

Primary cell models of HIV latency have been very useful to identify mechanisms contributing to HIV latency and to evaluate potential HIV cure strategies. However, the current models utilize in vitro infection with exogenous virus that does not fully recapitulate virus reactivation profiles of endogenous HIV in in vivo-infected CD4+ T cells. In contrast, obtaining sufficient amounts of CD4+ T cells from HIV-infected individuals to interrogate the HIV reservoir in vitro requires leukapheresis. In the model we propose here, in vitro expansion and extended culture of primary CD4+ T cells isolated from virally suppressed HIV-infected individuals enable obtaining large numbers of cells harboring endogenous latent HIV reservoirs without performing leukapheresis. This model captures the variability of HIV reservoirs seeded in different individuals and should be useful to evaluate future HIV cure strategies.
KEYWORDS: latency model, primary CD4 T cells, human immunodeficiency virus
ABSTRACT
The low frequency of latently HIV-infected cells in vivo limits the testing of potential HIV cure strategies using cells from successfully suppressed individuals. To date, primary cell models of latency use cells infected in vitro. Primary CD4+ T cell models carrying an individual’s endogenous HIV reservoir that recapitulate in vivo conditions of HIV latency are still outstanding. We developed a primary CD4+ T cell model of HIV latency derived from memory CD4+ T cells isolated from virally suppressed HIV-infected individuals that recapitulates HIV-1 latency and viral reactivation events. This model is based on the expansion of primary CD4+ T cells up to 300-fold in cell number. These cells reestablish a resting state without active virus production after extended culture and maintain a stable number of total HIV proviruses. The ability of these cells to respond to various classes of latency-reversing agents is similar to that of ex vivo CD4+ T cells directly isolated from blood. Importantly, viral outgrowth assays confirmed the ability of these expanded cells to produce replication-competent endogenous virus. In sum, this model recapitulates ex vivo viral reactivation conditions, captures the variability between individuals with different HIV reservoirs, and provides large numbers of cells for testing multiple agents from a single donor. The use of this novel model will allow accurate exploration of novel cure approaches aimed either at promoting viral reactivation or maintaining sustained latency.
IMPORTANCE Primary cell models of HIV latency have been very useful to identify mechanisms contributing to HIV latency and to evaluate potential HIV cure strategies. However, the current models utilize in vitro infection with exogenous virus that does not fully recapitulate virus reactivation profiles of endogenous HIV in in vivo-infected CD4+ T cells. In contrast, obtaining sufficient amounts of CD4+ T cells from HIV-infected individuals to interrogate the HIV reservoir in vitro requires leukapheresis. In the model we propose here, in vitro expansion and extended culture of primary CD4+ T cells isolated from virally suppressed HIV-infected individuals enable obtaining large numbers of cells harboring endogenous latent HIV reservoirs without performing leukapheresis. This model captures the variability of HIV reservoirs seeded in different individuals and should be useful to evaluate future HIV cure strategies.
INTRODUCTION
Currently, antiretroviral therapy (ART) controls viral replication in the majority of HIV-infected individuals. However, the virus rebounds once ART is interrupted even when ART was initiated at the earliest stage of acute HIV infection (1). HIV infects activated CD4+ T cells during acute HIV infection (AHI). Most of these infected cells die due to killing by HIV-specific CD8+ T cells, cytopathic effects of the virus, or their immune contraction (2). After AHI, HIV-infected cells that become resting memory T cells survive and are maintained by homeostatic proliferation without active virus production or antigen-driven expansion (3–5).
Since resting CD4+ T cells are quiescent, they do not express viral proteins, and hence HIV-specific CD8+ T cells are unable to recognize and kill them (6). This is one of the biggest obstacles for eradicating the HIV reservoir. Two mainstream approaches for the eradication of the viral reservoir are being considered. The “shock and kill” approach entails the use of latency-reversing agents (LRAs) under ART to trigger active viral production, potentially resulting in the elimination of these cells by HIV-specific CD8+ T cell killing or viral cytopathic effects (6–8). In contrast, the functional cure approach “block and lock” proposes the transcriptional silencing of HIV. The proof of concept of the latter has been demonstrated using the Tat inhibitor didehydro-cortistatin A (dCA) to maintain a state of sustained latency even upon treatment interruption (9, 10). However, the small number of latently HIV-infected cells in vivo has limited robust measurements to thoroughly investigate these HIV-1 cure approaches. Therefore, an in vitro HIV latency primary cell culture model that recapitulates the latent HIV reservoir in vivo is urgently needed.
Cell line models of latency have been very useful due to their tractability. However, they are unable to cycle between quiescent and active phases and have restricted viral integration sites due to their clonal nature (11). This has prompted the development of primary cell models of latency. Current primary cell models of latency utilize in vitro infection of exogenous virus (12–17), while using CD4+ T cells from HIV-infected individuals requires leukapheresis to obtain large amounts of cells (18). It has been reported that endogenous HIV in ex vivo CD4+ T cells from HIV-infected individuals can be reactivated by different classes of LRAs, including protein kinase C (PKC) agonist and P-TEFb modulator, as well as histone deacetylase inhibitors (HDACi) (13). In contrast, virus in CD4+ T cells infected in vitro can be reactivated predominantly by PKC agonists (13). Moreover, it has been recently shown that HIV-specific CD8+ T cells are not able to eliminate in vivo-infected CD4+ T cells harboring replication-competent virus, although the CD8+ T cells are able to kill CD4+ T cells infected in vitro with outgrown virus from the same donor (19). This suggests that in vivo-infected cells with replication-competent virus and in vitro-infected cells are intrinsically different and not targeted by HIV-specific CD8+ T cells in the same way. These data suggest that primary cell models of HIV latency utilizing in vitro infection do not completely recapitulate virus reactivation and antigen presentation profiles of endogenous HIV in in vivo CD4+ T cells. To overcome these limitations, we developed a novel primary CD4+ T cell model of HIV latency and reactivation. This model is derived from ex vivo memory CD4+ T cells isolated from virally suppressed HIV-infected individuals, thus harboring an endogenous HIV reservoir established in vivo. In order to obtain large numbers of cells without performing leukapheresis, primary CD4+ T cells are expanded twice in vitro in the presence of antiretrovirals (ARVs). Cells reestablish a resting state approximately 5 weeks after the secondary expansion and are maintained in culture for up to 8 weeks. This model should be useful not only to evaluate potential HIV cure strategies but also to understand the mechanisms responsible for the persistence of HIV latency in primary CD4+ T cells.
RESULTS
Primary memory CD4+ T cells from virally suppressed individuals successfully expand in vitro and reestablish a resting state.
To overcome the initial cell number limitation, memory CD4+ T cells isolated from freshly thawed peripheral blood mononuclear cells (PBMCs) from virally suppressed individuals were polyclonally expanded for 2 weeks with phytohemagglutinin (PHA) and irradiated feeder cells (PBMCs from healthy individuals) in media supplemented with ARVs (100 nM efavirenz, 180 nM zidovudine, and 200 nM raltegravir), recombinant human interleukin-2 (rhIL-2; 10 ng/ml), and natural human IL-2/T cell growth factor (TCGF; 5 Biological Response Modifiers Program [BRMP] U/ml). This last component has the same modification found in IL-2 in vivo and contains non-IL-2 cytokines (TCGF) produced from PHA-stimulated PBMCs at low levels to support better survival and proliferation (Fig. 1A). Allogenic EBV-immortalized B cells which express major histocompatibility complex class II were also included as irradiated feeder cells to provide a tonic T cell receptor signal to support the expansion and survival of primary CD4+ T cells. The feeder cells and PHA were added only on day 0 of the primary and secondary expansions, and the expanding CD4+ T cells were maintained without them after that. The CD4+ T cells were expanded an average of 21.0-fold (7.6- to 37.3-fold) in cell number, while viability remained over 70% (Fig. 1B and Table 1), and a stock was frozen down as primary expanded CD4+ T cells for posterior secondary expansions. During this initial expansion, the CD4+ T cells retained frequencies of cells harboring total HIV DNA comparable to those of cells directly isolated from the blood of HIV-infected individuals, termed ex vivo (Fig. 1C, P = 0.56 [Wilcoxon test]). Total HIV DNA was compared to integrated DNA (not shown), and the results were comparable; however, in patients’ cells with lower copy numbers, the total HIV DNA readout was more sensitive and reproducible. One concern would be that total HIV DNA could potentially detect nonintegrated HIV-DNA; however, since the cultures were always maintained on ART, novel infections should not occur, not interfering with the proviral number (20). We thus opted to use total DNA as a readout. The stock of primary expanded CD4+ T cells was then further expanded as in the initial expansion. The cells were expanded by 18.4-fold (11.3- to 24.8-fold) in cell number from the cell stock to reach a combined 369-fold (176- to 598-fold) from their initial input of ex vivo CD4+ T cells and maintained in culture for 56 more days, with a sustained cell viability over 70% and a stable total HIV DNA copy number over the course of the culture (Fig. 1D and E and Table 1). The T cell activation status was monitored by assessing expression of Ki-67 and CD25, with Ki-67+ and CD25+ cells peaking at day 7 after secondary stimulation with 63.2% (49.4 to 70.5%) and 77.7% (68.3 to 77.7%) positive cells, respectively (Fig. 1F and G). The expression levels of both T cell activation markers decreased around day 35 after secondary stimulation. Altogether, these results suggest that this in vitro primary T cell model provides more than 300-fold the number of resting memory CD4+ T cells from virally suppressed individuals, while retaining ex vivo proviral HIV reservoir frequencies.
FIG 1.

Reestablishment of a resting state of CD4+ T cells after in vitro expansion from primary memory CD4+ T cells. (A) Schematic of primary CD4+ T cell expansion, culture, and experimental design. (B) Purified CD45RA− CD4+ T cells from PBMCs were expanded with PHA and IL-2, and their fold expansion based on the initial input cell number was determined at 0, 7, and 14 days after stimulation. The frequency of live cells in the culture was determined by Aqua Live/Dead flow cytometric staining is shown. (C) Total HIV DNA copies/106 cells among ex vivo CD4+ T cells or CD4+ T cells 14 days after expansion. (D) CD4+ T cells from initial expansion were further expanded with PHA and IL-2. Their fold expansion and frequency of live cells was determined weekly until 56 days after secondary stimulation. (E) Total HIV DNA copies/106 cells among CD4+ T cells after secondary expansion. (F) Representative dot plot for coexpression of Ki-67 and CD25 in/on CD4+ T cells after secondary expansion. (G) Percentages of Ki-67+ and CD25+ cells in/on CD4+ T cells from six subjects after secondary expansion.
TABLE 1.
Initial input cell number and their fold expansion
| Donor ID | No. of PBMCs used for memory CD4+ T cell isolation (1 × 106) | No. of ex vivo memory CD4+ T cells expanded (1 × 106) | Fold expansion of CD4+ T cells after 2 wk of primary expansion | Fold expansion of CD4+ T cells after 5 wk of secondary expansion from ex vivo CD4+ T cells |
|---|---|---|---|---|
| ST045 | 56.0 | 1.6 | 37.3 | 597.7 |
| ST101 | 39.2 | 2.8 | 24.1 | 445.5 |
| ST104 | 76.9 | 3.4 | 17.9 | 444.8 |
| ST122 | 39.2 | 3 | 20.0 | 332.9 |
| ST137 | 60.6 | 4.2 | 7.6 | 176.0 |
| ST144 | 27.0 | 2.5 | 19.0 | 215.5 |
Expanded CD4+ T cells reestablish a resting state without active virus production after extended in vitro cell culture with ARVs and can be reactivated.
During the course of culture after secondary expansion, virus production was monitored weekly. High levels of virus production were detected 14 days after expansion, and virus production became undetectable in CD4+ T cell cultures from most of the participants at 35 days after secondary expansion (Fig. 2A), paralleling their T cell activation status (Fig. 1F and G). Expanded CD4+ T cells from two individuals actively produced virus beyond day 35, which may be explained by their higher frequency of cells harboring total HIV DNA (Fig. 2B). To confirm that HIV provirus in these expanded CD4+ T cells was capable of producing viral particles after reactivation, the expanded CD4+ T cells at time points with undetectable levels of viral production were treated with the LRA prostratin. Viral RNA was measured in the supernatant of the stimulated cultures, revealing that cells from all participants retained the capacity to produce virus upon prostratin treatment (Fig. 2C). These results indicate that the expanded CD4+ T cells maintain a pool of cells harboring latent and inducible viral genomes in this primary in vitro CD4+ T cell culture system.
FIG 2.

Virus production over the course of culture after secondary expansion. (A) HIV RNA in culture supernatants from seven subjects were evaluated by ultrasensitive RT-PCR over the course of culture. (B) Total HIV DNA copy number on day 35 between individuals with or without detectable HIV RNA on day 35 after secondary expansion. (C) After expanded CD4+ T cells stopped producing virus in the culture (days 35 to 56), they were treated with prostratin (1 μM) or DMSO (control) for 24 h, and the culture supernatants were collected to evaluate virus production. The HIV copy numbers are shown with the standard deviations. *, P < 0.05.
Resting latently HIV-infected CD4+ T cells in extended in vitro culture are reactivated similarly upon various classes of LRAs to ex vivo CD4+ T cells.
It has been reported that existing in vitro models of HIV latency show discrepancies in virus reactivation profile upon treatment with different classes of LRAs compared to ex vivo CD4+ T cells from virally suppressed HIV-infected individuals (13). To investigate this, ex vivo CD4+ T cells from freshly thawed PBMCs and expanded CD4+ T cells without active viral production were compared for their ability to produce virus upon treatment with LRAs. Latent HIV was reactivated in both ex vivo and expanded CD4+ T cells at similar levels upon treatment of the PKC agonist, prostratin (Fig. 3A, P > 0.99 [Wilcoxon test]). Virus reactivation profile was further investigated by treatment with HMBA (P-TEFb modulator), SAHA (HDACi), and bryostatin (PKC agonist), in addition to prostratin (Fig. 3B). The two PKC agonists, prostratin and bryostatin, triggered the highest virus reactivation in both ex vivo and expanded CD4+ T cells, observed in two independent secondary expansions from stock of primary expanded CD4+ T cells (experiments 1 and 2, Fig. 3B). The other classes of LRAs, HMBA and SAHA, also induced distinct virus reactivation in both cell conditions. Whereas HMBA does not reactivate viral production in most HIV latency models, here, even if at different magnitudes, it did reactivate the virus in both ex vivo and expanded CD4+ T cells. These results suggest that expanded CD4+ T cells retain a comparable ability than ex vivo cells to produce virus upon stimulation using common LRAs, although the frequency of these would not be exactly the same as in ex vivo CD4+ T cells after the expansions possibly inherent to the different provirus content in each sample. In addition, this model allowed us to test 15 conditions, including various classes of LRAs in parallel, and measure HIV production on 30 × 106 expanded CD4+ T cells from a single donor expanded from an initial input of only 0.05 × 106 to 0.2 × 106 primary memory CD4+ T cells (Fig. 3C). Based on the average yield of ex vivo memory CD4+ T cells from PBMCs (Table 1), this assay would have required approximately 470 × 106 PBMCs to perform it on ex vivo memory CD4+ T cells.
FIG 3.
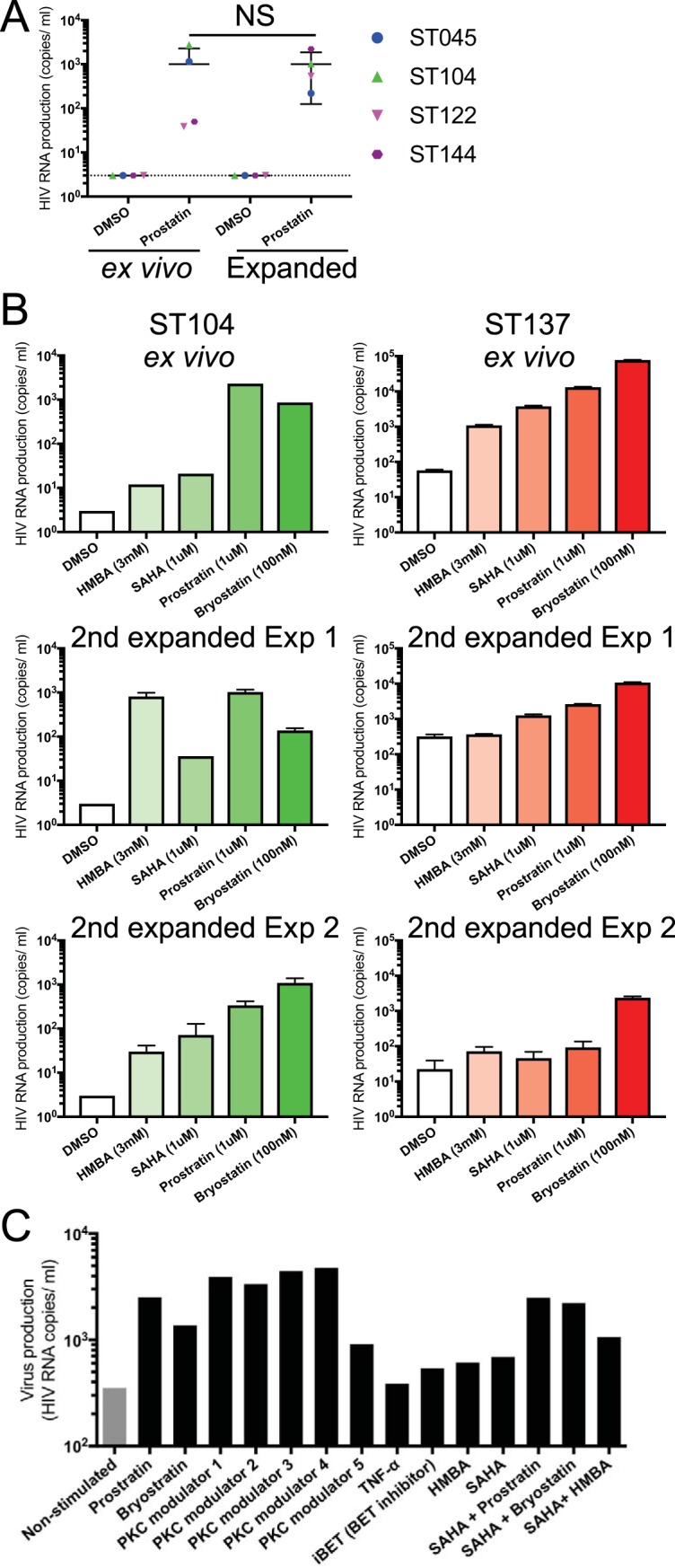
Virus reactivation in ex vivo memory CD4+ T cells and resting CD4+ T cells generated in vitro. (A) Purified ex vivo CD4+ T cells and expanded CD4+ T cells with more than 35 days in culture were treated with prostratin (1 μM) or DMSO (control) for 24 h, and the culture supernatants were collected to evaluate virus production. (B) Purified ex vivo CD4+ T cells and expanded CD4+ T cells in two independent expansions (experiments 1 and 2) from two representative donors were treated with different classes of LRAs for 24 h, and the culture supernatants were collected to evaluate virus production. The HIV copy numbers are shown with the standard deviations. (C) Cryopreserved expanded CD4+ T cells were recovered and treated for 3 days under 15 conditions, including 10 LRAs and their combinations. Culture supernatants were collected to evaluate virus production.
HIV latently infected CD4+ T cells generated in vitro retain the capacity to produce replication-competent viruses.
Next, we investigated whether the expanded cells retained the ability to produce replication competent viruses to the same degree as ex vivo CD4+ T cells, since the two rounds of expansion could have eliminated inducible HIV-infected cells due to viral cytopathic effects. Similar to ex vivo CD4+ T cells, the in vitro-expanded CD4+ T cells retained the ability to produce viral particles upon PHA stimulation (Fig. 4A). More importantly, the produced viruses from the expanded CD4+ T cells were replication competent based on a standard quantitative viral outgrowth assay (QVOA [21]), which also uses PHA as a stimulus and measures the frequency of cells harboring replication-competent virus (Fig. 4B). The variation in reactivation levels between ex vivo and expanded CD4+ T cells may stem from small differences in their proviral HIV content, as higher HIV DNA copy number resulted in higher reactivation levels (Fig. 4C). Altogether, these results suggest that this in vitro primary CD4+ T cell culture model is able to maintain endogenous replication-competent virus from HIV-infected individuals after two rounds of in vitro T cell expansion.
FIG 4.

Virus reactivation in resting CD4+ T cells generated in vitro. (A) Purified ex vivo CD4+ T cells and secondary expanded CD4+ T cells after 35 days of expansion were stimulated with PHA for 48 h, and virus production in the culture supernatants was evaluated. The HIV copy numbers are shown with the standard deviations. (B) The frequencies of latently infected CD4+ T cells harboring replication-competent virus were determined by using a standard quantitative viral outgrowth assay (QVOA). The frequencies are shown as median infectious units per million CD4+ T cells (IUPM) with 95% confidence intervals. (C) Total HIV DNA copy number in ex vivo memory CD4+ T cells and expanded CD4+ T cells 35 and 42 days after secondary expansion.
DISCUSSION
Here, we described a primary CD4+ T cell model of HIV latency that generated large numbers of latently HIV-infected CD4+ T cells and mirrors the original properties of cells directly isolated from the blood of people living with HIV. Namely, the latent HIV reservoir, the ability to respond to different classes of LRAs, and the production of replication-competent viruses are maintained. This model can also be utilized to test “block and lock” strategies as it was used recently to test the tat inhibitor dCA (9). The two rounds of primary memory CD4+ T cell expansions enabled us to generate >300-fold the initial cell number, which may allow the screening of a large number of LRAs without requiring leukapheresis, immortalized cell lines, in vitro HIV infection, or genetic modification of CD4+ T cells. Moreover, we were able to maintain these HIV latently infected CD4+ T cells in vitro for more than 8 weeks, cycling between activated and resting state as in vivo, and harboring endogenous HIV isolates as in genuine memory CD4+ T cells from virally suppressed HIV-infected individuals.
During the first 2 weeks after stimulation, we detected the production of viral particles in the supernatant that diminished concomitantly to the levels of cell activation and proliferation. However, this virus production did not lead to the depletion of cells carrying replication-competent virus, as we were able to recover replication-competent HIV after in vitro expansion. Our results suggest that in the absence of anti-HIV immunity, these virus-producing cells do not undergo apoptosis and are able to expand. This observation is in line with in vivo clonal expansion of HIV-infected cells, suggesting that these CD4+ T cells carrying HIV proviruses can also proliferate in vitro, most probably producing viral particles upon activation without dying (3, 4, 22, 23). It has been reported from multiple groups that HIV proviruses from ART-treated individuals were found in transcriptional units, especially in the BACH2 gene (4, 24–26). In contrast, proviruses from in vitro HIV-infected CD4+ T cells were found to not be limited to specific transcriptional units but were also present in the transcription start sites and GC regions of active genes (27–29). These data suggest that integration sites of endogenous HIV from HIV-infected individuals are distinct from integration sites of in vitro-infected CD4+ T cells (30), causing discordant virus reactivation profiles between these different sources of HIV-infected CD4+ T cells upon stimulation with different classes of LRAs (13, 31).
In this proposed primary CD4+ T cell culture model, we consistently observed that both ex vivo and expanded CD4+ T cells showed similar reactivation patterns upon treatment with the LRAs tested. Importantly, we observed distinct viral reactivation with HMBA and SAHA in our model (Fig. 3B), which is not the case with most HIV latency models that use exogenous in vitro HIV infection of either primary CD4+ T cells or Jurkat cell lines that show very weak or no virus reactivation (13). These results indicate that the in vivo HIV provirus is maintained in expanded CD4+ T cells in this model and that the retained HIV provirus landscape plays a critical role in HIV reactivation profiles. Some level of variation in proviral content is expected over the long-term length of these cultures, as cell clones with higher fitness may have a selective advantage. The observed variation in magnitude of virus reactivation between some ex vivo and expanded CD4+ T cells (Fig. 4A) probably stemmed from the variation in frequency of cells harboring replication-competent virus. This feature could be viewed as a limitation for this method, or a strength, as it captures the diversity of human samples containing variable HIV reservoirs.
In vivo replication-competent virus and its host CD4+ T cells might have undefined ways to evade immune-mediated killing by CD8+ T cells or NK cells that would not been observed with in vitro-infected CD4+ T cells (19). Therefore, our model could be preferentially used to test immune-mediated elimination of HIV reservoir cells and may better reflect the killing of reservoir cells compared to in vitro-infected cells with laboratory-adapted strains. Taken together, since this model recapitulates the nature of the reservoir and provides large amounts of cells, it should be useful not only to evaluate potential HIV latency-reversing agents but also to understand the mechanisms responsible for the persistence of latency in primary CD4+ T cells, including undefined immune escape mechanisms and testing immune strategies to eliminate these latently infected cells.
MATERIALS AND METHODS
Study participants and blood samples.
PBMCs were obtained from seven HIV-infected individuals on stable suppressive ART for more than 3 years at Martin Memorial Health Systems (Stuart, FL). None of the subjects had detectable plasma viremia at the time of blood sampling. All subjects provided signed informed consent approved by the Martin Health System Institutional review board prior to participation in the study. Human buffy coats used for the feeder cells were obtained from healthy donors on NCI IRB-approved NIH protocol 99-CC-0168. Research blood donors provided written informed consent, and blood samples were deidentified prior to distribution.
Primary CD4+ T cell expansion and culture maintenance.
PBMCs from seven HIV-infected individuals were isolated from a white blood cell fraction of leukapheresis by Ficoll Hypaque (GE Healthcare) density gradient centrifugation. Memory CD4+ T cells (CD45RA− CD4+ T cells) were purified from the PBMCs by using magnetic bead-based negative selection (StemCell Technologies, Canada) or fluorescence-activated cell sorting (FACS) of CD45RA− CD4+ T cells by a BD FACSAria II (BD Biosciences). The purity of the enriched CD4+ T cells was greater than 94% based on flow cytometry. The CD4+ T cells were expanded in RPMI 1640 medium supplemented with 8% human serum (Access Biologicals), 1 μg/ml of phytohemagglutinin-L (PHA-L; Sigma-Aldrich), 10 ng/ml of rhIL-2 (IL-2 improved sequence; Miltenyi Biotec), 5 BRMP U/ml of natural human IL-2/TCGF (ZeptoMetrix), ARVs (100 nM efavirenz, 180 nM zidovudine, and 200 nM raltegravir), 100 U/ml of penicillin-streptomycin (Quality Biological), and irradiated feeder cells (10 × 106 cells/plate of freshly isolated PBMCs from three different HIV-negative donors and 1 × 106 cells/plate of an allogeneic Epstein-Barr virus immortalized B cell line) (T cell expansion media). The purified primary CD4+ T cells (up to 3 × 106 CD4+ T cells/plate), along with feeder cells, were suspended in 15 ml of the T cell expansion media and plated in 96-well U bottom plates (150 μl of the cell suspension/well). The cells were cultured for 4 to 6 days until they became confluent. The expanding CD4+ T cells in 96-well plates were pooled and transferred from the plate to a 25-cm2 culture flask. Fresh RPMI 1640 medium containing 8% human serum, 10 ng/ml of rhIL-2, 5 BRMP U/ml of natural human IL-2/TCGF, the three-ARV cocktail (at the concentrations mentioned above), and 100 U/ml of penicillin-streptomycin (culture maintenance media) was added to the flask at a volume equal to that of the transferred CD4+ T cells. The cells were transferred from a 25-cm2 culture flask to a 75-cm2 culture flask once the cell number reached was more than 40 × 106 cells; they were then maintained at a cell density of 1 × 106 to 2 × 106 cells/ml over the course of the culture in culture maintenance media without adding feeder cells, changing half of media every 3 days or when the media turned too acidic before the 3-day mark. For the secondary expansion, 14 days after the primary expansion, 5 × 106 to 10 × 106 CD4+ T cells were further expanded similarly to the initial expansion in the T cell expansion media described earlier, and the culture was maintained for up to 8 weeks, as for the maintenance of primary expanded CD4+ T cells. The cell number in the culture was determined weekly. For quantification of HIV DNA, 1 × 106 cells were collected weekly and centrifuged twice in a 1.5-ml microtube at 16,000 × g for 5 min at 4°C, the supernatants were carefully removed, and the dry cell pellets were stored at –80°C until used for analyses. Another 2 × 106 cells were placed in a 5-ml round-bottom polypropylene tube at 1 × 106 cells/ml and cultured for 1 week. After a week, the tubes were centrifuged at 1,500 rpm, and the top 1 ml of each supernatant was collected for assessing virus production in the culture. Another 1 × 106 cells were stained for flow cytometric phenotyping weekly.
Antibodies and reagents for flow cytometry.
Alexa Fluor 700-labeled anti-CD3 (clone UCHT1), BV786-labeled anti-Ki-67 (clone B56), and BV786-labeled mouse IgG1 κ isotype control (clone X40) monoclonal antibodies (MAbs) were obtained from BD Biosciences. BV605-labeled anti-CD4 (clone RPA-T4), Pacific Blue-labeled anti-CD8 (clone RPA-T8), and FITC-labeled CD25 (clone BC96) MAbs were obtained from BioLegend. The Foxp3/transcription factor staining buffer set was from Thermo Fisher Scientific (eBioscience). Live/Dead Fixable Aqua dead cell stain kit (Live/Dead) was from Thermo Fisher Scientific (Molecular Probes).
Flow cytometry analysis.
CD4+ T cells from culture were first stained with Live/Dead at room temperature for 10 min then cell surface markers at 4°C for 20 min. Cells were washed with phosphate-buffered saline (PBS) containing 2% fetal bovine serum (washing buffer) twice and then fixed and permeabilized with the Foxp3/transcription factor staining buffer set for 30 min at 4°C. The cells were stained with anti-Ki-67 or mouse IgG1 κ isotype control antibodies at room temperature for 30 min, followed by two washes with Foxp3 permeabilization buffer and washing buffer, respectively. All the stained cells were resuspended in PBS containing 2% formaldehyde before analysis on LSRII (BD Biosciences). All the flow cytometry data were analyzed using FlowJo (v10; FlowJo, LLC), and Aqua Live/Dead+ dead cells were excluded from the data analysis.
Latent HIV reactivation with LRAs.
Expanded CD4+ T cells on day 35 or later in the culture, and ex vivo memory CD4+ T cells were placed in a 5-ml round-bottom polypropylene tube at 1 × 106 cells/ml for a total of 1.2 × 106 to 2 × 106 cells. The cells were treated with prostratin (1 μM; Sigma-Aldrich), HMBA (3 mM; Sigma-Aldrich), SAHA (1 μM; Sigma-Aldrich), bryostatin (100 nM; Sigma-Aldrich), or dimethyl sulfoxide (DMSO; negative control) for 24 h. The top 1 ml of each culture supernatant was harvested for quantification of the HIV RNA.
Quantification of HIV RNA.
Freshly collected culture supernatants were centrifuged at 30,000 × g for 1 h at 4°C to pellet the HIV particles. Viral RNA was extracted and subjected to ultrasensitive reverse transcription-PCR (RT-PCR) as previously described (5). Extracted viral RNA was reverse transcribed and amplified with primers and probes. Preamplified products were diluted 10-fold and subjected to a nested real-time PCR for 40 cycles on a Rotor-Gene Q instrument (Qiagen). In all experiments, serial dilutions of HIV particles (generated from ACH-2 cell culture supernatant; the cell line was obtained through the NIH AIDS Reagent Program from Thomas Folks [32, 33]) in culture medium were processed in parallel to experimental samples used as a standard. The absolute number of HIV RNA copies from ACH-2 cell culture supernatant used for the standard curve was quantified by digital-droplet PCR at the University of Miami.
Quantification of total HIV DNA.
The dry cell pellets from expanded CD4+ T cells and ex vivo memory CD4+ T cells were digested with lysis buffer containing proteinase K (Invitrogen), and the cell lysate was used to preamplify total HIV DNA and the human CD3 gene as previously described (20). Preamplified products were diluted 10-fold and subjected to a nested real-time PCR with specific primer sets for total HIV DNA and the human CD3 gene, using Rotor-Gene probe master mix (Qiagen) on a Rotor-Gene Q instrument (Qiagen) according to the manufacturer’s instructions. A standard curve was established using serially diluted ACH-2 cell lysates (ATCC) that contain a single copy of HIV per cell.
Quantitative viral outgrowth assay.
Purified CD4+ T cells from PBMCs or expanded CD4+ T cells were plated in 24-well plates at 2-fold dilutions of 8 to 12 replicates each and cultured in RPMI 1640 supplemented with 10% fetal bovine serum, penicillin-streptomycin, and 50 U/ml of IL-2. Cells were then stimulated with 2 μg/ml of PHA (Sigma) in the presence of 2 × 106/well irradiated feeder cells (PBMCs from HIV-negative donors) as previously described (19). After 24 h of culture, 106 MOLT-4/CCR5 cells were added to each well, along with a half medium change (21). Cultures were then incubated for 14 days, with partial medium changes every 3 to 4 days. On day 14, supernatants were collected, and p24+ wells were quantified by enzyme-linked immunosorbent assay (Perkin-Elmer) according to the manufacturer’s instructions. For each sample, values for cells/well, the number of positive wells, and the total wells plated for each condition were entered into a limiting-dilution analyzer (http://bioinf.wehi.edu.au/software/elda/) to calculate the infectious unit per million (IUPM) with 95% confidence intervals.
Statistical analysis.
Statistical analyses were performed using the Wilcoxon matched-pairs signed-rank test for comparisons of the HIV DNA copy number or HIV RNA production from the same participants between expanded cells and ex vivo cells, either prostatin stimulated and unstimulated. A P value of <0.05 was considered significant.
ACKNOWLEDGMENTS
We thank the study participants who donated blood samples at Martin Memorial Health Systems (Stuart, FL).
This research was funded by NIAID grant R01AI118432 (S.V. and L.T.) and cooperative agreement W81XWH-07-2-0067 (L.T.) between the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., and the U.S. Department of Defense. Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The views expressed are those of the authors and should not be construed to represent the positions of the U.S. Army or the Department of Defense. The investigators have adhered to the policies for protection of human subjects as prescribed in AR 70–25.
We thank Juyeon C. Kakazu and Shu-Wei Wu for technical assistance and the staff of the Blood Services Section of the NIH Department of Transfusion Medicine for the provision of human buffy coats from healthy blood donors.
The following reagents and cells were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: efavirenz (no. 4624), raltegravir (Merck & Company, Inc.; catalog no. 11680), zidovudine (DAIDS; no. 3485), and ACH-2 (Thomas Folks; catalog no. 379).
REFERENCES
- 1.Colby DJ, Trautmann L, Pinyakorn S, Leyre L, Pagliuzza A, Kroon E, Rolland M, Takata H, Buranapraditkun S, Intasan J, Chomchey N, Muir R, Haddad EK, Tovanabutra S, Ubolyam S, Bolton DL, Fullmer BA, Gorelick RJ, Fox L, Crowell TA, Trichavaroj R, O’Connell R, Chomont N, Kim JH, Michael NL, Robb ML, Phanuphak N, Ananworanich J. 2018. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat Med doi: 10.1038/s41591-018-0026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siliciano RF, Greene WC. 2011. HIV latency. Cold Spring Harb Perspect Med 1:a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel M-R, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy J-P, Haddad EK, Sékaly R-P. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, Spindler J, Ferris AL, Mellors JW, Kearney MF, Coffin JM, Hughes SH. 2014. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345:179–183. doi: 10.1126/science.1254194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vandergeeten C, Fromentin R, DaFonseca S, Lawani MB, Sereti I, Lederman MM, Ramgopal M, Routy JP, Sekaly RP, Chomont N. 2013. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood 121:4321–4329. doi: 10.1182/blood-2012-11-465625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. 2012. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trautmann L. 2016. Kill: boosting HIV-specific immune responses. Curr Opin HIV AIDS 11:409–416. doi: 10.1097/COH.0000000000000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, Zhang H, Margolick JB, Blankson JN, Siliciano RF. 2012. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kessing CF, Nixon CC, Li C, Tsai P, Takata H, Mousseau G, Ho PT, Honeycutt JB, Fallahi M, Trautmann L, Garcia JV, Valente ST. 2017. In vivo suppression of HIV rebound by didehydro-cortistatin A, a “block-and-lock” strategy for HIV-1 treatment. Cell Rep 21:600–611. doi: 10.1016/j.celrep.2017.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST. 2015. The Tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. mBio 6:e00465. doi: 10.1128/mBio.00465-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jordan A, Bisgrove D, Verdin E. 2003. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sáez-Cirión A, Shin SY, Versmisse P, Barré-Sinoussi F, Pancino G. 2010. Ex vivo T cell-based HIV suppression assay to evaluate HIV-specific CD8+ T-cell responses. Nat Protoc 5:1033–1041. doi: 10.1038/nprot.2010.73. [DOI] [PubMed] [Google Scholar]
- 13.Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, Mau M, Ruelas D, Saleh S, Shirakawa K, Siliciano RF, Singhania A, Soto PC, Terry VH, Verdin E, Woelk C, Wooden S, Xing S, Planelles V. 2013. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog 9:e1003834. doi: 10.1371/journal.ppat.1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang HC, Xing S, Shan L, O’Connell K, Dinoso J, Shen A, Zhou Y, Shrum CK, Han Y, Liu JO, Zhang H, Margolick JB, Siliciano RF. 2009. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest 119:3473–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lassen KG, Hebbeler AM, Bhattacharyya D, Lobritz MA, Greene WC. 2012. A flexible model of HIV-1 latency permitting evaluation of many primary CD4 T-cell reservoirs. PLoS One 7:e30176. doi: 10.1371/journal.pone.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saleh S, Solomon A, Wightman F, Xhilaga M, Cameron PU, Lewin SR. 2007. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood 110:4161–4164. doi: 10.1182/blood-2007-06-097907. [DOI] [PubMed] [Google Scholar]
- 17.Bosque A, Planelles V. 2009. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113:58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Archin NM, Eron JJ, Palmer S, Hartmann-Duff A, Martinson JA, Wiegand A, Bandarenko N, Schmitz JL, Bosch RJ, Landay AL, Coffin JM, Margolis DM. 2008. Valproic acid without intensified antiviral therapy has limited impact on persistent HIV infection of resting CD4+ T cells. AIDS 22:1131–1135. doi: 10.1097/QAD.0b013e3282fd6df4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang SH, Ren Y, Thomas AS, Chan D, Mueller S, Ward AR, Patel S, Bollard CM, Cruz CR, Karandish S, Truong R, Macedo AB, Bosque A, Kovacs C, Benko E, Piechocka-Trocha A, Wong H, Jeng E, Nixon DF, Ho YC, Siliciano RF, Walker BD, Jones RB. 2018. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J Clin Invest 128:876–889. doi: 10.1172/JCI97555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vandergeeten C, Fromentin R, Merlini E, Lawani MB, DaFonseca S, Bakeman W, McNulty A, Ramgopal M, Michael N, Kim JH, Ananworanich J, Chomont N. 2014. Cross-clade ultrasensitive PCR-based assays to measure HIV persistence in large-cohort studies. J Virol 88:12385–12396. doi: 10.1128/JVI.00609-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laird GM, Eisele EE, Rabi SA, Lai J, Chioma S, Blankson JN, Siliciano JD, Siliciano RF. 2013. Rapid quantification of the latent reservoir for HIV-1 using a viral outgrowth assay. PLoS Pathog 9:e1003398. doi: 10.1371/journal.ppat.1003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simonetti FR, Sobolewski MD, Fyne E, Shao W, Spindler J, Hattori J, Anderson EM, Watters SA, Hill S, Wu X, Wells D, Su L, Luke BT, Halvas EK, Besson G, Penrose KJ, Yang Z, Kwan RW, Van Waes C, Uldrick T, Citrin DE, Kovacs J, Polis MA, Rehm CA, Gorelick R, Piatak M, Keele BF, Kearney MF, Coffin JM, Hughes SH, Mellors JW, Maldarelli F. 2016. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc Natl Acad Sci U S A 113:1883–1888. doi: 10.1073/pnas.1522675113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Gurule EE, Brennan TP, Gerold JM, Kwon KJ, Hosmane NN, Kumar MR, Beg SA, Capoferri AA, Ray SC, Ho YC, Hill AL, Siliciano JD, Siliciano RF. 2018. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc Natl Acad Sci U S A 115:E2575–E2584. doi: 10.1073/pnas.1720665115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeda T, Shibata J, Yoshimura K, Koito A, Matsushita S. 2007. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. J Infect Dis 195:716–725. doi: 10.1086/510915. [DOI] [PubMed] [Google Scholar]
- 25.Imamichi H, Natarajan V, Adelsberger JW, Rehm CA, Lempicki RA, Das B, Hazen A, Imamichi T, Lane HC. 2014. Lifespan of effector memory CD4+ T cells determined by replication-incompetent integrated HIV-1 provirus. AIDS 28:1091–1099. doi: 10.1097/QAD.0000000000000223. [DOI] [PubMed] [Google Scholar]
- 26.Wagner TA, McLaughlin S, Garg K, Cheung CY, Larsen BB, Styrchak S, Huang HC, Edlefsen PT, Mullins JI, Frenkel LM. 2014. HIV latency: proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345:570–573. doi: 10.1126/science.1256304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brady T, Agosto LM, Malani N, Berry CC, O’Doherty U, Bushman F. 2009. HIV integration site distributions in resting and activated CD4+ T cells infected in culture. AIDS 23:1461–1471. doi: 10.1097/QAD.0b013e32832caf28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vatakis DN, Kim S, Kim N, Chow SA, Zack JA. 2009. Human immunodeficiency virus integration efficiency and site selection in quiescent CD4+ T cells. J Virol 83:6222–6233. doi: 10.1128/JVI.00356-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pace MJ, Graf EH, Agosto LM, Mexas AM, Male F, Brady T, Bushman FD, O’Doherty U. 2012. Directly infected resting CD4+ T cells can produce HIV Gag without spreading infection in a model of HIV latency. PLoS Pathog 8:e1002818. doi: 10.1371/journal.ppat.1002818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rezaei SD, Cameron PU. 2015. Human immunodeficiency virus (HIV)-1 integration sites in viral latency. Curr HIV/AIDS Rep 12:88–96. doi: 10.1007/s11904-014-0241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shan L, Xing S, Yang HC, Zhang H, Margolick JB, Siliciano RF. 2014. Unique characteristics of histone deacetylase inhibitors in reactivation of latent HIV-1 in Bcl-2-transduced primary resting CD4+ T cells. J Antimicrob Chemother 69:28–33. doi: 10.1093/jac/dkt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clouse KA, Powell D, Washington I, Poli G, Strebel K, Farrar W, Barstad P, Kovacs J, Fauci AS, Folks TM. 1989. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J Immunol 142:431–438. [PubMed] [Google Scholar]
- 33.Folks TM, Clouse KA, Justement J, Rabson A, Duh E, Kehrl JH, Fauci AS. 1989. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc Natl Acad Sci U S A 86:2365–2368. doi: 10.1073/pnas.86.7.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]