

Abstract
Wilson disease (WD) may present symptomatically at any age. There is great variability in the neurological symptoms present, in the clinical state of WD patients, and in the response to decoppering therapy. Early diagnosis and compliance with anti-copper therapy are essential. Here we present five different WD cases to illustrate different problems encountered during diagnosis and treatment. The first case demonstrates that decoppering therapy may be very effective even with severe neurological symptoms. In addition, we see the importance of family screening, especially among the proband’s siblings. Case 2 shows that we must be very careful during diagnosis. In the reported family, WD was diagnosed in the father of the proband although her brother had liver pathology but not caused by WD. Other cases teach us that decoppering therapy with d-penicillamine must be introduced slowly because of the high risk of neurological deterioration, especially in patients with typical WD brain changes even without neurological signs. We also have to consider concomitant therapies in WD patients. Neuroleptics may cause exacerbation and should be used at a low dose and for the shortest period possible. A full consideration for the issues surrounding the diagnosis and treatment of WD can lead to optimised care with reduced risk of progression and disability.
Keywords: Wilson disease (WD), diagnosis, family screening, clinical variability, brain imaging, anticopper therapy
Introduction
The mean age of symptoms onset in Wilson disease (WD) patients is usually between 20 and 30 years (1-3). However, the symptoms of WD may occur at any age (1,4,5). The initial signs and symptoms of WD can be classified as primarily hepatic (40%), neurological (40%) and psychiatric or asymptomatic (20%), although patients often develop combined hepatic and neurological, or psychiatric disease (1,2). Tremor, ataxia, dystonia and parkinsonism are the most characteristics disturbances, but we often observe a combination of these syndromes coexisting in one patient (1-3,6). As the primary metabolic defect of WD is located in the liver, brain injury occurs later, after the liver is overloaded with copper (1-3). However, in many cases liver injury has a cryptogenic course (without any clinical symptoms), so neurological features may be the first clinical manifestation (1-3,7). The clinical course can be variable: fast progression can lead rapidly to disability or WD may develop insidiously. Several months or even years can elapse between the first neurological symptoms and the diagnosis of WD (8). Neurological symptoms are often preceded by a history of liver disease, many years before, that was not diagnosed as WD (1,6,7). The clinical course after diagnosis and response to decoppering therapy is also variable and difficult to predict (3-9).
A full consideration of the issues surrounding the diagnosis and treatment of WD can lead to optimised care with reduced risk of progression and disability. In this paper we present five clinical cases to illustrate the typical and atypical course of WD in patients with neurological symptoms and to discuss some difficult aspects of diagnosis and treatment.
Case presentation
Case 1
A male, currently aged 68 years, was diagnosed with WD at 18 years old. He was healthy until he was 17 years old but he then began to complain of fatigue and slowness of movements. His speech became slower and a few months later he had marked rigidity. Tremor began in both hands, which was then seen in the head and whole body. A local neurologist observed the progression of symptoms and suspected WD. The patient was referred to our center in 1969. The diagnosis of WD was made based on the presence of Kayser-Fleischer (K-F) rings and low serum ceruloplasmin concentration (14 mg/dL, normal values 25–45 mg/dL). At the time of diagnosis, the patient was completely dependent for all daily living activities. Treatment began with a few injections of British anti-Lewisite (BAL) and later continued with d-penicillamine (DPA). After a few months, he started to improve and was nearly independent with minimal neurological signs after 2 years. After 5 years, he had no neurological signs and he was fully independent. He is a farmer driving agriculture machines and has two healthy children. He is still taking DPA regularly and makes routine visits to our clinic.
In 1969, we also examined his two brothers, who were then 12 and 16 years old. They both were without any symptoms or sign of WD. Both had low serum ceruloplasmin concentration (10 and 8.9 mg/dL, normal values 25–45 mg/dL). At that time, copper urinary excretion tests were not available in our laboratory and were performed 1 year later when they were taking DPA; they both had urinary copper excretion above 1,000 µg/24 hour (normal value <50 µg/24 hour). K-F rings were not detected. The younger brother is still without any hepatic or neurological symptoms. The older brother, despite drinking alcohol and not being fully compliant with DPA, did not develop neurological symptoms, although moderately increased liver enzymes have been observed on occasion.
In the 1980s, DNA analysis confirmed a diagnosed of WD in all 3 brothers: all had p.H1069Q in both alleles of the ATP7B gene. The last time they visited our center was in March 2018 (Figure 1).
Figure 1.

Three siblings with Wilson disease that were diagnosed at our center in 1969. With treatment, all three brothers are without neurological symptoms and are fully independent.
Lessons from this case:
Treatment with DPA may lead to full recovery, even in relatively severe WD cases (10). We started with BAL as DPA was not immediately available in our center. BAL is no longer recommended due to painful deep intramuscular injections, which sometimes led to hematomas. Hypertension, tachycardia, and fever have also been observed. However, BAL was used as a long-term rescue therapy in severe cases (11,12);
Screening for WD is necessary in all siblings (1). Regular decoppering treatment of asymptomatic patients may protect from developing the clinical signs of WD as a previous study has shown that survival of fully compliant asymptomatic patients is similar to that of the general population (13).
Case 2
A female, presently 31 years old, was diagnosed with WD at the age of 27 years. She developed severe hemolytic anemia (hemoglobin serum concentration 5.2 g/L, normal range 12–16 g/L), thrombocytopenia (90×109/L, normal range 150×109–400×109/L), and acute liver failure. Her medical history revealed that a few years before diagnosis she had menstrual irregularity, stomach pain and slightly increased liver enzymes. Based on decreased ceruloplasmin serum concentration and increased urinary copper excretion (Table 1), diagnosis of WD was made in a transplantation center when she developed liver failure and was a potential candidate for transplantation. However, she received DPA treatment and her liver function improved. When she visited our center, 3 years after treatment initiation, her liver function tests were normal, with no neurological signs, normal brain magnetic resonance imaging (MRI), and no K-F rings.
Table 1. Characteristics of an unusual family with Wilson disease diagnosed in the daughter (proband) and the father.
| Parameter | Proband | Brother* | Father |
|---|---|---|---|
| Neurological signs | No | No | Yes |
| Kayser-Fleischer ring | No | No | Yes |
| Brain magnetic resonance imaging | Normal | Not done | Typical changes |
| Serum ceruloplasmin concentration (normal range 25–45 mg/dL) | 3.4 | 18.8 | 3 |
| Total copper serum concentration (normal range 70–140 µg/dL) | 19.5 | 63.7 | 17.7 |
| Urinary copper excretion (normal range 0–50 µg/24 hours) | 74.8 | 189 | 571 |
| ATP7B mutation (direct sequencing method) | p.H1069Q/Q355Stop | Q355Stop/– | p.H1069Q/Q355Stop |
*, copper metabolism done after D-penicillamine treatment.
Pathogenic mutations were found in both alleles of ATP7B gene (Table 1). Her family history revealed that her brother was diagnosed with WD in another center and her father was suspected of having WD. Her brother was screened for WD at the age of 36 years old in a hepatology center, 1 year after his sister was diagnosed. In his local laboratory, he had decreased ceruloplasmin serum concentration (13 mg/dL, normal range 15–30 mg/dL), slightly higher copper urinary excretion (94 µg/24 hours, normal range 8–80 µg/24 hours), moderately increased liver enzymes [alanine aminotransferase (ALT) 151 IU/L; aspartate aminotransferase (AST) 58 IU/L] and low platelets count (120×109/L, normal range 150×109–400×109/L)]. Notably he was a heavy alcohol drinker. Liver ultrasonography revealed hyperechogenicity. He received DPA (750 mg daily). At admission to our center he had no K-F ring and no neurological signs. Laboratory tests performed in our center showed slightly decreased serum ceruloplasmin concentration and increased urinary copper excretion. However, at that time, he was already treated with DPA. DNA analysis revealed a mutation in only one allele of ATP7B gene (Table 1). We performed a test with radioactive copper (14), which did not confirm a diagnosis of WD. Treatment with DPA was stopped and he was referred for further care to his local physician.
Her father, then 62 years old, was diagnosed with WD 1 year after his daughter’s diagnosis. He was working as a plumber, drinking alcohol moderately, and was admitted to the internal medicine department due to pneumonia. During additional studies unexpected liver cirrhosis with esophageal varices were found. Low serum ceruloplasmin concentration suggested WD. He received DPA treatment 1,000 mg daily. In our clinic a year after diagnosis and DPA treatment, we diagnosed K-F rings which were not recognized before, probably due to concomitant arcus senilis (Figure 2). On neurological examination he had moderate hand tremor (intentional and postural) but this did not disturb his daily activities. Brain MRI revealed discrete hyperintensity in the midbrain and the pons. DNA analysis revealed mutations in both alleles of the ATP7B gene, similar to those detected in the proband. He died few months later in another hospital due to generalized cancer. An autopsy was not performed, and the primary source of the cancer was not found, but lung cancer was suspected.
Figure 2.
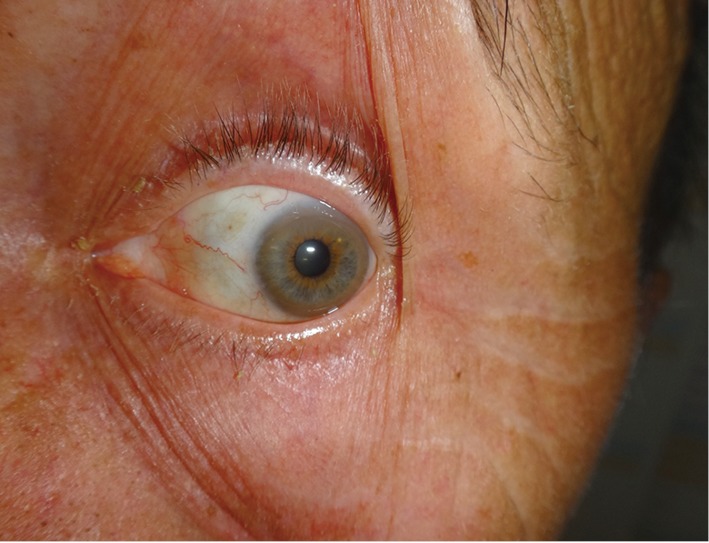
Kayser-Fleischer ring in a 62-year-old patient with Wilson disease diagnosed after family screening, with the father as the proband.
Lessons from this case:
Liver acute failure of WD can be accompanied by hemolysis. Erythrocytes injury is probably caused by high concentrations of free copper due to liver cell necrosis (15,16). Even in patients awaiting liver transplantation anti-copper therapy should be started, because in some cases, it can lead to clinical improvement and transplantation is no longer needed (17);
In siblings with clinical symptoms suggesting WD, careful clinical and laboratory diagnosis should be performed. Decreased serum ceruloplasmin concentration and slightly increased urinary copper excretion may be present in heterozygotes with a mutation in one allele of the ATP7B gene as well as in patients with liver disease not caused by WD (18). At the time of diagnosis in our center the patient had only a slight decrease in ceruloplasmin, urine was not diagnostic due to taking DPA and there was only one mutation in one allele of the ATP7B gene. Even taking into account the previously detected slight increase in copper excretion, he had only 3 points on the Leipzig scoring system. Four or more points are needed for the WD diagnosis and initiation of decoppering treatment (19).
The diagnosis of WD in the proband’s father was accidental. He had no overt liver disease, his neurological symptoms were mild and were connected with alcohol abuse. WD is an autosomal inherited condition, thus it usually occurs in siblings (1). However, we rarely observe a vertical (pseudo-dominant) type of inheritance, which is often first diagnosed in an offspring of the proband (20). At our center we diagnosed 9 such families (20). In unusual family of Case 2 WD diagnosis was first made in the offspring and next in her father. We have found only one such case in the literature (21).
Screening of the family of the proband is generally recommended among siblings and also among children (1). WD is regarded as a disease of young and middle-aged populations, so we did not routinely screen parents of WD cases. However, we have to remember that diagnosis of WD is more often presently done in older persons presenting both neurological and hepatic symptoms (4,5). We should more carefully investigate parents as it can change our approach. Some years ago, we thought that risk of WD among children was 0.5%, but recently we found that it is 4% (20).
Case 3
A 21-year-old man was admitted to our department to control anti-copper treatment and to confirm the diagnosis of WD. He was studying extramurally to become a policeman. One year before diagnosis he started heavy physical work, carrying and cutting metal constructions. The working hall was poorly ventilated and polluted with chemicals and he did not wear a mask. After 3 months he noticed brown spots on both calves and later, edema was also noted. He did not visit a physician despite having problems studying due to fatigue and poor concentration. Finally, after 10 months, he had to stop work. Basic laboratory tests results revealed low platelet count (30×109/L, normal range 150×109–400×109/L) and leukopenia (white blood cell count 2.5 109/L, normal range 4.5×109–11.0×109/L). He was admitted to his local hospital and transferred to the transplantation department. Liver cirrhosis was confirmed with the presence of portal hypertension, esophageal varices, and hypersplenism. WD was diagnosed based on low serum ceruloplasmin concentration (6.8 mg/dL, normal range 25–45 mg/dL) and increased urinary copper excretion (235 µg/24 hours, normal value <50 µg/24 hours). He had a brain MRI that showed hyperintense changes in the basal ganglia and WD was radiologically suspected. He received 750 mg of DPA and 160 mg of zinc sulfate. Ten days later, he was transferred to our department and bilateral K-F rings were detected. His neurological examination was normal (Figure 3). However, soon after starting DPA therapy, the patient reported slight slowness of speech and hypersalivation. MRI of his brain revealed potentially reversible symmetric solid hyperintensity changes in both caudate nuclei and putamen (Figure 4A,B). We decreased the dose of DPA to 250 mg daily and advised that the DPA dose should be slowly increased over 6 weeks to the final dose of 1,000 mg daily. Zinc sulfate was withdrawn. There was continued neurological deterioration over the next few months. His face was masked with oromandibular dystonia and he also developed hand and leg dystonia. General stiffness appeared and he had difficulties walking, but was mobile. He markedly deteriorated after having pneumonia and needed assistance with nearly all daily activities. He could walk only with the help of two people and a gastrostomy was implanted (Figure 3B). Brain MRI after 1 year of treatment showed modest hyperintense and hypointense signals in the putamen with markedly hyperintense signals in lateral putaminal margins (Figure 4C,D). Some putaminal changes as signs of a degeneration process are irreversible. Due to depression, he is taking escitalopram, verbal communication is impossible; however, he understands well, watches TV, reads, and can write slowly.
Figure 3.
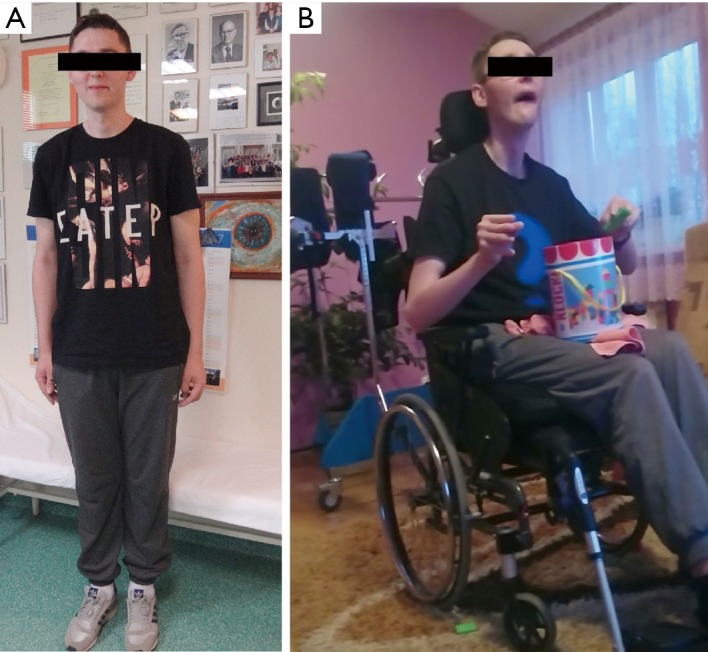
(A) A 21-year-old patient with the hepatic form of Wilson disease and normal neurological examination; (B) the patient deteriorated, exhibiting marked oromandibular dystonia, hipomimia, and dystonic posture of hands.
Figure 4.
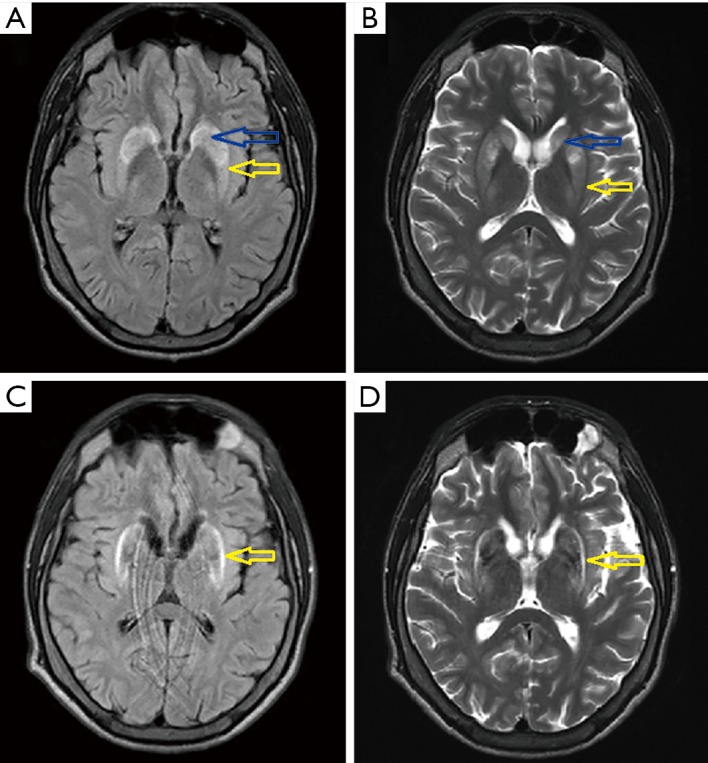
Magnetic resonance imaging of the brain of the 21-year-old patient without neurological signs revealed symmetric solid hyperintensity changes in both caudate nuclei (blue arrow) and putamen (yellow arrow) on FLAIR images (A) and T2-weighted (B). A follow-up study after deterioration of the neurological state shows modest hyperintense signals in the putamen (yellow arrow) with markedly hyperintense signals in lateral putaminal margins on FLAIR images (C) and T2-weighted (D). FLAIR, fluid attenuation inversion recovery.
Lessons from this case:
Heavy physical work and contact with toxins may accelerate liver damage (sometimes as fulminant liver failure) in patients with a preclinical form of WD (22,23);
Fast introduction of DPA may lead to rapid neurological deterioration or the appearance of neurological signs in patients without previous neurological signs. It is recommended to start DPA slowly and reach a dose of 1,000 mg over 6 weeks or longer (1);
Brain MR changes were already present when decoppering treatment was started. Around 50% of patients with the liver form of WD have brain pathology on MRI and such cases are more prone to early deterioration (24). We cannot, however, exclude that neurological deterioration was caused by a naturally aggressive form of the disease, but DPA could have accelerated this process;
We advise to perform MRI of the brain in all WD patients at diagnosis. It may serve as the marker of disease advancement and further, may help to monitor treatment. Normal MRI of the brain before starting decoppering therapy and typical WD changes later may indicate disease progression despite anti-copper treatment or poor compliance.
Case 4
A 29-year-old female with the neurological form of WD was admitted to our department in November 2017. She had been working physically abroad for several years, but she was healthy. In 2013, the patient was suffering from liver cirrhosis but despite many diagnostic procedures, the cause remained unknown and liver function was compensated. In October 2016, the patient’s family noticed her behavioral changes, she became calm and taciturn. In January 2017 paroxysmal dystonic-like involuntary movements occurred, lasting about 10 seconds, with involuntary tongue and mouth movements, especially sticking out the tongue, and there were abrupt cramps of the whole body. K-F rings were detected on slit lamp examination. Laboratory tests revealed thrombocytopenia and abnormal copper metabolism (low serum ceruloplasmin concentration). DNA analysis revealed p.H1069Q mutations in both alleles of ATP7B gene. DPA treatment was introduced with a low initial dose of 250 mg daily, but the dosage was increased to 1,000 mg daily in just 2 weeks. Her neurological status worsened as involuntary movements became more frequent with leg dystonia, marked dysarthria and dysphagia.
In November 2017 she was admitted to our department. Her neurological examination revealed very frequent and severe paroxysmal dystonic movements causing abnormal dystonic posture (Figure 5). During these attacks she was conscious and remembered words spoken to her. When she had no paroxysmal movements, she presented with marked oromandibular dystonia, extrapyramidal dysarthria and dysphagia with excessive salivation, increased muscle tone (extrapyramidal type), severe gait disturbances with freezing, and often falls. Assistance with some daily activities was needed. Her MRI showed hyperintensity changes in thalami and midbrain, modest hyperintense and hypointense signals in the putamen with markedly hyperintense signals in lateral putaminal margins (Figure 6).
Figure 5.
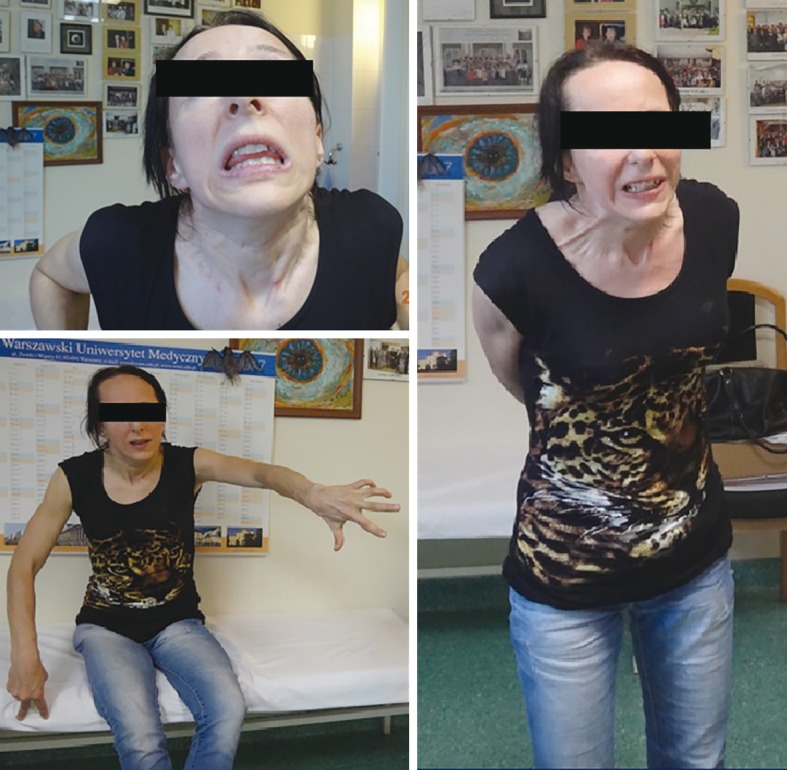
A 29-year-old patient with Wilson disease presented with severe paroxysmal dystonia.
Figure 6.

Magnetic resonance imaging of the brain of a 29-year-old patient. Fluid attenuation inversion recovery images (A,C) and T2- weighted (B,D) and shown solid hyperintensity changes in thalami (red arrow) and midbrain (green arrow), modest hyperintense and hypointense signals in the putamen (yellow arrow) with markedly hyperintense signals in lateral putaminal margins. Hyperintensities in thalami and midbrain are potentially reversible after treatment while some putaminal changes partly reflect the course of degeneration and are irreversible.
Electroencephalogram (EEG) examination did not reveal epileptic activity even during paroxysmal movements. Because of very frequent paroxysmal dystonic episodes, we administered diazepam, which did not bring any relief. Later we started oxcarbazepine 150 mg daily, increasing to 300 mg daily. We observed continuous improvement of the patient’s neurological status, particularly gait improvement, reduced freezing episodes and almost completely reduced paroxysmal dystonia. The other symptoms of WD remained unchanged. She was discharged at the end of December 2017. Unfortunately, few weeks later because of severe swallowing disturbances, the patient was diagnosed with pneumonia. She was admitted to her local hospital, and due to respiratory disturbances she was admitted to the intensive care unit. She needed a percutaneous endoscopic gastrostomy, she additionally developed liver failure and died at the end of March 2018. An autopsy was not performed.
Lessons from this case:
There are a few described cases suggesting that paroxysmal dystonia in WD may be induced by DPA (25). In our case the paroxysmal dystonia was the first neurological sign of WD but deterioration was observed relatively fast after the introduction of treatment. Similarly like in Case 3 we do not know whether deterioration was a result of the natural disease progression or caused by overly quick DPA introduction;
Diagnosis of WD is often delayed. In this case the first hepatic signs were 3 years before neurological signs and diagnosis. Hepatic signs may be mild and unspecific or liver injury is self-limiting clinically (1,7). At the time of diagnosis this case had normal liver function. The only clinical sign that might suggest abnormal liver function could have been undiagnosed problems with menstruation. Diagnosis delay is longer when hepatic signs occur than when neurological features are present (26,27).
Paroxysmal dystonia is a rare type of dyskinesia characterized by short episodic periods of dystonic movements that are only visible during attacks. It usually causes unilateral dystonic posture and is triggered by voluntary movement, tactile stimulation, startling noise or hyperventilation. It is not characteristic of WD and only a few cases of paroxysmal dystonia in WD have been described so far (28);
Oxcarbazepine is one of a few drugs that is effective as low-dose monotherapy in paroxysmal dystonia cases (29,30).
Case 5
A 23-year-old female was admitted to our center in 2016 to provide confirmation of a WD diagnosis and to continue decoppering therapy. Two years before diagnosis she started to have mild intentional and resting tremor that deteriorated and slurred speech occurred. She visited a neurologist who diagnosed WD and started therapy with DPA in slowly increasing doses. Her past medical history revealed that 7 years ago she had stomach pain. Abdominal ultrasonography revealed nodular structure of the liver, but liver function tests were normal and diagnostics were not continued. Additionally, 2 years before diagnosis she had rare menstruation.
On neurological examination at admission to our center she presented mild dysarthria, hypomimia, and mild postural and resting tremor in her hands, which was more evident on the right side. K-F rings were present on slit lamp examination. Brain MRI revealed solid hyperintensity changes in thalami and midbrain, and modest hyperintense and hypointense signals in the putamen with markedly hyperintense signals in lateral putaminal margins (Figure 7). Putaminal changes like those of case 3 (Figure 4) and 4 (Figure 6) partly reflect the course of degeneration and are irreversible.
Figure 7.

Magnetic resonance imaging of the brain of a 23-year-old female with Wilson disease revealed a typical variety of MRI changes during the course of the disease. On FLAIR images solid hyperintensity changes in thalami (red arrow) and midbrain (green arrow), and modest hyperintense and hypointense signals in the putamen (yellow arrow) with markedly hyperintense signals in lateral putaminal margins are seen. Putaminal changes partly reflect the course of degeneration and are irreversible. MRI, magnetic resonance imaging; FLAIR, fluid attenuation inversion recovery.
Ceruloplasmin serum concentration was reduced (17.9 mg/dL; normal values 25–45 mg/dL) and urinary copper was increased, but she was taking DPA at the time of the tests. She was found to have mutations in both alleles of ATP7B gene. She had mild symptoms of depression but did not want to take any treatment and continued with DPA therapy only. After 3 months, she visited our center and neurological improvement was noticed. Returning home, she experienced strong emotional stress (she took the wrong train) and after a few hours of traveling at night, she returned to our hospital. Because of the severe anxiety and even suicidal ideation, she was admitted to the psychiatric ward where she stayed for 6 weeks. She received aripiprazole (10 mg) and olanzapine (10 mg). Due to a skin rash DPA was changed to zinc sulfate (160 mg of zinc). After stabilization she was discharged home on the same psychiatric drugs as she was taking in hospital. Three months later, she returned to our center suffering from marked tremor of the head and arms that generalized to the whole body when she kept arms in a wing-beating position. Her speech was still understandable but was quiet and slow. At first, we thought that it was due to zinc therapy; however, she said that this deterioration started a few days after the dose of aripiprazole was increased to 20 mg daily following consultation with the local psychiatrist. The dose of aripiprazole was decreased to 10 mg and after 2 weeks she improved and after 4 weeks she had no more tremor and speech returned to normal. Due to the improvement of psychiatric symptoms, first aripiprazole and then olanzapine was withdrawn a few months later. She now feels healthy and she returned to work as a tailor. She has no neurological signs as well as no psychiatric symptoms.
Lessons from this case:
First hepatic symptoms occurred 7 years before diagnosis and neurological signs occurred 2 years before diagnosis. The problem of delayed diagnosis was discussed in Case 4;
Depression and anxiety are common presentations in patients with WD and may precede obvious neurological signs. In about one-third of WD patients, initial psychiatric symptoms are observed (1). Neuroleptics and antidepressants may cause neurological deterioration. Aripiprazole is regarded as a safe drug, but in our case, caused deterioration at higher doses. The general recommendation is to avoid using neuroleptics as far as possible and if needed, antidepressants are to be used at as low doses as possible (31);
Any deterioration of neurological symptoms must be fully investigated (24). In our case we initially considered that it was caused by poor response to zinc therapy; however, careful history of the disease led us to conclude that the cause of the exacerbation was aripiprazole.
Conclusions
WD is one of a few genetic metabolic diseases that is treatable with correct and early diagnosis; however, because of its rarity, diagnosis of WD is often missed or delayed. WD diagnosis should be taken into account in patients with liver impairment of unknown etiology, movement disorder and psychiatric disturbances in all patients, not only in young patients. As our cases show, inappropriate anti-copper treatment introduction may lead to irreversible neurological deterioration, while some patients may deteriorate because of an abruptly increased DPA dose. Concomitant therapy, especially neuroleptics may also be harmful in this group of patients. Thus, to prevent unnecessary treatment complication, unexperienced centers should consult specialists with knowledge of taking care of patients with WD.
Acknowledgements
None.
Informed Consent: Written informed consent was obtained from the patient for publication of this manuscript and any accompanying images.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.European Association for Study of Liver EASL Clinical Practice Guidelines: Wilson’s Disease. J Hepatol 2012;56:671-85. 10.1016/j.jhep.2011.11.007 [DOI] [PubMed] [Google Scholar]
- 2.Członkowska A, Litwin T, Dusek P, et al. Wilson Disease. Nat Rev Dis Primers 2018;4:21. 10.1038/s41572-018-0018-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfeiffer RF. Wilson’s disease. Semin Neurol 2007;27:123-32. 10.1055/s-2007-971173 [DOI] [PubMed] [Google Scholar]
- 4.Ferenci P, Członkowska A, Merle U, et al. Late-onset Wilson's disease. Gastroenterology 2007;132:1294-8. 10.1053/j.gastro.2007.02.057 [DOI] [PubMed] [Google Scholar]
- 5.Członkowska A, Rodo M, Gromadzka G. Late onset Wilson’s disease: therapeutic implications. Mov Disord 2008;23:896-8. 10.1002/mds.21985 [DOI] [PubMed] [Google Scholar]
- 6.Lorincz MT. Neurologic Wilson's disease. Ann N Y Acad Sci 2010;1184:173-87. 10.1111/j.1749-6632.2009.05109.x [DOI] [PubMed] [Google Scholar]
- 7.Steindl P, Ferenci P, Dienes HP, et al. Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology 1997;113:212-8. 10.1016/S0016-5085(97)70097-0 [DOI] [PubMed] [Google Scholar]
- 8.Członkowska A, Tarnacka B, Litwin T, et al. Wilson’s disease-cause of mortality in 164 patients during 1992-2003 observation period. J Neurol 2005;252:698-703. 10.1007/s00415-005-0720-4 [DOI] [PubMed] [Google Scholar]
- 9.Bruha R, Marecek Z, Pospisilova L, et al. Long-term follow-up of Wilson disease: natural history, treatment, mutations analysis and phenotypic correlation. Liver Int 2011;31:83-91. 10.1111/j.1478-3231.2010.02354.x [DOI] [PubMed] [Google Scholar]
- 10.Członkowska A, Litwin T. Treatment of Wilson’s disease – another point of view. Expert Opinion Orphan Drugs 2015;3:239-43. 10.1517/21678707.2015.1016907 [DOI] [Google Scholar]
- 11.Scheinberg IH, Sternlieb I. Wilson’s Disease. Philadelphia: WB Saunders, 1984. [Google Scholar]
- 12.Aggarwal A, Bhatt M. Advances in Treatment of Wilson Disease. Tremor Other Hyperkinet Mov (N Y) 2018;8:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dzieżyc K, Karliński M, Litwin T, et al. Compliant treatment with anti-copper agents prevents clinically overt Wilson's disease in pre-symptomatic patients. Eur J Neurol 2014;21:332-7. 10.1111/ene.12320 [DOI] [PubMed] [Google Scholar]
- 14.Członkowska A, Rodo M, Wierzchowska-Ciok A, et al. Accuracy of the radioactive copper incorporation test in the diagnosis of Wilson disease. Liver Int 2018;38:1860-6. 10.1111/liv.13715 [DOI] [PubMed] [Google Scholar]
- 15.Seth B, Gavhane J, Revathi N, et al. Haemolytic anaemia – Initial presentation of Wilson Disease. Indian J Basic Applied Med Res 2015;4:498-500. [Google Scholar]
- 16.Czlonkowska A. A study of haemolysis in Wilson’s disease. J Neurol Sci 1972;16:303-14. 10.1016/0022-510X(72)90194-3 [DOI] [PubMed] [Google Scholar]
- 17.Ahmad A, Torrazza-Perez E, Schilsky ML. Liver transplantation for Wilson disease. Handb Clin Neurol 2017;142:193-204. 10.1016/B978-0-444-63625-6.00016-1 [DOI] [PubMed] [Google Scholar]
- 18.Gromadzka G, Chabik G, Mendel T, et al. Middle-aged heterozygous carriers of Wilson’s disease do not present with significant phenotypic deviations related to copper metabolism. J Genet 2010;89:463-7. 10.1007/s12041-010-0065-3 [DOI] [PubMed] [Google Scholar]
- 19.Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int 2003;23:139-42. 10.1034/j.1600-0676.2003.00824.x [DOI] [PubMed] [Google Scholar]
- 20.Dzieżyc K, Litwin T, Chabik G, et al. Families with Wilson’s disease in subsequent generations: clinical and genetic analysis. Mov Disord 2014;29:1828-32. 10.1002/mds.26057 [DOI] [PubMed] [Google Scholar]
- 21.Brunet AS, Marotte S, Guillaud O, et al. Familial screening in Wilson’s disease: Think at the previous generation! J Hepatol 2012;57:1394-5. 10.1016/j.jhep.2012.07.011 [DOI] [PubMed] [Google Scholar]
- 22.Chen JD, Wang JD, Jang JP, et al. Exposure to mixtures of solvents among paint workers and biochemical alterations of liver function. Br J Ind Med 1991;48:696-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee WM, Squires RH, Nyberg SL, et al. Acute liver failure: Summary of a workshop. Hepatology 2008;47:1401-15. 10.1002/hep.22177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Litwin T, Dzieżyc K, Karliński M, et al. Early neurological worsening in patients with Wilson’s Diseases. J Neurol Sci 2015;355:162-7. 10.1016/j.jns.2015.06.010 [DOI] [PubMed] [Google Scholar]
- 25.Paliwal VK, Gupta PK, Pradhan S. Gabapentin as a rescue drug in D-penicillamine-induced status dystonicus in patients with Wilson disease. Neurol India 2010;58:761-3. 10.4103/0028-3886.72184 [DOI] [PubMed] [Google Scholar]
- 26.Walshe JM. Wilson's disease presenting with features of hepatic dysfunction: a clinical analysis of eighty-seven patients. Q J Med 1989;70:253-63. [PubMed] [Google Scholar]
- 27.Walshe JM, Yealland M. Wilson's disease: the problem of delayed diagnosis. J Neurol Neurosurg Psychiatry 1992;55:692-6. 10.1136/jnnp.55.8.692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HJ, Yoon JH. A case of Wilson's disease presenting with paroxysmal dystonia. Neurol Sci 2017;38:1881-2. 10.1007/s10072-017-3008-4 [DOI] [PubMed] [Google Scholar]
- 29.Micheli F, Tschopp L, Cersosimo MG. Oxcarbazepine-responsive paroxysmal kinesigenic dyskinesia in Wilson disease. Clin Neuropharmacol 2011;34:262-4. 10.1097/WNF.0b013e3182348964 [DOI] [PubMed] [Google Scholar]
- 30.Chillag KL, Deroos ST. Oxcarbazepine use in paroxysmal kinesigenic dyskinesia: report on four patients. Pediatr Neurol 2009;40:295-7. 10.1016/j.pediatrneurol.2008.09.024 [DOI] [PubMed] [Google Scholar]
- 31.Litwin T, Dusek P, Szafrański T, et al. Psychiatric manifestations in Wilson’s disease: possibilities and difficulties for treatment. Ther Adv Psychopharmacol 2018;8:199-211. 10.1177/2045125318759461 [DOI] [PMC free article] [PubMed] [Google Scholar]