

Abstract
The discovery of regulated cell death presents tantalizing possibilities for gaining control over the life–death decisions made by cells in disease. Although apoptosis has been the focus of drug discovery for many years, recent research has identified regulatory mechanisms and signalling pathways for previously unrecognized, regulated necrotic cell death routines. Distinct critical nodes have been characterized for some of these alternative cell death routines, whereas other cell death routines are just beginning to be unravelled. In this Review, we describe forms of regulated necrotic cell death, including necroptosis, the emerging cell death modality of ferroptosis (and the related oxytosis) and the less well comprehended parthanatos and cyclophilin D-mediated necrosis. We focus on small molecules, proteins and pathways that can induce and inhibit these non-apoptotic forms of cell death, and discuss strategies for translating this understanding into new therapeutics for certain disease contexts.
For many years, researchers have divided cell death processes into those that are regulated and those that are accidental. The first-discovered form of regulated cell death, apoptosis, was used as a synonym for regulated cell death and even for programmed cell death in the context of development and homeostasis, whereas the term necrosis was reserved as a synonym for accidental cell death. This paradigm was imbued with the notion that only apoptosis was considered therapeutically tractable, as the accidental and unregulated nature of its necrotic counterpart meant that it was deemed undruggable. This perspective led to the systematic neglect of the possibility that non-apoptotic cell death subroutines could represent causative processes in disease and a source of potentially pharmacologically tractable drug targets.
This long-standing paradigm in the field of cell death has recently been challenged and overturned by the recognition that tumour necrosis factor (TNF) can induce regulated cell death with apoptotic or necrotic features in a context-dependent manner1. Owing to its regulated nature, this necrotic form of cell death was termed necroptosis. So far, necroptosis is the best-studied form of regulated non-apoptotic cell death and has helped to illuminate two basic principles. First, regulated, and, indeed, developmentally programmed, cell death is not restricted to apoptosis, and second, cell death pathways can be interconnected. These factors need to be taken into consideration when pharmacological strategies for cytoprotective intervention are conceived and deployed.
The establishment of necroptosis as an alternative form of regulated cell death has resulted in several studies implicating necroptosis as the main contributor to cell death in diverse conditions. However, because other regulated forms of non-apoptotic cell death resulting in necrotic morphology (cytoplasmic swelling and loss of plasma membrane integrity) are interconnected and interdependent, a careful and critical re-evaluation of these studies is required to unequivocally determine which programmes are actually elicited under specific conditions2.
Necroptosis and other regulated non-apoptotic forms of cell death, such as ferroptosis, parthanatos and cyclophilin D-(CypD)-dependent necrosis, have attracted increasing consideration regarding their causative role in pathological settings, and there is already ongoing development to pharmacologically intervene in these pathways. Pharmacological modulation of other non-apoptotic forms of cell death — such as neutrophil extracellular-trap (NET)-associated cell death (termed NETosis), pyroptosis and autophagic cell death — are not the focus of this article, as these cell death modalities (with the exception of pyroptosis) lack a clear necrotic phenotype. We therefore refer the reader to excellent publications that cover these forms of cell death in detail3–6.
In this Review, we discuss the in vivo relevance of necroptosis, ferroptosis, parthanatos and CypD-dependent necrosis, and the opportunities for pharmacological modulation of these types of cell death, both positively and negatively. We anticipate that understanding the relevance of these pathways in vivo will help to lay the foundations for therapeutics that aim to trigger or prevent cell death in disease. As such, the triggering of alternative cell death programmes in tumours resistant to apoptosis has been proposed as a route for effective targeted therapy7. Moreover, exploring the prevention of these alternative regulated cell death modalities in pathological conditions in which anti-apoptosis approaches have not yielded encouraging outcomes — such as ischaemia–reperfusion injury (IRI) and neurodegenerative conditions, including Huntington disease — may open new avenues for the development of novel therapies. Based on the lessons learned from attempts to regulate apoptosis, we suggest that there is considerable therapeutic potential remaining for the pharmacological regulation of alternative cell death modalities.
Necroptosis
Necroptosis is characterized by cytoplasmic granulation and organelle and/or cellular swelling that together culminate in the leakage of intracellular contents from the cell8. Necroptosis induced by TNF has so far been the most thoroughly investigated form of regulated non-apoptotic cell death or regulated necrosis2,9 (FIG. 1). Necroptosis research surged when small molecules termed necrostatins were shown to be able to suppress this necrotic form of cell death10,11. One of these molecules, necrostatin-1 (FIG. 2; TABLE 1), was subsequently found to inhibit receptor-interacting serine/threonine kinase 1 (RIPK1), thus blocking the necrotic effect of TNF11. Necrostatin-1 and more-specific analogues have been used as tools to investigate the wide-ranging role of necroptosis in pathophysiological settings12.
Figure 1. The main signalling events downstream of TNFR activation.

Binding of tumour necrosis factor (TNF) to its cognate receptor TNFR1 triggers the assembly of complex I, which comprises TNFR1, TNFR1-associated death domain (TRADD), receptor-interacting serine/threonine-protein kinase 1 (RIPK1), TNFR-associated factor 2 (TRAF2), cellular inhibitor of apoptosis protein 1/2 (cIAP1/2), and linear ubiquitin chain assembly complex (LUBAC)22,160,161. Complex I provides the platform for Lys63-linked ubiquitylation (grey circles) or linear ubiquitylation (green circles) of RIPK1 by cIAP1/2 or LUBAC, respectively. This ubiquitylation is implicated in the decision between nuclear factor-κB (NF-κB) or survival signalling and cell death signalling. Ubiquitylation leads to the recruitment of other factors such as transforming growth factor-β (TGFβ)-activated kinase (TAK1), TAK1-binding protein 1 (TAB1) and TAB2, or NF-κB essential modulator (NEMO) and the inhibitor of the NF-κB kinase-α (IKKα)–IKKβ complex, usually triggering canonical NF-κB signalling. In the presence of cycloheximide (CHX), a translational inhibitor, TNF stimulation leads to the formation of a cytosolic complex IIa in which RIPK1 disappears, whereas TRADD and FAS-associated death domain (FADD) interaction leads to the activation of caspase 8 and effector caspases such as caspase 3 and caspase 7 and apoptotic cell death22. With cIAP1/2 inhibitors (SMAC (second mitochondria-derived activator of caspase) mimetics), knockdown of cIAPs23,162–165 or inhibition or depletion of TAK1 or NEMO24, complex IIb is formed. Complex IIb consists of RIPK1, RIPK3, FADD and caspase 8, and favours RIPK1-kinase-activity-dependent apoptosis. Heteromeric pro-caspase 8–FLICE-like inhibitory protein long isoform (FLIPL) inhibits necroptosis, probably by cleaving RIPK1, RIPK3 and cylindromatosis (CYLD)166–168. With sufficient expression of RIPK3 and concomitant inhibition or reduced expression of pro-caspase 8 and FLIPL (REF. 26), complex IIc (also known as the necrosome) is formed17,169,170. The formation of complex IIc entails the association of RIPK1 and RIPK3 followed by a series of auto-and transphosphorylation events of RIPK1 and RIPK3. Activated RIPK3 phosphorylates and recruits mixed lineage kinase domain-like protein (MLKL)49,171, eventually leading to the formation of a supramolecular protein complex at the plasma membrane and necroptosis27–30,34. SMAC mimetics are being developed to impair survival signalling and to sensitize cells to the triggering of cell death in the context of tumour treatment172. Z-VAD-FMK is a bona fide pan-caspase inhibitor. Necrostatin-1 (Nec-1)10 and the more-specific Nec-1s37, Cpd27 (REF. 41) and, more recently, a hybrid molecule consisting of Nec-1s and ponatinib called PN10 (REF. 42) all inhibit the kinase activity of RIPK1 and thus inhibit necroptotic signalling. Additional necroptosis inhibitors include the RIPK3 inhibitors GSKʹ840, GSKʹ843 and GSKʹ872 (REF. 33), as well as the MLKL inhibitors necrosulfonamide (NSA)49 and compound 1 (REF. 34). However, the specificity of these MLKL inhibitors is not restricted to inhibition of MLKL, and their efficacy is probably also due to effects on other steps in the necroptosis pathway. ActD, actinomycin D.
Figure 2. Chemical structures of inhibitors of non-apoptotic cell death.

The mechanisms of action, key functions and references for these inhibitors are provided in TABLE 1. CypD, cyclophilin D; RIPK1, receptor-interacting serine/threonine kinase 1.
Table 1 |.
Summary of small-molecule modulators of regulated necrotic cell death
| Compound name (alternative name) | Mechanism of action | Applications | Refs |
|---|---|---|---|
| RIPK1-dependent apoptosis | |||
| Necrostatin-1 | RIPK1 inhibitor | Protected mice and rats against neurodegenerative conditions (for example, TBI, stroke, HD and AD), retinal degeneration, inflammatory diseases (including sepsis, alcoholic or non-alcoholic liver diseases, pancreatitis and IBD) and microbial infections | 10,11,20 |
| Nec-1s (R-7-Cl-O-Nec-1) | RIPK1 inhibitor | Prevented TNF-induced lethality in a mouse model of SIRS | 37 |
| PN10 | RIPK1 inhibitor | Prevented TNF-induced lethality in a mouse model of SIRS | 42 |
| Cpd27 | RIPK1 inhibitor | Prevented TNF-induced lethality in a mouse model of SIRS | 41 |
| Necrosulfonamide | MLKL inhibitor | Prevented necroptosis induced by TNF plus Z-VAD-FMK in mouse fibroblasts | 49 |
| Necroptosis | |||
| GSKʹ840, GSKʹ843 and GSKʹ872 | RIPK3 inhibitors | Prevented LPS-induced death of mouse macrophages, cell death induced by TNF plus Z-VAD-FMK, and poly(I:C)-triggered death of interferon-β-sensitized cells in vitro | 33 |
| Compound 1 (SYN-1215 or GW806742X) | MLKL inhibitor | Inhibited necroptotic death of mouse dermal fibroblasts stimulated with a combination of caspase inhibitors, cIAP inhibitors and TNF | 34 |
| Inflammasome | |||
| MCC950 | NLRP3 inhibitor | Inhibited interleukin-1β production in vivo and attenuated the severity of EAE, a mouse model of multiple sclerosis | 151 |
| Parthanatos | |||
| MNNG | DNA-alkylating agent | Induced necrotic cell death through PARP overactivation | 182 |
| Benzamide | PARP inhibitor | Reduced neurodegeneration in a rat model of soman-induced seizure-related brain damage | 178,183 |
| Substituted benzamide derivatives; for example, INO-1001 (3-aminobenzamide) | Competitive PARP inhibitors | Widely used in various disease models, including TBI; reduced pulmonary injury and improved recovery of myocardial and endothelial function after hypothermic cardiac arrest; in a Phase II trial of INO-1001 in myocardial ischaemia, plasma from INO-1001-treated patients exhibited reduced in vitro PARP activity by >90% at all doses, with a trend towards blunting of inflammation | 112, 184–188 |
| 4-methoxyflavone | PARP inhibitor | Prevented cell death caused by the parthanatos-inducing DNA-alkylating agent MNNG | 179 |
| 3ʹ,4ʹ-dimethoxyflavone | PARP inhibitor | Prevented cell death caused by the parthanatos-inducing DNA-alkylating agent MNNG | 179 |
| PJ34 | PARP inhibitor | Reduced brain infarct size in a mouse model of transient focal ischaemia | 180 |
| DPQ | PARP inhibitor | Decreased cell death and prevented cardiac dysfunction after a non-lethal myocardial infarction in rats | 181 |
| Ferroptosis | |||
| Erastin | System Xc− inhibitor | Induced ferroptosis in diverse cell types | 56 |
| Piperazine erastin | System Xc− inhibitor | Reduced growth in a xenograft mouse tumour model (HT-1080 cells) | 60 |
| Erastin analogue 21 | System Xc− inhibitor | Induced ferroptosis in various cell types | 91 |
| Sorafenib | System Xc− inhibitor | Induced cell death in hepatocellular and renal cell carcinoma cell lines, as well as in other cancer cell lines | 91,93 |
| Liproxstatin-1 | Inhibits lipid peroxidation | Prevented Gpx4-knockout-induced acute renal failure and inhibited hepatic IRI in mice | 61 |
| Ferrostatin-1 | Inhibits lipid peroxidation | Prevented glutamate-induced neurotoxicity in cortical explants and IRI in a mouse model of kidney damage | 56,62 |
| Ferrostatin analogue SRS-11–92 | Inhibits lipid peroxidation | Efficacious in different cellular and slice models of HD, kidney tubule cell death and periventricular leukomalacia | 71 |
| Ferrostatin analogue SRS-16–86 | Inhibits lipid peroxidation | Inhibited tissue injury in a mouse model of renal IRI | 62 |
| (1S,3R)-RSL3 | GPX4 inhibitor | Reduced tumour growth in a mouse xenograft model | 60 |
| Altretamine | GPX4 inhibitor | Drug approved for use in ovarian cancer | 79 |
| Baicalein | LOX inhibitor | Protected against multiple degenerative disorders, including stroke | 157 |
| Ferroptosis (cont.) | |||
| LOXBlock-1 | LOX inhibitor | Reduced brain infarct size in a mouse model of transient focal ischaemia | 156 |
| LOXBlock-2 | LOX inhibitor | Protected a mouse hippocampal cell line from glutamate-induced oxytosis | 189 |
| LOXBlock-3 | LOX inhibitor | Protected a mouse hippocampal cell line from glutamate-induced oxytosis | 189 |
| α-tocopherol | Inhibits lipid peroxidation | In general, protects cells and tissues, particularly in the brain, muscles and blood cells. In the context of GPX4 deficiency, prevented thromboembolic events78 and impaired parasite infections and CD8+ T cell homeostasis77 | 190 |
| Compound 968 | Glutaminase inhibitor | Reduced infarct size after IRI in isolated hearts of mice | 85 |
| L-buthionine sulfoximine | γ-GCS inhibitor | Triggered glutathione depletion and GPX4 inhibition, and prevented malignant transformation | 191,192 |
| CypD-dependent or CypD-independent necrosis | |||
| Cyclosporin A | CypD inhibitor | Protective in several mouse and rat models of IRI in organs, such as retina, heart, brain and kidney | 115, 193–195 |
| Sanglifehrin A | CypD inhibitor | Protective in several mouse and rat models of IRI in organs, such as heart and kidney | 131,196 |
| Pifithrin-μ | Inhibits p53 translocation to mitochondria | Protected mice from gamma-radiation-induced lethal haematopoietic syndrome | 124,197 |
γ-GCS, γ-glutamylcysteine synthetase; AD; Alzheimer disease; baicalein, 5,6,7-trihydroxyflavone; cIAP, cellular inhibitor of apoptosis protein; CypD, cyclophilin D; DPQ, 3,4-dihydro-5[4-(1-piperindinyl)butoxy]-1(2H)-isoquinoline; EAE, experimental autoimmune encephalomyelitis; GPX4, glutathione peroxidase 4; HD, Huntington disease; IBD, inflammatory bowel disease; IRI, ischaemia–reperfusion injury; LOX, lipoxygenase; LPS, lipopolysaccharide; MLKL, mixed lineage kinase domain-like protein; MNNG, N-methyl-Nʹ-nitro-N-nitrosoguanidine; NLRP3, NOD-, LRR- and pyrin domain-containing 3; PARP, poly(ADP-ribose) polymerase; PJ34, N-(5,6-dihydro-6-oxo-2-phenanthridinyl)-2-acetamide hydrochloride); pol(I:C), polyinosinic-polycytidylic acid; RIPK, receptor-interacting protein kinase; SIRS, systemic inflammatory response syndrome; TBI, traumatic brain injury; TNF, tumour necrosis factor.
As a general feature, necroptosis is linked to pathological conditions that have an overt inflammatory signature; examples of such conditions include Crohn disease, IRI, amyotrophic lateral sclerosis, toxic epidermal necrolysis13 and multiple sclerosis, among others14–16. Although it is not yet fully known whether necroptosis is the primary culprit of these diseases or secondary to these pathological conditions, inhibition of the kinase activity of RIPK1 in these pathological settings generally slows down or blocks disease progression.
The majority of this knowledge of necroptosis was gained using RIPK1 inhibitors, such as necrostatin-1, and animal models lacking cognate members of the so-called complex IIc (also called the necrosome), including RIPK3 and mixed lineage kinase domain-like protein (MLKL). However, caution should be taken in interpreting the results of such studies, as in some disease models, such as cerulean-induced pancreatitis, RIPK1 inhibition and RIPK3 deficiency may not always be interchangeable17,18. Moreover, genetic studies of necroptotic mediators often generate unexpected findings. For example, the kinase-inactivating D161N mutation of Ripk3 in mice is embryonic lethal owing to enhanced RIPK1-mediated apoptosis. This result implies that RIPK3 negatively regulates this form of cell death19 and suggests that RIPK3 inhibitors may lead to increased RIPK1-dependent apoptosis (discussed below).
A clear assignment of a role for necroptosis in diseases is far from trivial because the necroptosis proteins themselves are pleiotropic. For these reasons, and despite genetic studies20 confirming several roles of necroptotic mediators in diseases (including in sepsis, discussed below), it has not been possible to implicate necroptosis in disease processes using only RIPK-inhibition studies. Current efforts at inhibiting necroptosis focus on blocking the activity of RIPK1, RIPK3 and MLKL. The feasibility of inhibiting these nodes as an approach to inhibit necroptosis has been validated in models of systemic inflammatory response syndrome (SIRS).
Molecular processes underlying necroptosis.
Necroptosis is thought to be one of the possible outcomes following exposure to TNF. The response of cells to TNF is complex and, in most cases, leads to the activation of nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signalling. However, depending on the cell type and context and the addition of cell death sensitizers, TNF may induce apoptosis or necroptosis (FIG. 1).
When TNF binds to its receptor TNFR1, a receptor-associated ‘complex I’ assembles. This complex consists of TNFR1, TNFR1-associated death domain (TRADD), RIPK1, TNFR-associated factor 2 (TRAF2), cellular inhibitor of apoptosis protein 1 (cIAP1), cIAP2 and the linear ubiquitin chain assembly complex (LUBAC). Complex I provides the platform for a series of ubiquitylation and deubiquitylation reactions that control the switching between NF-κB signalling, cell survival signals and cell-death-inducing signals (FIG. 1). Ubiquitylation of RIPK1 by cIAP1 and cIAP2 stabilizes complex I and enables further recruitment of additional factors, including transforming growth factor-β (TGFβ)-activated kinase (TAK1) in complex with TAK1-binding protein 2 (TAB2). Ubiquitylation of RIPK1 by LUBAC creates a linear ubiquitylation platform for the recruitment of the inhibitor of the NF-κB kinase (IKK) complex, leading to RIPK1 phosphorylation and sustaining a ‘survival’ mode and consequently igniting the canonical NF-κB pathway21.
Alternatively, complex I may instead give rise to cell death signalling. Destabilization of the receptor-associated complex I results in the formation of a cytosolic complex IIa, which contains TRADD, RIPK1, caspase 8 and FAS-associated death domain (FADD)22, leading to apoptosis. In some cell lines, this complex IIa-mediated apoptosis is sensitized in the presence of cycloheximide, an inhibitor of translation. By contrast, the formation of cytosolic complex IIb is promoted by the inhibition or knockdown of cIAPs23, the inhibition or knockdown of TAK1 (REF. 24), or the knockdown of NF-κB essential modulator (NEMO)25.
Complex IIb consists of RIPK1, RIPK3, FADD and caspase 8, and favours RIPK1-kinase-activity-dependent apoptosis, which can be inhibited by necrostatin-1. In conditions of sufficient expression of RIPK3 and MLKL, or inhibition or reduced expression of FLICE-like inhibitory protein long isoform (FLIPL), complex IIc (the necrosome) is formed. Heteromeric procaspase-8–FLIPL negatively controls necroptosis by cleaving RIPK1, RIPK3 and cylindromatosis, a deubiquitylating enzyme that contributes to the destabilization of complex I26. Whether complex I, IIa and IIb exist as separate entities or form dynamic transition states with changing composition depending on the availability and the post-translational modifications of their constituents remains to be determined.
The necrosome complex is formed through the association of RIPK1 and RIPK3 via their RIP homotypic interaction motif (RHIM) domains and through a series of RIPK1 and RIPK3 auto- and transphosphorylation events (although there is no evidence for heteromerictransphosphorylation between RIPK1 and RIPK3). Activated RIPK3 phosphorylates and recruits MLKL, creating a supramolecular protein complex at the plasma membrane27–30.
It should be noted that apparently neither RIPK1 nor RIPK3 is linked exclusively with necroptosis regulation. RIPK1 kinase activity is required for complex IIb-associated apoptosis induction (as discussed above) and for ripoptosome formation, which occurs as a result of DNA damage and a loss of expression of cIAP1 and cIAP2 (REFS 31,32). In some cases (discussed below), the loss or inhibition of RIPK3 kinase activity can lead to the enhanced formation of complex IIb19,24,33.
Formation of the necrosome leads to the recruitment of MLKL. The recently elucidated structure of MLKL sheds light on its mechanism of activation34. MLKL consists of a carboxy-terminal pseudokinase domain that is connected to an amino-terminal four-helix bundle (4HB) domain by a two-helix linker. The 4HB domain is the cell death executioner domain and is kept in an inactive state by the pseudokinase domain. In humans, upon engagement of MLKL by the kinase domain of RIPK3, RIPK3 phosphorylates MLKL at Thr357 and Ser358 — which are situated in the pseudokinase domain α-helix — thereby destabilizing the closed structure, releasing the N-terminal 4HB domain and allowing oligomerization of MLKL at the plasma membrane34. MLKL oligomers are reportedly able to bind to negatively charged phospholipids, such as phosphoinositides and possibly cardiolipin. The association of MLKL with the plasma membrane is required for cell death execution via two non-exclusive models of mode of action. First, these oligomers could act directly as a pore-forming complex, contributing to plasma membrane destabilization29,30 or, second, they could act indirectly by serving as a platform that deregulates Ca2+ or Na+ ion channels27,28. In both cases, the formation of MLKL oligomers is associated with an increase in intracellular osmotic pressure and thus contributes to cell death. It should be stressed that the necroptotic machinery can also activate the inflammasome machinery (BOX 1), which may also contribute to some of the effects observed in experimental disease models in vivo. It is therefore important to note that, in vivo, some RIPK-dependent effects may be related to inflammasome activation rather than to necroptotic signalling. However, in the context of inflammatory and tissue-degenerative diseases, the effects of RIPK1 inhibitors on the inflammasome may be advantageous.
Box 1 |. Interactions between the inflammasome and the cell death machinery.
Inflammasomes are molecular platforms that are activated by a wide range of stimuli and danger signals and that induce the processing of pro-interleukin-1β (pro-IL-1β) to IL-1β. Different types of inflammasome respond to different stimuli and engage different downstream machinery137. The NLRP3 (NOD-, LRR- and pyrin domain-containing 3) inflammasome is the most promiscuous, as it responds to a broad range of intracellular alterations and phagocytosed agents. The host-derived factor that mediates the activation of the NRLP3 inflammasome is a matter of debate, but candidate mechanisms and factors include K+ efflux141, mitochondrial translocation of the inflammasome142, mitochondria-derived reactive oxygen species (ROS)143, cardiolipin143 and cathepsins144.
Necroptosis functions as a ‘back-up’ cell death mechanism when caspase 8-mediated apoptosis is inhibited. Indeed, viruses such as murine cytomegalovirus use caspase 8 inhibition to escape the immune system145, indicating that necroptosis may have evolved as a safeguard mechanism against such viruses.
Evidence linking the apoptotic–necroptotic and inflammasome machineries and pathways is emerging. For instance, in dendritic cells, caspase 8 deficiency potentiates the lipopolysaccharide (LPS)-mediated assembly and function of the NLRP3 inflammasome — an effect that is dependent on receptor-interacting serine/ threonine-protein kinase 1 (RIPK1), RIPK3, mixed lineage kinase domain-like protein (MLKL) and phosphoglycerate mutase family member 5 (PGAM5)146. By contrast, stimulation of bone marrow-derived dendritic cells with LPS leads to the formation of a RIPK1–RIPK3–FAS-associated death domain (FADD)–caspase 8 complex (also known as a ripoptosome- or complex IIb-like complex) that contributes to IL-1β processing, independently of RIPK1 or RIPK3 kinase activity147. Strikingly, despite the RIPK3- and MLKL-induced IL-1β processing in these LPS-stimulated dendritic cells, no cell death was evident, suggesting that the involvement of these necroptosis-associated factors in an inflammasome-activation context does not induce necroptosis, confirming similar findings from earlier studies146.
Another example of a necrosome–inflammasome interaction has been revealed in LPS-triggered Toll-like receptor (TLR) signalling: signalling via the TLR adaptor protein TIR-domain-containing adaptor-inducing interferon-β (TRIF) leads to cellular inhibitor of apoptosis protein 1 (cIAP1)- or cIAP2-mediated ubiquitylation of RIPK3 and cell survival148. By contrast, LPS stimulation in the absence of IAPs leads to a RIPK3-mediated and kinase-independent activation of caspase 8 that culminates in NLRP3 inflammasome activation and apoptosis. When both of the IAPs and caspase 8 are absent, LPS stimulation promotes inflammasome activation, which requires RIPK3 kinase activity and MLKL148.
Together, these findings suggest that, depending on the cellular conditions, the same factors could be engaged as a scaffold or as a kinase. A better understanding of how cell-death-related protein signalling is directly intertwined with the innate immune responses will better inform strategies to inhibit secondary tissue damage that is inflicted by the immune compartment. NLRP3 is an important pharmacological node for the regulation of IL-1β production and the induction of pyroptosis, a form of cell death dependent on caspase 1 cleavage of gasdermin D149,150, and is normally associated with the antimicrobial response in macrophages and T cells. As such, NLRP3 inhibition is being recognized as an attractive strategy to counteract inflammatory conditions associated with tissue damage and infection. Recently, a new and specific NLRP3 inhibitor, MCC950 (FIG. 2; TABLE 1), was reported to block canonical and non-canonical activation of NLRP3, without having any effects on the absent in melanoma 2 (AIM2), NLR family, CARD domain-containing 4 (NLRC4) or NLRP1 inflammasomes151. MCC950 reduced caspase 1-mediated maturation of IL-1β in mice and attenuated the severity of experimental autoimmune encephalomyelitis, a model of multiple sclerosis.
RIPK1 inhibitors.
The first inhibitors of necroptosis were identified in 2005 in a phenotypic screen for small molecules able to inhibit the necrotic cell death induced by TNF in human monocytic U937 cells10,35. These studies led to the identification of necrostatin-1 (FIG. 2; TABLE 1), which was subsequently shown to inhibit the kinase activity of RIPK1 (REF. 11). Following its identification, necrostatin-1 has been extensively used to probe the involvement of RIPK1 in cell death. Nevertheless, critical issues concerning the use of necrostatin-1 in vivo have emerged. Among the concerns is the recognition that necrostatin-1 and the indoleamine-2,3-dioxygenase (IDO) enzyme inhibitor methyl-thiohydantoin-tryptophan are the same molecule36–38; indeed, the IDO–kynurenine pathway modulates the innate and adaptive immune system. Targeting IDO is used to interfere in inflammation-associated tumorigenesis to break tumour immunotolerance and to sensitize tumours to cell death39. Moreover, the dose–response curve of necrostatin-1 demonstrates that at low concentrations it sensitizes mice to the lethality of TNF-induced SIRS, suggesting that this compound is relatively toxic; nonetheless, survival in TNF-induced SIRS is achieved at higher doses36,37. Structure–activity relationship analysis informed chemical alterations to necrostatin-1 to yield Nec-1s (also called 7-Cl-O-Nec-1), which lacks IDO-inhibitory activity, has increased plasma stability and has increased specificity for RIPK over a broad range of kinases37. In addition, the more stable and specific Nec-1s lacks the toxic effect described for necrostatin-1 in the SIRS model, suggesting that Nec-1s is a preferred tool for targeting RIPK1 in vivo.
The high-resolution structure of RIPK1 bound to Nec-1s revealed that Nec-1s binds in a relatively hydrophobic pocket between the N and C lobes, in close proximity to the activation loop. This binding was proposed to shift the catalytic triad residues in RIPK1, making it unable to stabilize ATP. On the basis of this model, it was proposed that Nec-1s locks RIPK1 in an inactive conformation that is unable to phosphorylate RIPK3 and consequently unable to assemble the necrosome40. In addition, a GlaxoSmithKline group used a fluorescence polarization assay to assess molecules that can bind to the catalytic site of RIPK1 (REF. 41). This assay led to the discovery of three classes of compounds (1-aminoisoquinolines, pyrrolo[2,3-b]pyridines and furo[2,3-d]pyrimidines) that were all characteristic of the type II kinase inhibitor class, which targets the inactive, DFG-out conformation (DLG in the case of RIPK1). This class of inhibitors is believed to dislocate the phenylalanine side chains of the catalytic site inwards to obstruct the ATP-binding pocket. Moreover, one of the compounds of the furo[2,3-d]pyrimidine series, Cpd27, showed potent anti-RIPK1-kinase activity (FIG. 2; TABLE 1) and blocked TNF-induced lethality in a SIRS mouse model.
Recently, it was reported that the antileukaemic agents and BCR–ABL inhibitors ponatinib and pazopanib are also inhibitors of both RIPK1 and RIPK3 (REFS 42,43). Ponatinib and pazopanib were shown to inhibit RIPK1-and RIPK3-dependent cell death and transcription of TNF. Furthermore, fusion of the scaffold of ponatinib and Nec-1s generated a hybrid molecule that was a highly potent and selective RIPK1 inhibitor; this inhibitor, named PN10 (FIG. 2; TABLE 1), robustly protected against TNF-induced SIRS in vivo42.
RIPK3 inhibitors.
Interest in the development of RIPK3 inhibitors was stimulated by the observation that Ripk3-knockout mice are viable and without a spontaneous phenotype44; indeed, these animals were frequently used for studying the possible contributions of necroptotic cell death to pathological settings20. Also, the recognition that necroptosis could be engaged independently of RIPK1 spurred discussions that RIPK3 inhibitors might prove to be suitable for protection against a broad range of necroptotic pathologies that are independent of RIPK1 kinase function, such as viral infection, pancreatitis, aortic aneurism45 and Gaucher disease46–48.
Despite these data, studies using two different RIPK3-kinase-dead mice have yielded contradictory results. Mice expressing the kinase-dead variant (D161N) of RIPK3 die from increased apoptosis, suggesting that RIPK3 kinase activity has a crucial survival function19. Remarkably, another RIPK3-kinase-dead mutant (K51A) was viable, despite lower expression levels. The scaffold function of RIPK3 in stabilizing RIPK1 in complex IIb, thus propagating RIPK1-dependent apoptosis, is only revealed at higher protein expression levels. This observation is supported by studies of two RIPK3 kinase inhibitors, GSKʹ843 and GSKʹ872, which, at higher concentrations, promote TNF-induced RIPK1-dependent apoptosis and caspase 8 activation33.
The ability of the first-identified series of RIPK3 inhibitors to bind to the RIPK3 kinase domain was tested, and this led to the identification of compounds GSKʹ840, GSKʹ843 and GSKʹ872 (REF. 33) (FIG. 2; TABLE 1). These compounds inhibited RIPK3 with high specificity in a panel of 300 other human kinases; in particular, GSKʹ840 exhibited the most specific profile. GSKʹ840 is particularly interesting as, in contrast to GSKʹ843 and GSKʹ872 (REF. 33), it does not induce RIPK1-kinase-activity-dependent apoptosis at higher concentrations. Unfortunately, however, GSKʹ840 is unable to inhibit murine RIPK3, preventing its assessment in murine experimental disease models.
MLKL inhibitors.
The first compound reported to inhibit MLKL was (E)-N-(4-(N-(4,6-dimethylpyrimidin-2-yl) sulfamoyl)phenyl)-3-(5-nitrothiophene-2-yl)acrylamide (known as necrosulfonamide (NSA))49 (FIG. 2; TABLE 1). In fact, NSA was used as a probe to identify MLKL as a downstream target of RIPK3. Upon necroptosis induction by TNF, mCherry-labelled RIPK3 was observed to generate punctate structures in cells that resembled the amyloid-like structures described for the RIPK1–RIPK3 interaction50. As the formation of these structures could be prevented by necrostatin-1 but not by NSA, it was concluded that NSA does not affect RIPK3-dependent phosphorylation of MLKL. Moreover, NSA is able to protect human cells, but not murine cells, from necroptotic stimuli, precluding its use in murine preclinical models. This species specificity is because NSA alkylates the Cys86 of human MLKL (thus preventing MLKL oligomerization and translocation to the cell membrane), which is absent in murine MLKL49. As Cys86 in human MLKL is an exposed cysteine, NSA may potentially be promiscuous; therefore, it will probably be challenging to further improve on the NSA scaffold to create compounds that have improved potency, specificity and activity against murine MLKL9.
The potential promiscuity of NSA might be circumvented by a new class of MLKL inhibitors based on ‘compound 1’ (also known as GW806742X or SYN-1215) (FIG. 2; TABLE 1), which was recently described to target the pseudokinase domain of MLKL34. The 4HB domain of MLKL is kept inactive by its interaction with the pseudokinase domain, which is released upon RIPK3-mediated phosphorylation at Ser345 and Ser347 in the activation loop of MLKL. Thus, compounds that target the pseudokinase domain seem to block the switch that activates MLKL upon RIPK3-mediated phosphorylation, thus preventing MLKL oligomerization and translocation. Hence, targeting the catalytically dead, pseudokinase activity of MLKL is of potential therapeutic value to inhibit MLKL-induced necroptosis. Somewhat surprisingly, however, recent experiments have indicated that compound 1 also binds to RIPK1 (J. Silke, personal communication) and inhibits its kinase activity (D. Vucic, personal communication). Thus, some of the anti-necroptotic activity of compound 1 may be due to its inhibition of RIPK1, which means that compound 1 also cannot decisively be used to implicate MLKL in necroptosis, and that the specificity of compound 1 should be further investigated.
Inhibiting necroptosis in sepsis.
Sepsis is the leading cause of mortality in critically ill patients and is characterized by an acute, systemic immune response that culminates in multiple organ failure. Unrestrained cell death has been implicated as one of the major culprits during organ failure. A hallmark of sepsis is the oxidative burst and release of pro-inflammatory cytokines such as TNF, and lipopolysaccharide injections have been widely used as a mimic of the hyper-inflammatory phase of sepsis. Interestingly, inhibition of caspases by the bona fide pan-caspase inhibitor Z-VAD-FMK strongly sensitizes mice to TNF-induced SIRS and shock51, indicating that blocking apoptosis has a detrimental effect.
During TNF-induced SIRS and in the mild caecal ligation puncture model of septic shock, RIPK3 deficiency prevents the huge increase in circulating cell death markers and promotes survival52, suggesting that targeting RIPK3 in sepsis could be beneficial. Remarkably, caecal ligation puncture-mediated lethality was not restored in Mlkl-knockout mice53, suggesting that the functions of RIPK3 and MLKL may not completely overlap in this context. In a model of TNF-induced SIRS, pretreatment with necrostatin-1 or Nec-1s strongly inhibited lethality37, and this finding was supported by studies showing that RIPK1 kinase-inactive mice are also resistant to the lethality of TNF-induced SIRS and to the lethality of septic shock induced by a combination of TNF and Z-VAD-FMK19,54. Together, these data suggest that RIPK1 and RIPK3 inhibitors might be effective in treating SIRS and related diseases by targeting not only necroptosis but also other pro-inflammatory conditions regulated by these kinases.
Ferroptosis
Ferroptotic cell death was first described in a high-throughput screening campaign to identify molecules that could selectively induce cell death in isogenic cells carrying a RAS mutant isoform55. This campaign resulted in the identification of erastin (FIG. 3; TABLE 1); surprisingly, this compound was found to induce a regulated but non-apoptotic form of cell death that depended on cellular iron stores; overexpression of oncogenic HRAS sensitized the cells to this alternative form of cell death induced by erastin. Ferroptosis was ultimately found to be a form of cell death characterized by iron-dependent lipid peroxidation56. Lipid peroxidation is negatively regulated by the cystine–glutamate antiporter system Xc−, which provides cysteine that is used for glutathione and protein biosynthesis in the cell57,58. Glutathione is in turn crucial for the phospholipid peroxidase activity of glutathione peroxidase 4 (GPX4), which protects cells against lipid oxidation by reducing phospholipid hydroperoxides59,60 (FIG. 4). Therefore, the glutathione–GPX4 axis presents unique functions, as it is the sole cellular system responsible for the efficient repair of oxidized phospholipids. Although ferroptosis bears similarities to oxytosis (BOX 2), the morphological, biochemical and genetic uniqueness of ferroptosis has been demonstrated56.
Figure 3. Chemical structures of ferroptosis inducers and inhibitors.

The mechanisms of action, key functions and references for these inhibitors are provided in TABLE 1.
Figure 4. Upstream events in the control of ferroptosis.

The heterodimeric cystine–glutamate antiporter system Xc− exchanges one molecule of extracellular cystine (or cystathionine173) for one molecule of intracellular glutamate57. Once taken up by cells, cystine is reduced by reduced glutathione (GSH) or thioredoxin reductase 1 (TXNRD1)81 to cysteine, which is subsequently used for protein and GSH synthesis. Cysteine might also be provided by the transsulfuration pathway83. GSH is synthesized from cysteine, glutamate and glycine in two consecutive steps by γ-glutamylcysteine synthetase (γ-GCS) and glutathione synthase (GSS) at the expense of two molecules of ATP. Glutathione peroxidase 4 (GPX4) is one of the central upstream regulators of ferroptosis60: it prevents lipoxygenase (LOX) overactivation and lipid peroxidation63. GPX4 preferentially reduces phospholipid hydroperoxide (PL-OOH) to its corresponding alcohol phospholipid hydroxide (PL-OH), using two molecules of GSH59. Oxidized glutathione (GSSG) is then recycled back by glutathione reductase (GSR) using electrons from NADPH/H+. Concerted action of this pathway is essential to control the formation of oxidized phospholipids. A series of ferroptosis inducers have been developed and characterized that interfere at the different upstream events, either inhibiting cystine uptake (for example, erastin56, glutamate174, sulfasalazine175, (S)-4-carboxyphenylglycine176 (S-4-CPG) and sorafenib91), GSH biosynthesis (L-buthionine sulfoximine (BSO)) or GPX4 activity ((1S, 3R)-RSL3 (REF. 60) or altretamine79). Ferrostatins56, liproxstatin61, α-tocopherol and the iron chelator deferoxamine inhibit lipid peroxidation and signalling events downstream of GPX4 function. CBS, cystathionine β-synthase; GLS, glutaminase; CTH, cystathionase.
Box 2 |. Ferroptosis and oxytosis: lessons learned from a close relative.
Ferroptosis is related to another paradigm of regulated, non-apoptotic cell death: oxytosis. Both types of cell death are engaged by similar or, in some cases, even the same triggers of cell death, such as inhibition of the cystine–glutamate antiporter system Xc−. For example, similar to ferroptosis, oxytosis can be triggered in neuronal cell lines lacking N-methyl-D-aspartate (NMDA) receptors via the glutamate-induced inhibition of system Xc− and consequent depletion of glutathione152. Although ferroptosis and oxytosis share a common trigger mechanism (inhibition of the xCT light chain of Xc−) and a common execution mechanism (lipid peroxidation), lipoxygenases (LOXs) appear to have a prominent role in oxytosis, whereas their role in ferroptosis is not as clear.
During oxytosis, there is an increase in the so-called peroxide tone, an increase in the translocation of arachidonate 12-lipoxygenase (ALOX12) and ALOX15 to cellular membranes such as mitochondria followed by lipid peroxidation153, and an increase in the relocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus154,155. As such, it was suggested that LOXs might be integral players in neuronal cell damage following acute damage, such as following stroke or traumatic brain injury156. Indeed, putative pharmacological targeting of ALOX15 using the broad-range LOX inhibitor baicalein (5,6,7-trihydroxyflavone) (FIG. 3; TABLE 1) has been shown to attenuate damage after stroke in mice157. Moreover, Alox15-knockout mice have decreased lesions following experimental stroke158, although it is unclear whether this effect of ALOX15 deficiency on cell death occurs in a cell-autonomous manner or is due to altered immune function. Overall, the contribution of LOX enzymes in ferroptosis is not yet fully understood. Although LOX inhibitors, which are relatively promiscuous, can inhibit ferroptosis61,63, they seem to do so in a LOX-independent manner.
The fact that LOXs appear to be involved in oxytosis but not in ferroptosis may be because ferroptosis is associated with a more complex oxidizing environment, meaning that other sources of reactive oxygen species (ROS) such as NADPH oxidases may confer a more important contribution. In accordance with this notion, deletion of the ferroptosis regulator glutathione peroxidase 4 (GPX4) or combined deletion of the genes encoding ALOX15 and GPX4 led to ferroptotic cell death following injury61, whereas knockdown of Alox5 in Gpx4−/−;Alox15−/− lung fibroblasts led to partial protection from cell death. These findings suggest that more than one LOX is implicated in Gpx4-deletion-mediated ferroptosis, and indicate that there may be functional redundancy and cooperativity among LOX family members. Thus, it is plausible that oxytosis is a broader cell death programme that involves LOX, and that certain neuronal cells require certain ALOX isoforms to undergo oxytosis. In contrast to LOXs, AIF is involved in only oxytosis via a twofold mechanism: first, via its nuclease activity and, second, via a preconditioning effect, whereby the loss of AIF decreases levels of ROS159, although the exact contribution of each of these mechanisms is still under investigation.
Since its discovery, ferroptosis has not only been shown to be an attractive anticancer mechanism but has also been implicated in a broad range of pathological conditions, such as tissue IRI in kidney and liver. Moreover, pharmacological inhibition of ferroptosis has been shown to be feasible through distinct mechanisms by the use of potent inhibitors of lipid peroxidation, which have already provided encouraging results in the context of IRI in organs such as the liver, the kidney and the heart.
Disease relevance of ferroptosis.
Two recent studies have demonstrated that the pharmacological inhibition of ferroptosis can have a major impact during IRI. A ferroptosis inhibitor called liproxstatin-1 (FIG. 3; TABLE 1) increased overall survival by approximately 35% in a model of acute renal failure caused by the induced loss of Gpx4 in mice. Moreover, it improved the overall hepatic function of mouse subjected to hepatic IRI61. Concomitantly, third-generation ferrostatins, which also inhibit ferroptosis, were shown to decrease kidney damage upon ischaemia–reperfusion62. Collectively, these results suggest that ferroptosis could be a common denominator in IRI, independent of tissue origin.
Ferroptosis has also been implicated in neurological disorders. This suggestion is based on genetic studies demonstrating that GPX4 is essential for proper neuronal tissue development and homeostasis, as neuron-specific Gpx4 ablation causes neurodegeneration in mice63. In support of this observation, one study showed that the inducible loss of Gpx4 in adult mice leads to loss of neurons in the hippocampus and early lethality64. In addition, Gpx4+/− animals have an increased number of amyloid plaques owing to increased expression of β-secretase 1 (one of the enzymes that cleaves amyloid precursor protein into amyloid-β), supporting a potential role of GPX4 in the early stages of Alzheimer disease pathogenesis65. Similarly, in a mouse model of Alzheimer disease, whole-brain extracts showed a marked increase in oxidized lipid by-products, whereas Gpx4 expression was markedly decreased owing to the downregulation of the guanine-rich sequence-binding factor66, a factor known to control GPX4 synthesis67. The susceptibility of neurons to ferroptosis is further supported by other studies suggesting its involvement in other neurological conditions, including Parkinson disease68–70, Huntington disease71, cerebello-cortical atrophy72, traumatic brain injury73 and Smith–Lemli–Opiz syndrome74. Investigations using inducible Gpx4-null mice further demonstrated that GPX4 is essential for retinal protection75, hair follicle morphogenesis76, CD8+ T cell homeostasis77 and proper vascular functions78. These data therefore indirectly implicate the ferroptotic process in degenerative processes of the respective tissues.
Despite the recognition that blocking ferroptosis may be a valuable therapeutic approach for the above-mentioned disease conditions, it has also become evident that some tumours acquire a strong dependence on GPX4 and system Xc−, as reported for renal cell carcinomas and B cell-derived lymphomas and a subset of triple-negative breast cancer cell lines; thus, inducing ferroptosis could represent a new therapeutic anticancer strategy. A better understanding of the precise metabolic signature that leads to increased ferroptosis resistance will provide the basis for precision therapeutics aimed at triggering ferroptosis in such tumour entities60.
Ferroptotic mechanisms.
The ferroptotic pathway is illustrated in FIG. 4. Much of what is known about how to inhibit and trigger ferroptosis has been gained by the use of small-molecule inducers and inhibitors of this form of cell death. One of the ways in which ferroptosis can be triggered is by inhibiting system Xc− (REF. 56) (FIG. 4). This inhibition leads to cellular cysteine starvation and concomitant depletion of glutathione. Known small-molecule inhibitors of system Xc− include class I ferroptosis inducers such as erastin, sorafenib, sulfasalazine and the neurotransmitter glutamate. Recently, ferroptosis was also shown to be induced downstream of glutathione depletion via inhibition of GPX4 (REF. 60) by class II ferroptosis inducers, such as (1S, 3R)-RSL3, FIN56 (REF. 60), or altretamine (also known as hexamethylmelmine)79. Thus, the small molecules belonging to class I and class II ferroptosis-inducing agents trigger ferroptosis by inhibiting system Xc− and GPX4, respectively.
System Xc− and GPX4 are important players in the cellular antioxidant network. System Xc− is a heterodimeric 12-pass transmembrane cystine–glutamate anti-porter and consists of the xCT light chain (also known as SLC7A11), which mediates cystine transport specificity, and the 4F2 heavy chain (also known as SLC3A2), a sub-unit shared by different transporters57. Once taken up by cells via system Xc−, cystine is subsequently reduced, presumably by glutathione80 or thioredoxin reductase 1 (REF. 81), to cysteine.
Recently, it was reported that some of the tumour-suppressive activity of p53 may be attributable to its ability to inhibit system Xc−, which increases the sensitivity of the tumour cell to ferroptosis. Another tumour suppressor that has been linked to increased ferroptosis sensitivity is retinoblastoma protein, although its mechanism is unknown82. In addition, it was recently demonstrated that some cells can bypass their dependence on system Xc− but not on GPX4 (and therefore can be resistant to ferroptosis through inhibition of system Xc− but not of GPX4) by acquiring cysteine via the metabolism of methionine through the transsulfuration pathway83. In addition to its use in protein synthesis, cysteine is the rate-limiting substrate for the synthesis of the major cellular antioxidant glutathione by γ-glutamylcysteine synthetase (encoded by GGCS) and glutathione synthetase. In turn, glutathione directly links system Xc− and the transsulfuration pathway to GPX4 activity. Of all of the mammalian enzymes that rely on glutathione as an electron source, GPX4 is the rate-limiting enzyme that mediates the pro-survival function of glutathione; Gpx4-knockout mice die at the same embryonic stage as Ggcs-knockout mice84.
GPX4 is unique in its ability to directly reduce oxidized phospholipids and cholesterol hydroperoxides59. Where and how these oxidized species are formed are still under investigation. A recent study has demonstrated that ferroptosis requires the process of glutaminolysis85 for reactive oxygen species (ROS) production, and that this production could be mitigated by inhibiting glutaminase using compound 968 (FIG. 3; TABLE 1) or by knocking down a mitochondrial glutaminase isoform (GLS2) and glutamic-oxaloacetic transaminase 1. Moreover, ROS production could be restored by adding α-ketoglutarate, thus introducing speculation about a putative role of the α-ketoglutarate dehydrogenase complex as a source of ROS86. This role could be relevant, as the α-ketoglutarate dehydrogenase complex generates ROS that leak into the inner mitochondrial membrane, which could help to reconcile earlier observations: namely, that severe mitochondrial damage occurs during ferroptosis56,61 even though ferroptosis does not require a functional electron transport chain, and that targeting antioxidant to the mitochondrial matrix failed to prevent this form of cell death.
Another source of ROS that has been implicated in ferroptosis is the family of NADPH oxidases (NOXs); this family of enzymes has been shown to be essential for the sustained activation of proliferative signals derived from growth factors87. The activity of NOX has also been linked to an increased flow of carbohydrates through the pentose phosphate pathway (PPP), resulting in a constant supply of the NOX substrate NADPH88. Fructose-1,6-bisphosphatase (FBP1) restrains the PPP in a catalytic activity-independent manner by directly interacting with the ‘inhibitory domain’ of hypoxiainducible factor (HIF), thus inhibiting the function of HIF; a loss of FBP1 is found in almost all renal cell carcinomas and leads to increased PPP activity89. This mechanism might explain the high sensitivity of renal cell carcinomas to ferroptosis60.
Furthermore, ferroptosis is critically linked to lipid metabolism, as it requires polyunsaturated fatty acids (PUFAs) for oxidation. This association is highlighted by the vital function of GPX4 in negatively regulating ferroptosis. Lipid oxidation is essential for the ferroptotic process, although the precise mechanism of lipid oxidation is unknown. A recent screen in human haploid cells indicated that loss of lysophosphatidylcholine acyltransferase 3 (LPCAT3) or of acyl-CoA synthetase long-chain family member 4 (ACSL4) confers resistance to ferroptosis (possibly via the decrease of PUFAs in specific phospholipids)90. How exactly the genetic loss of either of these enzymes confers resistance to ferroptosis is unknown and warrants further investigation. However, because these enzymes have a substrate preference for arachidonic acid, the results from the screen in human haploid cells indicate that arachidonic acid oxidation products are likely to be essential for ferroptosis. This notion is consistent with other findings that arachidonic acid is oxidized during ferroptosis61.
Many questions remain to be answered. For example, what are the molecular events downstream of lipid peroxidation? Are there clearly defined molecular events during ferroptosis or just nonspecific lipid peroxidation and plasma membrane rupture? Does lipid peroxidation mark a point of no return? Indirect evidence points to the requirement of downstream events: cells resistant to ferroptosis express higher levels of members of the aldoketo reductase family, enzymes responsible for the detoxification of lipid breakdown products91. These species could potentially modify nucleophilic residues in proteins, such as cysteines, leading to the activation or loss of function of critical unrecognized signalling molecules and networks.
Finally, it is worth noting that ferroptosis was initially linked to its ability to target cells with activated RAS– RAF–MEK signalling, and was dependent on the expression of voltage-dependent anion channel 2 (VDAC2) and VDAC3 (REF. 92), although the roles of these channels or of RAS signalling in ferroptosis are not fully understood. RAS-activating mutations sensitized cells to ferroptosis when the gene was overexpressed, but the RAS mutational status in genuine tumour cells does not predict sensitivity to ferroptosis60, and its pharmacological inhibition does not always protect cells from ferroptosis93. By contrast, VDACs are only required for erastin-induced ferroptosis but not for ferroptosis induced by other compounds55. These discrepancies probably preclude the targeting of RAS and VDACs as potential cytoprotective therapeutic options to halt ferroptosis.
Inducers of ferroptosis.
The recognition that some tumours become highly sensitive to ferroptosis suggests that system Xc− and GPX4 inhibitors could have high therapeutic indices for cancer treatment94–96. Specifically, a recent study showed that a subset of triple-negative breast cancer cells that are auxotrophic for glutamine rely on the deamination of glutamine by glutaminase to glutamate to fuel xCT — linking a lack of glutamine metabolism to ferroptosis. Accordingly, inhibition of system Xc− in such cells with sulfasalazine (a drug that has been approved for more than 40 years for inflammatory bowel disease) was shown to be effective in reducing tumour growth in vivo, underscoring the importance of this pathway in breast tumours with poor prognosis97. Sulfasalazine is also currently marketed to treat rheumatoid arthritis, although its activity in this disease appears to be unrelated to system Xc− inhibition. Moreover, sulfasalazine is a relatively low-potency and metabolically unstable system Xc− inhibitor compared with erastin; therefore, stronger effects of erastin or its analogues should be expected in models in which sulfasalazine was efficacious. In addition, erastin-derived inhibitors of system Xc−, such as piperazine erastin, have been shown to be effective in other tumour xenograft models60. Recently, sorafenib, a multikinase inhibitor drug approved for treating primary kidney cancer, advanced primary liver cancer and radioactive iodine-resistant advanced thyroid carcinoma, was also found to induce ferroptosis in cancer cells93 and does so by inhibiting system Xc− (REF. 91). Therefore, it is tempting to speculate that ferroptosis may be partially responsible for the antitumour effect of sorafenib.
Ferroptosis can also be triggered independently from glutathione depletion; this effect was recently demonstrated by a second class of ferroptosis-inducing compounds. This class is exemplified by RSL3, which induces cell death with similar features as that caused by erastin, but without affecting glutathione levels. The target of RSL3 was shown to be GPX4 (REF. 60). Recently, altretamine98, which is proposed as a salvage therapy for relapsed ovarian cancer, was identified using network perturbation analysis as a novel GPX4 inhibitor79. At present, targeting GPX4 appears to be the preferred approach for the development of drugs that trigger ferroptosis. By contrast, system Xc− inhibitors present more pleiotropy; they lead to cysteine deprivation and concomitant endoplasmic reticulum stress.
Inhibitors of ferroptosis.
The first-characterized ferroptosis inhibitors — a group of arylalkylamines known as the ferrostatins that were identified in 2012 — were found to prevent erastin-induced cell death56. Ferrostatins prevent oxidative lipid degradation during ferroptosis, thereby preventing cell demise in cells and organotypic cultures71. Mechanistically, ferrostatins and related compounds may function by interrupting the radical chain reaction responsible for lipid peroxidation, or by donating two electrons to allylic lipid hydroperoxide substrates, thus preventing the oxidative decomposition of phospholipids71. Analysis of the structure–activity relationship associated with the ferrostatin scaffold revealed that a lipophilic anchor is needed for the ferrostatin-mediated inhibition of ferroptosis, presumably to target the compounds to a lipophilic membrane environment. Ferrostatin-1 (FIG. 3; TABLE 1) exhibits low plasma and metabolic stability, but a new series of ferrostatins with improved plasma stability and the ability to retard ferroptosis in the pathological setting of IRI in the kidney was recently described62.
A screening campaign to identify small molecules able to prevent ferroptosis was conducted in a GPX4-deficiency model and led to the discovery of a class of spiroquinoxalines termed liproxstatins. Liproxstatin-1 (FIG. 3; TABLE 1) inhibited ferroptosis in an in vivo model of Gpx4 ablation and in a preclinical model of hepatic IRI61. The compound exhibited good absorption, distribution, metabolism and excretion profiles, making it a good candidate with which to examine the ferroptotic contribution in diverse preclinical models. At present, the precise mechanism of how liproxstatin-1 prevents ferroptosis and lipid oxidation is unclear; however, owing to the presence of a secondary amine that allows an efficient stabilization of electron deficiency, it is plausible that, similar to ferrostatins, these molecules function through a mechanism that relies on the efficient reduction of lipid radicals.
Ferrostatins and liproxstatins do not behave like classical antioxidants. Indeed, α-tocopherol (one of the most efficient chain-breaking lipophilic antioxidants) is 10–100-fold less efficient than ferrostatin and liproxstatin at terminating lipid peroxidation61. A possible explanation for the superior efficiency of ferrostatins and liproxstatins is that they may reduce lipid hydroperoxides to inert alcohol, suggesting that they function as putative GPX4 mimetics. Another possibility might be that these compounds, in contrast to α-tocopherol, act catalytically owing to their potential to trap multiple radicals per molecule. This mechanism has been recently suggested for the diarylamines, which are closely structurally related to the ferrostatins71. It is therefore proposed that diarylamines would be highly efficient in preventing the oxidation of PUFAs during the course of ferroptosis. Moreover, it seems that ferrostatins and liproxstatins are highly selective for inhibiting ferroptosis triggered by GPX4 deletion or ferroptosis-inducing agents, such as erastin and the GPX4 inhibitor (1S, 3R)-RSL3 (FIG. 3; TABLE 1), as ferrostatins and liproxstatins failed to protect against other forms of cell death in vitro, including apoptosis and necroptosis56,61. One possible explanation for these findings is that these classes of compounds may be targeted to specific membranes, the oxidative modulation of which is required for ferroptotic signalling.
Other regulated forms of necrosis
Other forms of regulated necrosis that have been implicated in pathological conditions and that offer significant potential for drug development include parthanatos and CypD-mediated necrosis. Parthanatos has been mostly studied in the context of neuronal damage and neuro-degeneration, and its main hallmark is overactivation of poly (ADP-ribose) (PAR) polymerase 1 (PARP1). CypD-mediated necrosis is a general mechanism involved in IRI, and its pharmacological inhibition has already shown encouraging results.
Parthanatos.
Parthanatos is a form of cell death implicated in neuropathological conditions such as Parkinson disease99, and is characterized by the overactivation of the nuclear protein PARP1 by a wide array of stimuli, such as DNA damage and ROS production100. Three PARP proteins have been implicated in DNA damage repair: PARP1 and PARP2, which share a high degree of homology, and PARP3, which is less well characterized and appears to be functionally distinct from the other two PARPs. PARP1 and PARP2 have complex effects and are pleiotropic; they are known to be involved in DNA repair, chromosome stability and the inflammatory response101. However, in contrast to PARP2 and PARP3, PARP1 is sufficient for the process of parthanatos; specific inhibitors of the other PARP isoforms are unable to prevent this type of cell death102 (FIG. 5).
Figure 5. Key signalling events in parthanatos.
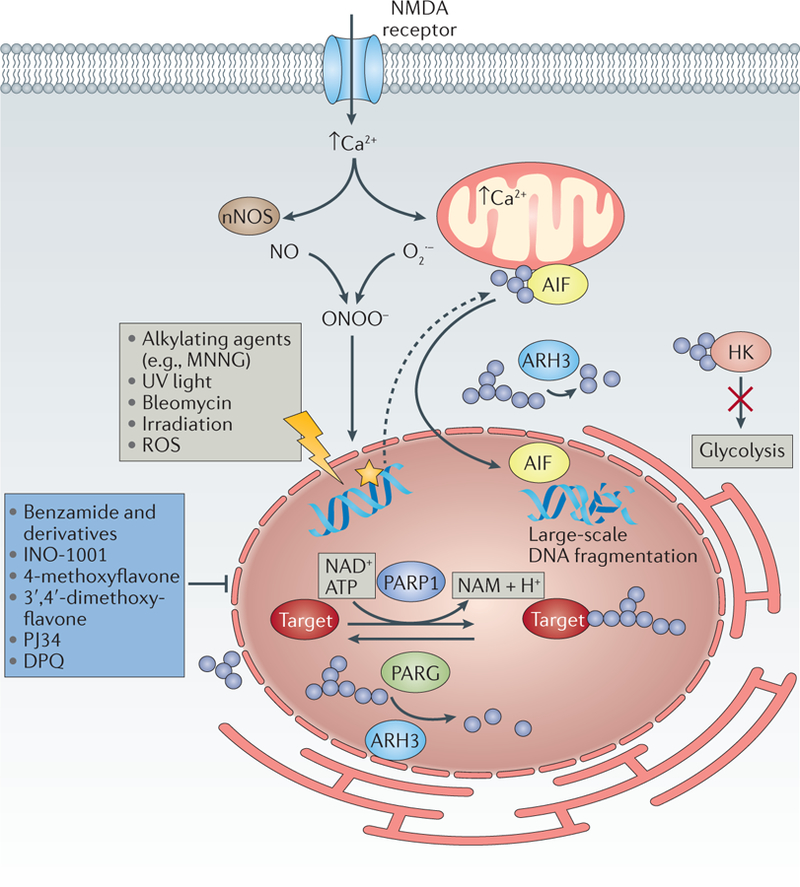
DNA-damaging agents trigger poly(ADP-ribose) (PAR) polymerase 1 (PARP1) activation, the DNA-damage response and the repair response. In neurons, sustained stimulation of N-methyl-D-aspartate (NMDA) receptors (for instance, following stroke or traumatic brain injury) also leads to parthanatos, through cellular increases in Ca2+ levels, activation of neuronal nitric oxide synthase (nNOS), increased superoxide production and generation of highly toxic peroxynitrite (ONOO–) ions177. PARP-mediated PARylation (blue circles) of target proteins, and of PARP1 itself, leads to recruitment of factors required for DNA repair (not shown). The increase in PARylated proteins is counterbalanced by poly(ADP-ribose) glycohydrolase (PARG), which cleaves glycosidic bonds in PAR polymers on target proteins and thereby regulates PAR length and releases PAR polymers. ADP-ribosyl-acceptor hydrolase 3 (ARH3) further degrades PAR polymers105. Excess DNA damage can elicit parthanatos by inducing PARylation of apoptosis-inducing factor (AIF)104 and relocation of AIF into the nucleus, as well as PARylation of hexokinase (HK)114, which impairs its glycolytic activity. AIF translocation to the nucleus is associated with large-scale fragmentation of DNA and cell death. Inhibitors of parthanatos include the PARP1 inhibitors benzamide178 and its derivatives, such as INO-1001, methoxyflavones179, PJ34 (REF. 180) and DPQ (3,4-dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2H)-isoquinolinone)181. MNNG, N-methyl-Nʹ-nitro-N-nitrosoguanidine; NAM, nicotinamide; NO, nitric oxide; O2–, superoxide anion; ROS, reactive oxygen species.
Activated PARP leads to parthanatos by transferring ADP-ribose groups from NAD+ to their targets (such as apoptosis inducing factor (AIF) or hexokinase (HK)), and subsequent translocation of AIF to the nucleus103. Reconstitution experiments with wild-type and mutant AIF in harlequin neuronal cells, which have reduced AIF expression104, showed that relocation of AIF from mitochondria to the nucleus depends on its direct interaction with PAR via a conserved region of AIF. This process is negatively regulated on at least at two levels: first, by the ADP-ribosyl-acceptor hydrolase 3 (ARH3), a protein able to lower PAR levels in the nucleus and cytoplasm102; and second, by the protein IDUNA (also known as E3 ubiquitin-protein ligase RNF146), a small cytosolic protein that binds PAR polymers and thus acts as a buffering protein, thereby preventing PARP1-induced AIF release and cell death105.
Parthanatos can be pharmacologically blocked by PARP1 inhibitors (FIG. 2; TABLE 1); however, the executioner mechanism for parthanatos is unclear, and so no other druggable node has been identified so far. Early studies of PARP were primarily focused on the importance of PARP in DNA repair, and nonspecific PARP inhibitors have entered clinical trials to treat specific tumours alone or in combination with selected anticancer drugs (reviewed in REF. 106). An increased recognition of the role of PARP in other biological processes was stimulated by the study of PARP1-deficient mice. These mice were shown to be protected from several stress conditions, such as stroke, kidney IRI107 and retinal degeneration108, which spurred the development of new classes of specific PARP1 inhibitors of various structural classes. These inhibitors include phenanthridinones such as PJ34 (N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-(N,N-dimethylamino)acetamide hydrochloride)109, isoquinolones, isoquinolinones110,111 and many other classes of PARP inhibitors that show half-maximal inhibitory concentrations in the low-micromolar to nanomolar range109. In addition to oncology indications, there have been two clinical trials with the aim of proving or disproving the validity of targeting parthanatos in degenerative disease scenarios: a small pilot trial testing the PARP1 inhibitor INO-1001 (also known as 3-aminobenzamide)in myocardial infarction112 and a recently completed Phase I study of JPI-289 in healthy participants as a basis for treating stroke113.
At present, it remains difficult to ascertain the contribution of activating PARP1 in triggering parthanatos in pathological conditions, as PARP1 has so many additional roles, including those in the DNA repair response and the immune response. Therefore, studies using PARP inhibitors and Parp-knockout animals should be interpreted with caution, and the discovery of downstream targets other than AIF should help to unequivocally assign the role of parthanatos in disease conditions.
A better characterization of parthanatos may result in alternative approaches to targeting this type of cell death. For instance, one study investigated the initial observation that PAR accumulation is correlated with a reduction of cellular NAD+ and an energetic collapse, which was assumed to be due to NAD+ depletion. It was reported that energetic collapse during parthanatos is instead mediated by defects in glycolysis that result from PAR-dependent inhibition of HK, before NAD+ depletion114. Thus, novel small molecules that can inhibit the PAR–HK interaction could therefore prevent bioenergetic collapse and parthanatos-mediated cell demise.
CypD-mediated necrosis.
Another form of regulated, non-apoptotic form of cell death that is amenable to pharmacological intervention and that has been implicated in some pathological conditions is CypD-dependent necrosis. CypD-dependent necrosis has been implicated in cardiac and renal IRI115, muscular dystrophy116, thrombosis117, diabetes118 and in a model of multiple sclerosis119. This type of cell death relies on the formation of the so-called mitochondrial permeability transition pore (MPTP), which depends on the presence of CypD. Candidates for causing the assembly of the MPTP include ions and metabolic intermediates such as ADP, ubiquinones and ROS120. Recent studies have suggested that the MPTP is composed of dimers of the ATP synthase complex, which can be opened by the interaction of CypD with the lateral stalk of the ATP synthase complex121. This finding highlights the fact that the metabolic and cell death machinery are tightly interconnected122,123.
An alternative proposal is that MPTP results from a ROS-driven association of p53 with CypD124. This association can be blocked by pifithrin-μ (FIG. 2; TABLE 1), which was found to inhibit the translocation of p53 to the mitochondria, although whether this mechanism is important for CypD-mediated necrosis is debated125. Interestingly, CypD-deficient mitochondria are still able to mount a delayed MPTP response to Ca2+ overload, suggesting that CypD is involved in, but not essential for, proper MPTP assembly126. Therefore, alternative mechanisms must exist that trigger MPTP in the absence of CypD.
Despite this lack of a known mechanism, CypD-deficient mice have been shown to be resistant to cardiac and renal IRI115,127,128. Pharmacological inhibition of CypD is achieved pharmacologically through the use of the immunophilin-binding ligands129 cyclosporin A or sanglifehrin A130, which have each been successfully used to inhibit the extent of IRI in the kidney131, the brain132 and the heart133. Moreover, cyclosporin A and sanglifehrin A are approved immunosuppressants that are currently used to prevent organ rejection following transplantation. Mechanistically, cyclosporin A binds to cyclophilin (an immunophilin) of T cells. The cyclosporin A–cyclophilin complex inhibits calcineurin and consequently reduces the transcription of interleukin-2. Conversely, sanglifehrin A does not target calcineurin130. It is therefore important to note that despite the encouraging results obtained using these compounds in models of IRI, one cannot exclusively attribute their protective effects to actions on CypD-dependent necrosis, as these molecules are also able to block the immune response.
Targeting multiple cell death pathways
Cell death pathways are predominantly studied as independent entities. However, their crosstalk and their interdependent engagement in pathological conditions are frequently overlooked, particularly in vivo. Nevertheless, there is increasing recognition that there is an intricate relationship between when and how cells die and how this affects the organism. Knowledge of this relationship is in its infancy, and it is currently only possible to speculate how these other forms of regulated non-apoptotic cell death are complementary to each other. Recent studies targeting multiple cell death paradigms as a cytoprotective therapeutic approach are teaching us that single therapy approaches may not ultimately be the most effective approach. For example, during early renal graft injury, remote pulmonary injury may occur owing to kidney–lung crosstalk, and this lung damage was inhibited by the dual targeting of parthanatos and necroptosis with INO-1001 and necrostatin-1, respectively134. Moreover, in kidney IRI, the combined targeting of necroptosis, ferroptosis and CypD-dependent necrosis resulted in an unforeseen resistance to an insult that was otherwise lethal for non-treated animals or for animals treated with a single inhibitor against each of these types of cell death62.
The rationale for an approach that targets several types of cell death follows the notion that tissue demise and subsequent organ failure results from an auto-amplification loop between cell death and inflammatory stimuli16. This hypothesis assumes that upon ischaemia–reperfusion, a first hit independent of an extrinsic signal — such as a ROS burst in response to transient dysfunction of electron flow in the mitochondrial respiratory chain — could ignite a pro-inflammatory cell death routine. Ferroptosis and CypD-dependent necrosis are both forms of cell death that can be triggered by ischaemia–reperfusion independently of an extrinsic signal. Ferroptosis and CypD-mediated necrosis are intimately linked to the cellular metabolic state, as increased mitochondrial ROS production and glutathione depletion are both established hallmarks of IRI. One of the characteristics of these forms of cell death is that they culminate with the release of cellular contents and several oxidized lipid mediators (such as prostaglandins in case of ferroptosis). Such molecules can engage an inflammatory response and can also trigger forms of cell death such as apoptosis and necroptosis that, in turn, would feed an amplification loop leading to increased cell death and a stronger inflammatory response.
Another lesson that we are beginning to learn is that regulated forms of cell death are not uniformly connected and regulated across different tissue and cell types. This notion is neatly exemplified by studies of genetic deletion of RIPK1. RIPK1 deficiency in intestinal endothelial cells leads to mortality owing to increased apoptosis135 that is rescued by caspase 8 deficiency but not by RIPK3 deficiency. By contrast, deletion of Ripk1 in keratinocytes leads to a psoriasis-like phenotype that is fully prevented by RIPK3 deficiency136. These findings imply that cell death pathways may not be uniformly regulated and that differences in such regulation should be taken into account when designing combinatorial approaches for concomitantly targeting these pathways.
Outlook
It has become clear that cells can die through several distinct ways not restricted to classical apoptosis. As such, efforts to induce or inhibit cell death by targeting apoptosis in different contexts have shown mixed outcomes, most probably owing to our currently poor knowledge of how cells die. There is an emerging list of non-apoptotic cell death modalities, including necroptosis, ferroptosis, parthanatos and CypD-mediated cell death. Other forms that are also implicated in pathological settings but that are not discussed here include the following: pyroptosis137, an inflammasome-dependent cell death modality that mainly occurs in monocytes138; autophagic cell death3,4; and NETosis, a granulocyte-induced death modality that is characterized by the release of chromatin as a platform for antimicrobial proteins during inflammation and infection5. Nonetheless, successes in targeting apoptosis, especially in cancer, encourage us to pursue a deeper understanding of cell death routines and to exploit this knowledge to modulate them efficiently in a clinical setting.
Recently, the question has been raised as to whether these non-apoptotic cell death routines evolved separately as responses to specific triggers or whether they represent parts of a biochemical network that shares common regulatory or executioner mechanisms, such as energy catastrophe, ROS production, reduced anti-oxidant activity or lipid peroxidation2. Such questions are now beginning to be answered, and it has become evident that some of these cell death routines are highly intertwined and that blocking only one road to cell death is not likely to yield the desired outcome. For example, the necroptotic machinery is readily engaged upon caspase 8 inhibition or inactivation15,139. Also, inhibition of the cancer cell survival factors IAPs results in a sensitization of an alternative pathway involving either RIPK1-dependent apoptosis or necroptosis, depending on the presence of caspase 8 and RIPK3 or MLKL. Nevertheless, whether and how these regulatory networks modulate shifts between other cell death routines is not as clear as for the better-characterized apoptosis–necroptosis link.
Another issue besides the intricate network that regulates several cell death modalities is the pleiotropy of some of the crucial regulators of cell death. In that respect, it is very likely that in vivo targeting of RIPK1 or RIPK3 in pathophysiological conditions that involve complex intercellular interactions may affect not only necroptosis but also apoptosis and activation of the inflammasome140. This complexity and pleiotropy should be taken into account when evaluating the therapeuticactivity of drugs in experimental disease models. That is, an unequivocal assignment of cell death to a specific form of death pathway should rely not only on pharmacologicalevidence but also on genetic confirmation — whenever possible — by using single- or even combination-knockout or transgenic animals and cells, with the ultimate goal of defining and guiding better treatment paradigms.
Novel therapeutic strategies aiming to target multiple cell death pathways under pathologically relevant conditions such as IRI are starting to emerge. Such approaches could be appointed for pathological conditions for which strategies to inhibit single modes of cell death have failed or were not very effective. It has become increasingly clear that, although in recent years we have accumulated knowledge on how cells die, we are still far from a requisite understanding of the much more challenging question of how these pathways converge or synergize in a relevant in vivo pathological setting. Such an understanding, however, will be instrumental in designing efficacious therapeutics in order to affect diseases that involve cell death.
Pseudokinase
A protein that contains a catalytically inactive kinase domain. The loss of activity is attributed to the lack of at least one of three motifs (namely, VAIK, HRD or DFG) that are normally required for catalysis.
Fluorescence polarization assay
An assay used to analyse macromolecular interactions: one of the studied molecules is labelled with a fluorophore, which allows the measurement of the ratio of bound to unbound molecule, thus directly providing an estimation of molecular affinity.
Type II kinase inhibitor
An inhibitor that binds competitively with ATP, using the Asp-Phe-Gly (DFG)-out conformation.
DFG-out conformation
A state in which the kinase adopts a catalytically inactive conformation whereby the Asp-Phe-Gly (DFG) motif at the amino terminus of the activation loop faces outwards.
Gaucher disease
A genetic disorder characterized by deficiency of the enzyme glucocerebrosidase, leading to the accumulation of sphingolipids in certain organs. The disease is also characterized by enlargement of the liver and the spleen, low blood cell count and anaemia.
Glutaminolysis
A series of metabolic reactions based on the use of glutamine to produce energy and substrates to replenish the tricarboxylic acid cycle.
Pentose phosphate pathway
(PPP). A series of metabolic reactions converting glucose into precursors for nucleotide biosynthesis and reducing equivalents in the form of NADPH/H+.
Network perturbation analysis
A hybrid computational and experimental approach for mode-of-action analysis. Compound-induced dysregulations in the expression of genes in a regulatory network are scored, allowing compounds with a similar mode of action to be clustered.
Peroxide tone
The steady-state level of peroxides or lipid peroxides in a given cell.
Harlequin neuronal cells
Neuronal cells derived from the Harlequin mouse. These animals contain a proviral insertion in the apoptosis inducing factor (Aif) gene, leading to approximately 70% reduction of AIF expression.
Acknowledgements
The authors are grateful to B. Proneth (Helmholtz Zentrum München) for help in the preparation of figures and to M. Lamkanfi (Flanders Institute for Biotechnology, Ghent University) for critically reading and comments on the inflammasome section of this manuscript. M.C. is partially supported by the Deutsche Forschungsgemeinschaft (DFG) research grant CO 291/2–3 and DFG Priority Programme 1710 (CO 291/5–1), the Human Frontier Science Program (HFSP) RGP0013/14, and the m4 Award (Bavarian Ministry of Economic Affairs). B.R.S. is an Early Career Scientist of the Howard Hughes Medical Institute and is supported by New York State Stem Cell Science (NYSTEM) contract No. C026715, and the US National Institutes of Health (NIH) (grants 5R01CA097061, 1R21CA177591 and R01CA161061). The authors thank J. Decatur and the Columbia Chemistry NMR core facility (US National Science Foundation grant CHE 0840451 and NIH grant 1S10RR025431–01A1). Research in the P.V. unit is supported by Belgian grants (Interuniversity Attraction Poles, IAP 7/32), Flemish grants (Research Foundation Flanders, FWO G.0875.11, FWO G.0973.11N, FWO G.0A45.12 N, FWO G.0172.12, FWO G.0787.13N, G0C3114N, FWO KAN 31528711, and Foundation against Cancer 2012–188), Ghent University grants (MRP, GROUP-ID consortium) and grants from the Flanders Institute for Biotechnology. P.V. holds a Methusalem grant (BOF09/01M00709) from the Flemish Government.
Footnotes
Competing interests statement
The authors declare competing interests: see Web version for details.
References
- 1.Laster SM, Wood JG & Gooding LR Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol 141, 2629–2634 (1988). [PubMed] [Google Scholar]
- 2.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H & Vandenabeele P Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol 15, 135–147 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Green DR & Levine B To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157, 65–75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galluzzi L, Pietrocola F, Levine B & Kroemer G Metabolic control of autophagy. Cell 159, 1263–1276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brinkmann V & Zychlinsky A Neutrophil extracellular traps: is immunity the second function of chromatin? J. Cell Biol 198, 773–783 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lupfer C, Malik A & Kanneganti TD Inflammasome control of viral infection. Curr. Opin. Virol 12, 38–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohammad RM et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol 35, S78–S103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vandenabeele P, Galluzzi L, Vanden Berghe T & Kroemer G Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol 11, 700–714 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Sun L & Wang X A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends Biochem. Sci 39, 587–593 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Degterev A et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol 1, 112–119 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Degterev A et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol 4, 313–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 10 and 11 describe the first series of small molecules able to prevent necrotic TNF-induced cell death via RIPK1 inhibition.
- 12.Pasparakis M & Vandenabeele P Necroptosis and its role in inflammation. Nature 517, 311–320 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Kim SK et al. Upregulated RIP3 expression potentiates MLKL phosphorylation-mediated programmed necrosis in toxic epidermal necrolysis. J. Invest. Dermatol 135, 2021–2030 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Ofengeim D et al. Activation of necroptosis in multiple sclerosis. Cell Rep 10, 1836–1849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gunther C et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linkermann A, Stockwell BR, Krautwald S & Anders HJ Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat. Rev. Immunol 14, 759–767 (2014). [DOI] [PubMed] [Google Scholar]
- 17.He S et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 137, 1100–1111 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Linkermann A et al. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-α-induced shock. Mol. Med 18, 577–586 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newton K et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Jouan-Lanhouet S et al. Necroptosis, in vivo detection in experimental disease models. Semin. Cell Dev. Biol 35, 2–13 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Dondelinger Y et al. NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Micheau O & Tschopp J Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Wang L, Du F & Wang X TNF-α induces two distinct caspase-8 activation pathways. Cell 133, 693–703 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Dondelinger Y et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ 20, 1381–1392 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Legarda-Addison D, Hase H, O’Donnell MA & Ting AT NEMO/IKKγ regulates an early NF-κB-independent cell-death checkpoint during TNF signaling. Cell Death Differ 16, 1279–1288 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oberst A et al. Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai Z et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol 16, 55–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24, 105–121 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dondelinger Y et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7, 971–981 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Wang H et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Tenev T et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43, 432–448 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Feoktistova M et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandal P et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates how small-molecule drugs targeting RIPK3 kinase activity indeed block necroptosis induction, but in some cases induce RIPK3-platform-mediated apoptosis involving caspase 8 and RIPK1 kinase activity.
- 34.Hildebrand JM et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl Acad. Sci. USA 111, 15072–15077 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teng X et al. Structure–activity relationship study of novel necroptosis inhibitors. Bioorg. Med. Chem. Lett 15, 5039–5044 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Vandenabeele P, Grootjans S, Callewaert N & Takahashi N Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ 20, 185–187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi N et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis 3, e437 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Degterev A, Maki JL & Yuan J Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ 20, 366 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prendergast GC, Chang MY, Mandik-Nayak L, Metz R & Muller AJ Indoleamine 2,3-dioxygenase as a modifier of pathogenic inflammation in cancer and other inflammation-associated diseases. Curr. Med. Chem 18, 2257–2262 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie T et al. Structural basis of RIP1 inhibition by necrostatins. Structure 21, 493–499 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Harris PA et al. Discovery of small molecule RIP1 kinase inhibitors for the treatment of pathologies associated with necroptosis. ACS Med. Chem. Lett 4, 1238–1243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Najjar M et al. Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep 10, 1850–1860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fauster A et al. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis 6, e1767 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Newton K, Sun X & Dixit VM Kinase RIP3 is dispensable for normal NF-κBs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol 24, 1464–1469 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Q et al. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ. Res 116, 600–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaiser WJ et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem 288, 31268–31279 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Upton JW, Kaiser WJ & Mocarski ES DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vitner EB et al. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med 20, 204–208 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Sun L et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012). [DOI] [PubMed] [Google Scholar]; In this paper, a pharmacoproteomics approach based on a necroptosis-inhibiting drug (NSA) led to the successful identification of the execution protein MLKL, which is regulated by RIPK3-dependent phosphorylation.
- 50.Li J et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cauwels A, Janssen B, Waeytens A, Cuvelier C & Brouckaert P Caspase inhibition causes hyperacute tumor necrosis factor-induced shock via oxidative stress and phospholipase A2. Nat. Immunol 4, 387–393 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Duprez L et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35, 908–918 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Wu J et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res 23, 994–1006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Polykratis A et al. Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol 193, 1539–1543 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang WS & Stockwell BR Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol 15, 234–245 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dixon SJ et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this paper, the term ferroptosis is coined, and it is shown that a functional system X – is required to c maintain glutathione levels to inhibit this form of cell death in a subset of cancer cell lines.
- 57.Sato H, Tamba M, Ishii T & Bannai S Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem 274, 11455–11458 (1999). [DOI] [PubMed] [Google Scholar]
- 58.Conrad M & Sato H The oxidative stress-inducible cystine/glutamate antiporter, system Xc–: cystine supplier and beyond. Amino Acids 42, 231–246 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Ursini F, Maiorino M, Valente M, Ferri L & Gregolin C Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 710, 197–211 (1982). [DOI] [PubMed] [Google Scholar]
- 60.Yang WS et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that GPX4 is the limiting glutathione-utilizing enzyme required for the prevention of ferroptosis in cancer cells.
- 61.Friedmann Angeli JP et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol 16, 1180–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that the ferroptosis inhibitor liproxstatin-1 protects mice from acute renal failure induced by inducible Gpx4 ablation as well as from hepatic IRI.
- 62.Linkermann A et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl Acad. Sci. USA 111, 16836–16841 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper indicates that in vivo inhibition of ferroptosis leads to a protective effect in a pathophysiological setting, such as IRI in the kidney.
- 63.Seiler A et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell. Metab 8, 237–248 (2008). [DOI] [PubMed] [Google Scholar]; This paper describes the first conditional-knockout model of the ferroptosis regulator GPX4 in cells and mice, which were further used for the development of liproxstatin-1 and for assessing the importance of ferroptosis in different tissues.
- 64.Yoo SE et al. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic. Biol. Med 52, 1820–1827 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen L, Na R, Gu M, Richardson A & Ran Q Lipid peroxidation up-regulates BACE1 expression in vivo: a possible early event of amyloidogenesis in Alzheimer’s disease. J. Neurochem 107, 197–207 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoo MH et al. Delineating the role of glutathione peroxidase 4 in protecting cells against lipid hydroperoxide damage and in Alzheimer’s disease. Antioxid. Redox Signal 12, 819–827 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ufer C et al. Translational regulation of glutathione peroxidase 4 expression through guanine-rich sequence-binding factor 1 is essential for embryonic brain development. Genes Dev 22, 1838–1850 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hauser DN, Dukes AA, Mortimer AD & Hastings TG Dopamine quinone modifies and decreases the abundance of the mitochondrial selenoprotein glutathione peroxidase 4. Free Radic. Biol. Med 65, 419–427 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bellinger FP et al. Changes in selenoprotein P in substantia nigra and putamen in Parkinson’s disease. J. Parkinsons Dis 2, 115–126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bellinger FP et al. Glutathione peroxidase 4 is associated with neuromelanin in substantia nigra and dystrophic axons in putamen of Parkinson’s brain. Mol. Neurodegener 6, 8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Skouta R et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc 136, 4551–4556 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Ferrostatins with improved properties are reported in this paper to be effective in several cellular disease models. These ferrostatins block lipid peroxidation, but not by inhibiting mitochondrial ROS production or preventing lysosomal membrane permeabilization.
- 72.Wirth EK et al. Cerebellar hypoplasia in mice lacking selenoprotein biosynthesis in neurons. Biol. Trace Elem. Res 158, 203–210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roth TL et al. Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Korade Z et al. Antioxidant supplementation ameliorates molecular deficits in Smith–Lemli–Opitz syndrome. Biol. Psychiatry 75, 215–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ueta T et al. Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J. Biol. Chem 287, 7675–7682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sengupta A et al. Targeted disruption of glutathione peroxidase 4 in mouse skin epithelial cells impairs postnatal hair follicle morphogenesis that is partially rescued through inhibition of COX-2. J. Invest. Dermatol 133, 1731–1741 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matsushita M et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med 212, 555–568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wortmann M et al. Combined deficiency in glutathione peroxidase 4 and vitamin E causes multiorgan thrombus formation and early death in mice. Circ. Res 113, 408–417 (2013). [DOI] [PubMed] [Google Scholar]
- 79.Woo JH et al. Elucidating compound mechanism of action by network perturbation analysis. Cell 162, 441–451 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bannai S Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem 261, 2256–2263 (1986). [PubMed] [Google Scholar]
- 81.Mandal PK et al. System Xc− and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem 285, 22244–22253 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang L et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper implicates ferroptosis as a non-canonical tumour suppressive function of p53 via p53-mediated transcriptional inhibition of system Xc−.
- 83.Hayano M, Yang WS, Corn CK, Pagano NC & Stockwell BR Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ 10.1038/cdd.2015.93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yant LJ et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med 34, 496–502 (2003). [DOI] [PubMed] [Google Scholar]
- 85.Gao M, Monian P, Quadri N, Ramasamy R & Jiang X Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper suggests that inhibiting glutaminolysis might be a putative anti-ferroptotic pharmacological approach and that cells that preferably use glutaminolysis for energy production are more sensitive to ferroptosis.
- 86.Tretter L & Adam-Vizi V Generation of reactive oxygen species in the reaction catalyzed by α-ketoglutarate dehydrogenase. J. Neurosci 24, 7771–7778 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bae YS et al. Platelet-derived growth factor-induced H2O2 production requires the activation of phosphatidylinositol 3-kinase. J. Biol. Chem 275, 10527–10531 (2000). [DOI] [PubMed] [Google Scholar]
- 88.Han CY et al. NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J. Biol. Chem 287, 10379–10393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li B et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513, 251–255 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dixon SJ et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol 10, 1604–1609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dixon SJ et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3, e02523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Here, inhibition of system Xc− by erastin or sorafenib is shown to be associated with endoplasmic reticulum stress and glutathione-specific γ-glutamylcyclotransferase 1 upregulation involved in glutathione degradation.
- 92.Yagoda N et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Louandre C et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 133, 1732–1742 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Chung WJ et al. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci 25, 7101–7110 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen RS et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/β-catenin pathway. Oncogene 28, 599–609 (2009). [DOI] [PubMed] [Google Scholar]
- 96.Nagano O, Okazaki S & Saya H Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene 32, 5191–5198 (2013). [DOI] [PubMed] [Google Scholar]
- 97.Timmerman LA et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24, 450–465 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Keldsen N, Havsteen H, Vergote I, Bertelsen K & Jakobsen A Altretamine (hexamethylmelamine) in the treatment of platinum-resistant ovarian cancer: a phase II study. Gynecol. Oncol 88, 118–122 (2003). [DOI] [PubMed] [Google Scholar]
- 99.Lee Y et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci 16, 1392–1400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Virag L, Robaszkiewicz A, Rodriguez-Vargas JM & Oliver FJ Poly(ADP-ribose) signaling in cell death. Mol. Aspects Med 34, 1153–1167 (2013). [DOI] [PubMed] [Google Scholar]
- 101.Curtin NJ & Szabo C Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol. Aspects Med 34, 1217–1256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mashimo M, Kato J & Moss J ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc. Natl Acad. Sci. USA 110, 18964–18969 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Andrabi SA, Dawson TM & Dawson VL Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. NY Acad. Sci 1147, 233–241 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Y et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal 4, ra20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Andrabi SA et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat. Med 17, 692–699 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this paper, the first endogenous inhibitor of parthanatos is described, indicating that interfering with PAR polymer signalling is a putative treatment for neurodegenerative disease.
- 106.Graziani G & Szabo C Clinical perspectives of PARP inhibitors. Pharmacol. Res 52, 109–118 (2005). [DOI] [PubMed] [Google Scholar]
- 107.del Moral RM et al. PARP inhibition attenuates histopathological lesion in ischemia/reperfusion renal mouse model after cold prolonged ischemia. ScientificWorldJournal 2013, 486574 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sahaboglu A et al. PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function. PLoS ONE 5, e15495 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jagtap P et al. Novel phenanthridinone inhibitors of poly (adenosine 5ʹ-diphosphate-ribose) synthetase: potent cytoprotective and antishock agents. Crit. Care Med 30, 1071–1082 (2002). [DOI] [PubMed] [Google Scholar]
- 110.Jagtap PG et al. The discovery and synthesis of novel adenosine substituted 2,3-dihydro-1H-isoindol-1-ones: potent inhibitors of poly(ADP-ribose) polymerase-1 (PARP-1). Bioorg. Med. Chem. Lett 14, 81–85 (2004). [DOI] [PubMed] [Google Scholar]
- 111.Jagtap PG et al. Discovery of potent poly(ADP-ribose) polymerase-1 inhibitors from the modification of indeno[1,2-c]isoquinolinone. J. Med. Chem 48, 5100–5103 (2005). [DOI] [PubMed] [Google Scholar]
- 112.Morrow DA et al. A randomized, placebo-controlled trial to evaluate the tolerability, safety, pharmacokinetics, and pharmacodynamics of a potent inhibitor of poly(ADP-ribose) polymerase (INO-1001) in patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of the TIMI 37 trial. J. Thromb. Thrombolysis 27, 359–364 (2009). [DOI] [PubMed] [Google Scholar]
- 113.US National Library of Medicine. ClinicalTrials.gov [online], https://www.clinicaltrials.gov/ct2/show/NCT01983358?term=NCT01983358&rank=1 (2013). [DOI] [PubMed]
- 114.Andrabi SA et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl Acad. Sci. USA 111, 10209–10214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Although parthanatos still lacks a clear core cell death pathway, this paper indicates that overactivation of PARP1 is caused by PAR-dependent inhibition of glycolysis through the inhibition of HK, explaining the bioenergetic collapse that results in necrotic cell death.
- 115.Devalaraja-Narashimha K, Diener AM & Padanilam BJ Cyclophilin D gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Renal Physiol 297, F749–F759 (2009). [DOI] [PubMed] [Google Scholar]
- 116.Millay DP et al. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med 14, 442–447 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jobe SM et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 111, 1257–1265 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fujimoto K, Chen Y, Polonsky KS & Dorn GW 2nd. Targeting cyclophilin D and the mitochondrial permeability transition enhances β-cell survival and prevents diabetes in Pdx1 deficiency. Proc. Natl Acad. Sci. USA 107, 10214–10219 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Forte M et al. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl Acad. Sci. USA 104, 7558–7563 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Javadov S & Kuznetsov A Mitochondrial permeability transition and cell death: the role of cyclophilin D. Front. Physiol 4, 76 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Giorgio V et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl Acad. Sci. USA 110, 5887–5892 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bonora M, Bravo-San Pedro JM, Kroemer G, Galluzzi L & Pinton P Novel insights into the mitochondrial permeability transition. Cell Cycle 13, 2666–2670 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Green DR, Galluzzi L & Kroemer G Cell biology. Metabolic control of cell death. Science 345, 1250256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vaseva AV et al. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Karch J & Molkentin JD Is p53 the long-sought molecular trigger for cyclophilin D-regulated mitochondrial permeability transition pore formation and necrosis? Circ. Res 111, 1258–1260 (2012). [DOI] [PubMed] [Google Scholar]
- 126.Nakagawa T et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658 (2005). [DOI] [PubMed] [Google Scholar]
- 127.Baines CP et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 (2005). [DOI] [PubMed] [Google Scholar]
- 128.Schinzel AC et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl Acad. Sci. USA 102, 12005–12010 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 126–128 characterize the requirement of CypD for the formation of the MPTP, and its inhibition as a pharmacologically amenable approach.
- 129.Zhang LH, Youn HD & Liu JO Inhibition of cell cycle progression by the novel cyclophilin ligand sanglifehrin A is mediated through the NFκB-dependent activation of p53. J. Biol. Chem 276, 43534–43540 (2001). [DOI] [PubMed] [Google Scholar]
- 130.Clarke SJ, McStay GP & Halestrap AP Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Biol. Chem 277, 34793–34799 (2002). [DOI] [PubMed] [Google Scholar]
- 131.Linkermann A et al. Two independent pathways of regulated necrosis mediate ischemia–reperfusion injury. Proc. Natl Acad. Sci. USA 110, 12024–12029 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nighoghossian N et al. Cyclosporine in acute ischemic stroke. Neurology 84, 2216–2223 (2015). [DOI] [PubMed] [Google Scholar]
- 133.Keogh A Calcineurin inhibitors in heart transplantation. J. Heart Lung Transplant 23, S202–S206 (2004). [DOI] [PubMed] [Google Scholar]
- 134.Zhao H et al. Necroptosis and parthanatos are involved in remote lung injury after receiving ischemic renal allografts in rats. Kidney Int 87, 738–748 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Takahashi N et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 513, 95–99 (2014). [DOI] [PubMed] [Google Scholar]
- 136.Dannappel M et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lamkanfi M & Dixit VM Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014). [DOI] [PubMed] [Google Scholar]
- 138.Bergsbaken T, Fink SL & Cookson BT Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol 7, 99–109 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ch’en IL, Tsau JS, Molkentin JD, Komatsu M & Hedrick SM Mechanisms of necroptosis in T cells. J. Exp. Med 208, 633–641 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Moriwaki K & Chan FK Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev 25, 167–174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Munoz-Planillo R et al. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Misawa T et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol 14, 454–460 (2013). [DOI] [PubMed] [Google Scholar]
- 143.Zhou R, Yazdi AS, Menu P & Tschopp J A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011). [DOI] [PubMed] [Google Scholar]
- 144.Hornung V et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol 9, 847–856 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Upton JW, Kaiser WJ & Mocarski ES Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7, 302–313 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kang TB, Yang SH, Toth B, Kovalenko A & Wallach D Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38, 27–40 (2013). [DOI] [PubMed] [Google Scholar]; This study puts forward for the first time the possible and complex interaction between the necrosome and inflammasome machinery.
- 147.Moriwaki K, Bertin J, Gough PJ & Chan FKA RIPK3-caspase 8 complex mediates atypical pro-IL-1β processing. J. Immunol 194, 1938–1944 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lawlor KE et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun 6, 6282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. 526, 666–671 Nature (2015). [DOI] [PubMed] [Google Scholar]
- 150.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. 526, 660–665 Nature (2015). [DOI] [PubMed] [Google Scholar]
- 151.Coll RC et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med 21, 248–255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tan S, Schubert D & Maher P Oxytosis: a novel form of programmed cell death. Curr. Top. Med. Chem 1, 497–506 (2001). [DOI] [PubMed] [Google Scholar]
- 153.Li Y, Maher P & Schubert D A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 19, 453–463 (1997). [DOI] [PubMed] [Google Scholar]
- 154.Pallast S et al. Increased nuclear apoptosis-inducing factor after transient focal ischemia: a 12/15-lipoxygenase-dependent organelle damage pathway. J. Cereb. Blood Flow Metab 30, 1157–1167 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pallast S, Arai K, Wang X, Lo EH & van Leyen K 12/15-lipoxygenase targets neuronal mitochondria under oxidative stress. J. Neurochem 111, 882–889 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yigitkanli K et al. Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann. Neurol 73, 129–135 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.van Leyen K et al. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke 37, 3014–3018 (2006). [DOI] [PubMed] [Google Scholar]; This study demonstrates that glutathione depletion leads to lipid peroxidation and cell death in neuronal cells — a phenomenon later termed as oxytosis, which is related to ferroptosis.
- 158.Jin G et al. Protecting against cerebrovascular injury: contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke 39, 2538–2543 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Oxler EM, Dolga A & Culmsee C AIF depletion provides neuroprotection through a preconditioning effect. Apoptosis 17, 1027–1038 (2012). [DOI] [PubMed] [Google Scholar]
- 160.Mahoney DJ et al. Both cIAP1 and cIAP2 regulate TNFα-mediated NF-κB activation. Proc. Natl Acad. Sci. USA 105, 11778–11783 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Haas TL et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36, 831–844 (2009). [DOI] [PubMed] [Google Scholar]
- 162.Varfolomeev E et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131, 669–681 (2007). [DOI] [PubMed] [Google Scholar]
- 163.Vince JE et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 131, 682–693 (2007). [DOI] [PubMed] [Google Scholar]
- 164.Petersen SL et al. Autocrine TNFα signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 12, 445–456 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bertrand MJ et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 (2008). [DOI] [PubMed] [Google Scholar]
- 166.Lin Y, Devin A, Rodriguez Y & Liu ZG Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13, 2514–2526 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Feng S et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 19, 2056–2067 (2007). [DOI] [PubMed] [Google Scholar]
- 168.O’Donnell MA et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol 13, 1437–1442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cho YS et al. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang DW et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009). [DOI] [PubMed] [Google Scholar]
- 171.Zhao J et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl Acad. Sci. USA 109, 5322–5327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fulda S Smac mimetics as IAP antagonists. Semin. Cell Dev. Biol 39, 132–138 (2015). [DOI] [PubMed] [Google Scholar]
- 173.Kobayashi S et al. Cystathionine is a novel substrate of cystine/glutamate transporter: implications for immune function. J. Biol. Chem 290, 8778–8788 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bannai S & Kitamura E Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J. Biol. Chem 255, 2372–2376 (1980). [PubMed] [Google Scholar]
- 175.Gout PW, Buckley AR, Simms CR & Bruchovsky N Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the Xc– cystine transporter: a new action for an old drug. Leukemia 15, 1633–1640 (2001). [DOI] [PubMed] [Google Scholar]
- 176.Patel SA, Warren BA, Rhoderick JF & Bridges RJ Differentiation of substrate and non-substrate inhibitors of transport system Xc–: an obligate exchanger of L-glutamate and L-cystine. Neuropharmacology 46, 273–284 (2004). [DOI] [PubMed] [Google Scholar]
- 177.Fatokun AA, Dawson VL & Dawson TM Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol 171, 2000–2016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shall S Proceedings: experimental manipulation of the specific activity of poly(ADP-ribose) polymerase. J. Biochem 77, 2p (1975). [PubMed] [Google Scholar]
- 179.Fatokun AA, Liu JO, Dawson VL & Dawson TM Identification through high-throughput screening of 4ʹ-methoxyflavone and 3ʹ,4ʹ-dimethoxyflavone as novel neuroprotective inhibitors of parthanatos. Br. J. Pharmacol 169, 1263–1278 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Greco R et al. Neuroprotection by the PARP inhibitor PJ34 modulates cerebral and circulating RAGE levels in rats exposed to focal brain ischemia. Eur. J. Pharmacol 744, 91–97 (2014). [DOI] [PubMed] [Google Scholar]
- 181.Wang J et al. Inhibition of poly (ADP-ribose) polymerase and inducible nitric oxide synthase protects against ischemic myocardial damage by reduction of apoptosis. Mol. Med. Rep 11, 1768–1776 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ha HC & Snyder SH Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl Acad. Sci. USA 96, 13978–13982 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Meier HL, Ballough GP, Forster JS & Filbert MG Benzamide, a poly(ADP-ribose) polymerase inhibitor, is neuroprotective against soman-induced seizure-related brain damage. Ann. NY Acad. Sci 890, 330–335 (1999). [DOI] [PubMed] [Google Scholar]
- 184.Purnell MR & Whish WJ Novel inhibitors of poly(ADP-ribose) synthetase. Biochem. J 185, 775–777 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Szabo G et al. INO-1001 a novel poly(ADP-ribose) polymerase (PARP) inhibitor improves cardiac and pulmonary function after crystalloid cardioplegia and extracorporal circulation. Shock 21, 426–432 (2004). [DOI] [PubMed] [Google Scholar]
- 186.d’Avila JC et al. Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. J. Neuroinflamm 9, 31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.US National Library of Medicine. ClinicalTrials.gov [online], https://clinicaltrials.gov/ct2/show/NCT00271765?term=NCT00271765&rank=1 (2006). [DOI] [PubMed]
- 188.US National Library of Medicine. ClinicalTrials.gov [online], https://clinicaltrials.gov/ct2/show/NCT00271167 (2005). [DOI] [PubMed]
- 189.van Leyen K et al. Novel lipoxygenase inhibitors as neuroprotective reagents. J. Neurosci. Res 86, 904–909 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Niki E & Traber MG A history of vitamin E. Ann. Nutr. Metab 61, 207–212 (2012). [DOI] [PubMed] [Google Scholar]
- 191.Griffith OW Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J. Biol. Chem 257, 13704–13712 (1982). [PubMed] [Google Scholar]
- 192.Harris IS et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 (2015). [DOI] [PubMed] [Google Scholar]
- 193.Kim SY et al. Inhibition of cyclophilin D by cyclosporin A promotes retinal ganglion cell survival by preventing mitochondrial alteration in ischemic injury. Cell Death Dis 5, e1105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Teixeira G et al. Synergistic protective effect of cyclosporin A and rotenone against hypoxia– reoxygenation in cardiomyocytes. J. Mol. Cell Cardiol 56, 55–62 (2013). [DOI] [PubMed] [Google Scholar]
- 195.Wang X et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J. Neurosci 29, 2588–2596 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hausenloy D, Wynne A, Duchen M & Yellon D Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation 109, 1714–1717 (2004). [DOI] [PubMed] [Google Scholar]
- 197.Strom E et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat. Chem. Biol 2, 474–479 (2006). [DOI] [PubMed] [Google Scholar]