MtpA and MtpC would comprise a two-component methylsulfide:CoM methyltransferase similar to MtsA/MtsB (9). Here, we explicitly test this hypothesis through biochemical characterization of MtpA purified from the native host. Contrary to expectations, our data support the conclusion that MtpA is a substrate-specific MMPA:cob(I)alamin MT1 methyltransferase. The copurification of the MtsF protein with an MtpA/MtpC complex suggests that it may be involved in MMPA metabolism, perhaps functioning as a switch that allows methyl transfer to both CoM and tetrahydromethanopterin.
RESULTS
Purification of MtpA.
MtpA was purified from the native host after expression from plasmids harboring an affinity-tagged allele of mtpA. When the tagged allele was expressed in an mtpA deletion mutant (WWM903) (Table 1), growth on MMPA was similar to that of the wild-type strain, showing that the tagged MtpA protein is fully functional in vivo. When MtpA was affinity purified from this host under strictly anoxic conditions, several additional proteins coeluted with the tagged protein (see Fig. S1 in the supplemental material). Mass spectrometric analysis identified two of these proteins as MtpC and MtsF. To eliminate the possibility that these coeluting proteins interfere with downstream assays, we expressed the tagged protein in a ΔmtsD ΔmtsF ΔmtsH ΔmtpCAP host (WWM998) (Table 1), which allowed isolation of MtpA that was free of additional proteins based on visual inspection of SDS-PAGE gels (see Fig. S2).
TABLE 1.
Strains used in this study
| Strain | Genotype or description | Purpose | Source |
|---|---|---|---|
| WWM902 | ΔmtpC Δhpt::pFH018 (mtpC C-terminal strep tag) | Overexpression of MtpC in ΔmtpC background | This study |
| WWM903 | ΔmtpA Δhpt::pFH020 (mtpA C-terminal strep tag) | Overexpression of MtpA in ΔmtpA background | This study |
| WWM973 | ΔmtsD ΔmtsF ΔmtsH ΔmtpCAP/pFH036 | Overexpression of MtpC and MtpA in ΔmtsD ΔmtsF ΔmtsH background | This study |
| WWM997 | ΔmtsD ΔmtsF ΔmtsH ΔmtpCAP/pFH041 | Overexpression of MtpC in ΔmtsD ΔmtsF ΔmtsH background | This study |
| WWM998 | ΔmtsD ΔmtsF ΔmtsH ΔmtpCAP/pFH042 | Overexpression of MtpA in ΔmtsD ΔmtsF ΔmtsH background | This study |
MtpA is a specific MMPA:cob(I)alamin methyltransferase.
The methyltransferase activity of purified MtpA was assessed using a spectrophotometric assay to monitor the methylation state of corrinoid cofactors and coupled gas chromatography-mass spectrometry (GC-MS) to measure the products and reactants. MtpA catalyzed a rapid methyl transfer reaction using free methylcob(III)alamin as the donor and mercaptopropionate (MPA) as the acceptor (Fig. 1). Methyltransferase activity was proportional to the protein concentration, while heat-denatured MtpA showed no detectable activity. The stoichiometry of MMPA production and methylcob(III)lamin and MPA consumption was 1:1:1. In the presence of 150 μM methylcob(III)alamin, MtpA displayed an apparent Km for MPA of 12.2 ± 1.3 μM, a Vmax of 491 ± 12 nmol · min−1 · mg−1 MtpA (kcat = 0.315 s−1), and kcat/Km of 2.6 × 104 M−1 · s−1. Significantly, CoM did not serve as a methyl acceptor under identical conditions (Fig. 2). Thus, MtpA does not catalyze an MT2-like reaction.
FIG 1.

MtpA-catalyzed methylation of MPA using methylcob(III)alamin. (Top left) Demethylation of methylcob(III)alamin monitored via UV-visible spectroscopy. The reaction catalyzed is shown in the inset. Spectra were collected at 30-s intervals from 0 min to 8 min. The diagnostic absorbance changes at 388 nm and 540 nm (arrow) and isosbestic points at 430 nm and 578 nm (arrowheads) demonstrate the transition from methyl-cob(III)alamin to cob(I)alamin. (Top right) Quantification of methylcob(III)alamin, MPA, and MMPA during the MtpA-catalyzed reaction. Methylcob(III)alamin was quantified by UV-visible spectroscopy. MPA and MMPA were measured by GC-MS. (Bottom left) Michaelis-Menten kinetics of MtpA. (Bottom right) Methyltransferase reaction is proportional to MtpA concentration. Rates were determined with 150 μM methylcob(III)alamin, 2.7 to 13.5 μg MtpA, 100 mM ZnCl2 and 5 mM Ti(III) citrate.
FIG 2.
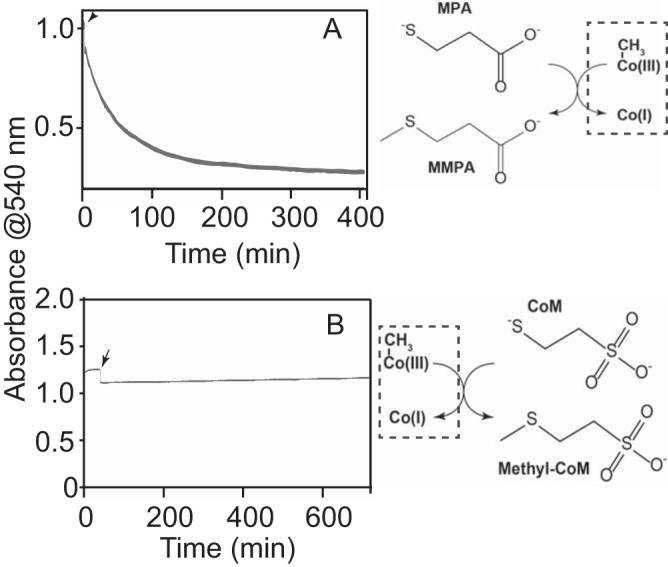
MtpA does not catalyze methylcob(III)alamin:CoM methyl transfer reaction. (A) MtpA-catalyzed methylcob(III)alamin:MPA methyltransferase activity monitored by UV-visible spectroscopy. The reaction being monitored is shown to the right of the plot. (B) A similar assay attempting to show MtpA-catalyzed methylcob(III)alamin:CoM methyltransferase activity. No activity was observed within the 12-h assay. Reactions were initiated by addition of MPA (A) or CoM (B) at 150 μM as indicated by the arrows.
Methylation of free cob(I)alamin by MMPA, which is the physiologically relevant direction, was observed only in the presence of very high levels of MMPA (Fig. 3). It should be noted that the requirement for high substrate concentrations was expected due to the unfavorable thermodynamics of similar reactions (ΔGo′ E 20 kJ/mol [14]). With 40 mM substrate, the initial rate of MMPA:cob(I)alamin methyl transfer was 32.7 ± 3.8 nmol · min−1 · mg−1 (n = 3). Methylation of cob(I)alamin was not observed using methanol, dimethylsulfide, or dimethylamine as methyl donors, despite prolonged incubations with high substrate concentrations (Fig. 4).
FIG 3.

MtpA-catalyzed methylation of cob(I)alamin methylation using MMPA. Methylation of cob(I)alamin monitored via UV-visible spectroscopy. The reaction catalyzed is shown. Spectra were collected at 10-min intervals from 0 min to 90 min. Arrows indicate the decrease/increase in absorbance over time.
FIG 4.

MtpA does not catalyze cob(I)alamin methylation using methyl-CoM, DMS, MeOH, or DMA. Methylation of cob(I)alamin monitored via UV-visible spectroscopy using a variety of potential methyl donors. Formation of methyl-cob(III)alamin would result in an increase in absorbance at 540 nm, as seen in Fig. 3, as opposed to the slight loss of absorbance, presumably caused by photobleaching, seen in these assays (indicated by arrows). (A) Spectra gathered of cob(I)alamin with 40 mM CH3-CoM at 30-min intervals for 10 h after the start of reaction. (B) Spectra gathered of cob(I)alamin with 40 mM DMS at 2-min intervals from 0 min to 40 min after the start of reaction. (C) Spectra gathered of cob(I)alamin with 40 mM MeOH at 2-min intervals from 0 min to 40 min after the start of reaction. (D) Spectra gathered of cob(I)alamin with 40 mM DMA at 2-min intervals from 0 min to 40 min after the start of reaction.
Purification and characterization of MtpC and an MtpC/MtpA complex.
Affinity-tagged alleles of MtpC were expressed in M. acetivorans mutants using plasmids similar to those used for MtpA. A ΔmtpC mutant carrying a plasmid expressing C-terminally tagged MtpC (WWM902) (Table 1) grew well in medium with MMPA as the sole growth substrate. Thus, tagged MtpC is also fully functional in vivo. Although the yield of MtpC was very low, we were able to isolate small amounts of affinity-tagged protein from this host. MtpA and MtsF coeluted with tagged MtpC purified from the ΔmtpC mutant (see Fig. S3). In contrast to our results with MtpA, we were unable to isolate any MtpC after expression in the ΔmtsD ΔmtsF ΔmtsH ΔmtpCAP host grown on trimethylamine (TMA) medium, suggesting that this protein is unstable in this strain background. However, when tagged versions of both MtpC and MtpA were expressed in the ΔmtsD ΔmtsF ΔmtsH ΔmtpCAP host, we were able to purify large amounts of both proteins. The resulting affinity-purified protein preparation contained only MtpA and MtpC, which formed a semistable complex, as evidenced by an additional band that appeared on the native PAGE gel compared to that on the SDS-PAGE gel (Fig. 5). Taken together, these data suggest that MtpC is unstable in the absence of MtpA.
FIG 5.
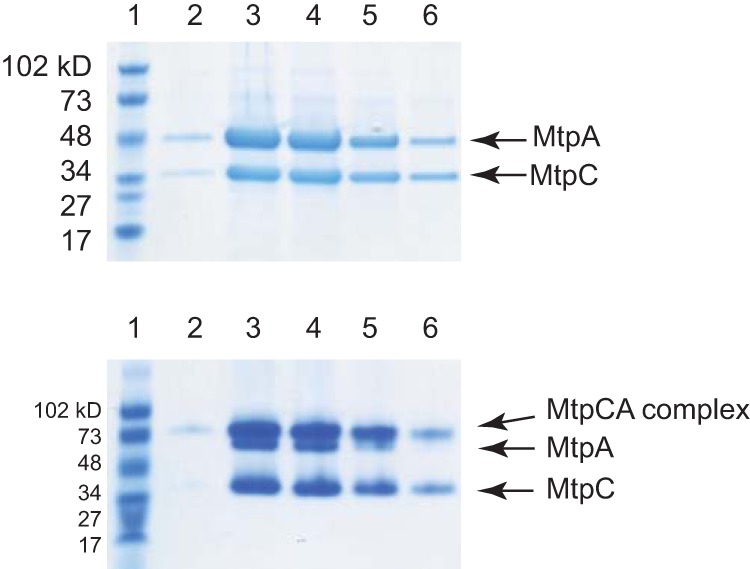
MtpA and MtpC form a complex. Cell-free extract from WWM973 was subjected to affinity purification. The eluted proteins were then separated by electrophoresis and stained with Coomassie blue. (Top) A denaturing SDS-PAGE gel (4% to 20%) was loaded with fractions from different steps of protein purification. Lane 1, protein standards; lanes 2 to 6, elution fractions. (Bottom) The same fractions were loaded on a nondenaturing PAGE gel (4% to 20%). At the pH of running buffer (pH 8.5), both MtpC (pI = 4.04) and MtpA (pI = 4.52) are negatively charged.
The purified MtpA/MtpC complex exhibited the characteristic spectrum of the corrinoid cofactor in the inactive Co(II) state (see Fig. S4) (15). Based on high-resolution mass spectrometry, the corrinoid cofactor in MtpC is 5-hydroxybenzimidazolyl cobamide (B12HBI) (Fig. 6), similar to other characterized Methanosarcina MT1 enzymes (16–18). We attempted to reactivate the MtpC protein by incubation with strong reducing agents using established protocols that work for some, but not all, corrinoid-dependent methyltransferases. The spectrophotometric analysis suggests that the cofactor remained in the Co(II) state throughout the 15-h incubation. Consistent with this observation, we were unable to demonstrate methyl transfer from MMPA to CoM, nor from methyl-CoM to MPA, using a sensitive 1H nuclear magnetic resonance (NMR) assay and the “reactivated” MtpA/MtpC complex (see Fig. S5).
FIG 6.

The cofactor of MtpC is 5-hydroxybenzimidazolyl cobamide. High-resolution matrix-assisted laser desorption ionization–mass spectrometry was used to characterize the corrinoid cofactor purified from the MtpC/MtpA complex. Several masses consistent with 5-hydroxybenzimidazolyl cobamide (shown in the inset) were identified. The predicted exact masses for C60H85CoN13O15P (m/z): 1,317.535 (100.0%), 1,318.539 (64.9%), 1,319.542 (20.7%), 1,318.532 (4.8%), 1,320.545 (3.5%), 1,319.539 (3.1%), 1,319.536 (2.9%), 1,320.543 (2.0%).
DISCUSSION
MMPA is an abundant molecule that plays an important role in the biogeochemical cycling of both carbon and sulfur. This naturally produced compound is largely derived from degradation of dimethylsulfoniopropionate (DMSP), which is produced in huge quantities by a variety of organisms, most notably, marine phytoplankton. Indeed, up to 10% of primary productivity can be directed toward DMSP production in such ecosystems (19). As such, a clear knowledge of the sources and sinks of DMSP is essential for understanding nutrient cycling and ecosystem function on a global scale. The catabolism of DMSP proceeds via two routes. In the first, the molecule is cleaved to release DMS; in the second, it is demethylated to produce MMPA. While the aerobic catabolism of MMPA and DMS has been well studied, our understanding of their fate under anoxic conditions has lagged (reviewed in references 20, to ,22). Nevertheless, a number of microbes, including methanogenic archaea, are known to catabolize both MMPA and DMS. We have been studying one such organism, M. acetivorans, in order to better understand the genes and enzymes required for the process.
Bioinformatic and genetic approaches suggested that MMPA catabolism in M. acetivorans proceeded via a bifunctional MT1/MT2 MMPA:CoM methyltransferase (9); however, the data presented here clearly show that this is incorrect. Because MtpA catalyzes an MMPA:cob(I)alamin methyltransferase reaction but not a methylcob(III)alamin:CoM methyltransferase reaction, the MtpA/MtpC complex must be considered an MMPA-specific MT1 enzyme. The high affinity of MtpA for MPA (apparent Km = 12 μM), coupled with its inability to utilize CoM, supports this conclusion. Indeed, the apparent Km of MtpA for MMPA is 10- to 1,000-fold lower than that of other substrate-specific MT1 methyltransferases: e.g., the Km for dimethylamine of MtbB is ca. 0.45 mM (23), that of MtgB for glycine betaine is 1.96 mM (24), and that of MtaB for methanol is ca. 40 mM (25). The affinity of MtpA for its substrate is also substantially lower than that of the bifunctional M. barkeri MtsA/MtsB complex, which is capable of using a variety of methylsulfide substrates, with apparent Km values of 10 μM for MMPA, 33 mM for DMS, and 11 mM for CoM (10, 11). Interestingly, the bifunctional MtsA/MtsB complex also has a much poorer affinity for CoM (11 mM) than the dedicated MT2 enzymes. For example, the MT2 enzymes MtbA and MtaA have Km values for CoM of 35 μM and or 20 μM, respectively, but very poor affinity for other sulfides such as MPA (Km of ca. 10 mM). Thus, it seems that the bifunctional complex has sacrificed affinity for broad substrate tolerance, whereas MtpA has retained high specificity for MMPA at the expense of being able to utilize CoM.
The lack of MT2 activity in purified MtpA implies that an as yet unidentified MT2 is required for MMPA metabolism. The observation that MtsF copurifies with both MtpA and MtpC raises the possibility that MtsF is this unidentified MT2. This idea is at least partially supported by genetic analysis of mts mutants, although the in vivo situation is considerably more complex. Thus, while there was no significant phenotype for single or double mts mutants, a triple mutant lacking mtsF and its two paralogs, mtsD and mtsH, shows significant defects in both growth rate and yield when grown in MMPA medium (9). These data show that any of the mts paralogs suffices for wild-type growth on MMPA. Moreover, the fact that the triple mutant retains some growth on MMPA shows that other MT2 enzymes are capable of fulfilling this function, albeit with lower efficiency than the Mts proteins. In this regard, many MT2 proteins display relaxed substrate specificity. For example, the “methanol-specific” MT2 protein MtaA can serve as the trimethylamine MT2 enzyme both in vitro and in vivo (26, 27). Similarly, MtsF was previously shown to be involved in the metabolism of both DMS (12) and MeSH (9), as well as the transfer of methyl groups from methyl-tetrahydromethanopterin (H4MPT) to CoM (13). Because the M. acetivorans C2A genome encodes thirteen putative MT2 proteins (8), the comprehensive genetic experiments that will be needed to establish the in vivo roles of each MT2 isoenyzme will be challenging.
In the near term, biochemical approaches may be more fruitful for identifying the MT2 protein(s) needed to transfer the methyl group of MMPA to CoM. The goal would be to reconstitute the full MT1/MT2 MMPA:CoM methyltransferase reaction by adding purified MT2 candidates to an active MtpA/MtpC complex. However, this approach will require development of a protocol to convert the inactive Co(II) form of MtaC into the active Co(I) form, which has eluded us to date. Although many corrinoid proteins can be chemically activated using Ti(III), a very-low-potential electron donor (25, 28), this was not possible for MtpC. Other corrinoid proteins, such as those involved in methylamine catabolism require both Ti(III) and a redox mediator, such as methyl viologen (5–7); however, this treatment also failed to generate the Co(I) form of MtpC. Thus, a different approach will be required to obtain active MtpC. A promising alternative would entail identification of a reductive activation protein, similar to the Methanosarcina RamA protein that can be used by to activate the methylamine corrinoid protein (7). We note that a homolog of RamA, denoted RamS, is highly induced by growth on MMPA (9), suggesting a potential path forward for these experiments.
While the data reported here clearly establish the biochemical function of MtpA in methylsulfide metabolism, numerous questions remain. Furthermore, it is becoming clear that the scale and diversity of methylsulfide-dependent methanogenesis have yet to be revealed. Accordingly, a widely distributed and novel order of hydrogen-dependent methylsulfide-metabolizing methanogens (“Candidatus Methanofastidiosa”) was only recently discovered (29). These intriguing observations suggest that investigations of methanogenic methylsulfide metabolism will remain fruitful for years to come.
MATERIALS AND METHODS
Strains, media, and growth conditions.
M. acetivorans strains were grown in single-cell morphology at 37°C using high-salt (HS) medium containing either 50 mM trimethylamine (TMA) or 20 mM MMPA (30). Cultures used to identify the corrinoid cofactor were grown is HS medium without added vitamins. Growth on medium solidified with 1.5% agar was as described previously (31). Puromycin was added from sterile anaerobic stocks at a final concentration of 2 μg/ml for selection of Methanosarcina strains carrying the puromycin transacetylase gene (pac) (32).
Chemicals.
Ti(III) citrate was prepared anaerobically from TiCl3 as described previously (33). Methylthiopropionate (MMPA; Tokyo Chemical Industry Co., Japan), and mercaptopropionic acid (MPA; Sigma, St. Louis, MO) were added from sterile anaerobic stocks to a final concentration of 20 mM. Methylcob(III)alamin (Sigma, St. Louis, MO) and hydroxocobalamin (Sigma, St. Louis, MO) were prepared as sterile anaerobic stocks at a final concentration of 1 mM.
Overexpression of proteins in M. acetivorans C2A.
Plasmids used for overexpression of MtpC or MtpA were constructed as described in Tables S1 and S2 in the supplemental material. A C-terminal strep tag was introduced into the MtpC and MtpA coding sequences before cloning into pJK027A to allow affinity purification. The recombinant mtpC or mtpA plasmids were integrated into the chromosome of corresponding deletion mutants as previously reported (9). To simultaneously overexpress MtpA and MtpC in their native host, a PCR product with both strep-tagged mtpC and mtpA was cloned into pJK026A, generating pFH035, which was further retrofitted with a pC2A derivative plasmid pAMG40 via the λ-att sites (34), generating the autonomous replicon pFH036.
Affinity purification of MtpA and MtpC from M. acetivorans C2A.
M. acetivorans strains carrying expression plasmids were grown to late exponential phase in high-salt medium containing either 20 mM MMPA or 50 mM TMA and 2 μg/ml puromycin. Cells were harvested by centrifugation at 5,000 × g for 15 min at 4°C in sealed anoxic bottles and then brought into an anaerobic chamber (4% H2, 96% N2; Coy Laboratory Products, Grass Lake, MI), where all subsequent manipulations were performed. All buffers used were adjusted to pH 7.2 (unless stated otherwise), made anaerobic by boiling under an N2 stream, and brought into the anaerobic chamber in advance. The cells were resuspended in 10 ml NPD buffer (50 mM NaH2PO4, 300 mM NaCl, and 2 mM dithiothreitol, pH 8.0) and then sonicated 2 times each for 30 s. The cell lysate was centrifuged at 15,000 × g at 4°C for 30 min to remove unbroken cells and debris using a microcentrifuge within the anaerobic chamber. One milliliter of Strep-Tactin Superflow Plus (Qiagen) resin was loaded onto Poly-Prep chromatography columns (Bio-Rad, Hercules, CA) and equilibrated with 20 ml anaerobic NPD buffer at 0.5 ml/min. The cell lysate was then loaded onto the column and eluted with 30 ml NPD buffer. The protein was eluted in five fractions with 0.5 ml elution buffer (50 mM NaH2PO4, 300 mM NaCl, 2 mM dithiothreitol, 2.5 mM desthiobiotin, pH 8.0). The protein concentration in eluted fractions was measured using a NanoDrop 2000 (Thermo Scientific). The purity of the resultant proteins was judged by a visual inspection of samples subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or native polyacrylamide gel electrophoresis (native PAGE) using precast gels (Bio-Rad, Hercules, CA).
Enzyme assays.
All assays were performed inside an anaerobic chamber at room temperature under an atmosphere of 96% N2 and 4% H2. Data were collected using a fiber optic lead connected to a Cary 50 UV-visible spectrophotometer. Purified proteins were used immediately after purification and never removed from the anaerobic chamber.
For characterizing the MPA methylation reaction, 1.0-ml reaction mixtures contained 50 mM MOPS (morpholinepropanesulfonic acid)-KOH (pH 7.2), 100 μM ZnCl2, 150 μM methylcob(III)alamin, 5 mM Ti(III) citrate, and 5 μg MtpA. The reaction was started by adding 150 μM MPA to the mixtures in a cuvette (10-mm light path). The transition from methylcob(III)alamin to cob(I)alamin was monitored by following the decrease in absorbance at 540 nm (ε = 4.4 mM−1 · cm −1) (15). The presence of two clear isosbestic points at 430 nm and 578 nm indicated that cob(II)alamin did not accumulate during the reaction. The apparent kinetic parameters of MtpA were investigated by varying the MPA concentration from 7.5 to 150 μM. Results were obtained in triplicates with independently purified batches of MtpA and analyzed using Origin (OriginLab) using nonlinear Michaelis-Menten parameters.
For methyltransferase assays using various methyl donors and cob(I)alamin as the methyl acceptor, 500-μl reaction mixtures were set up in a 10-mm length quartz cuvette (volume = 700 μl). The mixtures contained 50 mM MOPS-KOH (pH 7.2), 100 μM ZnCl2, 0.2 mM hydroxocob(III)alamin, 5 mM Ti(III) citrate, 40 mM methylated substrates (MMPA, DMS, methanol [MeOH], dimethylamine [DMA] or methyl-CoM), and 20 μg MtpA. Reactions were initiated by adding respective substrates once the cob(III)alamin was fully reduced to cob(I)alamin, as judged by the UV-visible spectrum. The enzyme-catalyzed transition from cob(I)alamin to methylcob(III)alamin was monitored by following the increase in absorbance at 540 nm. The detection limit of methylcob(III)alamin at 540 nm is 5 μM.
For 1H-NMR analysis of MtpC/MtpA activity, 1-ml reaction mixtures were set up in a test tube. The mixture contained 50 mM sodium phosphate (pH 7.2), 100 μM ZnCl2, 5 mM Ti(III) citrate, 0.42 mg MtpC/MtpA, 40 mM substrates (MMPA and CoM, or methyl-CoM and MPA). Negative controls used the same amounts of heat-killed proteins. The reaction mixtures were incubated at room temperature for 16 h. Samples were passed through 10-K Amicon Ultra filters before 1H-NMR analysis.
Identification of proteins by mass spectroscopy.
The protein samples were digested by trypsin as follows. Briefly, trypsin (proteomics grade; G-Biosciences) was dissolved in 25 mM ammonium bicarbonate and added at a ratio of 1:20 (trypsin/protein) and processed in a CEM microwave reactor at 55°C for 30 min. Digested peptides were lyophilized and separated using a Dionex Ultimate 3000 RSLCnano connected directly to a Thermo LTQ-Velos-ETD pro mass spectrometer. The column used was an Acclaim 300 C18 nano-column, 75 μm by 150 mm (particle size, 3 Å), with an Acclaim Guard column. The flow rate was 300 nl/ml. A typical sample load was 1 to 2 μg digested peptides, and a gradient from 100% A (water plus 0.1% formic acid) to 60% B (acetonitrile plus 0.1% formic acid) was used. Data collection was conducted using the “Big Five” protocol (Thermo, San Jose, CA), tandem mass spectrometry (MS/MS) data were collected using collision induced dissociation (CID). Raw data were collected by Xcalibur (Thermo, San Jose, CA) and processed using an in-house Mascot Distiller and Mascot Server (Cambridge, UK). Resultant peptides generated were searched against NCBI-NR or UniProt protein databases. Proteins identified that were below the ion score with extensive homology (P > 0.05, where P is the probability that the observed match is a random event) were discarded. For quantitation, the exponentially modified protein abundance index (emPAI) was obtained from Mascot Analysis (35).
Analysis of cobalamin by mass spectroscopy.
The corrinoid content of protein samples was determined as described previously (36). Briefly, a 0.5-ml protein sample was mixed with 0.7 ml of 95% ethanol and heated to 80°C for 10 min. The mixture was then placed in an ethanol-dry ice bath for 3 min before centrifugation at 10, 000 × g for 10 min. The supernatant was dried under vacuum using a rotary evaporator and resuspended in 20 μl of double-distilled water before subjecting to analysis using a Bruker UltrafleXtreme matrix-assisted laser desorption ionization (MALDI) system. The measurements were made using the reflection mode, and the positive ion was recorded. Samples were prepared by mixing 1 μl of samples and 10 μl of the dihydroxybenzoic acid (DHB) matrix, made from 20 mg of DHB with 1 ml of 50% acetone.
Determination of metabolites.
MMPA and MPA were converted to esters using ethanol before being subjected to GC-MS analysis. Briefly, 100-μl standard samples (10 μl pentanoic acid was added as an internal control) were mixed with a 40-μl pyridine-ethanol (4:1) mixture and 60 μl ethylchloroformate and incubated at ambient temperature for 45 min inside a fume hood. The bottom phase was extracted into 150 μl dichloromethane after the addition of 50 mg NaCl. The samples were dried under an N2 stream before being dissolved in 100 μl dichloromethane. Five microliters of the resultant samples was injected for GC-MS analysis. Standard samples were prepared on the same day experimental samples were collected and were run on the same equipment to generate standard curves. 1H-NMR spectra were recorded on an Agilent Technologies DD2 600-MHz spectrometer with D2O as the lock solvent (minimum 10% [vol/vol]) at the Carl R. Woese Institute for Genomic Biology.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dipti Nayak for constructive suggestions to this project, Peter Yau from the Roy J. Carver Biotechnology Center for protein identification, Alexander Vladimirovich Ulanov from the metabolomics center for help with quantification of MMPA and MPA, and Haijun Yao from the mass spectrometry laboratory for technical support with MALDI analysis of cobalamin structure.
NMR spectra were recorded on an instrument purchased with support from NIH grant S10 RR028833. This work was supported by National Science Foundation grant MCB-1022462 to W.W.M.
We declare no conflicts of interest.
H.F. performed all protein purification and enzyme assay experiments, analyzed results, and wrote the draft of the manuscript. M.N.G. performed 1H-NMR analysis. W.W.M. supervised the overall experimental design, provided critical suggestions, and revised the manuscript. All authors approved the final version of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00130-19.
REFERENCES
- 1.Keltjens JT, Vogels GD. 1993. Conversion of methanol and methylamines to methane and carbon dioxide, p 253–303. In Ferry JG. (ed), Methanogenesis: ecology, physiology, biochemistry & genetics. Chapman & Hall, New York, NY. [Google Scholar]
- 2.Costa KC, Leigh JA. 2014. Metabolic versatility in methanogens. Curr Opin Biotechnol 29:70–75. doi: 10.1016/j.copbio.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 3.van der Meijden P, Jansen L, van der Drift C, Vogels GD. 1983. Involvement of corrinoids in the methylation of coenzyme-M (2-mercaptoethanesulfonic acid) by methanol and enzymes from Methanosarcina barkeri. FEMS Microbiol Lett 19:247–251. doi: 10.1111/j.1574-6968.1983.tb00551.x. [DOI] [Google Scholar]
- 4.van der Meijden P, Heythuysen HJ, Pouwels A, Houwen F, van der Drift C, Vogels GD. 1983. Methyltransferases involved in methanol conversion by Methanosarcina barkeri. Arch Microbiol 134:238–242. doi: 10.1007/BF00407765. [DOI] [PubMed] [Google Scholar]
- 5.Ferguson DJ Jr, Gorlatova N, Grahame DA, Krzycki JA. 2000. Reconstitution of dimethylamine:coenzyme M methyl transfer with a discrete corrinoid protein and two methyltransferases purified from Methanosarcina barkeri. J Biol Chem 275:29053–29060. doi: 10.1074/jbc.M910218199. [DOI] [PubMed] [Google Scholar]
- 6.Ferguson DJ Jr, Krzycki JA. 1997. Reconstitution of trimethylamine-dependent coenzyme M methylation with the trimethylamine corrinoid protein and the isozymes of methyltransferase II from Methanosarcina barkeri. J Bacteriol 179:846–852. doi: 10.1128/jb.179.3.846-852.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burke SA, Krzycki JA. 1997. Reconstitution of monomethylamine:coenzyme M methyl transfer with a corrinoid protein and two methyltransferases purified from Methanosarcina barkeri. J Biol Chem 272:16570–16577. doi: 10.1074/jbc.272.26.16570. [DOI] [PubMed] [Google Scholar]
- 8.Galagan JE, Nusbaum C, Roy A, Endrizzi MG, Macdonald P, FitzHugh W, Calvo S, Engels R, Smirnov S, Atnoor D, Brown A, Allen N, Naylor J, Stange-Thomann N, DeArellano K, Johnson R, Linton L, McEwan P, McKernan K, Talamas J, Tirrell A, Ye W, Zimmer A, Barber RD, Cann I, Graham DE, Grahame DA, Guss AM, Hedderich R, Ingram-Smith C, Kuettner HC, Krzycki JA, Leigh JA, Li W, Liu J, Mukhopadhyay B, Reeve JN, Smith K, Springer TA, Umayam LA, White O, White RH, Conway de Macario E, Ferry JG, Jarrell KF, Jing H, Macario AJL, Paulsen I, Pritchett M, Sowers KR, et al. 2002. The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res 12:532–542. doi: 10.1101/gr.223902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu H, Metcalf WW. 2015. Genetic basis for metabolism of methylated sulfur compounds in Methanosarcina species. J Bacteriol 197:1515–1524. doi: 10.1128/JB.02605-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tallant TC, Krzycki JA. 1997. Methylthiol:coenzyme M methyltransferase from Methanosarcina barkeri, an enzyme of methanogenesis from dimethylsulfide and methylmercaptopropionate. J Bacteriol 179:6902–6911. doi: 10.1128/jb.179.22.6902-6911.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tallant TC, Paul L, Krzycki JA. 2001. The MtsA subunit of the methylthiol: coenzyme M methyltransferase of Methanosarcina barkeri catalyses both half-reactions of corrinoid-dependent dimethylsulfide: coenzyme M methyl transfer. J Biol Chem 276:4485–4493. doi: 10.1074/jbc.M007514200. [DOI] [PubMed] [Google Scholar]
- 12.Oelgeschlager E, Rother M. 2009. In vivo role of three fused corrinoid/methyl transfer proteins in Methanosarcina acetivorans. Mol Microbiol 72:1260–1272. doi: 10.1111/j.1365-2958.2009.06723.x. [DOI] [PubMed] [Google Scholar]
- 13.Vepachedu VR, Ferry JG. 2012. Role of the fused corrinoid/methyl transfer protein CmtA during CO-dependent growth of Methanosarcina acetivorans. J Bacteriol 194:4161–4168. doi: 10.1128/JB.00593-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiss DS, Gartner P, Thauer RK. 1994. The energetics and sodium-ion dependence of N5-methyltetrahydromethanopterin:coenzyme M methyltransferase studied with cob(I)alamin as methyl acceptor and methylcob(III)alamin as methyl donor. Eur J Biochem 226:799–809. doi: 10.1111/j.1432-1033.1994.00799.x. [DOI] [PubMed] [Google Scholar]
- 15.Kreft JU, Schink B. 1994. O-Demethylation by the homoacetogenic anaerobe Holophaga foetida studied by a new photometric methylation assay using electrochemically produced cob(I)alamin. Eur J Biochem 226:945–951. doi: 10.1111/j.1432-1033.1994.00945.x. [DOI] [PubMed] [Google Scholar]
- 16.Pol A, van der Drift C, Vogels GD. 1982. Corrinoids from Methanosarcina barkeri: structure of the alpha-ligand. Biochem Biophys Res Commun 108:731–737. doi: 10.1016/0006-291X(82)90890-7. [DOI] [PubMed] [Google Scholar]
- 17.Stupperich E, Kräutler B. 1988. Pseudo vitamin-B12 or 5-hydroxybenzimidazolyl-cobamide are the corrinoids found in methanogenic bacteria. Arch Microbiol 149:268–271. doi: 10.1007/BF00422016. [DOI] [Google Scholar]
- 18.Krautler B, Moll J, Thauer RK. 1987. The corrinoid from Methanobacterium thermoautotrophicum (Marburg strain). Spectroscopic structure-analysis and identification as Co beta-cyano-5'-hydroxybenzimidazolyl-cobamide (factor-Iii). Eur J Biochem 162:275–278. doi: 10.1111/j.1432-1033.1987.tb10596.x. [DOI] [PubMed] [Google Scholar]
- 19.Simo R, Archer SD, Pedros-Alio C, Gilpin L, Stelfox-Widdicombe CE. 2002. Coupled dynamics of dimethylsulfoniopropionate and dimethylsulfide cycling and the microbial food web in surface waters of the North Atlantic. Limnol Oceanogr 47:53–61. doi: 10.4319/lo.2002.47.1.0053. [DOI] [Google Scholar]
- 20.Moran MA, Reisch CR, Kiene RP, Whitman WB. 2012. Genomic insights into bacterial DMSP transformations. Annu Rev Mar Sci 4:523–542. doi: 10.1146/annurev-marine-120710-100827. [DOI] [PubMed] [Google Scholar]
- 21.Yoch DC. 2002. Dimethylsulfoniopropionate: its sources, role in the marine food web, and biological degradation to dimethylsulfide. Appl Environ Microbiol 68:5804–5815. doi: 10.1128/AEM.68.12.5804-5815.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bullock HA, Luo H, Whitman WB. 2017. Evolution of dimethylsulfoniopropionate metabolism in marine phytoplankton and bacteria. Front Microbiol 8:637. doi: 10.3389/fmicb.2017.00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wassenaar RW, Keltjens JT, van der Drift C, Vogels GD. 1998. Purification and characterization of dimethylamine:5-hydroxybenzimidazolyl-cobamide methyltransferase from Methanosarcina barkeri Fusaro. Eur J Biochem 253:692–697. doi: 10.1046/j.1432-1327.1998.2530692.x. [DOI] [PubMed] [Google Scholar]
- 24.Ticak T, Kountz DJ, Girosky KE, Krzycki JA, Ferguson DJ Jr. 2014. A nonpyrrolysine member of the widely distributed trimethylamine methyltransferase family is a glycine betaine methyltransferase. Proc Natl Acad Sci U S A 111:E4668–E4676. doi: 10.1073/pnas.1409642111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sauer K, Harms U, Thauer RK. 1997. Methanol:coenzyme M methyltransferase from Methanosarcina barkeri. Purification, properties and encoding genes of the corrinoid protein MT1. Eur J Biochem 243:670–677. doi: 10.1111/j.1432-1033.1997.t01-1-00670.x. [DOI] [PubMed] [Google Scholar]
- 26.Bose A, Pritchett MA, Metcalf WW. 2008. Genetic analysis of the methanol- and methylamine-specific methyltransferase 2 genes of Methanosarcina acetivorans C2A. J Bacteriol 190:4017–4026. doi: 10.1128/JB.00117-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferguson DJ Jr, Krzycki JA, Grahame DA. 1996. Specific roles of methylcobamide:coenzyme M methyltransferase isozymes in metabolism of methanol and methylamines in Methanosarcina barkeri. J Biol Chem 271:5189–5194. doi: 10.1074/jbc.271.9.5189. [DOI] [PubMed] [Google Scholar]
- 28.Naidu D, Ragsdale SW. 2001. Characterization of a three-component vanillate O-demethylase from Moorella thermoacetica. J Bacteriol 183:3276–3281. doi: 10.1128/JB.183.11.3276-3281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nobu MK, Narihiro T, Kuroda K, Mei R, Liu WT. 2016. Chasing the elusive Euryarchaeota class WSA2: genomes reveal a uniquely fastidious methyl-reducing methanogen. ISME J 10:2478–2487. doi: 10.1038/ismej.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sowers KR, Boone JE, Gunsalus RP. 1993. Disaggregation of Methanosarcina spp. and growth as single cells at elevated osmolarity. Appl Environ Microbiol 59:3832–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metcalf WW, Zhang JK, Wolfe RS. 1998. An anaerobic, intrachamber incubator for growth of Methanosarcina spp. on methanol-containing solid media. Appl Environ Microbiol 64:768–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Metcalf WW, Zhang JK, Apolinario E, Sowers KR, Wolfe RS. 1997. A genetic system for Archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc Natl Acad Sci U S A 94:2626–2631. doi: 10.1073/pnas.94.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zehnder A, Wuhrmann K. 1976. Titanium(III) citrate as a nontoxic oxidation-reduction buffering system for the culture of obligate anaerobes. Science 194:1165–1166. doi: 10.1126/science.793008. [DOI] [PubMed] [Google Scholar]
- 34.Guss AM, Rother M, Zhang JK, Kulkarni G, Metcalf WW. 2008. New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea 2:193–203. doi: 10.1155/2008/534081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M. 2005. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics 4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Kremer JD, Cao X, Krzycki J. 1993. Isolation of two novel corrinoid proteins from acetate-grown Methanosarcina barkeri. J Bacteriol 175:4824–4833. doi: 10.1128/jb.175.15.4824-4833.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.