

HIV-1 infection of macrophages leads to the sequestration of newly formed viruses in virus-containing compartments (VCCs), where virions remain infectious and hidden from immune surveillance. The restriction factor BST2, which prevents HIV-1 dissemination by tethering budding viral particles, can be found in VCCs. The HIV-1 Vpu protein counteracts BST2. This study explores the interplay between Vpu and BST2 in the viral protein functions on HIV-1 release and viral particle sequestration in VCCs in macrophages. The results show that Vpu controls the volume of VCCs and favors viral particle release. These Vpu functions partly depend on Vpu’s ability to antagonize BST2. This study highlights that the transmembrane domain of Vpu and two motifs of the Vpu cytoplasmic domain are required for these functions. These motifs were notably involved in the control of the volume of VCCs by Vpu but were dispensable for the prevention of the specific accumulation of BST2 in these structures.
KEYWORDS: BST2/tetherin, VCC, Vpu, human immunodeficiency virus, macrophage, restriction factor, vesicular trafficking, viral release
ABSTRACT
HIV-1 infection of macrophages leads to the sequestration of newly formed viruses in intracellular plasma membrane-connected structures termed virus-containing compartments (VCCs), where virions remain infectious and hidden from immune surveillance. The cellular restriction factor bone marrow stromal cell antigen 2 (BST2), which prevents HIV-1 dissemination by tethering budding viral particles at the plasma membrane, can be found in VCCs. The HIV-1 accessory protein Vpu counteracts the restriction factor BST2 by downregulating its expression and removing it from viral budding sites. Numerous studies described these Vpu countermeasures in CD4+ T cells or model cell lines, but the interplay between Vpu and BST2 in VCC formation and HIV-1 production in macrophages is less explored. Here, we show that Vpu expression in HIV-1-infected macrophages enhances viral release. This effect is related to Vpu’s ability to circumvent BST2 antiviral activity. We show that in absence of Vpu, BST2 is enriched in VCCs and colocalizes with capsid p24, whereas Vpu expression significantly reduces the presence of BST2 in these compartments. Furthermore, our data reveal that BST2 is dispensable for the formation of VCCs and that Vpu expression impacts the volume of these compartments. This Vpu activity partly depends on BST2 expression and requires the integrity of the Vpu transmembrane domain, the dileucine-like motif E59XXXLV64 and phosphoserines 52 and 56 of Vpu. Altogether, these results highlight that Vpu controls the volume of VCCs and promotes HIV-1 release from infected macrophages.
IMPORTANCE HIV-1 infection of macrophages leads to the sequestration of newly formed viruses in virus-containing compartments (VCCs), where virions remain infectious and hidden from immune surveillance. The restriction factor BST2, which prevents HIV-1 dissemination by tethering budding viral particles, can be found in VCCs. The HIV-1 Vpu protein counteracts BST2. This study explores the interplay between Vpu and BST2 in the viral protein functions on HIV-1 release and viral particle sequestration in VCCs in macrophages. The results show that Vpu controls the volume of VCCs and favors viral particle release. These Vpu functions partly depend on Vpu’s ability to antagonize BST2. This study highlights that the transmembrane domain of Vpu and two motifs of the Vpu cytoplasmic domain are required for these functions. These motifs were notably involved in the control of the volume of VCCs by Vpu but were dispensable for the prevention of the specific accumulation of BST2 in these structures.
INTRODUCTION
Macrophages are one of the major cellular targets of HIV-1 and play a critical role in HIV-1-mediated disease. Infected macrophages are distributed throughout the tissues and organs and are able to produce a large number of viral particles over an extended period. Moreover, macrophages are more resistant to HIV-1 cytopathic effect than CD4+ T cells and appear to act as a pharmacologic sanctuary for HIV-1 when patients are under combination antiretroviral therapy (1–4). Macrophages could thus participate in HIV-1 propagation in infected individuals, notably by enabling virus spread through direct cell-to-cell contact with HIV-1 target cells via the so-called virological synapse (5) or by the selective capture of HIV-1-infected T cells (6).
In contrast to CD4+ T lymphocytes, HIV-1 replication in macrophages leads to the accumulation of mature viral particles in intracellular structures termed virus-containing compartments (VCCs) or intracellular plasma membrane-connected compartments (IPMCs) (7–9). Several studies showed by electron microscopy that the accumulation of viral particles in VCCs results directly from the assembly and budding of viral particles at the limiting membrane of VCCs (10–14). However, other studies revealed that viral assembly occurs at the cell surface of the macrophage prior to the internalization of the newly budded virions and, subsequently, to the sequestration of virus in VCCs (15–17). These discrepancies could be explained by the complex architecture of VCCs: these structures are composed of both a tubular network connected to the cell surface by narrow channels and enclosed vesicular compartments (18–20). Several evidences suggest that VCCs are derived from an invagination of the plasma membrane (12, 19) with which they share specific markers, like the cell surface glycoprotein CD44 or phosphatidylinositol 4,5-biphosphate (20). The major histocompatibility complex class II molecules (10) and tetraspanins (such as CD9, CD53, CD63, CD81, and CD82) (11, 12), which are located at the plasma membrane as well as in endosomal compartments, are also found in VCCs. However, VCCs seem distinct from endosomes by harboring a neutral pH and being hardly reached by endocytic tracers (12, 13, 16). Altogether, these advances led to a consensus in which HIV-1 assembly takes place predominantly at specialized plasma membrane domains, sequestered internally in macrophages, where newly formed particles are retained and subsequently transferred to target cells via the virological synapse.
The limited accessibility of the VCC lumen to antibodies and small molecules suggests that the sequestered viruses may be hidden from immune surveillance and protected from drug treatments (12, 16, 21). However, infected host cells possess intrinsic defense mechanisms aimed at blocking viral spread. Among them, the restriction factor bone marrow stromal cell antigen 2 (BST2) (also known as tetherin, HM1.24, or CD317) has been identified as a key protein that prevents the dissemination of enveloped viruses by tethering viral particles at the cell surface (22–29) and then reducing virus release. The ability of BST2 to tether virions at the surface of infected cells is due to its unusual conformation, allowing it to be anchored simultaneously to the plasma membrane and to the envelope of nascent viruses. BST2 is a type II membrane protein with a C-terminal glycophosphatidylinositol anchor. It resides in lipid rafts at the plasma membrane, where the budding of several enveloped viruses occurs (30–33). BST2 is constitutively expressed in a wide variety of human tissues and cell types (34) and is induced by the interferon (IFN) response, notably in CD4+ T cells and macrophages (35–38).
The HIV-1 accessory protein Vpu antagonizes the restriction activity of BST2 on HIV-1 release in different cell types (28, 29). Vpu activities against BST2 restriction are multiple and involve (i) the downregulation of BST2 and (ii) its removal from the viral budding sites. Vpu was shown to downregulate BST2 at the cell surface by accelerating restriction factor sorting for lysosomal degradation (39–41). This activity depends on the increased ubiquitination of BST2 through the recruitment of β-TrCP, a subunit of the Skp1-Cullin-F box E3 ubiquitin ligase, via Vpu phosphorylated serine residues (S52 and S56) (42–44) and on increased endosomal sorting complexes required for transport (ESCRT)-mediated targeting of ubiquitinated BST2 for lysosomal degradation (41). Vpu also favors HIV-1 release by removing BST2 from viral budding sites; this Vpu function is independent of its degradative activity on BST2 (44–46). We recently described that Vpu favors the displacement of BST2 from viral budding sites through the recruitment of the microtubule-associated protein 1 light chain 3 (LC3)-associated phagocytosis (LAP) machinery, involving the LC3C protein. Indeed, Vpu interacts via its L63VEM66 motif, present on the second alpha helix of its cytoplasmic tail, with LC3C to promote the phagocytosis of BST2 (47). Interestingly, the Vpu E59XXXLV64 dileucine-like motif, which overlaps the L63VEM66 motif, was also shown to be important for efficient HIV-1 release. This dileucine-like motif is involved in the interaction of Vpu with clathrin adaptor protein complex 1 (AP-1) and AP-2, two key regulators of the vesicular trafficking system that are required for Vpu-induced removal of BST2 from viral budding sites (48–50).
In macrophages, it has been shown that Vpu is required for efficient HIV-1 release. However, its ability to downregulate BST2 levels and the role of BST2 in the formation of VCCs are subjects of controversy (36, 38). In the present study, we deciphered the interplay between Vpu and BST2 in the viral protein functions in HIV-1 release and viral particle sequestration in VCCs in monocyte-derived macrophages. We report that Vpu dramatically reduces the amount of BST2 detected in VCCs and decreases the volume of these compartments in a BST2-dependent manner. Notably, we observed that mutations of the Vpu transmembrane (TM) domain, necessary for the interaction with BST2, leads to an increased volume of VCCs and an enrichment of BST2 in these compartments. Interestingly, we also highlight that phosphoserines 52 and 56 and the E59XXXLV64 motif of Vpu are involved in the control of the volume of VCCs but are dispensable for the prevention of the specific accumulation of BST2 in these structures.
RESULTS
Vpu promotes HIV-1 release in macrophages.
To clarify the role of the interplay between Vpu and BST2 in HIV-1 infection in macrophages, we confirmed in our infectious model that Vpu promotes viral release, as previously reported (36, 38, 51). In that respect, we used monocyte-derived macrophages (MDMs) obtained by culturing healthy donors’ blood monocytes in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) for 6 days (52). After MDM differentiation, the cells were infected at a high multiplicity of infection (MOI) of 2 with vesicular stomatitis virus G (VSV-G)-pseudotyped wild-type (WT) NL4-3 HIV-1 or its counterpart that is unable to express Vpu (Udel) (53). The percentage of infected cells varies from 5 to 60%, depending on the donor (data not shown). Of note, VSV-G pseudotyping of the non-macrophage-tropic NL4-3 strain enabled us to assess HIV-1 production in a single round of infection. At 5 days postinfection, supernatants were recovered, and cells were lysed to monitor HIV-1 release by quantifying the viral capsid protein (CAp24) by an enzyme-linked immunosorbent assay (ELISA). The ELISA used in this study detects only mature CAp24 (Fig. 1A and B). Indeed, ELISA quantification of CAp24 in infected saquinavir-treated cells showed a very weak signal in cell lysates and released viral particles produced compared to those in control cells, whereas only the Pr55gag precursor was detected by Western blotting of the cell lysates. This is consistent with a block in Gag maturation upon saquinavir treatment, indicating that the ELISA kit used specifically detects CAp24 but not the immature forms of Gag. CAp24 quantification showed an accumulation of cell-associated CAp24 in MDMs infected with the Udel virus compared to WT-infected cells (Fig. 1C and D, black bars); the level of accumulation strongly varied among the donors. The increased amount of HIV-1 CAp24 present in producer cells infected with Udel virus was confirmed by Western blotting (Fig. 1E). The Western blot analysis did not show an increased amount of the precursor proteins Pr55Gag and p41 when MDMs were infected with Udel virus compared to WT-infected cells, suggesting that the increased amount of cell-associated CAp24 is not due to a higher infection level of the mutant virus but is due to the accumulation of mature viral particles in Udel virus-infected MDMs. These observations were consistent with the CAp24 quantification in supernatants, since a 2- to 5-fold decrease of released CAp24 was observed in Udel HIV-1-infected cells in comparison to the WT virus (Fig. 1C and D, gray bars). Calculation of the HIV-1 release index, corresponding to the ratio of extracellular CAp24 to total CAp24 produced in cell culture (cell-associated CAp24 plus released CAp24), showed that Vpu expression promotes a 40 to 75% increase in the amount of the viral particles released from MDMs (Fig. 1F).
FIG 1.

Vpu enhances HIV-1 release in macrophages. HeLa cells infected with VSV‐G-pseudotyped NL4‐3 HIV‐1 were treated with saquinavir (0.01 μM) for 24 h or with DMSO (dimethyl sulfoxide). Cell were then harvested and lysed. (A) Western blot analysis of profiles of Gag expression in cell lysates. (B) Graphical representation of ELISA quantification of CAp24 in cell lysates (cell-associated CAp24) and released into the cell supernatants (released CAp24). (C to F) MDMs from different donors were infected with VSV-G-pseudotyped WT NL4-3 (WT) or Vpu-deficient NL4-3 (Udel) HIV-1. (C and D) CAp24 present within the cells (cell-associated CAp24) and CAp24 released into the supernatant (released CAp24) of infected cells were quantified by an ELISA at 5 days postinfection. Data normalized to value for HIV-1 NL4-3 WT-infected cells in panel C and raw data are presented in panel D. Bars represent the means ± SEM from replicates done on the same donor. (E) Western blot analysis of HIV-1-infected MDMs from donor 4 performed using anti-CAp24, anti-BST2, anti-Vpu, and anti-tubulin antibodies. (F) HIV-1 release was determined as the ratio of released CAp24 to total CAp24 production (cell-associated CAp24 plus released CAp24). The values were normalized to the value for HIV-1 NL4-3 WT-infected cells. Statistical significance was determined using unpaired two-tailed Student’s t test (means ± SEM; n = 4 donors). **, P ≤ 0.01.
Vpu promotes virus release by counteracting BST2 antiviral functions in infected cells (36, 38–41, 44, 47, 48, 54). We thus monitored BST2 expression in WT or Udel HIV-1-infected MDMs by Western blot analysis. The immunoblot analysis of BST2 revealed a decreased expression of this protein in WT HIV-1-infected cells compared to Udel HIV-1-infected cells, with the level of BST2 in WT- and Udel-infected cells being higher than that detected in noninfected (NI) cells (Fig. 1E). This is consistent with previous reports showing that HIV-1 infection of macrophages induced interferon-mediated BST2 expression (37, 38). However, the upregulation of BST2 observed in infected primary macrophages was proposed by Chu et al. to be Nef dependent and not the consequence of type I IFN induction (36). To address this question, we first confirmed that BST2 expression was inducible by type I interferon. MDMs from two donors were stimulated by increasing doses of IFN-α (20 to 500 U/ml) for 24 h. Consistent with data from previous studies (37, 38), Western blotting against BST2 showed a clear increase of the BST2 expression level, even at the lowest concentration of IFN used (20 U/ml) (Fig. 2A), supporting the notion that BST2 expression is induced by type I IFN in primary macrophages. We next assessed whether HIV-1 infection induces BST2 expression and whether this upregulation depends on Nef expression. MDMs were infected with VSV-G-pseudotyped WT NL4-3 and Nef-deficient NL4-3 (ΔNef) HIV-1. The course of infection was monitored by ELISA quantification of the CAp24 released in the culture supernatant (Fig. 2B), and BST2 expression was assayed by reverse transcription-quantitative PCR (RT-qPCR) and Western blotting (Fig. 2C and D) at different time points. Our result showed that HIV-1 infection upregulates BST2 expression at the mRNA level during the course of infection, independently of Nef expression. Furthermore, the BST2 protein level was increased at early time points of infection and then downregulated. The patterns of BST2 expression were similar in WT- and ΔNef-infected MDMs, suggesting that BST2 upregulation is not dependent on HIV-1 Nef expression.
FIG 2.

Type I interferon upregulates BST2 expression in macrophages. (A) MDMs from two donors were stimulated for 24 h with 0, 20, 50, 100, 200, or 500 U/ml IFN-α. The cells were then harvested, and the BST2 protein level was determined by Western blot analysis. (B to D) MDMs were infected with VSV-G-pseudotyped WT NL4-3 (WT) or Nef-deficient NL4-3 (ΔNef) HIV-1, and the cells were harvested at different days postinfection. (B) CAp24 released into the supernatant of infected cells was quantified by an ELISA. (C and D) BST2 expression was assayed by RT-qPCR (C) and Western blotting (D) during the course of infection. For RT-qPCR analysis, BST2 mRNA expression was normalized with the GAPDH gene expression level, and data are presented as the fold increase relative to the mRNA level at day 0, for each virus. Western blot analysis was performed on cell lysates using anti-BST2, anti-Nef, and anti-actin antibodies. Actin is the loading control. Data shown in panels B to D are representative of results from two experiments done on two different donors.
Together, these data show that BST2 expression is induced by type I IFN in primary macrophages. HIV-1 infection also induces an upregulation of BST2, an effect that is independent of HIV-1 Nef expression. Moreover, we confirm that Vpu expression during HIV-1 infection of MDMs enhances viral particle release and leads to the downregulation of BST2 expression.
Vpu counteracts BST2 to promote HIV-1 release in macrophages.
To investigate whether the Vpu-induced enhanced release of viral particles depends on Vpu’s ability to counteract the restriction activity of BST2, we carried out small interfering RNA (siRNA)-mediated knockdown of BST2 in MDMs. For this purpose, cells were treated with control siRNA (siCtrl) or siRNA targeting BST2 (siBST2) and infected 2 days later with VSV-G-pseudotyped WT or Udel NL4-3 HIV-1 (MOI = 2). At 5 days postinfection, supernatants and cell lysates were collected to confirm the knockdown of BST2 by Western blotting (Fig. 3A) and monitor HIV-1 production by a CAp24 ELISA (Fig. 3B and C). Whereas the amount of released CAp24 was decreased by ∼70% in the absence of Vpu expression (Fig. 3B, compare released CAp24 in siCtrl-treated MDMs infected with HIV-1 Udel versus that in MDMs infected with the HIV-1 WT), BST2 silencing resulted in a similar release of CAp24 in the absence and presence of Vpu (compare CAp24 released in siBST2-treated MDMs infected with HIV-1 Udel versus that in MDMs infected with the HIV-1 WT). Calculation of the HIV-1 release index from 3 donors confirms that BST2 depletion rescued Udel virus release to a level similar to that of WT virus-infected MDMs (Fig. 3D). Interestingly, no difference in WT HIV-1 particle release was observed after siBST2 treatment, compared to siCtrl treatment, suggesting that Vpu is sufficient to overcome the BST2-imposed restriction on HIV-1 release in macrophages. Overall, in accordance with previous observations (36, 38), our results indicate that Vpu promotes virus release from macrophages by counteracting the restriction factor BST2.
FIG 3.

BST2 expression inhibits HIV-1 release in macrophages. MDMs from different donors transfected with control siRNA (siCtrl) or siRNA targeting BST2 (siBST2) were infected with VSV-G-pseudotyped WT NL4-3 (WT) or Vpu-deficient NL4-3 (Udel) HIV-1. (A) At 5 days postinfection, Western blot analysis of HIV-1-infected MDMs from donor 5 was performed using anti-BST2, anti-CAp24, anti-Vpu, and anti-actin antibodies. (B and C) CAp24 present within the cells (cell-associated CAp24) and CAp24 released into the supernatant (released CAp24) of infected cells were quantified by an ELISA. Data normalized to the value for WT NL4-3-infected siCtrl cells in panel B and raw data are presented in panel C. Bars represent the means ± SEM from replicates done on the same donor. (D) HIV-1 release was determined as the ratio of released CAp24 to total CAp24 production (cell-associated CAp24 plus released CAp24). Data were normalized to the value for WT NL4-3-infected siCtrl cells. Statistical significance was determined using one-way ANOVA combined with Holm-Sidak’s post hoc test (means ± SEM; n = 3 donors). **, P ≤ 0.01; ***, P ≤ 0.001; ns, not significant.
Vpu downregulates cellular and cell surface levels of BST2.
Vpu enhances HIV-1 release by antagonizing BST2 restriction in macrophages (Fig. 3). It has been shown that Vpu counteracts BST2 by downregulating cellular and cell surface expression levels of BST2 and by excluding BST2 from viral budding sites (36, 38–41, 44, 47, 48, 54). A previous study indicated that Vpu is able to counteract the restriction activity of BST2 without necessarily downregulating cell surface and intracellular levels of BST2 in a cell type-specific manner (37). In macrophages, the ability of Vpu to downregulate the cell surface level of BST2 remains controversial: two studies report that Vpu is unable to decrease the level of BST2 present at the surface of infected MDMs (36, 55), whereas another shows a clear downregulation of the BST2 level at the surface of HIV-1-infected MDMs expressing Vpu (38). Thus, we first quantitatively measured cellular and cell surface levels of BST2 in MDMs infected with VSV-G-pseudotyped WT NL4-3 virus or its Vpu-deficient counterpart. The expression level of BST2 was analyzed 5 days later by flow cytometry of CAp24-positive cells (Fig. 4). Quantification of cellular (Fig. 4A and B) and cell surface (Fig. 4C and D) levels of BST2 was in line with the data obtained by Western blot analysis (Fig. 1E and Fig. 3A): Vpu-induced downregulation of BST2 was observed in HIV-1 WT-infected cells. Indeed, HIV-1 Udel-infected cells displayed a statistically higher BST2 mean fluorescence intensity (MFI) (1.2- to 1.8-fold increase in cells and 1.2- to 2.3-fold increase at the cell surface) than HIV-1 WT-infected cells at the cellular level and at the cell surface level (Fig. 4). Of note, both the upregulation of BST2 in response to Udel HIV-1 infection and the capacity of Vpu to downregulate this restriction factor differed widely between donors. Furthermore, in agreement with the Western blot results (Fig. 1E and Fig. 3A), flow cytometry analysis indicated that Vpu expression does not completely downregulate BST2 expression (Fig. 4A and C). Indeed, we noticed a higher level of BST2 in MDMs infected with the WT virus than in noninfected cells. Overall, these results suggest that, under our experimental conditions, Vpu can downregulate intracellular and cell surface levels of BST2 in HIV-1-infected MDMs, with an intensity that differs between donors.
FIG 4.

Cellular and cell surface expressions of BST2 in infected macrophages. MDMs from different donors were infected with VSV-G-pseudotyped WT NL4-3 (WT) or Vpu-deficient NL4-3 (Udel) HIV-1. Five days later, the intracellular (A and B) and cell surface (C and D) levels of BST2 in noninfected or VSV-G-pseudotyped WT NL4-3 (WT) or Vpu-deficient NL4-3 (Udel) HIV-1-infected MDMs were analyzed by flow cytometry using anti-BST2 and anti-CAp24 antibodies. Mean fluorescence intensity (MFI) values for BST2 in CAp24-positive cells were normalized to that for noninfected cells. For panels B and D, each symbol represents the normalized BST2 MFI values of MDMs for each donor. Statistical significance was determined using unpaired two-tailed Student’s t test (means ± SEM; n = 3 donors). *, P ≤ 0.05.
The integrity of the Vpu transmembrane domain and of two motifs of the Vpu cytoplasmic domain is necessary for Vpu to promote HIV-1 release in macrophages.
Previous studies in HeLa, HEK293T, and T cells have reported that Vpu counteraction of BST2 restriction requires the integrity of the Vpu transmembrane domain and of two motifs of the Vpu cytoplasmic domain: phosphoserine residues 52 and 56 and the dileucine-like motif E59XXXLV64. While it has been shown that phosphoserine residues 52 and 56 are essential for β-TrCP recruitment and lysosomal targeting of BST2, the E59XXXLV64 motif is involved in the trafficking of BST2 via its interaction with the clathrin adaptors AP-1 and AP-2 and the removal of the restriction factor from viral budding sites (44, 48, 49). We therefore analyzed the impact of mutations of these motifs on the beneficial effect of Vpu on HIV-1 release in macrophages (Fig. 5). We also examined the release of an HIV-1 Vpu mutant (A14W22) that is unable to physically interact through its transmembrane domain with BST2 (42). MDMs were infected at an MOI of 2 with VSV-G-pseudotyped HIV-1 NL4-3 viruses mutated in either Vpu phosphoserine residues 52 and 56 (S52-S56/N-N) (2.6 virus), the Vpu dileucine-like motif E59XXXLV64 (ELV/AAA) (ELV virus), or Vpu residues A14 and W22 (A14-W22/L-A) (A14W22 virus). The percentage of infected cells varies from 5 to 60%, depending on the donor (data not shown). ELISA quantification of HIV-1 CAp24 and calculation of the HIV-1 release index revealed a significant decrease of viral particle release from ELV and 2.6 virus-infected MDMs compared to WT virus-infected cells (Fig. 5A to C). This decrease was slightly less pronounced than that observed with Udel and A14W22 viruses. Moreover, this decrease was associated with an accumulation of cell-associated CAp24 (Fig. 5A, black bars), which we also observed in our Western blot analysis (Fig. 5D, lanes 3 to 6). We also noted that Vpu expression levels were similar in WT, ELV, and 2.6 HIV-1-infected MDMs and that mutation of the A14 and W22 residues in Vpu led to a protein with a higher apparent molecular weight and reduced expression (Fig. 5D), as previously shown (42, 47). In addition, we observed that mutated Vpu proteins are less efficient in downregulating BST2 than WT Vpu (Fig. 5D). Thus, the integrity of the Vpu TM domain is crucial for efficient HIV-1 production in MDMs. The Vpu dileucine-like motif E59XXXLV64 and the S52 and S56 residues are, in turn, equally necessary for Vpu to promote HIV-1 release.
FIG 5.

Impact of mutations at Vpu E59/L63/V64, S52/S56, and A14/W22 residues on HIV-1 release in macrophages. MDMs were infected with VSV-G-pseudotyped WT, Udel, ELV, 2.6, or A14W22 NL4-3 HIV-1. (A and B) Five days later, CAp24 present within the cells (cell-associated CAp24) and CAp24 released into the supernatant (released CAp24) of infected cells were quantified by an ELISA. Data were normalized to the values for WT NL4-3-infected cells in panel A, and raw data are presented in panel B. (C) HIV-1 release was determined as the ratio of released CAp24 to total CAp24 production (cell-associated CAp24 plus released CAp24). Data were normalized to value for NL4-3 WT-infected cells. Statistical analysis was done using one-way ANOVA combined with Holm-Sidak’s post hoc test (means ± SEM; n = 3 donors). The P values are indicated above each comparison. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. (D) Western blot analysis of HIV-1-infected MDMs from three donors was performed on cell lysates using anti-CAp24, anti-BST2, anti-Vpu, and anti-actin antibodies. Quantification of BST2 and actin bands was done using ImageJ, and ratios between BST2 and actin are shown above Western blot lines.
Vpu impairs BST2 enrichment in VCCs independently of its S52 and S56 residues and the E59XXXLV64 motif.
We observed that Vpu-mutated viruses are less efficient in downregulating BST2 in MDMs (Fig. 5D). We thus examined whether mutations of the Vpu dileucine-like motif E59XXXLV64 or the Vpu S52 and S56 residues modify the subcellular distribution of BST2 and viral proteins in infected MDMs, particularly in VCCs. It was previously reported that BST2 is present in VCCs in the absence of Vpu expression in HIV-1-infected MDMs (38). Interestingly, BST2 was proposed to contribute to the formation of VCCs (36), although this hypothesis has been controversial (38). To analyze the distribution of BST2 in VCCs, MDMs were plated on coverslips and infected with HIV-1 NL4-3 WT, Udel, 2.6, or ELV. At 5 days postinfection, cells were costained for CAp24, BST2, and CD81 (a marker of VCCs) and analyzed by confocal microscopy (Fig. 6A). In noninfected cells, BST2 labeling was detected as puncta dispersed throughout the cell, in a perinuclear compartment and at the cell surface; some BST2-positive puncta colocalized with CD81 (Fig. 6A), a plasma membrane and endosomal marker. In the absence of Vpu, a subset of BST2 molecules clearly colocalized with CAp24-positive compartments, as confirmed by the calculation of Mander’s coefficient (0.20 ± 0.05 in WT HIV-1-infected cells versus 0.79 ± 0.03 in Udel HIV-1-infected cells) (Fig. 6B), revealing the degree of colocalization between CAp24 and BST2. Conversely, the expression of Vpu during HIV-1 infection dramatically reduced the presence of BST2 in VCCs since we failed to observe significant colocalization between BST2 and CAp24 (Fig. 6A and B). Moreover, we did not detect any noticeable colocalization between BST2 and CAp24 in ELV or 2.6 virus-infected cells (Fig. 6A and B), although we detected an increased level of BST2 in MDMs infected with these mutated viruses by Western blotting (Fig. 5D).
FIG 6.

Impact of mutations at Vpu E59/L63/V64 and S52/S56 residues on the subcellular distribution of BST2. MDMs were infected with VSV-G-pseudotyped (WT, Udel, ELV, or 2.6) NL4-3 HIV-1. (A) At 5 days postinfection, cells were fixed; immunolabeled for BST2 (green), CAp24 (red), and CD81 or control isotype antibodies; and analyzed by confocal microscopy (bars, 20 μm). CD81 labeling is shown in gray in the monochrome panels to facilitate the visibility and in blue in the merge panels. Higher magnifications of virus-containing compartments (VCCs) are shown on the bottom left corner of each image. (B) BST2/CAp24 colocalization was evaluated by calculating Mander’s coefficient on at least 15 cells under each condition using the JACoP plug-in with ImageJ. Statistical analysis was done using one-way ANOVA combined with Bonferroni’s post hoc test (means ± SEM). ***, P ≤ 0.001. (C and D) The BST2 mean fluorescence intensity (MFI) in VCCs (C) and in the cytoplasm (D) was measured using ImageJ on the confocal optical section that presented the largest and highest number of VCCs under each infection condition. Statistical significance was determined using one-way ANOVA combined with Bonferroni’s post hoc test (means ± SEM; n = 3 donors). *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
To exclude that the lack of BST2 enrichment in the VCCs of MDMs infected with the ELV or 2.6 virus is due to a reduced expression of BST2 in these cells, we quantified, on our confocal images, the MFI of cytosolic BST2 staining and BST2 associated with the VCCs by creating specific masks for the VCCs and the cytoplasm. These data showed similar expression levels of BST2 in the cytoplasm of ELV and 2.6 HIV-infected MDMs, compared to that detected in Udel HIV-infected MDMs (Fig. 6C). We also found that BST2 strongly accumulated on the VCCs in Udel-infected MDMs, compared to the WT HIV-infected ones (Fig. 6D). A slight increase of the BST2 MFI associated with VCCs was also observed on ELV- and 2.6-infected MDMs, to a much lesser extent than that detected on VCCs found in Udel-infected MDMs and consistent with a lack of enrichment of BST2 in the VCCs observed in cells infected by these mutants (Fig. 6D).
Altogether, the S52 and S56 residues and the E59XXXLV64 motif do not seem to be the main determinants of Vpu by which the viral protein impairs BST2 enrichment in VCCs.
The integrity of the Vpu transmembrane domain is required to impair BST2 enrichment in VCCs.
We next examined the localization of BST2, CAp24, and CD81 in MDMs infected with the Vpu TM HIV-1 mutant by confocal microscopy, as described above. Mutation of the A14 and W22 residues of the Vpu TM domain (A14W22) led to a striking enrichment of BST2 molecules in VCCs labeled for CAp24 and CD81 (Fig. 7A), as observed for MDMs infected with a virus deficient in Vpu expression (Udel). This redistribution of BST2 in the CAp24-positive compartments was confirmed by the increase of the CAp24/BST2 colocalization coefficient (Mander’s coefficient) found in A14W22 HIV-1-infected cells (0.67 ± 0.03), compared to that for WT HIV-1-infected cells (0.21 ± 0.01) (Fig. 7B). This increase is similar to those observed for Udel HIV-1-infected MDMs and indicates that the integrity of the Vpu transmembrane domain is required to impair BST2 enrichment in VCCs.
FIG 7.

Impact of mutations at Vpu A14/W22 residues on the subcellular distribution of BST2. MDMs were infected with VSV-G-pseudotyped (WT, Udel, or A14W22) NL4-3 HIV-1. (A) At 5 days postinfection, cells were fixed; immunolabeled for BST2 (green), CAp24 (red), and CD81; and analyzed by confocal microscopy (bars, 20 μm). CD81 labeling is shown in gray in the monochrome panels to facilitate visibility and in blue in the merge panels. (B) BST2/CAp24 colocalization was evaluated by calculating Mander’s coefficient on 29 cells under each condition using the JACoP plug-in with ImageJ. Statistical analysis was done using one-way ANOVA combined with Bonferroni’s post hoc test (means ± SEM; n = 1 donor). ***, P ≤ 0.001; ns, not significant.
Mutations of Vpu cytoplasmic residues S52 and S56 or the E59XXXLV64 motif increase the volume of VCCs.
Previous work showed a correlation between the presence of BST2 in VCCs and an increased size of these compartments sequestering viruses (36). We thus determined the sum of the VCC volume per cell using three-dimensional (3D) reconstruction. WT and mutated HIV-1-infected MDMs were fixed and labeled for CAp24, CD81, and BST2. Series of confocal images were then 3D reconstructed using Imaris software (Fig. 8A). The volumetric analyses of VCCs revealed a clear increase of the sum of the VCC volume per infected cell in Vpu A14W22 mutant HIV-1-infected MDMs (81 ± 11.3 μm3), similar to what we observed in Vpu-deficient HIV-1-infected macrophages (97.4 ± 14.4 μm3), in comparison to WT HIV-1-infected cells (18.3 ± 3.1 μm3) (Fig. 8B). Interestingly, in a second set of experiments, we also observed a significant increase of the overall volume of these compartments in Vpu-mutated ELV (142.7 ± 18.7 μm3) or 2.6 (102 ± 16.9 μm3) HIV-1-infected MDMs in comparison to WT HIV-1-infected cells (26.9 ± 4.5 μm3), without reaching the size of those compartments that are present in Udel HIV-1-infected macrophages (224.8 ± 21.5 μm3) (Fig. 8C). Together, our data revealed that mutations of the Vpu S52 and S56 residues or the Vpu E59XXXLV64 motif increase the VCC volume per cell without a visible enrichment of BST2 in these compartments.
FIG 8.

Impact of mutations at Vpu E59/L63/V64 and S52/S56 residues on the sum of the VCC volume. MDMs were infected with VSV-G-pseudotyped WT, Udel, ELV, or 2.6 NL4-3 HIV-1. (A) At 5 days postinfection, cells were fixed and immunolabeled for BST2, CD81, and CAp24. The volumetric analysis of VCCs was performed using Imaris 7.2 software (Bitplane) by 3D reconstruction of MDMs. An example of 3D reconstruction of MDMs is shown. The merge presented on the right is the overlap of the three two-dimensional (2D) images corresponding to the 3D panels shown. Arrows show structures considered VCCs. (B and C) Histograms showing the sum of the VCC volume per cell obtained by thresholding fluorescence intensity values on 62 cells under each condition from three independent donors. Statistical analysis was done using one-way ANOVA combined with Bonferroni’s post hoc test (means ± SEM; n = 3 donors). ***, P ≤ 0.001; *, P ≤ 0.05. Bars, 20 μm.
BST2 is dispensable for the formation of VCCs.
Finally, we examined the correlation between the volume of VCCs in HIV-1-infected macrophages and BST2 expression. To address this issue, MDMs were treated with siBST2 or siCtrl and infected with WT or Udel VSV-G-pseudotyped NL4-3 HIV-1. At 5 days postinfection, the cells were colabeled with anti-CAp24 and anti-BST2 antibodies and observed by confocal microscopy. SiCtrl-treated MDMs infected with WT and Udel HIV-1 displayed VCCs accompanied by a strong reduction of BST2 accumulation in these structures when Vpu was expressed (Fig. 9A). Interestingly, BST2 depletion did not prevent the formation of VCCs in HIV-1-infected MDMs, suggesting that BST2 is dispensable for the formation of VCCs (Fig. 9A, siBST2 treatment of WT-infected MDMs). We next assessed the sum of the volume of VCCs per infected cell using 3D reconstruction of siRNA-treated HIV-1-infected MDMs. As described above (Fig. 8), Vpu expression significantly reduced the total volume of VCCs by 2-fold (83.4 ± 1.2 μm3 in WT HIV-1-infected siCtrl-treated cells compared to 210.4 ± 34.2 μm3 in Udel HIV-1-infected siCtrl-treated cells) (Fig. 9B). On the contrary, in siBST2-treated MDMs, the total volume of VCCs was not significantly different in WT (86.8 ± 11.0 μm3) and Udel (119.6 ± 10.6 μm3) HIV-1-infected cells and close to that of WT HIV-1-infected siCtrl-treated MDMs, suggesting that WT HIV-1 Vpu might restrict the volume of VCCs by preventing BST2 accumulation in these structures and/or by counteracting BST2 functions.
FIG 9.
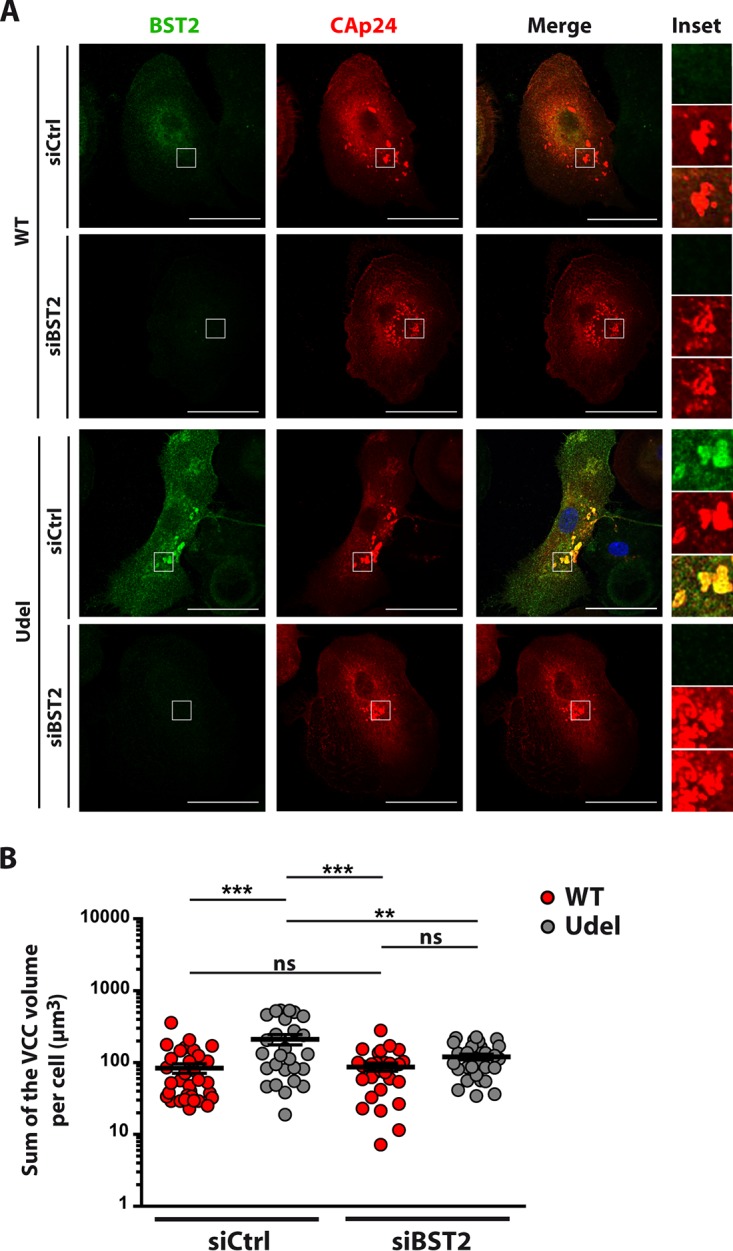
Vpu reduces the sum of the VCC volume in a BST2-dependent manner. MDMs transfected with control siRNA (siCtrl) or siRNA targeting BST2 (siBST2) were infected with VSV-G-pseudotyped WT NL4-3 (WT) or Vpu-deficient NL4-3 (Udel) HIV-1. (A) At 5days postinfection, cells were fixed, immunolabeled for BST2 (green) and CAp24 (red), and analyzed by confocal microscopy. Bars, 20 μm. Higher magnifications (inset) of VCCs are shown on the right of each merge image. (B) Volumetric analysis of VCCs was performed using Imaris 7.2 software (Bitplane) by 3D reconstruction of MDMs. The histogram shows the sum of the VCC volume per cell obtained by thresholding fluorescence intensity values on at least 26 cells under each condition. Statistical analysis was done using one-way ANOVA combined with Bonferroni’s post hoc test (means ± SEM; n = 3 donors). **, P ≤ 0.01; ***, P ≤ 0.001; ns, not significant.
Taken together, our results indicate that the formation of VCCs is BST2 independent. However, in the absence of Vpu, these structures are enriched in BST2 and display an increased volume. This volume relies on the lack of Vpu counteraction against BST2 activities. Finally, the increase of the VCC volume is not necessarily related to a strong enrichment of BST2 in these structures. This is consistent with the notion that BST2 does not contribute to the formation of VCCs but impacts their volume, depending on its capacity to tether viruses.
DISCUSSION
In HIV-1-infected macrophages, newly synthesized virions are sequestered into subcellular virus-containing compartments (VCCs) (56–58). During cell-to-cell contacts, VCCs reach the cell surface and transfer viral particles to HIV-1 target cells, allowing HIV-1 to escape the host immune surveillance system (59). The efficient spread of viral particles also depends on the ability of HIV-1 to circumvent intrinsic immunity, particularly the restriction factor BST2, which tethers fully formed viruses to the plasma membrane. Therefore, HIV-1 has evolved to encode the accessory protein Vpu to counteract BST2. Molecular mechanisms driving Vpu activities against BST2 restriction of HIV-1 release were mostly described for CD4+ T lymphocytes and cell lines. In macrophages, there have been conflicting reports regarding the effects of Vpu on BST2 as well as a potential contribution of BST2 to the formation of VCCs (36, 38). How VCCs are formed and persist in macrophages is a major concern to develop treatments that block HIV-1 dissemination in patients, preventing disease progression toward AIDS. In this study, we investigated the role played by Vpu in HIV-1 production and in the formation of VCCs in macrophages. Through the generation of Vpu-mutated HIV-1, we show that Vpu controls the volume of VCCs and favors viral particle release in macrophages. We notably highlight that the transmembrane domain of Vpu, as well as the E59XXXLV64 motif and phosphoserine residues S52 and S56 in the cytoplasmic tail, is necessary for these functions.
Using an HIV-1 mutant that is unable to express Vpu (Udel virus), we confirmed previous reports (36–38, 51, 60) revealing that Vpu is required for the efficient release of viral particles in primary human monocyte-derived macrophages (MDMs) (Fig. 1). Silencing of BST2 expression in Udel HIV-1-infected MDMs restored HIV-1 release to a level similar to that of WT HIV-1, indicating that Vpu favors HIV-1 release by counteracting BST2 restriction activity in macrophages (Fig. 3). In addition, our data indicated that HIV-1 infection of MDMs, in the presence or absence of Vpu, induces an increased level of BST2 (Fig. 4). Interestingly, this upregulation of the BST2 protein level is observed at early time points of infection and is independent of HIV-1 Nef expression (Fig. 2). Our results did not sustain those presented by Chu and colleagues (36) and support a scheme in which the upregulation of BST2 expression is a consequence of the induction of type I IFN by infection rather than the expression of Nef protein (Fig. 2D). In accordance with the results of Giese and Marsh, we also show that Vpu is able to downregulate BST2 (38) (Fig. 4). In these experiments, we noticed a significant variation of the HIV-1 Vpu response against BST2 restriction among the six donors analyzed. This singularity could explain the controversy observed in the literature regarding the impact of Vpu on the cell surface expression of BST2 in MDMs (36, 38, 55). We also noted in our flow cytometry analysis that Vpu is unable to downregulate BST2 at the same level as that detected in noninfected macrophages in the majority of donors (Fig. 4). Nevertheless, we did not observe any increase of viral release in WT-infected cells depleted for BST2 (Fig. 3), suggesting that Vpu is able to efficiently counteract BST2 restriction activity in macrophages without inducing a complete degradation of HIV-1-induced BST2 expression. These results agreed with those reported by Giese and Marsh (38) and are in line with previous reports indicating that Vpu-induced degradation of BST2 is not necessarily coupled with the ability of Vpu to promote HIV-1 release (37, 44, 61).
Our analysis by confocal microscopy also revealed that the absence of Vpu leads to a strong enrichment of BST2 in VCCs (Fig. 6) and increases the total volume of these intracellular compartments (Fig. 8). Previous studies have proposed that the volume of VCCs may correlate with the amount of accumulated viral particles (19) and that BST2 may indirectly favor the enlargement of these compartments by tethering viruses to the limiting membrane (38). Our data obtained by 3D reconstruction of VCCs support these notions. Indeed, we observed that Vpu expression induces a pronounced reduction of the total volume of these structures in HIV-1-infected macrophages (Fig. 8). Moreover, the analysis of the volume and characteristics of VCCs in MDMs infected with HIV-1 mutated in Vpu transmembrane residues A14 and W22, which makes Vpu unable to interact with BST2 and to mediate its cell surface downregulation, demonstrated that the integrity of the Vpu TM domain is essential for removing BST2 from VCCs (Fig. 7) and for limiting the size of these compartments (Fig. 8). Furthermore, we show that this domain is crucial for antagonizing BST2 antiviral activities and favoring HIV-1 release from MDMs (Fig. 5). Our analyses also indicated that BST2 is not required for the formation of VCCs. Indeed, VCCs are still observed in the absence of BST2, and their volumes are similar in HIV-1 WT-infected MDMs in absence and presence of BST2 expression (Fig. 9B). These data corroborate the findings of Giese and Marsh but contradict the results reported by Chu et al. (36, 38).
Mutation of the Vpu dileucine-like motif E59XXXLV64 (ELV virus) or phosphoserines 52 and 56 (2.6 virus), which are involved in the degradation or trafficking of BST2, leads to a decreased release of viral particles with respect to WT virus (Fig. 5C), as described previously for infection of CD4+ T cells and model cell lines (29, 39, 40, 44, 48–50, 62). These mutations alter Vpu-mediated degradation of BST2, leading to accumulation of BST2 in infected MDMs (Fig. 5D). Moreover, mutations of these motifs also decrease Vpu’s ability to reduce the size of VCCs in infected MDMs. Infections of these cells with ELV or 2.6 virus lead to the formation of intermediate-sized VCCs (Fig. 6A), compared to the small and large compartments formed by WT and Udel viruses, respectively (Fig. 8B and C). All types of VCCs contained CD81 (Fig. 6A) as well as CD9, Siglec-1, and CD63 (data not shown). Surprisingly, microscopy analyses revealed that the increased volume of the VCCs is not necessarily associated with a distinctive accumulation of BST2 in these compartments (Fig. 6 and 8). Consequently, these experiments point to a more complex contribution of Vpu functions to VCC formation and composition.
Therefore, we postulate that Vpu limits the size of VCCs by reducing the number of viruses tethered at the cell surface and, subsequently, sequestered into these structures once the plasma membrane invaginates. This phenomenon would be the result of the downregulation of BST2 molecules present at viral budding sites, through the interaction between the TM domains of Vpu and BST2, leading to the exclusion of BST2 from the budding sites. Furthermore, Vpu-induced disturbance of BST2 trafficking (in favor of its degradation) via its E59XXXLV64 and phosphoserine S52/S56 motifs might also contribute to some extent to the regulation of the restriction factor’s expression at the viral budding site. Indeed, Vpu-induced degradation of BST2 is not strictly required for its induced direct exclusion of BST2 from viral budding sites (44–46). Nevertheless, the global increase of BST2 expression in cells expressing the ELV or 2.6 Vpu mutant might indirectly cause its increase at viral budding sites (Fig. 6C and D); this might ultimately lead to restriction of HIV-1 release by entrapping newly formed viruses at the cell surface, leading to their sequestration in invaginated membranes and their retention in VCCs (Fig. 8C). However, this would not significantly enhance the presence of BST2 in VCCs, thus limiting the detection of BST2 molecules in our immunofluorescence assay.
Moreover, the intermediate volume of VCCs observed in HIV-1 ELV- and 2.6-infected MDMs (compared to those detected in the presence of WT and Udel viruses, respectively) might suggest that BST2 is not the only factor involved in virus sequestration in VCCs. As BST2 is excluded from VCCs when cells are infected with these mutants, additional factors may contribute to virus entrapment. Moreover, BST2 depletion in the absence of Vpu results in a decrease of the total volume of VCCs, similar to what is observed in siCtrl-treated macrophages infected with WT HIV-1 (Fig. 9B), suggesting that Vpu partly regulates the size of these compartments by antagonizing BST2 function. Thus, we could not exclude the hypothesis that Vpu interferes with another cellular partner to regulate the VCCs’ size. Interestingly, several additional factors, such as Siglec-1 and mannose receptor 1, have been described to tether virus and favor their sequestration in MDMs (17, 63). Moreover, several studies report that Vpu downregulates the tetraspanins CD63, CD81, and CD82 that are found enriched in VCCs (64–66). Such factors could participate in virus sequestration in VCCs, and Vpu could alter the cell surface expression of these factors and, in this way, modify virus sequestration in VCCs. Thus, future work will be required to explore the role of additional cellular factors in virus sequestration in MDMs.
Our study provides new insights into the mechanism regulating HIV-1 compartmentalization in infected macrophages. The control of the VCC volume during the course of the infection is a process achieved by Vpu and requires the integrity of the Vpu TM domain, the Vpu E59XXXLV64 motif, and Vpu phosphoserines S52 and S56. This process is based on the ability of Vpu to reduce the level of the restriction factor BST2 at viral budding sites. By this activity, Vpu reduces the accumulation of tethered virions in VCCs, preventing the expansion of these compartments. Moreover, our study highlights that Vpu, through additional activities, limits the enrichment of BST2 in VCCs in HIV-1-infected macrophages.
MATERIALS AND METHODS
Purification, culture, and characterization of macrophages.
Monocytes were obtained from HIV-seronegative human buffy coats. Peripheral blood mononuclear cells (PBMCs) were purified by density centrifugation using Ficoll-Paque Plus (density, 1.077 g/ml; GE Healthcare) at 400 × g for 30 min at 20°C. Monocytes were negatively selected from PBMCs using magnetic-activated cell sorting (MACS) monocyte isolation kit II (Miltenyi Biotec) according to the manufacturer’s recommendations. Monocytes were then plated in RPMI 1640 medium (Gibco, Life Technologies) supplemented with 10% decomplemented fetal calf serum (FCS), 1% antibiotic-antimycotic cocktail, 1% HEPES, 1% sodium pyruvate, 1% minimal essential medium (MEM) nonessential amino acid solution (Gibco, Life Technologies), 20 ng/ml recombinant human M-CSF (Eurobio, Paris, France), and 2 ng/ml recombinant human GM-CSF (Eurobio) to induce differentiation into monocyte-derived macrophages (MDMs), as previously described (52). Seven days later, the purity of the MDM preparation was assessed by flow cytometry analysis of the following cell surface markers: CD3, CD11b, CD11c, CD14, CD163, CD206, and HLA-DR. MDMs were detached by gentle scraping in cold phosphate-buffered saline (PBS) and incubated for 30 min in FcR-blocking reagent (Miltenyi Biotec) in PBS at 4°C. Cells were stained for 1 h at 4°C with the appropriate fluorophore-conjugated antibodies, fixed in 4% paraformaldehyde (PFA) in PBS, and analyzed on a BD Accuri C6 cytometer (BD Biosciences).
Cell lines.
HEK293T and HeLa cells were propagated and subcultured in Dulbecco’s modified Eagle’s medium (DMEM) with GlutaMAX (Life Technologies) supplemented with 10% decomplemented FCS and 1% antibiotic-antimycotic cocktail and were maintained at 37°C in a 5% CO2 incubator.
Viral stocks.
The following CXCR4-tropic NL4-3 HIV-1 proviral clones were used: WT NL4-3 (NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID), NL4-3 Udel (from K. Strebel [53]), NL4-3 ELV (a virus with the E59, L63, and V64 residues of the Vpu protein mutated to alanine), NL4-3 2.6 (a virus with the S52 and S56 residues of the Vpu protein mutated to asparagine [from F. Margottin-Goguet]), and NL4-3 A14W22 (a virus with the A14 and W22 residues of Vpu mutated to leucine and alanine, respectively) (47). To generate mutations in an NL4-3 HIV-1 proviral clone, the original BamHI-EcoRI fragment containing the Vpu open reading frame (ORF) of WT NL4-3 HIV-1 proviral DNA was cloned into the pEGFP-N1 vector, and site-directed mutagenesis was performed using the QuikChange II XL kit (Stratagene, France). Mutagenesis and subclonings were verified by DNA sequencing. Vpu ELV NL4-3 HIV-1 proviral DNA was obtained by replacing the original BamHI-EcoRI fragment containing the Vpu ORF of WT NL4-3 HIV-1 proviral DNA with the corresponding Vpu-mutated fragment of NL4-3 HIV-1.
Viral stocks of VSV-G-pseudotyped (WT or Vpu-mutated) NL4-3 HIV-1 were obtained by transfection of HEK293T cells (2 × 106 cells) with 4.5 μg of HIV-1 proviral DNA along with 1.5 μg of a VSV-G expression vector (pMD.G), using X-tremeGENE HP DNA transfection reagent (Roche), according to the manufacturer’s instructions. Supernatants of the transfected cells were collected 48 h later, filtered, ultracentrifuged, and quantified for the HIV-1 CAp24 antigen by an ELISA (Perkin-Elmer). Viral titers were determined by infection of HeLa cells with serial dilutions of the stocks, followed 24 h later by flow cytometry analyses of CAp24 antigen expression on fixed and permeabilized cells labeled with KC57-fluorescein isothiocyanate (FITC) (anti-CAp24 antibody; Beckman Coulter).
Western blot analysis.
Cells were washed twice and lysed for 20 min at 4°C with radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 10 mM Tris [pH 8], 1 mM EDTA, 0.1% [vol/vol] sodium deoxycholate, 1% Triton X-100) containing cOmplete protease inhibitor cocktail (Roche). Lysates were spun at 12,000 × g at 4°C for 10 min, and the supernatant was recovered. The protein concentration was determined using a Bradford protein assay (Bio-Rad), and equal amounts of protein for each sample were mixed with 2× Laemmli buffer (Sigma-Aldrich), boiled for 5 min at 95°C, and subjected to SDS-PAGE. After protein separation, samples were transferred onto hydrophobic polyvinylidene difluoride (PVDF) membranes, followed by blocking in milk buffer (Tris-buffered saline [TBS] [0.5 M Tris {pH 8.4}, 9% {wt/vol} NaCl], 5% [wt/vol] nonfat dry milk, 0.05% [vol/vol] Tween 20) for 30 min at room temperature (RT). Membranes were incubated overnight at 4°C with primary antibodies in milk buffer. Blots were washed with TBS containing 0.05% (vol/vol) Tween 20 and probed with secondary antibodies in milk buffer for 1 h at RT. After washing, protein bands were detected by using Amersham ECL Prime Western blotting detection reagent (GE Healthcare).
siRNA treatment.
siRNA oligonucleotide duplexes targeting BST2 (SMARTpool ON-TARGETplus BST2 siRNA, catalog number L-011817-00) and negative-control siRNA (ON-TARGETplus nontargeting siRNA, catalog number D-001810-01) were purchased from Dharmacon. MDMs (1 × 106 cells) were transfected twice, on the day when the cells were plated, using the forward transfection protocol, and 24 h later, using the reverse transfection protocol, with 30 nM siRNA using Lipofectamine RNAiMAX (Life Technologies) according to previously described protocols (52). Forty-eight hours later, the cells were washed and infected.
HIV-1 production.
MDMs (1 × 106 cells/well) were plated in 6-well plates and infected 2 days later for 3 h with VSV-G-pseudotyped (WT or mutated) NL4-3 HIV-1 at an MOI of 2. siRNA-treated MDMs were infected similarly with VSV-G-pseudotyped (WT or mutated) NL4-3 HIV-1 24 h after the forward transfection. Four days later, the cells were washed twice with prewarmed medium (37°C) and incubated for a further 24 h. Cell culture supernatants were then collected and filtered on 0.45-μm membranes. A part of the supernatant was recovered for HIV-1 CAp24 quantification by an ELISA (released CAp24). The remaining supernatant was purified by ultracentrifugation on a 20% sucrose cushion for 90 min at 150,000 × g, resuspended in Laemmli buffer, and analyzed by Western blotting. MDMs were pelleted and lysed with RIPA buffer as indicated above. Cell lysates were analyzed by Western blotting and quantified using a CAp24 ELISA (cell-associated CAp24). The HIV-1 release index was assessed as the ratio of released CAp24 to total CAp24 production in the cell culture (released CAp24 plus cell-associated CAp24).
Flow cytometry analysis.
At 5 days postinfection, MDMs were washed twice with ice-cold PBS, harvested by scraping, and pelleted by centrifugation. For cell surface staining of BST2, the cells were blocked for 45 min at 4°C in ice-cold PBS with 1% bovine serum albumin (BSA) supplemented with FcR-blocking reagent according to the manufacturer’s instructions. The cells were then washed twice in ice-cold PBS–1% BSA and stained for 1 h at 4°C with an Alexa 647-conjugated anti-BST2 or an Alexa 647-conjugated control antibody (BioLegend) in PBS–1% BSA. MDMs were washed twice with ice-cold PBS–1% BSA, fixed with 4% PFA in PBS for 20 min, and permeabilized for 30 min at room temperature with PBS–0.5% BSA–0.1% saponin. The cells were then incubated with FITC-conjugated anti-CAp24 (KC57-FITC) for 1 h at room temperature, washed three times, and fixed in PBS–1% PFA. Finally, cells were analyzed on a BD Accuri C6 Plus flow cytometer (BD Biosciences). For intracellular staining of BST2, MDMs were fixed for 20 min in PBS–4% PFA and permeabilized for 30 min at room temperature with PBS–0.5% BSA–0.1% saponin supplemented with FcR-blocking reagent. The cells were then stained for 1 h with FITC-conjugated anti-CAp24 and an allophycocyanin (APC)-conjugated anti-BST2 or a control-APC antibody (Miltenyi Biotec). MDMs were washed three times, fixed, and analyzed as described above.
Immunofluorescence.
MDMs (1 × 106 cells/well) were plated on glass coverslips in a 6-well plate and infected 2 days later for 3 h with VSV-G-pseudotyped (WT or mutated) NL4-3 HIV-1 at an MOI of 1. siRNA-treated MDMs were infected similarly with VSV-G-pseudotyped NL4-3 (WT or Udel) virus 24 h after the forward transfection. Five days later, cells were washed twice with prewarmed (37°C) medium and fixed for 30 min by adding an equal volume of 8% PFA to prewarmed medium. After two washes with PBS, cells were permeabilized for 15 min in PBS–0.1% Triton supplemented with human IgG at 6 μg/ml. The cells were then immunolabeled for 1 h with primary antibodies diluted in PBS–0.5% BSA, washed 3 times in PBS–0.5% BSA, and incubated for 1 h with the appropriate fluorescent conjugated secondary antibodies. For triple labeling, the cells were blocked using mouse serum, washed, and incubated with a mouse monoclonal antibody against CD81 coupled to FITC during 30 min in PBS–0.5% BSA supplemented with human IgG at 6 μg/ml to avoid unspecific labeling. The glass coverslips were then washed, mounted on glass slides using Fluoromount-G (SouthernBiotech), and analyzed by using a 63× objective and a spinning-disk (catalog number CSU-X1; Yokogawa) confocal microscope (catalog number DMI6000; Leica Microsystems, Wetzlar, Germany).
CAp24/BST2 colocalization in infected cells was assessed by calculating Mander’s overlap coefficient using the JACoP plug-in of ImageJ software. ImageJ was also used to determine the BST2 mean fluorescence intensity (MFI) in the virus-containing compartments (VCCs) and the cytoplasm. To do so, 39 cells (from 3 healthy donors) under each infection condition were manually segmented by creating specific masks for the VCCs and the cytoplasm. For each MDM, CAp24/BST2 colocalization and BST2 MFI measurements were performed on the confocal optical section that presented the largest and highest number of VCCs.
Measurement of the volume of the VCCs was allowed by 3D reconstruction of these compartments using Imaris 7.2 software (Bitplane). For each MDM, a z-stack of optical sections (0.3-μm step size; 30 to 50 slices) was acquired. Next, the 3D reconstruction of the VCCs was displayed using Imaris software, and the surface segmentation was manually adjusted to accurately select the VCCs. The VCCs’ total volume was assessed by adding the individual compartments’ volume values provided by the software for each cell. The values were obtained for at least 26 cells under each condition and from up to 3 independent donors (depending on the experiment).
Antibodies.
The following antibodies were used in this study for immunofluorescence or Western blotting: rabbit polyclonal anti-human BST2 (NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID), rabbit polyclonal anti-Vpu (NIH), mouse monoclonal anti-CAp24 (ARP366; National Institute for Biological Standards and Control) for Western blotting, mouse monoclonal anti-CAp24 (25A; Hybridolab) for immunofluorescence, sheep polyclonal anti-TGN46 (AbD Serotec), mouse FITC-conjugated anti-human CD81 (BD Pharmingen), mouse monoclonal anti-α-tubulin (Sigma-Aldrich), and goat monoclonal anti-actin (Santa Cruz Biotechnology). Alexa Fluor 488-, Alexa Fluor 594-, or Alexa Fluor 647-conjugated secondary antibodies against mouse or rabbit (purchased from Invitrogen) and Cy5-conjugated anti-sheep antibody (purchased from Jackson ImmunoResearch) were used for immunofluorescence. Horseradish peroxidase (HRP)-conjugated secondary antibodies against mouse, rabbit, or goat immunoglobulin (Dako) were used for immunoblot experiments.
RT-qPCR.
Total cellular RNA was extracted by using the RNeasy kit (Qiagen) according to the manufacturer’s instructions and was treated with the RNase-Free DNase set (Qiagen) to remove residual genomic DNA. RNA concentrations were determined using a Nanodrop system. For each sample, cDNA synthesis was performed using 100 ng of total RNA with oligo(dT) and a high-capacity cDNA reverse transcription kit (Applied Biosystems). The mRNA levels of BST2 and IFN-β were assayed by using LightCycler 480 SYBR green I master (Roche) in a LightCycler instrument (Roche). The PCR conditions and cycles were as follows: an initial DNA denaturation step at 95°C for 10 min, 45 cycles at 95°C for 10 s, an annealing step at 59°C for 30 s, and an extension step at 72°C for 10 s, followed by a melting-curve analysis cycle. Analysis of each point was performed in technical duplicate. The relative abundance of BST2 mRNA (sense primer 5′-CACACTGTGATGGCCCTAATG-3′ and antisense primer 5′-GTCCGCGATTCTCACGCTT-3′) was calculated by the comparative ΔΔCT method with normalization to the mRNA of the housekeeping gene product glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (sense primer 5′-TGCACCACCAACTGCTTAGC-3′ and antisense primer 5′-GCATGGACTGTGGTCATGAG-3′) and comparison to one reference sample, indicated on each graph. Data presented are normalized to those obtained under each condition at day 0 of infection, set at 100%.
Statistical analysis.
All average values are presented as the means ± standard errors of the means (SEM). Data were analyzed by either unpaired two-tailed Student’s t test, when only two groups were compared, or one-way analysis of variance (ANOVA) combined with Bonferroni’s or Holm-Sidak’s post hoc test, when at least three groups were compared.
ACKNOWLEDGMENTS
We thank Stéphane Emiliani, Sarah Gallois-Montbrun, and Florence Margottin-Goguet for helpful discussions and critical readings of the manuscript. We thank Thomas Guilbert from the Imaging Facility and the Immunobiology Facility of the Cochin Institute for technical assistance. Mouse antibodies against CAp24 (38:96K, EF7, EVA365, and 366) were obtained from B. Wahren through the NIBCS, and the following reagents were obtained through the NIH AIDS reagent program: anti-human BST2 from K. Strebel and A. Andrew, HIV-1 NL4-3 Vpu antiserum from K. Strebel and F. Maldarelli, and pNL4-3 from M. Martin. We thank K. Strebel for the gift of pNL4-3/Udel proviral DNA.
L.L. holds a fellowship from the Ministère Français de l’Enseignement Supérieur et de la Recherche. O.L. holds a fellowship from SIDACTION and one from the ANRS. This work is funded by SIDACTION and the ANRS.
REFERENCES
- 1.Aquaro S, Bagnarelli P, Guenci T, De Luca A, Clementi M, Balestra E, Caliò R, Perno C-F. 2002. Long-term survival and virus production in human primary macrophages infected by human immunodeficiency virus. J Med Virol 68:479–488. doi: 10.1002/jmv.10245. [DOI] [PubMed] [Google Scholar]
- 2.Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS. 1988. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med 167:1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orenstein JM, Meltzer MS, Phipps T, Gendelman HE. 1988. Cytoplasmic assembly and accumulation of human immunodeficiency virus types 1 and 2 in recombinant human colony-stimulating factor-1-treated human monocytes: an ultrastructural study. J Virol 62:2578–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zalar A, Figueroa MI, Ruibal-Ares B, Baré P, Cahn P, de Bracco MM, Belmonte L. 2010. Macrophage HIV-1 infection in duodenal tissue of patients on long term HAART. Antiviral Res 87:269–271. doi: 10.1016/j.antiviral.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Groot F, Welsch S, Sattentau QJ. 2008. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 111:4660–4663. doi: 10.1182/blood-2007-12-130070. [DOI] [PubMed] [Google Scholar]
- 6.Baxter AE, Russell RA, Duncan CJA, Moore MD, Willberg CB, Pablos JL, Finzi A, Kaufmann DE, Ochsenbauer C, Kappes JC, Groot F, Sattentau QJ. 2014. Macrophage infection via selective capture of HIV-1-infected CD4+ T cells. Cell Host Microbe 16:711–721. doi: 10.1016/j.chom.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan J, Sattentau QJ. 2013. The HIV-1-containing macrophage compartment: a perfect cellular niche? Trends Microbiol 21:405–412. doi: 10.1016/j.tim.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Benaroch P, Billard E, Gaudin R, Schindler M, Jouve M. 2010. HIV-1 assembly in macrophages. Retrovirology 7:29. doi: 10.1186/1742-4690-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pelchen‐Matthews A, Marsh M. 2007. Electron microscopy analysis of viral morphogenesis. Methods Cell Biol 79:515–542. doi: 10.1016/S0091-679X(06)79020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raposo G, Moore M, Innes D, Leijendekker R, Leigh-Brown A, Benaroch P, Geuze H. 2002. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic 3:718–729. doi: 10.1034/j.1600-0854.2002.31004.x. [DOI] [PubMed] [Google Scholar]
- 11.Pelchen-Matthews A, Kramer B, Marsh M. 2003. Infectious HIV-1 assembles in late endosomes in primary macrophages. J Cell Biol 162:443–455. doi: 10.1083/jcb.200304008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deneka M, Pelchen-Matthews A, Byland R, Ruiz-Mateos E, Marsh M. 2007. In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J Cell Biol 177:329–341. doi: 10.1083/jcb.200609050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jouve M, Sol-Foulon N, Watson S, Schwartz O, Benaroch P. 2007. HIV-1 buds and accumulates in “nonacidic” endosomes of macrophages. Cell Host Microbe 2:85–95. doi: 10.1016/j.chom.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Nkwe DO, Pelchen-Matthews A, Burden JJ, Collinson LM, Marsh M. 2016. The intracellular plasma membrane-connected compartment in the assembly of HIV-1 in human macrophages. BMC Biol 14:50. doi: 10.1186/s12915-016-0272-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jouvenet N, Neil SJD, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol 4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welsch S, Keppler OT, Habermann A, Allespach I, Krijnse-Locker J, Kräusslich H-G. 2007. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog 3:e36. doi: 10.1371/journal.ppat.0030036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammonds JE, Beeman N, Ding L, Takushi S, Francis AC, Wang JJ, Melikyan GB, Spearman P. 2017. Siglec-1 initiates formation of the virus-containing compartment and enhances macrophage-to-T cell transmission of HIV-1. PLoS Pathog 13:e1006181. doi: 10.1371/journal.ppat.1006181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett AE, Narayan K, Shi D, Hartnell LM, Gousset K, He H, Lowekamp BC, Yoo TS, Bliss D, Freed EO, Subramaniam S. 2009. Ion-abrasion scanning electron microscopy reveals surface-connected tubular conduits in HIV-infected macrophages. PLoS Pathog 5:e1000591. doi: 10.1371/journal.ppat.1000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welsch S, Groot F, Kräusslich H-G, Keppler OT, Sattentau QJ. 2011. Architecture and regulation of the HIV-1 assembly and holding compartment in macrophages. J Virol 85:7922–7927. doi: 10.1128/JVI.00834-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mlcochova P, Pelchen-Matthews A, Marsh M. 2013. Organization and regulation of intracellular plasma membrane-connected HIV-1 assembly compartments in macrophages. BMC Biol 11:89. doi: 10.1186/1741-7007-11-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koppensteiner H, Banning C, Schneider C, Hohenberg H, Schindler M. 2012. Macrophage internal HIV-1 is protected from neutralizing antibodies. J Virol 86:2826–2836. doi: 10.1128/JVI.05915-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jouvenet N, Neil SJD, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD. 2009. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol 83:1837–1844. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. 2009. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A 106:2886–2891. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radoshitzky SR, Dong L, Chi X, Clester JC, Retterer C, Spurgers K, Kuhn JH, Sandwick S, Ruthel G, Kota K, Boltz D, Warren T, Kranzusch PJ, Whelan SPJ, Bavari S. 2010. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J Virol 84:10569–10580. doi: 10.1128/JVI.00103-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, Früh K. 2009. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J Virol 83:9672–9681. doi: 10.1128/JVI.00597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weidner JM, Jiang D, Pan X-B, Chang J, Block TM, Guo J-T. 2010. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J Virol 84:12646–12657. doi: 10.1128/JVI.01328-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu F, Tan J, Liu R, Xu D, Li Y, Geng Y, Liang C, Qiao W. 2011. Tetherin inhibits prototypic foamy virus release. Virol J 8:198. doi: 10.1186/1743-422X-8-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neil SJD, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 29.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell SM, Crowe SM, Mak J. 2001. Lipid rafts and HIV-1: from viral entry to assembly of progeny virions. J Clin Virol 22:217–227. doi: 10.1016/S1386-6532(01)00193-7. [DOI] [PubMed] [Google Scholar]
- 31.Rollason R, Korolchuk V, Hamilton C, Jepson M, Banting G. 2009. A CD317/tetherin-RICH2 complex plays a critical role in the organization of the subapical actin cytoskeleton in polarized epithelial cells. J Cell Biol 184:721–736. doi: 10.1083/jcb.200804154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Billcliff PG, Rollason R, Prior I, Owen DM, Gaus K, Banting G. 2013. CD317/tetherin is an organiser of membrane microdomains. J Cell Sci 126:1553–1564. doi: 10.1242/jcs.112953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rollason R, Dunstan K, Billcliff PG, Bishop P, Gleeson P, Wise H, Digard P, Banting G. 2013. Expression of HIV-1 Vpu leads to loss of the viral restriction factor CD317/tetherin from lipid rafts and its enhanced lysosomal degradation. PLoS One 8:e75680. doi: 10.1371/journal.pone.0075680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erikson E, Adam T, Schmidt S, Lehmann-Koch J, Over B, Goffinet C, Harter C, Bekeredjian-Ding I, Sertel S, Lasitschka F, Keppler OT. 2011. In vivo expression profile of the antiviral restriction factor and tumor-targeting antigen CD317/BST-2/HM1.24/tetherin in humans. Proc Natl Acad Sci U S A 108:13688–13693. doi: 10.1073/pnas.1101684108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Homann S, Smith D, Little S, Richman D, Guatelli J. 2011. Upregulation of BST-2/tetherin by HIV infection in vivo. J Virol 85:10659–10668. doi: 10.1128/JVI.05524-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu H, Wang JJ, Qi M, Yoon JJ, Chen X, Wen X, Hammonds J, Ding L, Spearman P. 2012. Tetherin/BST-2 is essential for the formation of the intracellular virus-containing compartment in HIV-infected macrophages. Cell Host Microbe 12:360–372. doi: 10.1016/j.chom.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyagi E, Andrew AJ, Kao S, Strebel K. 2009. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc Natl Acad Sci U S A 106:2868–2873. doi: 10.1073/pnas.0813223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giese S, Marsh M. 2014. Tetherin can restrict cell-free and cell-cell transmission of HIV from primary macrophages to T cells. PLoS Pathog 10:e1004189. doi: 10.1371/journal.ppat.1004189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dubé M, Roy BB, Guiot-Guillain P, Binette J, Mercier J, Chiasson A, Cohen ÉA. 2010. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog 6:e1000856. doi: 10.1371/journal.ppat.1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitchell RS, Katsura C, Skasko MA, Fitzpatrick K, Lau D, Ruiz A, Stephens EB, Margottin-Goguet F, Benarous R, Guatelli JC. 2009. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog 5:e1000450. doi: 10.1371/journal.ppat.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janvier K, Pelchen-Matthews A, Renaud J-B, Caillet M, Marsh M, Berlioz-Torrent C. 2011. The ESCRT-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog 7:e1001265. doi: 10.1371/journal.ppat.1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vigan R, Neil SJD. 2010. Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J Virol 84:12958–12970. doi: 10.1128/JVI.01699-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skasko M, Wang Y, Tian Y, Tokarev A, Munguia J, Ruiz A, Stephens EB, Opella SJ, Guatelli J. 2012. HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J Biol Chem 287:58–67. doi: 10.1074/jbc.M111.296772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McNatt MW, Zang T, Bieniasz PD. 2013. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog 9:e1003299. doi: 10.1371/journal.ppat.1003299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lewinski MK, Jafari M, Zhang H, Opella SJ, Guatelli J. 2015. Membrane anchoring by a C-terminal tryptophan enables HIV-1 Vpu to displace bone marrow stromal antigen 2 (BST2) from sites of viral assembly. J Biol Chem 290:10919–10933. doi: 10.1074/jbc.M114.630095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jafari M, Guatelli J, Lewinski MK. 2014. Activities of transmitted/founder and chronic clade B HIV-1 Vpu and a C-terminal polymorphism specifically affecting virion release. J Virol 88:5062–5078. doi: 10.1128/JVI.03472-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madjo U, Leymarie O, Frémont S, Kuster A, Nehlich M, Gallois-Montbrun S, Janvier K, Berlioz-Torrent C. 2016. LC3C contributes to Vpu-mediated antagonism of BST2/tetherin restriction on HIV-1 release through a non-canonical autophagy pathway. Cell Rep 17:2221–2233. doi: 10.1016/j.celrep.2016.10.045. [DOI] [PubMed] [Google Scholar]
- 48.Kueck T, Neil SJD. 2012. A cytoplasmic tail determinant in HIV-1 Vpu mediates targeting of tetherin for endosomal degradation and counteracts interferon-induced restriction. PLoS Pathog 8:e1002609. doi: 10.1371/journal.ppat.1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kueck T, Foster TL, Weinelt J, Sumner JC, Pickering S, Neil SJD. 2015. Serine phosphorylation of HIV-1 Vpu and its binding to tetherin regulates interaction with clathrin adaptors. PLoS Pathog 11:e1005141. doi: 10.1371/journal.ppat.1005141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jia X, Weber E, Tokarev A, Lewinski M, Rizk M, Suarez M, Guatelli J, Xiong Y. 2014. Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. Elife 3:e02362. doi: 10.7554/eLife.02362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schubert U, Clouse KA, Strebel K. 1995. Augmentation of virus secretion by the human immunodeficiency virus type 1 Vpu protein is cell type independent and occurs in cultured human primary macrophages and lymphocytes. J Virol 69:7699–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bauby H, Lopez-Vergès S, Hoeffel G, Delcroix-Genête D, Janvier K, Mammano F, Hosmalin A, Berlioz-Torrent C. 2010. TIP47 is required for the production of infectious HIV-1 particles from primary macrophages. Traffic 11:455–467. doi: 10.1111/j.1600-0854.2010.01036.x. [DOI] [PubMed] [Google Scholar]
- 53.Klimkait T, Strebel K, Hoggan MD, Martin MA, Orenstein JM. 1990. The human immunodeficiency virus type 1-specific protein Vpu is required for efficient virus maturation and release. J Virol 64:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roy N, Pacini G, Berlioz-Torrent C, Janvier K. 2017. Characterization of E3 ligases involved in lysosomal sorting of the HIV-1 restriction factor BST2. J Cell Sci 130:1596–1611. doi: 10.1242/jcs.195412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schindler M, Rajan D, Banning C, Wimmer P, Koppensteiner H, Iwanski A, Specht A, Sauter D, Dobner T, Kirchhoff F. 2010. Vpu serine 52 dependent counteraction of tetherin is required for HIV-1 replication in macrophages, but not in ex vivo human lymphoid tissue. Retrovirology 7:1. doi: 10.1186/1742-4690-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharova N, Swingler C, Sharkey M, Stevenson M. 2005. Macrophages archive HIV-1 virions for dissemination in trans. EMBO J 24:2481–2489. doi: 10.1038/sj.emboj.7600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jolly C, Sattentau QJ. 2004. Retroviral spread by induction of virological synapses. Traffic 5:643–650. doi: 10.1111/j.1600-0854.2004.00209.x. [DOI] [PubMed] [Google Scholar]
- 58.Herbein G, Coaquette A, Perez-Bercoff D, Pancino G. 2002. Macrophage activation and HIV infection: can the Trojan horse turn into a fortress? Curr Mol Med 2:723–738. doi: 10.2174/1566524023361844. [DOI] [PubMed] [Google Scholar]
- 59.Duncan CJA, Williams JP, Schiffner T, Gärtner K, Ochsenbauer C, Kappes J, Russell RA, Frater J, Sattentau QJ. 2014. High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. J Virol 88:2025–2034. doi: 10.1128/JVI.03245-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schubert U, Bour S, Willey RL, Strebel K. 1999. Regulation of virus release by the macrophage-tropic human immunodeficiency virus type 1 AD8 isolate is redundant and can be controlled by either Vpu or Env. J Virol 73:887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tervo H-M, Homann S, Ambiel I, Fritz JV, Fackler OT, Keppler OT. 2011. β-TrCP is dispensable for Vpu’s ability to overcome the CD317/tetherin-imposed restriction to HIV-1 release. Retrovirology 8:9. doi: 10.1186/1742-4690-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Früh K, Moses AV. 2009. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a βTrCP-dependent mechanism. J Virol 83:7931–7947. doi: 10.1128/JVI.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sukegawa S, Miyagi E, Bouamr F, Farkašová H, Strebel K. 2018. Mannose receptor 1 restricts HIV particle release from infected macrophages. Cell Rep 22:786–795. doi: 10.1016/j.celrep.2017.12.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matheson NJ, Sumner J, Wals K, Rapiteanu R, Weekes MP, Vigan R, Weinelt J, Schindler M, Antrobus R, Costa ASH, Frezza C, Clish CB, Neil SJD, Lehner PJ. 2015. Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe 18:409–423. doi: 10.1016/j.chom.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lambelé M, Koppensteiner H, Symeonides M, Roy NH, Chan J, Schindler M, Thali M. 2015. Vpu is the main determinant for tetraspanin downregulation in HIV-1-infected cells. J Virol 89:3247–3255. doi: 10.1128/JVI.03719-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haller C, Müller B, Fritz JV, Lamas-Murua M, Stolp B, Pujol FM, Keppler OT, Fackler OT. 2014. HIV-1 Nef and Vpu are functionally redundant broad-spectrum modulators of cell surface receptors, including tetraspanins. J Virol 88:14241–14257. doi: 10.1128/JVI.02333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]