

In this study, we targeted the development and evaluation of broad-host-range CRISPR/Cas9 gene-editing tools in order to enhance the genetic-engineering capabilities of an industrially relevant methanotrophic biocatalyst. The CRISPR/Cas9 system developed in this study expands the genetic tools available to define molecular mechanisms in methanotrophic bacteria and has the potential to foster advances in the generation of novel biocatalysts to produce biofuels, platform chemicals, and high-value products from natural gas- and biogas-derived methane. Further, due to the broad-host-range applicability, these genetic tools may also enable innovative approaches to overcome the barriers associated with genetically engineering diverse, industrially promising nonmodel microorganisms.
KEYWORDS: CRISPR/Cas9, Methylococcus capsulatus, gene editing, methane biocatalyst, methane monooxygenase, methanotroph
ABSTRACT
Methanotrophic bacteria play a crucial role in the Earth’s biogeochemical cycle and have the potential to be employed in industrial biomanufacturing processes due to their capacity to use natural gas- and biogas-derived methane as a sole carbon and energy source. Advanced gene-editing systems have the potential to enable rapid, high-throughput methanotrophic genetics and biocatalyst development. To this end, we employed a series of broad-host-range expression plasmids to construct a conjugatable clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene-editing system in Methylococcus capsulatus (Bath). Heterologous coexpression of the Streptococcus pyogenes Cas9 endonuclease and a synthetic single guide RNA (gRNA) showed efficient Cas9 DNA targeting and double-stranded DNA (dsDNA) cleavage that resulted in cell death. We demonstrated effective in vivo editing of plasmid DNA using both Cas9 and Cas9D10A nickase to convert green fluorescent protein (GFP)- to blue fluorescent protein (BFP)-expressing cells with 71% efficiency. Further, we successfully introduced a premature stop codon into the soluble methane monooxygenase (sMMO) hydroxylase component-encoding mmoX gene with the Cas9D10A nickase, disrupting sMMO function. These data provide proof of concept for CRISPR/Cas9-mediated gene editing in M. capsulatus. Given the broad-host-range replicons and conjugation capability of these CRISPR/Cas9 tools, they have potential utility in other methanotrophs and a wide array of Gram-negative microorganisms.
IMPORTANCE In this study, we targeted the development and evaluation of broad-host-range CRISPR/Cas9 gene-editing tools in order to enhance the genetic-engineering capabilities of an industrially relevant methanotrophic biocatalyst. The CRISPR/Cas9 system developed in this study expands the genetic tools available to define molecular mechanisms in methanotrophic bacteria and has the potential to foster advances in the generation of novel biocatalysts to produce biofuels, platform chemicals, and high-value products from natural gas- and biogas-derived methane. Further, due to the broad-host-range applicability, these genetic tools may also enable innovative approaches to overcome the barriers associated with genetically engineering diverse, industrially promising nonmodel microorganisms.
INTRODUCTION
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) prokaryotic immune system from Streptococcus pyogenes has revolutionized genetic-engineering capabilities in a wide array of organisms. CRISPR/Cas9 gene editing is mediated by the programmable Cas9 endonuclease, which targets a genomic locus with high specificity and induces a double-stranded DNA (dsDNA) break (1–4). Target specificity is controlled by a coexpressed single guide RNA (gRNA) that contains a 20-base protospacer complementary to the target sequence, which is adjacent to a 5′-NGG-3′ protospacer-adjacent motif (PAM) site (2, 3).
Cas9-induced dsDNA breaks can result in repair via error-prone nonhomologous end joining (NHEJ) or high-fidelity homology-directed repair (HDR) (5). Although a subset of bacteria contain Ku- and DNA ligase-catalyzed NHEJ systems, most rely on RecA/RecBCD-dependent HDR to repair Cas9-induced dsDNA breaks (5, 6). In bacteria in which dsDNA breaks have been reported to be lethal, the Cas9D10A nickase has been successfully employed for CRISPR-based gene editing (7–9). This variant contains a disabled RuvC1 nuclease domain that can induce single-stranded DNA (ssDNA) nicks in order to induce single-nick-assisted HDR (3, 7–9). In some microbes tested, Cas9D10A mediated higher gene editing efficiencies than wild-type Cas9 (7–9). Combined, these CRISPR/Cas9 tools can enable robust multiplex and high-throughput gene-editing strategies (10–12) and may fast track the development of nonmodel microorganisms with limited genetic tractability for industrial applications.
Methanotrophic bacteria are key players in Earth’s biogeochemical carbon cycle and are of increasing industrial interest for their capacity to utilize methane as a sole carbon and energy source (13). The model gammaproteobacterial methanotroph Methylococcus capsulatus has been extensively studied for decades and is currently used for the industrial production of single-cell protein. Broad-host-range replicative plasmids containing RP4/RK2, RSF1010, and pBBR1 replicons are functional in M. capsulatus and have enabled the development of promoter-probe vectors and heterologous gene expression in the organism (14, 15). Further, chromosomal insertions and unmarked genetic mutations using allelic-exchange vectors and sucrose or p-chlorophenylalanine counterselection have been reported (15–19). These tools have served as a basis for the recent expansion of the methanotroph genetic toolbox that has enabled several proteobacterial methanotrophs to be engineered to convert methane-rich natural gas and aerobic-digestion-derived biogas into high-value products (20–25). Notably, these tools also lay the foundation for the development of advanced CRISPR genome-editing systems that facilitate multiplex or high-throughput gene-editing strategies. The development of advanced genome-editing tools offers a means to enable rapid evaluation of fundamental methanotrophic governing mechanisms while expanding metabolic engineering capabilities in these hosts for methane sequestration, bioremediation, and biomanufacturing.
In this study, we developed broad-host-range CRISPR/Cas9 gene-editing tools and evaluated their efficacy in the methanotroph M. capsulatus. Using the CRISPR/Cas9 system, we demonstrated editing of methanotroph-harbored plasmid DNA by introducing in vivo point mutations in a gene encoding green fluorescent protein (GFP) to generate a blue fluorescent protein (BFP) variant. Further, we successfully achieved chromosomal editing by generating a soluble methane monooxygenase (sMMO) mutant strain via the introduction of a premature stop codon in the mmoX open reading frame using the Cas9D10A nickase. The CRISPR/Cas9 tools developed here will facilitate the development of advanced methanotrophic biocatalysts and have potential utility in an array of nonmodel, industrially promising bacteria.
RESULTS AND DISCUSSION
Development of a broad-host-range CRISPR/Cas9 gene-editing system.
We employed the superfolder GFP (26) reporter to evaluate the functionality and strength of heterologous and native M. capsulatus promoters to be used for expression of Cas9 nuclease and gRNA components. We tested whether pCAH01, an RK2-based broad-host-range expression plasmid that contains the inducible tetracycline promoter/operator (PtetA), previously demonstrated to function in the related gammaproteobacterial methanotroph Methylomicrobium buryatense 5GB1 (21, 23), was also functional in M. capsulatus. The PtetA promoter exhibited strong inducible activation in M. capsulatus, as indicated by an ∼10-fold increase of GFP fluorescence in pCAH01::GFP-harboring cells after exposure to the anhydrotetracycline (aTc) inducer (Fig. 1A). Based on the ability to temporally control gene expression, Cas9- and the Cas9D10A nickase variant-encoding genes were cloned downstream of PtetA in pCAH01SpR to generate pCas9 (Fig. 2A) and pCas9D10A, respectively (see Fig. S1A in the supplemental material).
FIG 1.

Promoter activity in M. capsulatus. (A) GFP fluorescence in M. capsulatus expressing GFP from the tetracycline promoter/operator (PtetA) in pCAH01::GFP. Where indicated, GFP was induced by plating on NMS agar supplemented with 500 ng/ml aTc for 72 h. The empty pCAH01 plasmid was used as a negative control. (B) GFP fluorescence in M. capsulatus with GFP expression controlled by the indicated gene promoters in pQCH::GFP. Fluorescence intensity was measured from cells grown on NMS agar with or without 5 µM CuSO4 for 72 h. (C) GFP fluorescence in E. coli expressing GFP from the indicated promoters in pQCH::GFP. Fluorescence intensity was measured from cells grown to an OD600 of 0.5 in LB liquid medium. In panels B and C, the promoterless pQCH::GFP plasmid was used as a negative control. The data represent the fluorescence intensity normalized to OD600 and are depicted as mean RFU and standard deviations (SD) from 3 independent replicates. ***, P < 0.001; *, P < 0.05; ns, not significant.
FIG 2.

Broad-host-range CRISPR/Cas9 gene-editing system. (A) Plasmid map of pCAH01SpR::Cas9 (pCas9). Inducible expression of Cas9 is driven from the tetracycline promoter/operator (PtetA). (B) Plasmid map of pBBR1-gRNA (pgRNA) containing a 1-kb DNA repair template. Expression of the gRNA is driven from the M. capsulatus mxaF promoter (Pmxa). (C) Experimental design schematic of the CRISPR/Cas9 gene-editing system. M. capsulatus harboring pCas9 was conjugated with E. coli S17 harboring pgRNA on NMS mating agar supplemented with 500 ng/ml aTc. After 48 h of conjugation, the biomass was spread onto NMS selection agar containing 500 ng/ml aTc, gentamicin, and spectinomycin until colonies appeared. (D) OD600 of bacterial cultures 24 h postinoculation with (+aTc) or without (−aTc) Cas9 or Cas9D10A induction. The cultures were inoculated at an OD600 of 0.1. (E) CFU of Cas9- or Cas9D10A-expressing M. capsulatus after conjugation with pgRNA-mmoX. Empty pBBR1 plasmid was used as a negative control. The data are depicted as mean CFU and SD from 3 independent replicates. *, P < 0.05; ns, not significant.
Next, we evaluated the activity of M. capsulatus promoters to select a suitable promoter to drive constitutive gRNA expression. Promoters from the gamma subunit of particulate methane monooxygenase 1 (PpmoC1), particulate methane monooxygenase 2 (PpmoC2), the sMMO hydroxylase component mmoX (PmmoX), and methanol dehydrogenase mxaF (Pmxa) genes were cloned upstream of a GFP coding sequence in the RSF1010-derived vector pQCH (see Fig. S1B), and promoter activity was assessed via fluorescence. As previously demonstrated via transcriptomic studies (19, 27, 28), PpmoC1, PpmoC2, and Pmxa were highly active in M. capsulatus, driving significant GFP transcription under both copper-replete and copper-depleted conditions (Fig. 1B). As expected, we detected limited PmmoX promoter activity under copper-replete growth conditions; however, an ∼7.5-fold increase in GFP expression was observed when cells were grown under copper-depleted conditions (Fig. 1B) (19, 27, 28). Notably, the copper switch-regulated PmmoX promoter may be employed for the inducible expression of heterologous genes in M. capsulatus. Both PpmoC1 and PpmoC2 were functional in Escherichia coli, but, interestingly, Pmxa and PmmoX promoter activity was not detected in the organism (Fig. 1C), presumably due to the absence of required regulatory factors unique to M. capsulatus (15). Based on these results, we chose the Pmxa promoter to express the gRNA due to the undetectable levels of GFP expression from this promoter in E. coli in order to avoid gRNA expression during cloning or conjugation procedures. A schematic of the broad-host-range plasmid pBBR1 containing a Pmxa-expressing gRNA and a 1-kb DNA repair template (pgRNA) is depicted in Fig. 2B.
Initial experiments evaluating Cas9 or Cas9D10A expression in M. capsulatus indicated that nuclease expression did not affect bacterial growth in the absence of gRNA expression (Fig. 2D). Next, Cas9 or Cas9D10A was coexpressed with an mmoX-targeting gRNA to evaluate nuclease targeting and activity using cell viability as a phenotypic readout. Transformants coexpressing Cas9 and pgRNA-mmoX exhibited ∼99% cell death compared to cells expressing Cas9 without a gRNA (Fig. 2E). The surviving ∼1% represent the background of the system and may serve as a putative limiting factor for gene-editing efficiency (29). Conversely, DNA digestion by Cas9D10A did not result in cell death (Fig. 2E) and, assuming Cas9D10A is functional under these experimental conditions, indicates that dsDNA breaks have a high degree of lethality while ssDNA nicks are more efficiently repaired by native M. capsulatus systems.
In vivo plasmid editing.
To evaluate CRISPR/Cas9 gene editing in M. capsulatus, we employed a screening assay with a direct fluorescent readout that shifts the emission and excitation of GFP to that of BFP following targeted gene editing (30). We constructed a vector (pgRNA-GFPBFP) with a gfp-targeting gRNA and a 1-kb DNA repair template containing the GFP point mutations 194C→G, 196T→C, and 201T→G, which introduces the missense codon substitutions T64S and Y65H to convert GFP to BFP while concurrently abolishing the Cas9 PAM site (Fig. 3A; see Fig. S2 in the supplemental material). No detectable BFP fluorescence, only GFP fluorescence, was observed in cells expressing pgRNA-GFPBFP and GFP, demonstrating that the native homologous-recombination machinery did not integrate the DNA repair template into the gfp locus in the absence of Cas9 and that BFP is not expressed from the pgRNA-GFPBFP repair template (Fig. 3B). Positive transformants coexpressing Cas9, pgRNA-GFPBFP, and GFP were analyzed for BFP fluorescence to determine editing efficiency. In the absence of Cas9 induction, we observed ∼5% BFP-positive transformants after selection (Fig. 3B), indicating that leaky Cas9 expression from PtetA is sufficient for gene editing. The induction of Cas9 during the conjugal transfer of pgRNA-GFPBFP significantly increased plasmid DNA-editing efficiency, with 71% of the transformants originally encoding GFP now expressing BFP (Fig. 3B). Fluorescence microscopy of isolated BFP-expressing colonies showed that all the cells uniformly expressed BFP, with no GFP-expressing cells observed in the colony population (Fig. 3C). Sequence analysis of the fluorescent-protein-encoding loci from transformants identified as BFP positive confirmed the incorporation of the p.T64S and p.Y65H GFP-to-BFP mutations (Fig. 3D).
FIG 3.

CRISPR/Cas9-targeted editing of plasmid DNA converting GFP to BFP. (A) Amino acid (boldface) and nucleotide (underlined) substitutions required to convert GFP to BFP with the Cas9 PAM site (italicized). (B) BFP intensity of Cas9-expressing M. capsulatus after conjugation with pgRNA-GFPBFP. Where indicated, Cas9 was induced by plating on NMS agar supplemented with 500 ng/ml aTc during mating and selection. Each data point represents the fluorescence intensity of a unique colony as RFU normalized to the OD600. The horizontal line represents the median fluorescence intensity. (C) Representative fluorescence micrograph of GFP-expressing and Cas9-edited BFP-expressing M. capsulatus. (D) Representative sequencing chromatogram of a Cas9-edited pQCH::PpmoC1-BFP locus. ***, P < 0.001.
Similar to the experimental design with Cas9 described above, we tested whether the Cas9D10A nickase could also be utilized for targeted DNA editing. In the absence of Cas9D10A induction, no BFP fluorescence was detected in pgRNA-GFPBFP-expressing transformants (Fig. 4A). However, when Cas9D10A and pgRNA-GFPBFP were coexpressed, we found that 71% of the transformants exhibited BFP fluorescence at varying intensities (Fig. 4A). Intriguingly, many BFP-expressing transformants were also positive for GFP fluorescence, with the degree of BFP fluorescence intensity positively correlated with a decrease in GFP fluorescence intensity (Fig. 4B). Fluorescence microscopy showed that transformant colonies consisted of both GFP- and BFP-expressing cells (Fig. 4C), presumably due to the expansion of plasmid copies that either were not cleaved by Cas9D10A or were repaired after cleavage without incorporation of the homologous DNA repair template.
FIG 4.
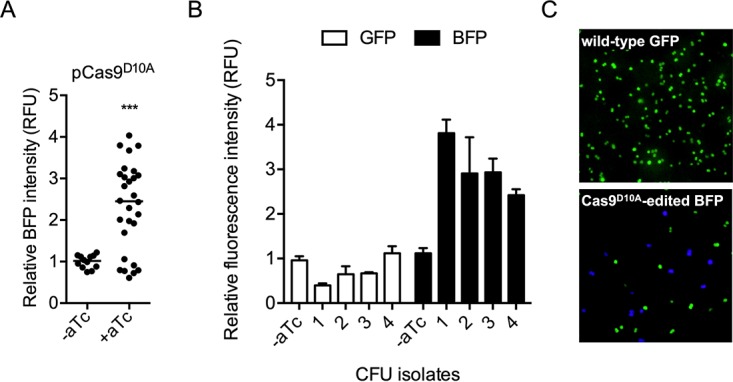
CRISPR/Cas9D10A nickase-targeted editing of plasmid DNA converting GFP to BFP. (A) BFP fluorescence in Cas9D10A-expressing M. capsulatus after conjugation with pgRNA-GFPBFP. Where indicated, Cas9D10A was induced by plating on NMS agar supplemented with 500 ng/ml aTc during mating and selection. Each data point represents the fluorescence intensity of a unique colony as RFU normalized to the OD600. The horizontal line represents the median fluorescence intensity. (B) BFP and GFP fluorescence in representative transformants coexpressing Cas9D10A and pgRNA-GFPBFP. The data represent mean RFU and SD from 3 independent replicates. (C) Representative fluorescence micrograph of Cas9D10A-edited BFP-expressing M. capsulatus cells. ***, P < 0.001.
In vivo genome editing.
We next evaluated Cas9- and Cas9D10A-mediated chromosomal editing by targeting the mmoX gene encoding the sMMO hydroxylase component. We constructed a vector that harbored an mmoX-targeting gRNA and a DNA repair template containing an HpaI endonuclease restriction site, GTTAAC, which concurrently introduces a nonsense mmoX C151X (mmoXTAA) mutation (pgRNA-mmoXTAA) (Fig. 5A; see Fig. S3 in the supplemental material). Transformants coexpressing pgRNA-mmoXTAA and Cas9 or Cas9D10A were screened by colony PCR and subsequent digestion with HpaI endonuclease. We were unable to isolate an mmoXTAA transformant in Cas9- and pgRNA-mmoXTAA-expressing tranformants. In contrast, we identified the targeted mmoXTAA edit in 2% (5/250) of the Cas9D10A nickase- and pgRNA-mmoXTAA-expressing transformants, as verified by HpaI digestion (Fig. 5B). Incorporation of the mmoXTAA nonsense mutation into the chromosome was also verified by sequence analysis (Fig. 5C). Further, the colorimetric sMMO activity assay demonstrated that the mmoXTAA strain was unable to convert naphthalene to naphthol after copper-depleted growth, confirming disruption of sMMO functionality (Fig. 5D).
FIG 5.

CRISPR/Cas9D10A-mediated editing of the M. capsulatus sMMO hydroxylase mmoX chromosomal locus. (A) Nonsense codon (boldface) and nucleotide (underlined) substitutions for generation of the mmoX p.C151X (mmoXTAA) mutation with the Cas9 PAM site shown (italicized). wt, wild type. (B) Representative agarose gel showing HpaI-digested mmoX PCR products from the wild type or a positive mmoXTAA transconjugant. (C) Representative sequencing chromatogram of a Cas9D10A-edited mmoXTAA locus. (D) Wild-type and mmoXTAA biomass on NMS agar with or without 5 µM CuSO4. The activity of sMMO was assessed by naphthalene and o-dianisidine colorimetric assay. Positive sMMO activity is indicated by development of red coloration.
Inducing single-nick-assisted HDR by the Cas9D10A nickase has been employed in other microbes when wild-type Cas9-based editing has been unsuccessful (7–9). Previous studies have demonstrated that single-nick-assisted HDR undergoes repair via an independent mechanism with higher fidelity than dsDNA break-induced repair (31). Our data indicate that the Cas9D10A-mediated nick induces higher chromosome recombination efficiency than a wild-type Cas9 dsDNA break, leading to increased numbers of transformants and enhanced mmoX locus-editing efficiency (Fig. 2D and 5). Intriguingly, contrary to the differential observed for chromosomal editing, plasmid-editing efficiencies were identical whether Cas9 or Cas9D10A was utilized. Future studies are thus needed to understand the differences in plasmid- and chromosome-editing efficiencies observed during the course of these investigations. Notably, successful incorporation of exogenous DNA into plasmid or genomic DNA by M. capsulatus was achieved via native recombination machinery. Evaluating the efficacy of heterologous recombinases, such as those employed in lambda red recombineering, may offer a potential means to enhance Cas9-mediated editing while also decreasing repair template size requirements (32, 33).
Conclusions.
Rational metabolic-engineering pursuits in methanotrophic bacteria require several metabolic mutations in order to enhance metabolic flux, end products, stress tolerances, and substrate utilization for the improved production of bio-based products (20). The tools developed here may enable CRISPR-based multiplex gene knockout strategies that can accelerate this time- and labor-intensive process. Further, CRISPR-based gene editing, together with high-throughput oligonucleotide synthesis, presents a path to genetic-library construction targeting rate-limiting metabolic enzymes to isolate strains with enhanced or altered characteristics (10, 12). The ability to utilize Cas9 for DNA targeting makes it possible to also leverage the suite of available Cas9 variants (e.g., dCas9) (34–41) for transcriptional control, development of novel regulatory circuits, or optimization of CRISPR/Cas editing efficiency to enable advanced synthetic biology applications in methanotrophic bacteria.
In this study, we developed dual-plasmid, broad-host-range CRISPR/Cas9 tools and demonstrated targeted plasmid and chromosomal DNA editing with Cas9 and Cas9D10A in the methanotroph M. capsulatus. This genetic system represents an advance in methanotroph molecular microbiology via expansion of the genetic toolbox. These advanced genetic tools may facilitate innovative strain engineering strategies that enable the development of methanotrophic biocatalysts for the production of biofuels, platform chemicals, and high-value products from methane. Further, novel molecular mechanisms underlying methanotroph biology can be probed with the addition of CRISPR/Cas9 to the methanotroph genetic toolbox. The replicons utilized in the CRISPR/Cas9 system developed here are recognized by phylogenetically diverse bacteria; thus, they have the potential to facilitate facile genetic querying and innovative strain-engineering strategies for the development of industrial biocatalysts in an array of nonmodel microbes.
MATERIALS AND METHODS
Bacterial strains and cultivation conditions.
The strains used in this study are described in Table 1. Methylococcus capsulatus (Bath) was cultured on modified nitrate mineral salts (NMS) agar supplemented with 5 µM CuSO4 (the formulation is shown in Table S1 in the supplemental material), unless otherwise indicated, at 37°C inside stainless-steel gas chambers (Schuett-biotec GmbH) containing 20% (vol/vol) methane in air (42). The NMS agar was supplemented with 100 µg/ml kanamycin, 100 µg/ml spectinomycin, and/or 30 µg/ml gentamicin for selection and cultivation of the respective M. capsulatus strains. E. coli strains were cultured on lysogeny broth (LB) agar or in LB liquid medium at 37°C at 200 rpm. LB liquid medium was supplemented with 50 µg/ml kanamycin, 50 µg/ml spectinomycin, 10 µg/ml gentamicin, and/or 100 µg/ml ampicillin for selection and cultivation of the respective E. coli strains. Broad-host-range plasmids were transferred to M. capsulatus via biparental mating using E. coli S17-1 cells on NMS mating agar (the formulation is shown in Table S1), as described previously (25). Prior to conjugation, M. capsulatus biomass harboring pCas9 or pCas9D10A was spread on NMS mating medium supplemented with 500 ng/ml aTc and incubated at 37°C inside stainless-steel gas chambers (Schuett-biotec GmbH) containing 20% (vol/vol) methane (99.97% purity) in air for 24 h. A schematic of the experimental design for using the CRISPR/Cas9 system in M. capsulatus is shown in Fig. 2C.
TABLE 1.
Strains and plasmids
| Name | Genotype or description | Source |
|---|---|---|
| Strains | ||
| Methylococcus capsulatus Bath | Wild type | ATCC 33009 |
| E. coli Zymo 10B | F− mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara leu) 7697 galU galK rpsL nupG λ− | Zymo Research |
| E. coli S17-1 | Tpr Smr recA thi pro hsd(r− m+)RP4-2-Tc::Mu::Km Tn7 | ATCC 47055 |
| Plasmids | ||
| pCAH01 | PtetA bla tetR CoE1 ori F1 oriV oriT trfA ahp | 21 |
| pCAH01SpR | ahp CDS exchanged with aadA CDS | This study |
| pCAH01::GFP | GFP cloned downstream of PtetA | 21 |
| pCAH01SpR::Cas9 | Cas9 cloned downstream of PtetA | This study |
| pAWP78 | Source of ahp locus used to generate pQCH | 25 |
| RSF1010 | oriV oriT mobABC repABC sul2 strAB | 44 |
| pQCH | RSF1010Δ[7.626-2.200 kb] ahp | This study |
| pQCH::GFP | pQCH with promoterless superfolder GFP | This study |
| pQCH::PmmoX-GFP | PmmoX cloned upstream of GFP in pQCH::GFP | This study |
| pQCH::Pmxa-GFP | Pmxa cloned upstream of GFP in pQCH::GFP | This study |
| pQCH::PpmoC1-GFP | PpmoC1 cloned upstream of GFP in pQCH::GFP | This study |
| pQCH::PpmoC2-GFP | PpmoC2 cloned upstream of GFP in pQCH::GFP | This study |
| pQCH::PpmoC1-BFP | PpmoC1-BFP cloned from PpmoC1-GFP | This study |
| pBBR1MCS-5 | pBBR oriT aacC1 | 45 |
| pBBR1-GFP | Pmxa-gRNA-GFP cloned into pBBR1 | This study |
| pBBR1-GFPBFP | Pmxa-gRNA-GFPBFP cloned into pBBR1 | This study |
| pBBR1-mmoX | Pmxa-gRNA-mmoX cloned into pBBR1 | This study |
| pBBR1-mmoXTAA | Pmxa-gRNA-mmoXTAA cloned into pBBR1 | This study |
To evaluate promoter activity, M. capsulatus harboring GFP reporter plasmids was spread onto NMS agar supplemented with 0 µM or 5 µM CuSO4. GFP expression from the tetracycline promoter/operator (PtetA) in pCAH01 was induced by plating on NMS agar supplemented with 500 ng/ml aTc. Strains were incubated at 37°C inside stainless-steel gas chambers (Schuett-biotec GmbH) in 20% (vol/vol) methane in air for 72 h, and the GFP fluorescence intensity was quantified as described below. Promoter activity was determined in E. coli subcultured 1/100 in LB liquid medium and incubated for ∼3 h to an optical density at 600 nm (OD600) of 0.5 at 37°C at 200 rpm. A 200-µl volume of E. coli cell suspension was transferred to a 96-well plate for quantification of the GFP fluorescence intensity as described below.
Cloning and genetic manipulation.
The plasmids used in this study are described in Table 1. The primers and synthetic DNA fragments used in this study were synthesized by Integrated DNA Technologies, Inc. (IDT), and are described in Table 2 and Table S2 in the supplemental material, respectively. Plasmids and DNA inserts were amplified using Q5 High-Fidelity 2× Master Mix (NEB), assembled using Gibson NEBuilder HiFi DNA assembly (New England Biolabs), and transformed into Mix and Go competent E. coli strain Zymo 10B (Zymo Research), according to the manufacturers’ instructions. Genetic constructs were verified by Sanger sequencing (Genewiz). The Cas9 open reading frame was amplified, using primers TT16 and CAH537 (Table 2), from Addgene plasmid no. 42876 (29) and cloned into pCAH01SpR via Gibson assembly. The Cas9D10A nickase variant was generated by site-directed mutagenesis with primers TT143 and TT144 (Table 2) using the QuikChange primer design program and protocol (Agilent). Single gRNAs containing a 20-mer adjacent to the PAM site on the target DNA and editing cassettes were synthesized by IDT (see Table S2) and cloned into the pBBR1MCS-5 vector via Gibson assembly. pQCH was constructed by replacing a 3,312-bp region of RSF1010 containing the sulfR, smrA, and smrB genes with the kan2 locus from pAWP78 (25). To evaluate promoter activity, native M. capsulatus promoters were cloned upstream of the superfolder green fluorescent protein-encoding gene (26) into pQCH.
TABLE 2.
Primers
| Purpose | Primer namea | Sequenceb |
|---|---|---|
| Insert aadA into pCAH01 | CAH520 aadA F | cagtgttacaaccaattaaccaattctgatTTATTTGCCGACTACCTTG |
| CAH521 aadA R | cttacataaacagtaatacaaggggtgttaATGGCTTGTTATGACTGTTTTTTTG | |
| CAH522 pCAH01 F | TAACACCCCTTGTATTACTG | |
| CAH519 pCAH01 R | ATCAGAATTGGTTAATTGGTTG | |
| Clone Cas9 into pCAH01 | TT16 Cas9 F | cactccctatcagtgatagagaaaagtgaaATGGATAAGAAATACTCAATAGGC |
| CAH537 Cas9 R | cttcacaggtcaagcttTTTTAGGAGGCAAAAATGGATAAG | |
| CAH152 pCAH01 F | AAGCTTGACCTGTGAAGTG | |
| CAH149 pCAH01 R | TTCACTTTTCTCTATCACTGATAG | |
| Construct Cas9D10A | TT143 Cas9D10A F | GAAATACTCAATAGGCTTAGCCATCGGCACAAATAGCGTCG |
| TT144 Cas9D10A R | CGACGCTATTTGTGCCGATGGCTAAGCCTATTGAGTATTTC | |
| Construct pQCH | CAH1032 ahp F | ttatattcaatggcttatttGCTCGGGACGCACGGCGC |
| CAH1033 ahp R | cggaacatgcctcatgtggcGCGTGATCTGATCCTTCAACTCAGCAAAAGTTCGATTTAT | |
| CAH1034 RSF1010 F | GCCACATGAGGCATGTTCCG | |
| CAH1031 RSF1010 R | AAATAAGCCATTGAATATAAAAGATAAAAATGTC | |
| Construct pQCH::GFP | CAH1044 pQCH F | CATACAGTCTATCGCTTAGCG |
| CAH1037 pQCH R | TATTGCAAGGACGCGGAAC | |
| CAH1045 GFP F | ATGAGCAAAGGAGAAGAAC | |
| Amplify pQCH::GFP parts | CAH1040 GFP F | gaggaaacaagtaATGAGCAAAGGAGAAGAAC |
| CAH1041 GFP R | ttatttgatgcctTTATTTGTAGAGCTCATCC | |
| CAH1042 rrnBT1T2 F | gctctacaaataaAGGCATCAAATAAAACGAAAGGC | |
| CAH1043 rrnBT1T2 R | tttccgctaagcgatagactgtatgCATCCGTCAGGATGGCCTTC | |
| Clone promoters into pQCH::GFP | CAH1046 Pmxa F | aggcatgttccgcgtccttgcaataGAGGTTCAGGCGAAACCG |
| CAH1047 Pmxa R | ctcctttgctcatGTGTCTCCTCCAAGAATGATTG | |
| CAH1048 PpmoC1 F | aggcatgttccgcgtccttgcaataAACGTCACGATGGGTGTTC | |
| CAH1049 PpmoC1 R | ctcctttgctcatTGTTTGTTCCTCCTAAAGTGATG | |
| CAH1050 PpmoC2 F | aggcatgttccgcgtccttgcaataCCCTCGTGTCCGGCGTAC | |
| CAH1051 PpmoC2 R | ctcctttgctcatTTTTACCTCCAACTGTTATATCGATGTGAACAC | |
| CAH1038 PmmoX F | aggcatgttccgcgtccttgcaataTCCGCAGTGGTCGGATCG | |
| CAH1039 PmmoX R | ctcctttgctcatTACTTGTTTCCTCCGTAACACATTCTATG | |
| Construct pgRNA-GFP and pgRNA-mmoX | TT254 Pmxa gRNA F | tccaattcgccctatagtgaGAGGTTCAGGCGAAACCG |
| TT272 gRNA only R | gcaatagacataagcggctaGGATCAGATCACGCATCTTC | |
| TT256 pBBR1 F | TAGCCGCTTATGTCTATTGCTG | |
| TT253 pBBR1 R | TCACTATAGGGCGAATTGGAG | |
| Construct GFPBFP editing template | TT207 GFP F | atctgatccttcggaccgacggattGGACCGACGGATTTTATG |
| TT208 BFP R | cattgaaccccatggcTCAGAGTAGTGACAAGTGTTG | |
| TT209 BFP F | ctactctgagccatgggGTTCAATGCTTTTCCCGTT | |
| TT255 GFP R | gcaatagacataagcggctaTGCCATGTGTAATCCCAG | |
| Check GFP/BFP editing locus | TT288 GFP check F | TCCGCGTCCTTGCAATAAAC |
| TT289 GFP check R | CCGCTAAGCGATAGACTGTATG | |
| TT271 GFP seq R | GTACATAACCTTCGGGCATG | |
| Check mmoX editing locus | TT290 mmoX check F | CCAGTACGTCACCGTTATG |
| TT291 mmoX check R | AGATCTTGCCGTAGTGGTC | |
| TT292 mmox seq F | CTGGAAGTGGGCGAATAC |
F, forward; R, reverse.
Lowercase indicates homologous sequence for Gibson assembly.
GFP and BFP expression quantification.
To evaluate Cas9- and Cas9D10A-mediated plasmid editing, fluorescence intensity was measured in a Fluostar Omega microplate reader (BMG Labtech) at an excitation wavelength (λex) of 485 nm and an emission wavelength (λem) of 520 nm (GFP) or a λex of 355 nm and a λem of 460 nm (BFP). For GFP-to-BFP gene-editing experiments, the data represent relative fluorescence units (RFU) of the measured BFP intensity relative to pQCH::PpmoC1-GFP control intensity normalized to the cell density.
Verification of mutations by colony PCR and HpaI digestion.
To evaluate Cas9- and Cas9D10A-mediated genomic editing of the mmoX locus, colony PCR was performed with primers TT290 and TT291 (Table 2) using Taq 2× Master Mix (NEB) according to the manufacturer’s instructions. The PCR mixture was used directly as the template for HpaI endonuclease (NEB) digestion, and the edited strains were identified by positive DNA digestion visualized by DNA electrophoresis. Targeted editing of the mmoX locus in positive transformants was verified by sequence analysis.
Colorimetric sMMO assay.
M. capsulatus sMMO activity was tested with a colorimetric assay as previously described (43). Briefly, ∼1e6 cells were spotted onto NMS agar with or without 5 µM CuSO4 and cultured at 37°C. After 96 h of growth, ∼300 to 400 mg naphthalene (Sigma-Aldrich) crystals was placed into the petri dish lid and incubated with the bacteria for 1 h at 37°C to allow conversion of naphthalene to naphthol. After incubation, 20 µl of freshly prepared 5-mg/ml o-dianisidine (Sigma-Aldrich), which turns purple in the presence of naphthol, was added directly to the M. capsulatus biomass. Color development was allowed to occur for 15 min at 37°C.
Statistical analysis.
Statistical analysis of data was performed and graphical representations were created using GraphPad Prism 6.0 software. Determination of statistical significance between two comparisons was achieved using an unpaired t test. Determination of statistical significance between multiple comparisons was achieved using a one-way analysis of variance (ANOVA) followed by Dunnett’s test with the appropriate controls. Normal distribution and equal variance between test groups were assumed prior to performing statistical tests using Prism software.
Supplementary Material
ACKNOWLEDGMENTS
We thank Bryon Donahoe for providing fluorescence micrographs and Ellsbeth Webb for technical assistance.
This work was conducted at the National Renewable Energy Laboratory (NREL), operated by the Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under contract no. DE-AC36-08GO28308.
The work was supported in part by the Laboratory Directed Research and Development (LDRD) and Director’s Fellowship Programs at NREL.
The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00340-19.
REFERENCES
- 1.Garneau JE, Dupuis M-È, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S. 2010. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 2.Gasiunas G, Barrangou R, Horvath P, Siksnys V. 2012. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. 2006. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui L, Bikard D. 2016. Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res 44:4243–4251. doi: 10.1093/nar/gkw223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta R, Barkan D, Redelman-Sidi G, Shuman S, Glickman MS. 2011. Mycobacteria exploit three genetically distinct DNA double-strand break repair pathways. Mol Microbiol 79:316–330. doi: 10.1111/j.1365-2958.2010.07463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu T, Li Y, Shi Z, Hemme CL, Li Y, Zhu Y, Van Nostrand JD, He Z, Zhou J. 2015. Efficient genome editing in Clostridium cellulolyticum via CRISPR-Cas9 nickase. Appl Environ Microbiol 81:4423–4431. doi: 10.1128/AEM.00873-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li K, Cai D, Wang Z, He Z, Chen S. 2018. Development of an efficient genome editing tool in Bacillus licheniformis using CRISPR-Cas9 nickase. Appl Environ Microbiol 84:e02608-17. doi: 10.1128/AEM.02608-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song X, Huang H, Xiong Z, Ai L, Yang S. 2017. CRISPR-Cas9D10A nickase-assisted genome editing in Lactobacillus casei. Appl Environ Microbiol 83:e01259-17. doi: 10.1128/AEM.01259-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garst AD, Bassalo MC, Pines G, Lynch SA, Halweg-Edwards AL, Liu R, Liang L, Wang Z, Zeitoun R, Alexander WG, Gill RT. 2017. Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat Biotechnol 35:48. doi: 10.1038/nbt.3718. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Lin Z, Huang C, Zhang Y, Wang Z, Tang Y-J, Chen T, Zhao X. 2015. Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab Eng 31:13–21. doi: 10.1016/j.ymben.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Liang L, Liu R, Garst AD, Lee T, Nogué VSi, Beckham GT, Gill RT. 2017. CRISPR EnAbled Trackable genome Engineering for isopropanol production in Escherichia coli. Metab Eng 41:1–10. doi: 10.1016/j.ymben.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Hanson RS, Hanson TE. 1996. Methanotrophic bacteria. Microbiol Rev 60:439–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ali H, Murrell JC. 2009. Development and validation of promoter-probe vectors for the study of methane monooxygenase gene expression in Methylococcus capsulatus Bath. Microbiology 155:761–771. doi: 10.1099/mic.0.021816-0. [DOI] [PubMed] [Google Scholar]
- 15.Csaki R, Bodrossy L, Klem J, Murrell JC, Kovacs KL. 2003. Genes involved in the copper-dependent regulation of soluble methane monooxygenase of Methylococcus capsulatus (Bath): cloning, sequencing and mutational analysis. Microbiology 149:1785–1795. doi: 10.1099/mic.0.26061-0. [DOI] [PubMed] [Google Scholar]
- 16.Welander PV, Summons RE. 2012. Discovery, taxonomic distribution, and phenotypic characterization of a gene required for 3-methylhopanoid production. Proc Natl Acad Sci U S A 109:12905–12910. doi: 10.1073/pnas.1208255109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishikawa M, Yokoe S, Kato S, Hori K. 2018. Efficient counterselection for Methylococcus capsulatus (Bath) by using a mutated pheS gene. Appl Environ Microbiol 84:e01875-18. doi: 10.1128/AEM.01875-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stolyar S, Costello AM, Peeples TL, Lidstrom ME. 1999. Role of multiple gene copies in particulate methane monooxygenase activity in the methane-oxidizing bacterium Methylococcus capsulatus Bath. Microbiology 145:1235–1244. doi: 10.1099/13500872-145-5-1235. [DOI] [PubMed] [Google Scholar]
- 19.Stolyar S, Franke M, Lidstrom ME. 2001. Expression of individual copies of Methylococcus capsulatus bath particulate methane monooxygenase genes. J Bacteriol 183:1810. doi: 10.1128/JB.183.5.1810-1812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henard CA, Guarnieri MT. 2018. Metabolic engineering of methanotrophic bacteria for industrial biomanufacturing, p 117–132. In Kalyuzhnaya MG, Xing X-H (ed), Methane biocatalysis: paving the way to sustainability. Springer International Publishing, New York, NY. [Google Scholar]
- 21.Henard CA, Smith H, Dowe N, Kalyuzhnaya MG, Pienkos PT, Guarnieri MT. 2016. Bioconversion of methane to lactate by an obligate methanotrophic bacterium. Sci Rep 6:21585. doi: 10.1038/srep21585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen AD, Hwang IY, Lee OK, Kim D, Kalyuzhnaya MG, Mariyana R, Hadiyati S, Kim MS, Lee EY. 2018. Systematic metabolic engineering of Methylomicrobium alcaliphilum 20Z for 2,3-butanediol production from methane. Metab Eng 47:323–333. doi: 10.1016/j.ymben.2018.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Garg S, Clomburg JM, Gonzalez R. 2018. A modular approach for high-flux lactic acid production from methane in an industrial medium using engineered Methylomicrobium buryatense 5GB1. J Ind Microbiol Biotechnol 45:379–391. doi: 10.1007/s10295-018-2035-3. [DOI] [PubMed] [Google Scholar]
- 24.Garg S, Wu H, Clomburg JM, Bennett GN. 2018. Bioconversion of methane to C-4 carboxylic acids using carbon flux through acetyl-CoA in engineered Methylomicrobium buryatense 5GB1C. Metab Eng 48:175–183. doi: 10.1016/j.ymben.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Puri AW, Owen S, Chu F, Chavkin T, Beck DAC, Kalyuzhnaya MG, Lidstrom ME. 2015. Genetic tools for the industrially promising methanotroph Methylomicrobium buryatense. Appl Environ Microbiol 81:1775. doi: 10.1128/AEM.03795-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pédelacq J-D, Cabantous S, Tran T, Terwilliger TC, Waldo GS. 2006. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24:79. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 27.Larsen Ø, Karlsen OA. 2016. Transcriptomic profiling of Methylococcus capsulatus (Bath) during growth with two different methane monooxygenases. Microbiologyopen 5:254–267. doi: 10.1002/mbo3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen AK, Gerdes K, Murrell JC. 1997. Copper-dependent reciprocal transcriptional regulation of methane monooxygenase genes in Methylococcus capsulatus and Methylosinus trichosporium. Mol Microbiol 25:399–409. doi: 10.1046/j.1365-2958.1997.4801846.x. [DOI] [PubMed] [Google Scholar]
- 29.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glaser A, McColl B, Vadolas J. 2016. GFP to BFP conversion: a versatile assay for the quantification of CRISPR/Cas9-mediated genome editing. Mol Ther Nucleic Acids 5:e334. doi: 10.1038/mtna.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metzger MJ, McConnell-Smith A, Stoddard BL, Miller AD. 2011. Single-strand nicks induce homologous recombination with less toxicity than double-strand breaks using an AAV vector template. Nucleic Acids Res 39:926–935. doi: 10.1093/nar/gkq826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bassalo MC, Garst AD, Halweg-Edwards AL, Grau WC, Domaille DW, Mutalik VK, Arkin AP, Gill RT. 2016. Rapid and efficient one-step metabolic pathway integration in E. coli. ACS Synth Biol 5:561–568. doi: 10.1021/acssynbio.5b00187. [DOI] [PubMed] [Google Scholar]
- 33.Corts AD, Thomason LC, Gill RT, Gralnick JA. 2019. A new recombineering system for precise genome-editing in Shewanella oneidensis strain MR-1 using single-stranded oligonucleotides. Sci Rep 9:39. doi: 10.1038/s41598-018-37025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. 2016. Rationally engineered Cas9 nucleases with improved specificity. Science 351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, Doudna JA. 2017. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550:407–410. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, Liu DR. 2018. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. 2016. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. 2017. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guilinger JP, Thompson DB, Liu DR. 2014. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32:577–582. doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. 2016. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whittenbury R, Phillips KC, Wilkinson JF. 1970. Enrichment, isolation and some properties of methane-utilizing bacteria. Microbiology 61:205–218. doi: 10.1099/00221287-61-2-205. [DOI] [PubMed] [Google Scholar]
- 43.Graham DW, Korich DG, LeBlanc RP, Sinclair NA, Arnold RG. 1992. Applications of a colorimetric plate assay for soluble methane monooxygenase activity. Appl Environ Microbiol 58:2231–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guerry P, van Embden J, Falkow S. 1974. Molecular nature of two nonconjugative plasmids carrying drug resistance genes. J Bacteriol 117:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.