

Abstract
Traumatic brain injuries (TBI) lead to dramatic changes in the surviving brain tissue. Altered ion concentrations, coupled with changes in the expression of membrane-spanning proteins, create a post-TBI brain state that can lead to further neuronal loss caused by secondary excitotoxicity. Several GABA receptor agonists have been tested in the search for neuroprotection immediately after an injury, with paradoxical results. These drugs not only fail to offer neuroprotection, but can also slow down functional recovery after TBI. Here, using computational modeling, we provide a biophysical hypothesis to explain these observations. We show that the accumulation of intracellular chloride ions caused by a transient upregulation of Na+-K+-2Cl- (NKCC1) co-transporters as observed following TBI, causes GABA receptor agonists to lead to excitation and depolarization block, rather than the expected hyperpolarization. The likelihood of prolonged, excitotoxic depolarization block is further exacerbated by the extremely high levels of extracellular potassium seen after TBI. Our modeling results predict that the neuroprotective efficacy of GABA receptor agonists can be substantially enhanced when they are combined with NKCC1 co-transporter inhibitors. This suggests a rational, biophysically principled method for identifying drug combinations for neuroprotection after TBI.
Keywords: chloride, depolarization block, GABA, neuroprotection, potassium
Introduction
The concentration of ions inside and outside neurons varies considerably across brain states.1–4 The ratio of extracellular to intracellular ion concentrations determines the reversal potential of a given ion species (Eion;5–7). Changes in this ratio alter neuronal excitability and can have a significant impact on the oscillatory states that a neural circuit is capable of producing, as well as on the computations it can perform.4,8–11 Behaviorally important examples include the rapid changes in practically all relevant ions during transitions between sleep and wake states,2 and the long-term decreases in intracellular chloride concentrations ([Cl-]i) seen over the initial stages of normal development.12–16
The blood–brain barrier has numerous specializations to keep the concentration of ions in the extracellular space within certain physiological bounds.17 However, large changes in the concentrations of key ions are often seen in many neurological disorders. Epileptic seizures are accompanied by a two-to fourfold increase in extracellular potassium concentration ([K+]o;1,18–20). By moving the reversal potential of potassium (EK) and the resting potential closer to the threshold for action potentials, this dramatic increase in [K+]o makes cells even more excitable, further exacerbating the runaway excitation seen during most types of seizures.21
Traumatic brain injuries (TBI) also lead to dramatic increases in [K+]o,22–27 and these increases can often far exceed even those seen during seizures.1,18–20 Such massive increases in [K+]o can lead to cells entering depolarization block (DB),28–31 a biophysical state characterized by the buildup of sodium channel inactivation. This leads to an inability of the cell to fire subsequent action potentials until and unless it is first hyperpolarized enough to de-inactivate some of the sodium channels.32 The electrical silencing of neuronal activity caused by depolarization block is sometimes reflected as spreading depression across injured brain regions.33,34 Such prolonged depolarization can exacerbate the secondary injury cascade induced by injury and hypoxia,33,35,36 and greatly increases the chances of cell death.36–39 Drugs that hyperpolarize neurons can suppress spreading depression40,41 and potentially offer neuroprotective benefits if delivered immediately following an injury. However, a variety of GABAA receptor agonists have proven to be ineffective in TBI, and can often exacerbate TBI-related symptoms.42–50
Here, we use biophysical modeling to try to understand why GABAA receptor agonists are not effective in treating TBI, and to suggest biophysically principled drug combinations51 that may offer greater neuroprotection. TBI causes unique changes in the expression of chloride transporters: transient upregulation of Na+-K+-2Cl- (NKCC1) co-transporters52–56 and/or transient downregulation of KCC2 co-transporters is seen immediately after the injury.54,57 These transient changes in the expression of NKCC1 after an injury can lead to increases in the concentration of intracellular chloride, depolarizing the reversal potential for chloride.54,57,58 The net effect of these changes makes GABA, a typically inhibitory neurotransmitter, excitatory, potentially explaining why GABAA receptor agonists are ineffective in TBI. With our model, we show that, in TBI, GABAA receptor agonists alone do not rescue neurons from depolarization block. This means that neurons in the secondary injury site continue to remain in an excitotoxic state even in the presence of GABAA receptor agonists. However, we show that that a combination of GABAA receptor agonists and NKCC1 co-transporter blockers may confer large neuroprotective benefits, and suggests a path toward a biophysically rational selection of combination drug therapies in TBI.
Methods
Model motivation
Our primary goals for this study were to mechanistically understand why GABAA receptor agonists are not effective in treating TBI, and to test the efficacy of physiologically motivated drug combinations for neuroprotection. For this purpose, we built a Hodgkin–Huxley59 based compartmental model of a regular spiking (RS) neuron60 based on realistic and known physiology. Specifically, we chose to model a prototypical RS pyramidal neuron, given its abundance and critical involvement in both the normal and pathological function of neocortical circuits.61–63 The properties of the model will be described in detail subsequently.
RS neuron model
Morphology and passive properties
Based on the morphology of a prototypical neocortical pyramidal cell,64 we constructed a multi-compartmental model with one somatic compartment and seven dendritic compartments. Our model has a specific membrane resistance of 25 kΩcm2 and a specific membrane capacitance of 1 μF/cm2 resulting in a membrane time constant of 25 ms.65 The model's input resistance is 399 MΩ, closely matching the input resistance of neocortical pyramidal neurons.65 The model has a resting membrane potential of −71.87 mV.64,66 The model was simulated at a temperature of 30°C. To scale the temperature dependence of time constants, we used a q10 value of 3.67
Active properties
The model consists of two ionic currents: fast sodium current and delayed rectifier potassium current. The channel kinetics and properties of these two currents will be described in detail subsequently. The model has a spike width of 1.03 ms68 and a threshold of −42.32 mV.66,69 Spike threshold was calculated as the membrane potential when 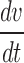 crosses 10 V/s for the first time in the positive direction.70
crosses 10 V/s for the first time in the positive direction.70
Fast sodium current
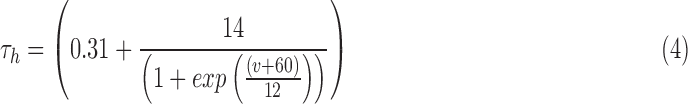
RS neurons exhibit fast sodium currents that are necessary for action potential generation.71 The fast sodium current was modeled based on the Hodgkin–Huxley formalism59 and consists of three activation gates and a single inactivation gate. This channel was distributed in both the somatic and dendritic compartments. Utilizing experimental data from a previous study,71 we modeled these fast sodium currents based on sodium channel gating properties commonly found in RS neurons. The maximal channel conductance (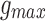 ) of the channel is shown in Table 1. The equations for voltage dependence of steady state activation/inactivation
) of the channel is shown in Table 1. The equations for voltage dependence of steady state activation/inactivation  their respective time constants
their respective time constants  and channel currents
and channel currents 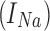 are provided here.
are provided here.
Table 1.
List of Parameters Used in the Model
| Parameter description | Somatic compartment | Dendritic compartments |
|---|---|---|
| Specific membrane resistance (kΩcm2) | 25 | 25 |
| Specific membrane capacitance (μF/cm2) | 1 | 1 |
| Membrane time constant (ms) | 25 | 25 |
| Axial resistivity (Ω-cm) | 200 | 200 |
| Fast sodium current gmax (S/cm2) | 0.45 | 0.1125 |
| Delayed rectifier potassium current (S/cm2) | 0.1 | 0.03 |
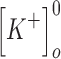 (mM) (mM) |
3.5 | 3.5 |
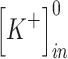 (mM) (mM) |
140 | 140 |
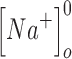 (mM) (mM) |
140 | 140 |
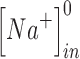 (mM) (mM) |
20 | 20 |
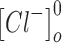 (mM) (mM) |
110 | 110 |
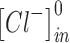 (mM) (mM) |
6 | 6 |
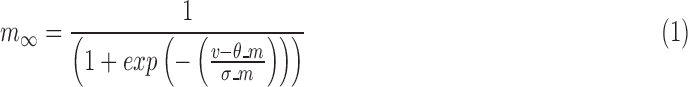 |
 |
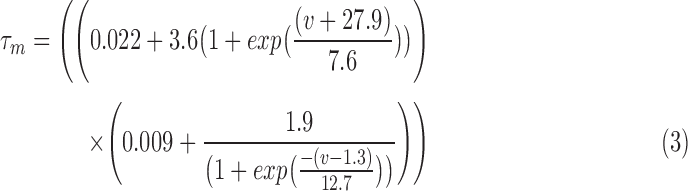 |
 |
where 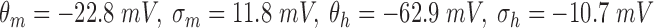 .
.
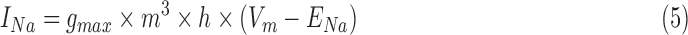 |
Delayed rectifier potassium current
RS neurons of the neocortex and the hippocampus express delayed rectifier potassium currents,72–76 which are known to significantly contribute to action potential repolarization.72,76 The delayed rectifier potassium channel was modeled based on Hodgkin–Huxley formalism 59 and has two activation gates.77 Similarly to the sodium channel, the delayed rectifier potassium channel was distributed in both the somatic and dendritic compartments. By using data from a previous experimental study on pyramidal neurons,72 we modeled the potassium current by simulating the voltage dependence of steady state activation and time constant. The equations for voltage dependence of steady state activation 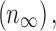 its time constant
its time constant 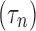 , and channel currents
, and channel currents 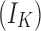 are provided here.
are provided here.
 |
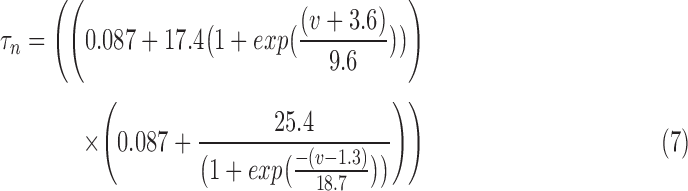 |
where 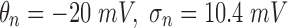
 |
Background inputs
RS neurons are known to receive phasic excitatory, phasic inhibitory, and tonic inhibitory background inputs in vivo.78–80 Taking these aspects of the circuit into account, we modeled these inputs in the following way: 30 phasic excitatory (AMPA) inputs were modeled with a firing rate of 1 Hz to mimic the background firing rate of pyramidal neurons in vivo.81–83 The time course of AMPAergic synaptic conductance was modeled using the following equation84,85:
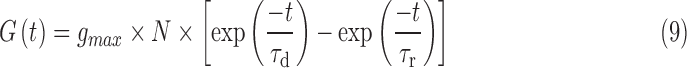 |
where 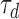 and
and 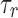 are decay and rise time constants respectively, and
are decay and rise time constants respectively, and 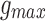 is the maximal synaptic conductance. N is a normalization factor that makes maximum of
is the maximal synaptic conductance. N is a normalization factor that makes maximum of 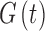 equal to
equal to 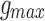 . The values of
. The values of 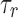 and
and 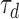 were 0.9 ms and 5.1 ms respectively, based on AMPAergic currents from pyramidal neurons of the rat prefrontal cortex.78
were 0.9 ms and 5.1 ms respectively, based on AMPAergic currents from pyramidal neurons of the rat prefrontal cortex.78
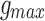 for phasic excitatory inputs was set to a value of 200 pS.
for phasic excitatory inputs was set to a value of 200 pS.
Two hundred phasic inhibitory inputs were modeled using Equation (9). The firing frequency of phasic inhibitory input was set to 5 Hz.86 These inputs had a rise time constant of 0.88 ms and a decay time constant of 9.4 ms, which were taken from inhibitory post-synaptic current (IPSC) recordings of pyramidal neurons of the rat visual cortex.79 Phasic inhibitory inputs were modeled with a 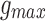 of 600 pS, threefold greater than that of phasic excitatory inputs, closely replicating the excitatory-IPSC ratio of layer 2/3 pyramidal neurons.87
of 600 pS, threefold greater than that of phasic excitatory inputs, closely replicating the excitatory-IPSC ratio of layer 2/3 pyramidal neurons.87
Tonic inhibitory background inputs were modeled with a conductance value of 350 pS 80,88 using the following equation89:
 |
where 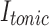 is the tonic inhibitory current,
is the tonic inhibitory current, 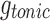 is the tonic inhibitory conductance, and
is the tonic inhibitory conductance, and 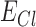 is the chloride reversal potential.
is the chloride reversal potential.
Modeling of increase in extracellular potassium and intracellular chloride
TBI is characterized by increases in the concentration of both extracellular potassium and intracellular chloride.22,57,58 The effect of changes in the concentration of these ionic species on the excitability of the neuron were modeled using the Nernst and Goldman equations.5–7,84 Increase in extracellular potassium affects the Nernst potential of potassium ions as well as the resting membrane potential. The effect of extracellular potassium increase on the potassium reversal potential was modeled according to the following equation5–7,84:
 |
where R is the gas constant (8.314 J K−1 mol−1), T is the temperature in Kelvin, F is Faraday's constant (96485 C mol−1), Z is the valence of the ion, 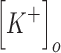 is the extracellular potassium concentration,
is the extracellular potassium concentration, 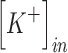 is the intracellular potassium concentration, and EK is the reversal potential of potassium.
is the intracellular potassium concentration, and EK is the reversal potential of potassium.
The effect of increase in extracellular potassium on the resting membrane potential was modeled according to the Goldman equation84,90:
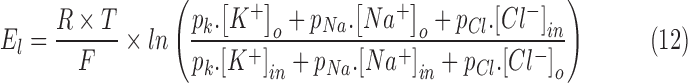 |
where pk, 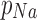 and
and 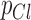 are the relative permeability of potassium, sodium, and chloride ions respectively (1 : 0.04 : 0.45).90
are the relative permeability of potassium, sodium, and chloride ions respectively (1 : 0.04 : 0.45).90
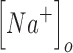 and
and 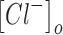 are the extracellular sodium and chloride ion concentration respectively, and El is the leak reversal potential.
are the extracellular sodium and chloride ion concentration respectively, and El is the leak reversal potential. 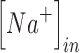 and
and 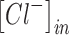 are the intracellular sodium and chloride ion concentration respectively. Extracellular and intracellular concentration of various ions at resting conditions are given as follows:
are the intracellular sodium and chloride ion concentration respectively. Extracellular and intracellular concentration of various ions at resting conditions are given as follows: 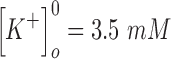 ,
,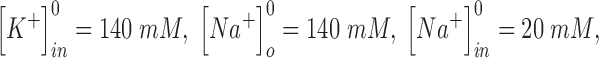
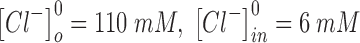 . R, T, Z, and F follow the same schema as described previously.
. R, T, Z, and F follow the same schema as described previously.
Increases in the concentration of intracellular chloride ions affect the reversal potential of chloride through the following equation:
 |
where ECl is the reversal potential of chloride ions.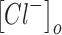 ,
, 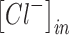 , R, T, Z, and F follow the same schema as listed previously. Increases in intracellular chloride also affects the resting membrane potential as shown in Equation (12).
, R, T, Z, and F follow the same schema as listed previously. Increases in intracellular chloride also affects the resting membrane potential as shown in Equation (12).
Modeling of drug conditions
In this study, we tested the efficacy of GABAA receptor agonists alone and in combination with an NKCC1 co-transporter blocker to determine if these drugs can rescue the RS neuron model from depolarization block. GABAA receptor agonists were modeled as twofold increases in the maximal synaptic conductance of phasic inhibitory inputs based on experiments that show that a GABAA agonist increases phasic inhibitory single channel conductance by two- to sevenfold.91
NKCC1 co-transporter blocker was modeled as a 95% decrease in the elevated levels of intracellular chloride according to the following equation:
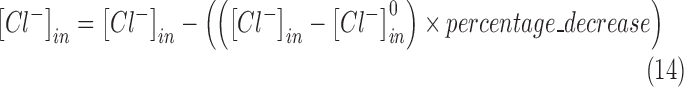 |
where 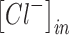 is the concentration of intracellular chloride,
is the concentration of intracellular chloride, 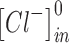 is the initial concentration of intracellular chloride (6 mM), and the value of percentage decrease is 0.95. All simulations were run using a range of these parameters, with no change in the qualitative results.
is the initial concentration of intracellular chloride (6 mM), and the value of percentage decrease is 0.95. All simulations were run using a range of these parameters, with no change in the qualitative results.
We also probed the mechanisms behind the neuroprotective efficacy of inverse agonists of GABAA receptors in cerebral ischemia.92 GABAA receptor inverse agonist was modeled as an 80% reduction in the maximal synaptic conductance of phasic GABAergic inputs.93
Modeling of bicarbonate dependent GABAA conductance
We separately modeled the ionic mechanisms responsible for activity dependent membrane depolarization mediated by GABAergic activity.94 The GABAA current in these simulations was modeled using the following equations95:
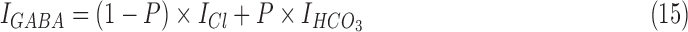 |
 |
 |
where 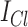 is the chloride ion mediated GABAergic current,
is the chloride ion mediated GABAergic current, 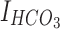 is the bicarbonate ion mediated GABAergic current, and
is the bicarbonate ion mediated GABAergic current, and 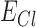 ,
, 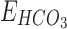 are their respective reversal potentials. P is the relative permeability of bicarbonate ion through the GABAA receptor. P was set to a value of 0.2, consistent with published estimates.96.
are their respective reversal potentials. P is the relative permeability of bicarbonate ion through the GABAA receptor. P was set to a value of 0.2, consistent with published estimates.96. 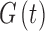 follows the same schema as in equation (9).
follows the same schema as in equation (9).
Modeling of GABAergic activity induced change in the concentration of anions
The anionic concentrations (chloride and bicarbonate) in the intra- and extracellular compartments were updated from their respective currents at each time step of the simulation.
The change in the concentration of chloride ions as a result of GABAergic inputs and their decay back to resting values were modeled according to the following equations.30,95 In this schema, 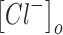 and
and 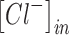 were updated at each time step as follows:
were updated at each time step as follows:
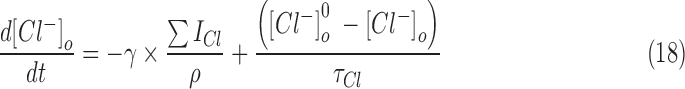 |
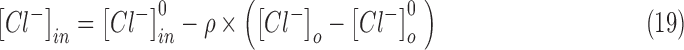 |
Similarly, 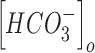 and
and 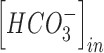 were updated at each time step of the simulation using the following equations:
were updated at each time step of the simulation using the following equations:
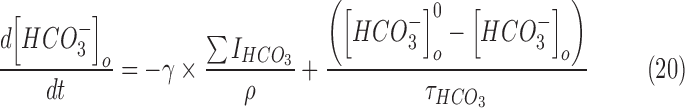 |
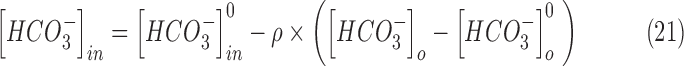 |
where 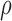 is the ratio of extracellular volume to intracellular volume, also known as the volume fraction.97 The value of
is the ratio of extracellular volume to intracellular volume, also known as the volume fraction.97 The value of 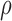 was set to 0.1429,30,40,97,98 mimicking the relative proportion of extracellular volume (14.29% of intracellular volume) under normal conditions when osmotic forces are non-existent.
was set to 0.1429,30,40,97,98 mimicking the relative proportion of extracellular volume (14.29% of intracellular volume) under normal conditions when osmotic forces are non-existent. 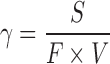 aids the conversion from current units to concentration units,30 where S is the surface area of the neuron, V is its volume, and F is the Faraday's constant.
aids the conversion from current units to concentration units,30 where S is the surface area of the neuron, V is its volume, and F is the Faraday's constant. 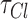 was set to 3 s to mimic the physiological chloride ion decay to resting levels (extrusion).95,99 Regeneration of
was set to 3 s to mimic the physiological chloride ion decay to resting levels (extrusion).95,99 Regeneration of 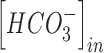 in the simulations was facilitated by setting
in the simulations was facilitated by setting 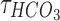 to 0.1 ms, while the regeneration was blocked by setting
to 0.1 ms, while the regeneration was blocked by setting 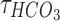 to 10 s.
to 10 s. 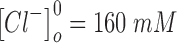 and
and 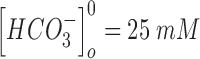 are the extracellular concentrations of the respective ionic species at resting conditions.100 Similarly,
are the extracellular concentrations of the respective ionic species at resting conditions.100 Similarly, 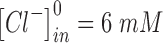 and
and 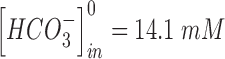 are the intracellular concentrations of the respective ionic species at rest.100
are the intracellular concentrations of the respective ionic species at rest.100
The anionic reversal potentials were also updated at each time step using the Nernst equation (Eqn [13]). Equations (18)–(21) ensure that the total number of chloride (and bicarbonate ions) inside and outside the neuron is conserved throughout the duration of the simulation.30
Data analysis
All simulations were performed using the NEURON 7.5 simulation environment.101 Spike times and membrane potential were recorded with a sampling frequency of 40,000 Hz and analyzed using custom routines written in MATLAB 2017B (www.mathworks.com).
We considered the model neuron to have entered depolarization block when the mean peak height of its spikes was < −20 mV during a 1 sec simulation run. To determine if the model reached our threshold of depolarization block, we injected direct current (DC) in steps of 20 pA amplitude up to 4 nA. The minimum current that produced spikes below −20 mV was categorized as IDB. Other values of depolarization block thresholds (0 mV, 5 mV, 10 mV) yielded results that were qualitatively similar. Firing rate was calculated as the number of spikes during a 1 sec simulation run of the model. Membrane potential was calculated by taking the median of all membrane potential data points during a 1 sec simulation run of the model. The percentage change in IDB of the neuron model in the presence of the drug(s) was computed from seven different model runs according to the following equation:
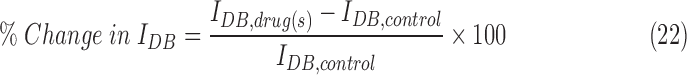 |
where 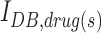 is the IDB of the neuron model in the presence of the drug(s) and
is the IDB of the neuron model in the presence of the drug(s) and 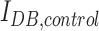 is the IDB of the neuron model under drug-free conditions. Similarly, the percentage change in the median membrane potential of the neuron model in the presence of the drug(s) was computed from seven different model runs according to the following equation:
is the IDB of the neuron model under drug-free conditions. Similarly, the percentage change in the median membrane potential of the neuron model in the presence of the drug(s) was computed from seven different model runs according to the following equation:
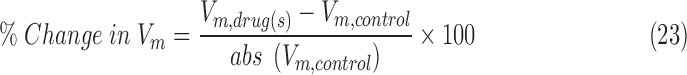 |
where Vm represents the median membrane potential.
Results
Depolarization block characteristics of the neocortical RS neuron model
We first characterized the depolarization block dynamics of the RS neuron model in response to progressively increasing current injections. Unsurprisingly, increased current led to more depolarization and increased spike rates, eventually resulting in depolarization block (Fig. 1). The current that resulted in depolarization block is easily visible in the firing frequency versus current plot (F-I curve) as the firing rate rapidly falls to 0 at and above this value (Fig. 1C). We define this minimal current value necessary to reach depolarization block as IDB.
FIG. 1.

Depolarization block in the regular spiking (RS) neuron model. (A) Membrane potential traces plotted for three values of injected current. Note the lack of action potentials when the cell is injected with a large 2700 pA current. (B) Median membrane potential increases as a function of injected current, as expected. (C) Frequency–current relationship (F–I curve) of the RS neuron model. The minimum current injection that results in the cell entering depolarization block (IDB) is apparent as a dramatic decrease in firing rate.
Post-TBI ion concentrations are very likely to result in depolarization block
The normal concentration of potassium in the interstitial space is ∼3–5 mM,1,4,102,103 and normal physiological levels of chloride inside a mature neuron are ∼6–10 mM (Fig. 2).58,104–107 Although numerous homeostatic mechanisms15–17,108,109 maintain these physiologically normal ion concentrations in the brain, large deviations from this range are often seen during pathological brain states. We modeled the conditions seen in epilepsy and TBI. During seizures, potassium levels in the interstitial space can reach values of up to 8.5–15 mM (Fig. 2).1,18–20 TBI is characterized by potentially much larger changes in ion concentration, not only of extracellular potassium (reaching values as high as 50 mM),22,23 but also of intracellular chloride levels (reaching values up to 39 mM) (Fig. 2).54,57,58 In addition, we differentiated between two subtypes of TBI, based on the changes in extracellular potassium concentration reported after mild versus severe injuries.22,23
FIG. 2.

Ionic concentration ranges for extracellular potassium and intracellular chloride in normal, epileptic, and mild versus severe post-traumatic brain injury (TBI) brain states. This schematic is derived from values reported in the following references.1,4,18–20,22,23,54,58,102,106
We determined the response of the RS neuron model in the presence of healthy and pathological ion concentrations. Figure 3 captures the firing response of the RS neuron model when exposed to three ionic concentration regimes corresponding to normal (Fig. 3A), epileptic (Fig. 3B), and post-TBI brain states (Fig. 3C). The RS neuron model, which normally fired ∼1–2 Hz in normal conditions (Fig. 3A), increased its mean firing rate to >9 Hz in the presence of the high potassium levels seen during seizures (12.5 mM; Fig. 3B). Note that this increased firing was purely the result of the increased reversal potential of potassium caused by the increase in its extracellular concentration, as neither synaptic inputs nor injected current were increased. Under post-TBI-like conditions, with high extracellular potassium and high intracellular chloride, the RS neuron model entered depolarization block (Fig. 3C). Increases in intracellular chloride led to depolarization of the reversal potential of GABAergic inputs (Fig. S1) and this, combined with elevated levels of extracellular potassium, accelerated the entry of the neuron into depolarization block in post-TBI brain states (see online supplementary material at http://www.liebertpub.com). We systematically altered both extracellular potassium and intracellular chloride, and observed the expected increases in membrane potential as a function of increase in either ion (Fig. 3D). The firing rate of cells, however, decreased dramatically when both extracellular potassium and intracellular chloride were high (Fig. 3E), highlighting the concentrations that result in the neuron entering depolarization block. Note that under all three conditions the amount of injected current was identical, and only the ion concentrations were altered as illustrated.
FIG. 3.

The impact of potassium and chloride ion concentrations on regular spiking (RS) neuronal firing and depolariziaton block. (A) The membrane potential of the RS neuron model under normal physiological levels of extracellular potassium and intracellular chloride is shown in the lower panel. The cell maintains a low firing rate, with occasional spikes driven by the stochastic synaptic inputs. The precise values of extracellular potassium and intracellular chloride are shown in the upper panel. (B) Same as in A, but for high extracellular potassium conditions similar to those seen during epileptic seizures. Note the increase in firing rate as a function of increased extracellular potassium. (C) Same as in A, but for high extracellular potassium plus high intracellular chlroide conditions similar to those seen after a traumatic brain injury. The cell first increases its firing rate, but then enters depolariziation block under these conditions. (D) Median membrane potential of the RS neuron model as a function of both extracellular potassium and intracellular chloride. (E) Same as in D, but for firing rate of the RS neuron model. Note the lack of firing in the upper right quadrant, corresponding to depolariziaton block of the RS neuron under high extracellular potassium and high intracellular chloride conditions.
GABAA agonists, only when combined with NKCC1 co-transporter blockers, can rescue RS neurons from depolarization block after TBI
Having established a model that captures the conditions seen in the immediate aftermath of TBI, we next sought to understand why GABAA receptor agonists have been unsuccessful as neuroprotective agents after TBI.42–46 We modeled GABAA receptor agonists as leading to a twofold increase in the peak conductance of phasic synaptic inputs91 (see the Methods section). In epileptic conditions, GABAA receptor agonists successfully reduced the firing rate of the neuron (from 9.2 to 2.5 Hz in the example shown in Fig. 4A). Under post-TBI conditions, however, GABAA receptor agonists failed to rescue the neuron from depolarization block (Fig. 4B). This is because of the increased levels of intracellular chloride in post-TBI conditions, triggered by the upregulation of the NKCC1 co-transporter in TBI.52–55 The increased intracellular chloride increases the reversal potential of chloride, making GABA excitatory instead of inhibitory (EGABA changes from −75.9 mV to −31.4 mV in this example). We hypothesized that depolarizing chloride gradients can be reversed by blockers of NKCC1 co-transporters, which would decrease intracellular chloride levels. Indeed, we found that the application of a combination of GABAA receptor agonists and NKCC1 co-transporter blocker was able to rescue the model RS neuron from depolarization block (Fig. 4C).
FIG. 4.

Effect of GABAA receptor agonists on the response of regular spiking (RS) neuron model in pathological brain states, in the presence and absence of Na+-K+-2Cl- (NKCC1) co-transporter blockers. (A) Response of the RS neuron model to GABAA receptor agonists in an epileptic brain state. The values of extracellular potassium and intrcellular chloride are shown in the upper panel. Under these epileptic conditions, GABAA receptor agonists can still help hyperpolarize the neuron, as would be expected from their common use as anti-epileptic drugs. (B) Same as in A, but for a post-traumatic brain injury (TBI) brain state. The GABAA agonist is unable to rescue the neuron from depolarizaiton block. (C) Response of the RS neuron model to a combination of GABAA receptor agonist and NKCC1 co-transporter blocker in a post-TBI brain state. Dotted line signifies the change in intracellular chloride caused by the effect of the NKCC1 co-transporter blocker. The combination of the two drugs is able to rescue the model neuron from depolariziation block.
We quantified the efficacy of the drug(s) (GABAA receptor agonists with or without NKCC1 co-transporter blockers) by computing the propensity of the neuron to enter depolarization block under a wide range of ionic conditions spanning both epileptic and post-TBI regimes (see the Methods section). We computed the change in IDB of the neuron model in the presence of drug(s) (IDB, drug(s)) compared with drug free conditions (IDB, control) (see the Methods section). A positive % change (increase in IDB in the presence of the drug[s]) implies a therapeutic benefit, as it means that the neuron is less susceptible to depolarization block. A negative % change (decrease in IDB in the presence of the drug[s]) implies a pathological worsening, with the cell being even less likely to be rescued from depolarization block. Figure 5A shows that under the high chloride conditions typical of the post-TBI state, GABAA agonists either had no effect or increased the propensity of the neuron to enter depolarization block (% change in IDB < 0). However, when combined with an NKCC1 co-transporter blocker (Fig. 5B), GABAA agonists reduced the propensity of the neuron to enter depolarization block (% change in IDB > 0) under the same high chloride conditions. The same results were seen under two pathologically high extracellular potassium conditions, reflective of mild and severe TBI subtypes respectively (compare Fig. 5A and B in 12.5 mM extracellular potassium with Fig. 5C and D in 18.5 mM extracellular potassium). We obtained qualitatively similar results when the neuroprotective efficacy of drug(s) was quantified using median membrane potential (Fig. S2), affirming the robustness of our results (see online supplementary material at http://www.liebertpub.com). These results suggest that a combination of GABAA receptor agonists and NKCC1 co-transporter blockers would lead to increased hyperpolarization and enhanced neuroprotection after a TBI.
FIG. 5.

GABAA agonists, only in combination with Na+-K+-2Cl- (NKCC1) co-transporter blockers, can rescue neurons from depolariziaton block across a range of pathological ion concentations. (A) Effect of GABAA receptor agonists on the percent change in IDB of the regular spiking (RS) neuron model when Koutside = 12.5 mM, reflecting mild traumatic brain injury (TBI). Note that values >0 reflect a therapeutic effect, whereas values <0 reflect a pathological worsening. Under these potassium conditions, GABAA receptor agonists can reduce the propensity to depolarization block when intracellular chloride concentrations are close to the normal physiological range, but exert little or negative effect when chloride concentrations inside the cell exceed the physiological range. (B) Same as in A, but for the combination of a GABAA receptor agonist and NKCC1 co-transporter blocker. By blocking NKCC1 co-transporter and hence reducing intracellular chloride concentration, this combination of drugs can rescue the model neuron from depolariziation block across a wide range of intracellular chloride concentrations. This suggests that this combination of drugs can have a therapeutic benefit in post-TBI brain states. (C, D) Same as A, B, but for Koutside = 18.5 mM, reflecting more severe TBI.
Relative neuroprotective efficacies of GABAAR agonists and inverse agonists in ischemic brain injury
To understand a related set of pathological observations, we next sought to understand why diazepam (a GABAAR agonist) fails to provide neuroprotection when administered post-ischemia, whereas an inverse agonist of GABAA receptors is successful in reducing post-ischemic hippocampal cell death.92 Ischemic conditions are accompanied by increases in the concentration of potassium110,111 and GABA112,113 in the extracellular space. Although the effect of increased extracellular potassium is known to result in membrane depolarization (as shown in the above simulations), we wanted to characterize the effect of increases in extracellular GABA levels on neuronal excitability. For this purpose, we conducted simulations in which GABAARs in the model (Fig. S3A) were subjected to strong stimulation (200 Hz input train for 200 ms) (see online supplementary material at http://www.liebertpub.com). This mimics the increased extracellular GABA levels seen post-ischemia (but not seen after TBI114,115). The GABA synapses in these simulations were modeled as a combined chloride and bicarbonate conductance with a relative permeability of 0.2.96 The concentration of the anions (chloride and bicarbonate) were updated at each time step from their respective currents (see the Methods section). EGABA was set to −72 mV and resting potential was set to −68 mV, so that GABA was hyperpolarizing at rest. Bicarbonate ionic gradients were not allowed to break down (see the Methods section) as intracellular bicarbonate levels are known to be tightly regulated by pH buffers.99 Under these conditions, GABA elicits an initial short hyperpolarizing response followed by a large depolarizing response (Fig. S3B), as seen in experiments.94 This is because of the depolarization of EGABA in the dendritic compartment (Fig. S3C) as a result of accumulation of intracellular chloride (Fig. S3D). However, if the bicarbonate ionic gradients were allowed to collapse (Fig. S3E), GABA mediated depolarization was significantly reduced (Fig. S3A) (see online supplementary material at http://www.liebertpub.com). Therefore, increases in extracellular GABA levels (as seen during ischemia) would result in depolarization of neurons, and this effect is dependent on the maintenance of the bicarbonate ionic gradient.
Having elucidated the ionic mechanisms behind activity-dependent GABA- mediated depolarization, we modeled ischemia-induced excitotoxicity in the RS neuron model (Fig. S4) (see online supplementary material at http://www.liebertpub.com). Under ischemic conditions (increased extracellular potassium and continuous stimulation of GABAARs by increased extracellular GABA levels), the RS neuron model entered depolarization block (Fig. S4A). GABAAR agonists failed to rescue the neuron from depolarization block (Fig. S4A). However, inverse agonists partially rescued the neuron from the excitotoxic depolarization block (Fig. S4B) (see online supplementary material at http://www.liebertpub.com). Thus, our model can help to explain why diazepam fails to provide neuroprotection when administered post-ischemic injury, whereas inverse GABAAR agonists are more successful in preventing cell death under these same post-ischemic conditions.92
Discussion
Our results provide a biophysical explanation for why GABAA receptor agonists fail to offer neuroprotection after TBI. Both epilepsy and TBI are characterized by increases in the levels of extracellular potassium, increasing the excitability of neurons.1,18–20,22–27 However, because of post-TBI elevations in the expression of NKCC1 co-transporter,52–54,56 levels of intracellular chloride increase54,57,58 and further aggravate post-TBI excitotoxicity by reversing the polarity of the normally inhibitory neurotransmitter GABA. Our modeling work shows that as a result of this excitatory nature of GABA after TBI,58 GABAA receptor agonists fail to help neurons recover from depolarization block and therefore would fail to prevent secondary cell death. However, we have shown that if GABAA receptor agonists are combined with NKCC1 co-transporter blockers, the combination of drugs is likely to rescue neurons from depolarization block, even in pathological, post-TBI brain states. Therefore, our modeling results predict that GABAA receptor agonists will be effective in providing neuroprotection in TBI when co-administered with NKCC1 co-transporter inhibitors. This is a biophysically principled prediction that our laboratory is now testing experimentally.
Depolarization block as an indicator of cell death in an energy-compromised tissue
TBI is characterized by the widespread depolarization block of neuronal activity.33,36,37,116 The depression of neuronal firing (often called spreading depression34,37) from depolarization block is caused by spontaneous spreading waves of depolarization that have also been seen in electrocorticography (ECoG) recordings after TBI.33,36,37 Spreading depression is accompanied by changes in ionic gradients, cell swelling, decreased input resistance, and other changes that result in the massive depolarization of neurons.37 This hyperexcitable brain state eventually leads to the silencing of neuronal activity through depolarization block33,36,37 because neurons are unable to fire any action potentials because of complete inactivation of voltage-gated sodium channels.
Spreading depression can propagate at a speed of 1–5 mm/min33,36 between brain regions and may indicate progression of secondary brain injuries.35,36 There are many studies that support this observation: an experiment utilizing the fluid percussion injury model in rats found a positive correlation among the number of spreading depression cycles, intracranial pressure, and mortality rates.35 In a similar study involving human subjects, intracranial pressure was found to be higher in patients who experienced episodes of spreading depression.33 Additionally, in a rat model of cerebral ischemia, spreading depression in the peri-infarct zone was associated with increased infarct volume and cell death.36,38
Rescuing neurons from depolarization block is therefore an important neuroprotective goal, and would help recovery from TBI in multiple ways. First, it would reduce the activity of Na+/K+ pumps that are constantly working to restore ionic gradients during depolarization block and hence reduce adenosine triphosphate (ATP) consumption.31,116–118 This would help to ease the metabolic distress that the neurons are under119 and gradually restore the physiological ionic gradients. Second, N-methyl-d-aspartate (NMDA) receptors would be active during depolarization block, as the pore of the receptor would be relieved from magnesium ion block at these depolarized membrane potentials.117,118,120–122 Calcium entry via NMDA receptors is a major source of post-TBI excitotoxicity123; therefore, rescuing the neuron from depolarization block is likely to reduce the net calcium entry into the neurons via NMDA channels. Therefore, drugs that decrease the propensity of a neuron to reach depolarization block would represent promising therapeutic candidates for neuroprotection in TBI.
GABAA receptor agonists and their history in treating TBI
Several studies have established that GABAA receptor agonists are ineffective in helping functional recovery after TBI.42–47 In a study involving lesions to the anterior-medial areas of the cortex, administration of diazepam (a benzodiazepine) immediately after the injury impaired recovery from sensory asymmetry (until 22 days post-injury) in rats and caused no change to lesion size.44 Similarly, when rats were administered phenobarbital following injury, the drug impeded functional recovery by up to 4 weeks and did not exert any significant effect on the lesion size compared with controls.45 Importantly, in a model of hemiplegia in rats, local infusion of GABA to the injured motor cortex impeded recovery of motor function.47 In a recent study involving a controlled cortical impact (CCI) model of TBI in rats, pentobarbital, diazepam, and propofol all failed to reduce the contusional volume when administered immediately after the impact.42 Additionally, rats treated with propofol exhibited poor performance on motor tasks, whereas those treated with diazepam performed worse on cognitive tasks than those injured rats that were not treated with any drugs.42 Isoflurane, an anesthetic agent that is commonly used in TBI experiments, caused increased cell death and impaired functional recovery at 48 h post-CCI-induced injury in rats.49 Similar effects were observed with two other novel GABA potentiating anticonvulsant drugs, topiramate48,124 and vigabatrin,125 following brain injury. An emerging consensus from these studies illustrates that GABA or GABAA receptor agonists, when administered on their own after a brain injury, actually impede the recovery of function and have no beneficial effect on the lesion/contusion volume.
Changes in expression of chloride transporters after TBI
The expression levels of NKCC1 proteins in the cortex and hippocampus post-TBI have been quantified in numerous studies using the Western blot method.52,54,56 In a study involving CCI in mice,52 NKCC1 co-transporter upregulation in the cortex started at 6 h and persisted until 24 h post-injury. The expression levels in this study peaked at 12 h following injury (showing a fourfold increase over baseline).52 Similarly, NKCC1 proteins in the hippocampus were upregulated as early as 2 h following the injury and continued to be upregulated for 24 h post-injury in rats that were injured using the weight drop method (twofold peak upregulation compared to sham).56 Additionally, mice that received closed head injury exhibited increases in NKCC1 expression levels at 1 day post-injury and the upregulation continued for 7 days (twofold peak upregulation compared with sham).54 From these studies, it becomes evident that irrespective of the injury method, NKCC1 co-transporter is upregulated rapidly following TBI (as early as 2 h) and the upregulation may persist until a week following the injury. As a direct consequence of this upregulation, the concentration of chloride ion inside the neuron increases, resulting in depolarized values of the reversal potential of GABA currents (EGABA).54,57,58 Elevated levels of EGABA following brain trauma have been reported in multiple electrophysiological studies.54,57,58 Therefore, as shown by our model, the paradoxical effect of GABAA receptor agonists following brain trauma reported in earlier studies42,44,45,47 is likely caused by the upregulation of NKCC1 co-transporters. Confirming this hypothesis, blockers of NKCC1 co-transporters (e.g., bumetanide) mildly reduced edema formation, contusional volume, and incidence of post-traumatic seizures following TBI.52–56,126,127
Changes in the expression of NKCC1 co-transporters post-TBI were modeled as an increase in the intracellular concentration of chloride ions. The intracellular concentration of chloride ions in the simulation was varied to capture the extent of change in the reversal potential of GABAergic currents post-trauma, as reported in the literature.54,58 The change in GABA reversal potential reported in these studies54,58 spans a wide range from 3 mV to 47 mV. Accordingly, we varied the intracellular chloride levels in the model (in steps of 3 mM, up to 39 mM) to capture the magnitude of the corresponding depolarizing shift in the reversal potential of GABAergic currents.
A few studies54,57 have suggested that KCC2 co-transporters are downregulated after TBI, in addition to the upregulation of NKCC1. Inhibition of NKCC1 co-transporters is still likely to be beneficial and would help to restore the inhibitory efficacy of GABAA receptor agonists for the following reasons: NKCC1 co-transporter is expressed not only in neurons but also in glial cells.128 The combined blockade of NKCC1 co-transporters in both neurons and glia would help to prevent the transport of chloride ions from the extracellular space to the intracellular space. This results in an increase in the levels of chloride in the extracellular space. Therefore, the increase in the extracellular chloride levels would (partially) offset the effect of increase in the levels of intracellular chloride that might be caused by the downregulation of KCC2 co-transporters. The literature is supportive of this reasoning: in certain pathological conditions that result in a concomitant change in the expression of NKCC1 (upregulation) and KCC2 (downregulation) co-transporters, bumetanide, a specific blocker of NKCC1 co-transporters, is still sufficient for abolishing the excitatory action of GABA and restoring its hyperpolarizing effect.129–131
The combination of GABAA receptor agonists and blockers of NKCC1 co-transporter is likely to be far more efficacious than either drug on its own
Our modeling results suggest that the therapeutic efficacy of GABAA receptor agonists in TBI could be enhanced when they are combined with blockers of NKCC1 co-transporters. Because the protein expression of NKCC1 is upregulated a few hours after injury (as early as 2 h), GABAAR agonists would start to lose their inhibitory efficacy immediately following TBI as a result of altered chloride transport, and may continue to be in that state for up to 7 days following injury. In order to prevent cell death, our modeling suggests that these drugs should be combined with NKCC1 co-transporter blockers as soon as possible following injury, and that the drug combination may be continued until 7 days post-TBI to achieve optimal neuroprotection. However, if the upregulation continues beyond the 7 day time period in particularly severe TBI cases, continuation of the drug combination might be necessary to maintain the inhibitory effect of GABAAR agonists. Supporting our hypothesis, in rodent models of neonatal epilepsies, the combination of a GABAA receptor agonist (phenobarbital) and an NKCC1 co-transporter blocker (bumetanide) has been shown to be effective in combating seizures.132,133 Similarly, in a model of hypoxia-ischemia in neonatal rats, bumetanide enhances the combined neuroprotective efficacy of phenobarbital and hypothermia.134 Further, in adult mice, bumetanide reinstates the efficacy of diazepam in reducing seizures by reversing the altered chloride gradients following development of status epilepticus.135,136 Our modeling work suggests that such combination of drugs will also be neuroprotective in TBI.
Future work
In this study, we used a computational model of a neocortical RS neuron to identify efficacious drug combinations for neuroprotection in TBI. The neocortex is endowed with a diversity of neurons,137–140 and it is highly likely that these neuronal subtypes, by virtue of their different membrane and synaptic properties and morphology, exhibit varying propensities to depolarization block. For example, inhibitory interneurons (specifically fast spiking [FS] neurons) might be at an increased risk of potassium- mediated excitotoxicity because of the dense expression of voltage-gated potassium channels in these neurons.141 This could lead to a buildup of local microdomains of extracellular potassium during periods of intense neuronal activity as seen during brain trauma. Therefore, future work will focus on extending this study to other subtypes of cortical neurons including intrinsically burst-firing,142 fast-spiking inhibitory,137,143 and many other inhibitory neuronal types of the neocortex and hippocampus.137,138
Our single-neuron model is likely to underestimate the amount of excitotoxicity that happens in a brain state post-TBI.144–147 TBI is characterized by hypoxia, ionic imbalance, cell swelling, and glutamate excitotoxicity culminating in cell death.144,148,149 Therefore, detailed pathophysiology of TBI must be implemented in network models of different brain regions at multiples levels of complexity to mimic TBI-induced excitotoxicity. Multi-scale simulation of TBI requires careful modeling of oxygen and glucose metabolism, potassium diffusion in the extracellular space, glutamate diffusion in the synaptic cleft, various pumps and transporters on neurons and glial cells, cytotoxic edema, volume dynamics, and various modes of calcium entry into the neuron.28–30,123,150 Given the heterogeneity of injury types in TBI,151,152 the multi-scale modeling approach would greatly help with the discovery of rational, biophysically principled drug combinations for neuroprotection in TBI.152
Conclusion
Our results provide a simple mechanistic explanation for why GABAA receptor agonists have failed to work in TBI treatment, and suggest that combining these drugs with an NKCC1 co-transporter blocker is likely to offer significantly improved neuroprotection. Work in progress in our laboratory is now testing the possible therapeutic efficacy of this drug combination in a rodent model of TBI.
Supplementary Material
Acknowledgments
We thank Scott Cruikshank, Tibin John, and Ellen Wixted for a careful reading of this manuscript. This work was supported by grants to O.J.A. from the National Institutes of Health (NIH) (R03MH111316), the American Epilepsy Society (Junior Investigator Award), Michigan Institute for Computational Discovery & Engineering (MICDE) (Catalyst Grant) and the Joyce and Don Massey Family Foundation/Michigan Center for Integrative Research in Critical Care (MCIRCC) (TBI Grand Challenge Award).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Fröhlich F., Bazhenov M., Iragui-Madoz V., and Sejnowski T.J. (2008). Potassium dynamics in the epileptic cortex: new insights on an old topic. Neuroscientist 14, 422–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ding F., O'Donnell J., Xu Q., Kang N., Goldman N., and Nedergaard M. (2016). Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science 352, 550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sulis Sato S., Artoni P., Landi S., Cozzolino O., Parra R., Pracucci E., Trovato F., Szczurkowska J., Luin S., Arosio D., Beltram F., Cancedda L., Kaila K., and Ratto G.M. (2017). Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc. Natl. Acad. Sci. 114, E8770–E8779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Somjen G.G. (2004). Ions in the Brain: Normal Function, Seizures And Stroke, 2nd ed Oxford University Press: New York [Google Scholar]
- 5.Nernst W. (1888). Zur Kinetik der in Losung befindlichen Korper. Erste Abhandlung. Theorie der Diffusion. Z. Phys. Chem 2, 613–637 [Google Scholar]
- 6.Nernst W. (1889). Zur Theorie umkehrbarer galvanischer Elemente. Sitzungsber. Preuss. Akad. Wiss. 1889, 83–95 [Google Scholar]
- 7.Nernst W. (1889). Die elektromotorische Wirksamkeit der Jonen. Z. Phys. Chem. 4, 129–181 [Google Scholar]
- 8.Forsythe I.D., and Redman S.J. (1988). The dependence of motoneurone membrane potential on extracellular ion concentrations studied in isolated rat spinal cord. J. Physiol. 404, 83–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ritter P., Jirsa V.K., McIntosh A.R., and Breakspear M. (2015). State-dependent brain computation. Editorial. Front. Comput. Neurosci. 9, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krishnan G.P., González O.C., and Bazhenov M. (2018). Origin of slow spontaneous resting-state neuronal fluctuations in brain networks. Proc. Natl. Acad. Sci. 115, 6858–6863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bazhenov M., Timofeev I., Steriade M., and Sejnowski T.J. (2004). Potassium model for slow (2–3 Hz) in vivo neocortical paroxysmal oscillations. J. Neurophysiol. 92, 1116–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Ari Y., Cherubini E., Corradetti R., and Gaiarsa J.L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirmse K., Kummer M., Kovalchuk Y., Witte O.W., Garaschuk O., and Holthoff K. (2015). GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat. Commun. 6, 1–13 [DOI] [PubMed] [Google Scholar]
- 14.Ben-Ari Y., Khalilov I., Kahle K.T., and Cherubini E. (2012). The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18, 467–486 [DOI] [PubMed] [Google Scholar]
- 15.Plotkin M.D., Snyder E.Y., Hebert S.C., and Delpire E. (1997). Expression of the Na – K – 2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol. 33, 781–795 [DOI] [PubMed] [Google Scholar]
- 16.Lu J., Karadsheh M., and Delpire E. (1999). Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J. Neurobiol. 39, 558–568 [PubMed] [Google Scholar]
- 17.Daneman R., and Prat A. (2015). The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 7, a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sypert G.W., and Ward A.A. (1974). Changes in extracellular potassium activity during neocortical propagated seizures. Exp. Neurol. 45, 19–41 [DOI] [PubMed] [Google Scholar]
- 19.Jensen M.S., and Yaari Y. (1997). Role of intrinsic burst firing, potassium accumulation, and electrical coupling in the elevated potassium model of hippocampal epilepsy. J. Neurophysiol. 77, 1224–1233 [DOI] [PubMed] [Google Scholar]
- 20.Traynelis S.F., and Dingledine R. (1988). Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J.Neurophysiol. 59, 259–276 [DOI] [PubMed] [Google Scholar]
- 21.Trevelyan A.J., Sussillo D., Watson B.O., and Yuste R. (2006). Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J. Neurosci. 26, 12,447–12,455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katayama Y., Becker D.P., Tamura T., and Hovda D.A. (1990). Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 73, 889–900 [DOI] [PubMed] [Google Scholar]
- 23.Takahashi H., Manaka S., and Sano K. (1981). Changes in extracellular potassium concentration in cortex and brain stem during the acute phase of experimental closed head injury. J. Neurosurg. 55, 708–717 [DOI] [PubMed] [Google Scholar]
- 24.Madikians A., and Giza C.C. (2006). A clinician's guide to the pathophysiology of traumatic brain injury. Indian J. Neurotrauma 3, 9–17 [Google Scholar]
- 25.Reinert M., Khaldi A., Zauner A., Doppenberg E., Choi S., and Bullock R. (2000). High level of extracellular potassium and its correlates after severe head injury: relationship to high intracranial pressure. J. Neurosurg. 93, 800–807 [DOI] [PubMed] [Google Scholar]
- 26.Choi D.W. (1987). Ionic dependence of glutamate neurotoxicity. J. Neurosci. 7, 369–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen A.J. (1977). Extracellular potassium concentration in juvenile and adult rat brain cortex during anoxia. Acta Physiol. Scand. 99, 412–420 [DOI] [PubMed] [Google Scholar]
- 28.Ullah G., Cressman J.R., Barreto E., and Schiff S.J. (2009). The influence of sodium and potassium dynamics on excitability: II network and glial dynamics. J. Comput. Neurosci. 26, 171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cressman J.R., Ullah G., Ziburkus J., Schiff S.J., and Barreto E. (2009). The influence of sodium and potassium dynamics on excitability, seizures, and the stability of persistent states: I. single neuron dynamics. J. Comput. Neurosci. 26, 159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei Y., Ullah G., and Schiff S.J. (2014). Unification of neuronal spikes, seizures, and spreading depression. J. Neurosci. 34, 11,733–11,743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bikson M., Hahn P.J., Fox J.E., and Jefferys J.G.R. (2003). Depolarization block of neurons during maintenance of electrographic seizures. J. Neurophysiol. 90, 2402–2408 [DOI] [PubMed] [Google Scholar]
- 32.Morozova E.O., Zakharov D., Gutkin B.S., Lapish C.C., and Kuznetsov A. (2016). Dopamine neurons change the type of excitability in response to stimuli. PLoS Comput. Biol. 12, e1005233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hartings J.A., Strong A.J., Fabricius M., Manning A., Bhatia R., Dreier J.P., Mazzeo A.T., Tortella F.C., and Bullock M.R. (2009). Spreading depolarizations and late secondary insults after traumatic brain injury. J. Neurotrauma 26, 1857–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leão A.A. (1944). Spreading depression of activity in the cerebral cortex. J. Neurophysiol. 7, 359–390 [DOI] [PubMed] [Google Scholar]
- 35.Rogatsky G.G., Sonn J., Kamenir Y., Zarchin N., and Mayevsky A. (2003). Relationship between intracranial pressure and cortical spreading depression following fluid percussion brain injury in rats. J. Neurotrauma 20, 1315–1325 [DOI] [PubMed] [Google Scholar]
- 36.Lauritzen M., Dreier J.P., Fabricius M., Hartings J.A., Graf R., and Strong A.J. (2011). Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J. Cereb. Blood Flow Metab. 31, 17–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dreier J.P. (2011). The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 17, 439–447 [DOI] [PubMed] [Google Scholar]
- 38.Hartings J.A., Rolli M.L., Lu X.-C.M., and Tortella F.C. (2003). Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: relation to infarct growth and neuroprotection. J. Neurosci. 23, 11,602–11,610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mies G., Iijima T., and Hossmann K.A. (1993). Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat. Neuroreport 4, 709–711 [DOI] [PubMed] [Google Scholar]
- 40.Kager H., Wadman W., and Somjen G.G. (2002). Conditions for the triggering of spreading depression studied with computer simulations. J. Neurophysiol. 88, 2700–2712 [DOI] [PubMed] [Google Scholar]
- 41.Somjen G.G. (2001). Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev. 81, 1065–1096 [DOI] [PubMed] [Google Scholar]
- 42.Statler K.D., Alexander H., Vagni V., Dixon C.E., Clark R.S.B., Jenkins L., and Kochanek P.M. (2006). Comparison of seven anesthetic agents on outcome after experimental traumatic brain injury in adult, male rats. J. Neurotrauma 23, 97–108 [DOI] [PubMed] [Google Scholar]
- 43.Tecoult E., Mesenge C., Stutzmann J.-M., Plotkine M., and Wahl F. (2000). Influence of anesthesia protocol in experimental traumatic brain injury. J. Neurosurg. Anesthesiol. 12, 255–261 [DOI] [PubMed] [Google Scholar]
- 44.Schallert T., Hernandez T.D., and Barth T. (1986). Recovery of function after brain damage: severe and chronic disruption by diazepam. Brain Res. 379, 104–111 [DOI] [PubMed] [Google Scholar]
- 45.Hernandez T.D., and Holling L.C. (1994). Disruption of behavioral recovery by the anti-convulsant phenobarbital. Brain Res. 635, 300–306 [DOI] [PubMed] [Google Scholar]
- 46.Goldstein L.B. (1995). Prescribing of potentially harmful drugs to patients admitted to hospital after head injury. J Neurol. Neurosurg. Psychiatry 58, 753–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brailowsky S., Knight R.T., Blood K., and Scabini D. (1986). γ-Aminobutyric acid-induced potentiation of cortical hemiplegia. Brain Res. 362, 322–330 [DOI] [PubMed] [Google Scholar]
- 48.Hoover R.C., Motta M., Davis J., Saatman K.E., Fujimoto S.T., Thompson H.J., Stover J.F., Dichter M.A., Twyman R., White H.S., and McIntosh T.K. (2004). Differential effects of the anticonvulsant topiramate on neurobehavioral and histological outcomes following traumatic brain injury in rats. J. Neurotrauma 21, 501–512 [DOI] [PubMed] [Google Scholar]
- 49.Hertle D., Beynon C., Zweckberger K., Vienenkötter B., Jung C.S., Kiening K., Unterberg A., and Sakowitz O.W. (2012). Influence of isoflurane on neuronal death and outcome in a rat model of traumatic brain injury, in: Intracranial Pressure and Brain Monitoring XIV. M.U Schuhmann, and M. Czosnyka (eds.). Springer: Vienna, pps. 383–386 [DOI] [PubMed] [Google Scholar]
- 50.Kennardt M.A. (1945). The effect of anticonvulsant drugs on recovery of function following cerebral cortical lesions. J. Neurophysiol. 8, 221–231 [Google Scholar]
- 51.[No authors listed] (2017). Rationalizing combination therapies. Editorial. Nat. Med. 23, 1113. [DOI] [PubMed] [Google Scholar]
- 52.Hui H., Rao W., Zhang L., Xie Z., Peng C., Su N., Wang K., Wang L., Luo P., Hao Y.L., Zhang S., and Fei Z. (2016). Inhibition of Na+-K+-2Cl- cotransporter-1 attenuates traumatic brain injury-induced neuronal apoptosis via regulation of erk signaling. Neurochem. Int. 94, 23–31 [DOI] [PubMed] [Google Scholar]
- 53.Lu K.T., Wu C.Y., Cheng N.C., Wo Y.Y.P., Yang J.T., Yen H.H., and Yang Y.L. (2006). Inhibition of the Na+-K+-2Cl–cotransporter in choroid plexus attenuates traumatic brain injury-induced brain edema and neuronal damage. Eur. J. Pharmacol. 548, 99–105 [DOI] [PubMed] [Google Scholar]
- 54.Wang F., Wang X., Shapiro L.A., Cotrina M.L., Liu W., Wang E.W., Gu S., Wang W., He X., Nedergaard M., and Huang J.H. (2017). NKCC1 up-regulation contributes to early post-traumatic seizures and increased post-traumatic seizure susceptibility. Brain Struct. Funct. 222, 1543–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ben-Ari Y. (2017). NKCC1 chloride importer antagonists attenuate many neurological and psychiatric disorders. Trends Neurosci. 40, 536–554 [DOI] [PubMed] [Google Scholar]
- 56.Lu K.T., Cheng N.C., Wu C.Y., and Yang Y.L. (2008). NKCC1-mediated traumatic brain injury-induced brain edema and neuron death via raf/mek/mapk cascade. Crit. Care Med. 36, 917–922 [DOI] [PubMed] [Google Scholar]
- 57.Bonislawski D.P., Schwarzbach E.P., and Cohen A.S. (2007). Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol. Dis. 25, 163–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pol van den A.N., Obrietan K., and Chen G. (1996). Excitatory actions of GABA after neuronal trauma. J. Neurosci. 16, 4283–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hodgkin A.L., and Huxley A.F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCormick D.A., Connors B.W., Lighthall J.W., and Prince D.A. (1985). Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. J. Neurophysiol. 54, 782–806 [DOI] [PubMed] [Google Scholar]
- 61.Masland R.H. (2004). Neuronal cell types. Curr. Biol. 14, R497–R500 [DOI] [PubMed] [Google Scholar]
- 62.Maxwell W., Dhillon H.K., Espin J., MacIntosh T., Smith D., and Graham D. (2003). There is differential loss of pyramidal cells from the human hippocampus with survival after blunt head injury. J. Neuropathol. Exp. Neurol. 62, 272–279 [DOI] [PubMed] [Google Scholar]
- 63.MacDonald V., and Halliday G.M. (2002). Selective loss of pyramidal neurons in the pre-supplementary motor cortex in Parkinson's disease. Mov. Disord. 17, 1166–1173 [DOI] [PubMed] [Google Scholar]
- 64.Vierling-Claassen D., Cardin J.A., Moore C.I., and Jones S.R. (2010). Computational modeling of distinct neocortical oscillations driven by cell-type selective optogenetic drive: separable resonant circuits controlled by low-threshold spiking and fast-spiking interneurons. Front. Hum. Neurosci. 4, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cruikshank S.J., Lewis T.J., and Connors B.W. (2007). Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat. Neurosci. 10, 462–468 [DOI] [PubMed] [Google Scholar]
- 66.Guan D., Lee J.C.F., Higgs M.H., Spain W.J., and Foehring R.C. (2007). Functional roles of Kv1 channels in neocortical pyramidal neurons. J. Neurophysiol. 97, 1931–1940 [DOI] [PubMed] [Google Scholar]
- 67.Bertil Hille. (2001). Ion Channels of Excitable Membranes, 3rd ed Sinauer Associates Inc.: Sunderland, MA [Google Scholar]
- 68.Beierlein M., Gibson J.R., and Connors B.W. (2003). Two dynamically distinct inhibitory networks in layer 4 of the neocortex. J. Neurophysiol. 90, 2987–3000 [DOI] [PubMed] [Google Scholar]
- 69.Levy R.B., and Reyes A.D. (2012). Spatial profile of excitatory and inhibitory synaptic connectivity in mouse primary auditory cortex. J. Neurosci. 32, 5609–5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Farries M.A., Kita H., and Wilson C.J. (2010). Dynamic spike threshold and zero membrane slope conductance shape the response of subthalamic neurons to cortical input. J. Neurosci. 30, 13180–13191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martina M., and Jonas P. (1997). Functional differences in Na+ channel gating between fast-spiking interneurons and principal neurones of rat hippocampus. J. Physiol. 505, 593–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu P.W., and Bean B.P. (2014). Kv2 channel regulation of action potential repolarization and firing patterns in superior cervical ganglion neurons and hippocampal CA1 pyramidal neurons. J. Neurosci. 34, 4991–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Murakoshi H., and Trimmer J.S. (1999). Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J. Neurosci. 19, 1728–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guan D., Tkatch T., Surmeier D.J., Armstrong W.E., and Foehring R.C. (2007). Kv2 subunits underlie slowly inactivating potassium current in rat neocortical pyramidal neurons. J. Physiol. 581, 941–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kihira Y., Hermanstyne T.O., and Misonou H. (2010). Formation of heteromeric Kv2 channels in mammalian brain neurons. J. Biol. Chem. 285, 15,048–15,055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Locke R.E., and Nerbonne J.M. (1997). Role of voltage-gated K+ currents in mediating the regular-spiking phenotype of callosal-projecting rat visual cortical neurons. J. Neurophysiol. 78, 2321–2335 [DOI] [PubMed] [Google Scholar]
- 77.Golomb D., Donner K., Shacham L., Shlosberg D., Amitai Y., and Hansel D. (2007). Mechanisms of firing patterns in fast-spiking cortical interneurons. PLoS Comput. Biol. 3, e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang H., Stradtman G.G., Wang X., and Gao W. (2008). A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc. Natl. Acad. Sci. 105, 5–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiang Z., Huguenard J.R., Prince D.A., Huguenard J.R., and Synaptic D.A.P. (2002). Synaptic inhibition of pyramidal cells evoked by different interneuronal subtypes in layer V of rat visual cortex. J. Neurophysiol. 88, 740–750 [DOI] [PubMed] [Google Scholar]
- 80.Ransom C.B., Wu Y., and Richerson G.B. (2010). Postdepolarization potentiation of GABAA receptors: a novel mechanism regulating tonic conductance in hippocampal neurons. J. Neurosci. 30, 7672–7684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nakamura S., Baratta M. V, Pomrenze M.B., Dolzani S.D., and Cooper D.C. (2012). High fidelity optogenetic control of individual prefrontal cortical pyramidal neurons in vivo. F1000Res. 1, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dégenètais E., Thierry A.-M., Glowinski J., and Gioanni Y. (2002). Electrophysiological properties of pyramidal neurons in the rat prefrontal cortex: an in vivo intracellular recording study. Cereb. Cortex 12, 1–16 [DOI] [PubMed] [Google Scholar]
- 83.Koga K., Li X., Chen T., Steenland H.W., Descalzi G., and Zhuo M. (2010). In vivo whole-cell patch-clamp recording of sensory synaptic responses of cingulate pyramidal neurons to noxious mechanical stimuli in adult mice. Mol. Pain 6, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sterratt D., Graham B., Gillies A., and Willshaw D. (2011). Principles of Computational Modeling in Neuroscience. Cambridge University Press: Cambridge [Google Scholar]
- 85.Sudhakar S.K., Torben-Nielsen B., and De Schutter E. (2015). Cerebellar nuclear neurons use time and rate coding to transmit purkinje neuron pauses. PLoS Comput. Biol. 11, e1004641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tseng K.Y., Mallet N., Toreson K.L., Moine C.L.E., Gonon F., and Donnell P.O. (2006). Excitatory response of prefrontal cortical fast-spiking interneurons to ventral tegmental area stimulation in vivo. Synapse 59, 412–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Adesnik H., and Scanziani M. (2010). Lateral competition for cortical space by layer-specific horizontal circuits. Nature 464, 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Urban-Ciecko J., Kossut M., and Mozrzymas J.W. (2010). Sensory learning differentially affects GABAergic tonic currents in excitatory neurons and fast spiking interneurons in layer 4 of mouse barrel cortex. J. Neurophysiol. 104, 746–754 [DOI] [PubMed] [Google Scholar]
- 89.Sudhakar S.K., Hong S., Raikov I., Publio R., Lang C., Close T., Guo D., Negrello M., and De Schutter E. (2017). Spatiotemporal network coding of physiological mossy fiber inputs by the cerebellar granular layer. PLoS Comput. Biol. 13, e1005754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hodgkin A.L., and Katz B. (1949). The effect of sodium ions on the electrical activity of the giant axon of the squid. J. Physiol. 108, 37–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eghbali M., Curmi J.P., Birnir B., and Gage P.W. (1997). Hippocampal GABAA channel conductance increased by diazepam Nature 388, 71–75 [DOI] [PubMed] [Google Scholar]
- 92.Hall E.D., Fleck T.J., and Oostveen J.A. (1998). Comparative neuroprotective properties of the benzodiazepine receptor full agonist diazepam and the partial agonist PNU-101017 in the gerbil forebrain ischemia model. Brain Res. 798, 325–329 [DOI] [PubMed] [Google Scholar]
- 93.Im H.K., Im W. B.in, Von Voigtlander P.F., Carter D.B., Murray B.H., and Jacobsen E.J. (1996). Characterization of U-101017 as a GABAA receptor ligand of dual functionality. Brain Res. 714, 165–168 [DOI] [PubMed] [Google Scholar]
- 94.Staley K.J., Soldo B.L., and Proctor W.R. (1995). Ionic mechanisms of neuronal excitation by inhibitory GABA(A) receptors. Science 269, 977–981 [DOI] [PubMed] [Google Scholar]
- 95.Jedlicka P., Deller T., Gutkin B.S., and Backus K.H. (2011). Activity-dependent intracellular chloride accumulation and diffusion controls GABAA receptor-mediated synaptic transmission. Hippocampus 21, 885–898 [DOI] [PubMed] [Google Scholar]
- 96.Kaila K., Voipio J., Paalasmaa P., Pasternack M., and Deisz R.A. (1993). The role of bicarbonate in GABAA receptor-mediated IPSPs of rat neocortical neurones. J. Physiol. 464, 273–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sykova E., and Nicholson C. (2008). Diffusion in brain extracellular space. Physiol. Rev. 88, 1277–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nicholson C., and Sykova E. (1998). Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 21, 207–215 [DOI] [PubMed] [Google Scholar]
- 99.Staley K.J., and Proctor W.R. (1999). Modulation of mammalian dendritic GABA(A) receptor function by the kinetics of Cl-and HCO3-transport. J. Physiol. 519, 693–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Farrant M., and Kaila K. (2007). The cellular, molecular and ionic basis of GABAA receptor signalling. Prog. Brain Res. 160, 59–87 [DOI] [PubMed] [Google Scholar]
- 101.Hines M.L. and Carnevale N.T. (2001). Neuron: a tool for neuroscientists. Neuroscientist 7, 123–135 [DOI] [PubMed] [Google Scholar]
- 102.Somjen G.G. (1979). Extracellular potassium in the mammalian central nervous system. Annu. Rev. Physiol. 41, 159–77 [DOI] [PubMed] [Google Scholar]
- 103.Lothman E., Lamanna J., Cordingley G., Rosenthal M., and Somjen G.G. (1975). Responses of electrical potential, potassium levels, and oxidative metabolic activity of the cerebral neocortex of cats. Brain Res. 88, 15–36 [DOI] [PubMed] [Google Scholar]
- 104.Houston C.M., Bright D.P., Sivilotti L.G., Beato M., and Trevor G. (2014). Intracellular chloride ions regulate the time-course of GABA- mediated inhibitory synaptic transmission. J. Neurosci. 29, 10,416–10,423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lodish H., Arnold B., Zipursky L.S., Paul M., David B., and James D. (2000). Molecular Cell Biology, 4th ed W.H.Freeman: New York [Google Scholar]
- 106.Berglund K., Schleich W., Krieger P., Loo L.S., Wang D., Cant N.B., Feng G., Augustine G.J., and Kuner T. (2006). Imaging synaptic inhibition in transgenic mice expressing the chloride indicator, Clomeleon. Brain Cell Biol. 35, 207–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pathak H.R., Weissinger F., Terunuma M., Carlson G.C., Hsu F.-C., Moss S.J., and Coulter D.A. (2007). Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J. Neurosci. 27, 14,012–14,022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kofuji P., and Newman E. (2004). Potassium buffering in the central nervous system. Neuroscience 129, 1045–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., and Kurachi Y. (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366 [DOI] [PubMed] [Google Scholar]
- 110.Hansen A.J., and Zeuthen T. (1981). Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol. Scand. 113, 437–445 [DOI] [PubMed] [Google Scholar]
- 111.Hansen A.J., Gjedde A., and Siemkowicz E. (1980). Extracellular potassium and blood flow in the post-ischemic rat brain. Pflugers Arch. 389, 1–7 [DOI] [PubMed] [Google Scholar]
- 112.Schwartz-Bloom R.D., and Sah R. (2001). g -Aminobutyric acid neurotransmission and cerebral ischemia. J. Neurochem. 77, 353–371 [DOI] [PubMed] [Google Scholar]
- 113.Hutchinson P.J., O'Connell M.T., Al-Rawi P.G., Kett-White C.R., Gupta A.K., Maskell L.B., Pickard J.D., and Kirkpatrick P.J. (2002). Increases in GABA concentrations during cerebral ischaemia: a microdialysis study of extracellular amino acids. J. Neurol. Neurosurg. Psychiatry 72, 99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Goodrich G.S., Kabakov A.Y., Hameed M.Q., Dhamne S.C., Rosenberg P.A., and Rotenberg A. (2013). Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J. Neurotrauma 30, 1434–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Harris J.L., Yeh H.W., Choi I.Y., Lee P., Berman N.E., Swerdlow R.H., Craciunas S.C., and Brooks W.M. (2012). Altered neurochemical profile after traumatic brain injury: 1 H-MRS biomarkers of pathological mechanisms. J. Cereb. Blood Flow Metab. 32, 2122–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dreier J.P., and Reiffurth C. (2015). The stroke-migraine depolarization continuum. Neuron 86, 902–922 [DOI] [PubMed] [Google Scholar]
- 117.Muller M., and Somjen G.G. (2000). Na(+) dependence and the role of glutamate receptors and Na(+) channels in ion fluxes during hypoxia of rat hippocampal slices. J. Neurophysiol. 84, 1869–80 [DOI] [PubMed] [Google Scholar]
- 118.Jing J., Aitken P.G., and Somjen G.G. (1993). Role of calcium channels in spreading depression in rat hippocampal slices. Brain Res. 604, 251–259 [DOI] [PubMed] [Google Scholar]
- 119.Engl E., and Attwell D. (2015). Non-signalling energy use in the brain. J. Physiol. 593, 3417–3429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marie B.L., and VanDongen A.M. (2009). Activation mechanisms of the NMDA receptor, in: Biology of the NMDA Receptor. A.M. VanDongen (ed.). CRC Press/Taylor & Francis: Boca Raton, FL. [PubMed] [Google Scholar]
- 121.Daniela B., Addolorata M., Alessandro L., Cristina M., Helene M., Tirozzi B., and Michele M. (2012). On the mechanisms underlying the depolarization block in the spiking dynamics of CA1 pyramidal neurons. J. Comput. Neurosci. 33, 207–225 [DOI] [PubMed] [Google Scholar]
- 122.Tucker K.R., Huertas M.A., Horn J.P., Canavier C.C., and Levitan E.S. (2012). Pacemaker rate and depolarization block in nigral dopamine neurons: a somatic sodium channel balancing act. J. Neurosci. 32, 14,519–14,531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weber J.T. (2012). Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 3, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Perucca E. (1997). A pharmacological and clinical review on topiramate, a new antiepileptic drug. Pharmacol. Res. 35, 241–256 [DOI] [PubMed] [Google Scholar]
- 125.Wallace A.E., Kline A.E., Montanez S., and Hernandez T.D. (1999). Impact of the novel anti-convulsant vigabatrin on functional recovery following brain lesion. Restor. Neurol. Neurosci. 14, 35–45 [PubMed] [Google Scholar]
- 126.Lu K.-T., Wu C.-Y., Yen H.-H., Peng J.-H.F., Wang C.-L., and Yang Y.-L. (2007). Bumetanide administration attenuated traumatic brain injury through IL-1 overexpression. Neurol. Res. 29, 404–409 [DOI] [PubMed] [Google Scholar]
- 127.Kahle K.T., Gerzanich V., and Simard M.J. (2010). Molecular mechanisms of microvascular failure in CNS injury – synergistic roles of NKCC1 and SUR1/TRPM4. J. Neurosurg. 113, 611–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kanaka C., Ohno K., Okabe A., Kuriyama K., Itoh T., Fukuda A., and Sato K. (2001). The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KCC1,2) and Na-K-2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience 104, 933–946 [DOI] [PubMed] [Google Scholar]
- 129.Huberfeld G., Wittner L., Clemenceau S., Baulac M., Kaila K., Miles R., and Rivera C. (2007). Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 27, 9866–9873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dargaei Z., Bang J.Y., Mahadevan V., Khademullah C.S., Bedard S., Parfitt G.M., Kim J.C., and Woodin M.A. (2018). Restoring GABAergic inhibition rescues memory deficits in a Huntington's disease mouse model. Proc. Natl. Acad. Sci. 115, E1618–E1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Palma E., Amici M., Sobrero F., Spinelli G., Di Angelantonio S., Ragozzino D., Mascia A., Scoppetta C., Esposito V., Miledi R., and Eusebi F. (2006). Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc. Natl. Acad. Sci. 103, 8465–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dzhala V.I., Brumback A.C., and Staley K.J. (2008). Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann. Neurol. 63, 222–235 [DOI] [PubMed] [Google Scholar]
- 133.Cleary R.T., Sun H., Huynh T., Manning S.M., Li Y., Rotenberg A., Talos D.M., Kahle K.T., Jackson M., Rakhade S.N., Berry G., and Jensen F.E. (2013). Bumetanide enhances phenobarbital efficacy in a rat model of hypoxic neonatal seizures. PLoS One 8, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liu Y., Shangguan Y., Barks J.D.E., and Silverstein F.S. (2012). Bumetanide augments the neuroprotective efficacy of phenobarbital plus hypothermia in a neonatal hypoxia-ischemia model. Pediatr. Res. 71, 559–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sivakumaran S., and Maguire J. (2016). Bumetanide reduces seizure progression and the development of pharmacoresistant status epilepticus. Epilepsia 57, 222–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Deeb T.Z., Maguire J., and Moss S.J. (2012). Possible alterations in GABAA receptor signaling that underlie benzodiazepine-resistant seizures. Epilepsia 53, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Markram H., Toledo-Rodriguez M., Wang Y., Gupta A., Silberberg G., and Wu C. (2004). Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 5, 793–807 [DOI] [PubMed] [Google Scholar]
- 138.Kawaguchi Y. (1995). Physiological subgroups of nonpyramidal cells with specific morphological characteristics in layer II/III of rat frontal cortex. J. Neurosci. 15, 2638–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cruikshank S.J., Ahmed O.J., Stevens T.R., Patrick S.L., Amalia N., Elmaleh M., and Connors B.W. (2012). Thalamic control of layer 1 circuits in prefrontal cortex. J. Neurosci. 32, 17,813–17,823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Muralidhar S., Wang Y., and Markram H. (2014). Synaptic and cellular organization of layer 1 of the developing rat somatosensory cortex. Front. Neuroanat. 7, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rudy B., and McBain C.J. (2001). Kv3 channels: Voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 24, 517–526 [DOI] [PubMed] [Google Scholar]
- 142.Agmon A., and Connors B.W. (1989). Repetitive burst-firing neurons in the deep layers of mouse somatosensory cortex. Neurosci. Lett. 99, 137–141 [DOI] [PubMed] [Google Scholar]
- 143.Martina M., Schultz J.H., Ehmke H., Monyer H., and Jonas P. (1998). Functional and molecular differences between voltage-gated K+ channels of fast-spiking interneurons and pyramidal neurons of rat hippocampus. J. Neurosci. 18, 8111–8125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Werner C., and Engelhard K. (2007). Pathophysiology of traumatic brain injury. Br. J. Anaesth. 99, 4–9 [DOI] [PubMed] [Google Scholar]
- 145.Algattas H., and Huang J.H. (2013). Traumatic brain injury pathophysiology and treatments: early, intermediate, and late phases post-injury. Int. J. Mol. Sci. 15, 309–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xiong Y., Mahmood A., and Chopp M. (2009). Emerging treatments for traumatic brain injury. Expert Opin. Emerg. Drugs 14, 67–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Guerriero R.M., Giza C.C., and Rotenberg A. (2015). Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 15, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cantu D., Walker K., Andresen L., Taylor-Weiner A., Hampton D., Tesco G., and Dulla C.G. (2015). Traumatic brain injury increases cortical glutamate network activity by compromising GABAergic control. Cereb. Cortex 25, 2306–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dulla C.G., Coulter D.A., and Ziburkus J. (2015). From molecular circuit dysfunction to disease: case studies in epilepsy, traumatic brain injury, and alzheimers disease. Neuroscientist 22, 295–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Durand D.M., Park E.-H., and Jensen A.L. (2010). Potassium diffusive coupling in neural networks. Philos. Trans. R. Soc. 365, 2347–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Maas A. (2016). Traumatic brain injury: changing concepts and approaches. Chin. J. Traumatol. 19, 3–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Saatman K.E., Duhaime A.-C., Bullock R., Maas A.I.R., Valadka A., and Manley G.T. (2008). Classification of traumatic brain injury for targeted therapies. J. Neurotrauma 25, 719–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.