

Persistent HBV infection relies on stable maintenance and proper functioning of a nuclear episomal form of the viral genome called cccDNA, the most stable HBV replication intermediate. One of the major reasons for the failure of currently available antiviral therapeutics to cure chronic HBV infection is their inability to eradicate or inactivate cccDNA. We report here a chemical genetics approach to identify host cellular factors essential for the biosynthesis and maintenance of cccDNA and reveal that cellular DNA topoisomerases are required for both de novo synthesis and intracellular amplification of cccDNA. This approach is suitable for systematic screening of compounds targeting cellular DNA metabolic enzymes and chromatin remodelers for their ability to disrupt cccDNA biosynthesis and function. Identification of key host factors required for cccDNA metabolism and function will reveal molecular targets for developing curative therapeutics of chronic HBV infection.
KEYWORDS: DNA topoisomerase, hepatitis B virus, antiviral agents, cccDNA
ABSTRACT
In order to identify host cellular DNA metabolic enzymes that are involved in the biosynthesis of hepatitis B virus (HBV) covalently closed circular (ccc) DNA, we developed a cell-based assay supporting synchronized and rapid cccDNA synthesis from intracellular progeny nucleocapsid DNA. This was achieved by arresting HBV DNA replication in HepAD38 cells with phosphonoformic acid (PFA), a reversible HBV DNA polymerase inhibitor, at the stage of single-stranded DNA and was followed by removal of PFA to allow the synchronized synthesis of relaxed circular DNA (rcDNA) and subsequent conversion into cccDNA within 12 to 24 h. This cccDNA formation assay allows systematic screening of the effects of small molecular inhibitors of DNA metabolic enzymes on cccDNA synthesis but avoids cytotoxic effects upon long-term treatment. Using this assay, we found that all the tested topoisomerase I and II (TOP1 and TOP2, respectively) poisons as well as topoisomerase II DNA binding and ATPase inhibitors significantly reduced the levels of cccDNA. It was further demonstrated that these inhibitors also disrupted cccDNA synthesis during de novo HBV infection of HepG2 cells expressing sodium taurocholate cotransporting polypeptide (NTCP). Mechanistic analyses indicate that whereas TOP1 inhibitor treatment prevented the production of covalently closed negative-strand rcDNA, TOP2 inhibitors reduced the production of this cccDNA synthesis intermediate to a lesser extent. Moreover, small interfering RNA (siRNA) knockdown of topoisomerase II significantly reduced cccDNA amplification. Taking these observations together, our study demonstrates that topoisomerase I and II may catalyze distinct steps of HBV cccDNA synthesis and that pharmacologic targeting of these cellular enzymes may facilitate the cure of chronic hepatitis B.
IMPORTANCE Persistent HBV infection relies on stable maintenance and proper functioning of a nuclear episomal form of the viral genome called cccDNA, the most stable HBV replication intermediate. One of the major reasons for the failure of currently available antiviral therapeutics to cure chronic HBV infection is their inability to eradicate or inactivate cccDNA. We report here a chemical genetics approach to identify host cellular factors essential for the biosynthesis and maintenance of cccDNA and reveal that cellular DNA topoisomerases are required for both de novo synthesis and intracellular amplification of cccDNA. This approach is suitable for systematic screening of compounds targeting cellular DNA metabolic enzymes and chromatin remodelers for their ability to disrupt cccDNA biosynthesis and function. Identification of key host factors required for cccDNA metabolism and function will reveal molecular targets for developing curative therapeutics of chronic HBV infection.
INTRODUCTION
Hepatitis B virus (HBV), the prototype member of Hepadnaviridae family, chronically infects 257 million people worldwide (1), and approximately one-third of these individuals will die from severe liver diseases, such as cirrhosis and hepatocellular carcinoma (HCC), if left untreated (2, 3). Therapies with currently available antiviral regimens, including pegylated interferon alpha (IFN-α) and nucleos(t)ide analog viral DNA polymerase inhibitors, can improve liver diseases and reduce hepatocellular carcinoma morbidity and mortality in a portion of treated patients (4, 5). However, HBV surface antigen (HBsAg) loss or seroconversion, the hallmark of a successful immunological response to HBV with complete and durable control of infection, or a “functional cure,” is rarely achieved with the current therapies (6, 7), and a life-long antiviral therapy is thus required to maintain the therapeutic benefits (8, 9).
HBV contains a relaxed circular (rc) partially double-stranded DNA (3.2 kb in length) genome but replicates its genomic DNA via reverse transcription of an RNA intermediate called pregenomic RNA (pgRNA) (10, 11). However, unlike classical retroviruses where viral RNAs are transcribed from integrated proviral DNA within host cellular chromosomes, HBV RNAs are transcribed from episomal covalently closed circular DNA (cccDNA) minichromosomes in the nuclei of infected hepatocytes (12). Briefly, HBV infects hepatocytes by binding to its cellular receptor, sodium taurocholate cotransporting polypeptide (NTCP), on the cell surface and delivers nucleocapsid into the cytoplasm via endocytosis (13, 14). The viral rcDNA genome in nucleocapsid is then transported into the nucleus and converted into cccDNA to serve as a template for transcription of viral RNA. Binding of viral DNA polymerase to the stem-loop structure at the 5′ terminus of pgRNA initiates the packaging by core protein dimers to form a nucleocapsid whereby viral DNA polymerase converts the pgRNA first to a single-stranded DNA (ssDNA) and then to rcDNA. The rcDNA-containing mature nucleocapsid can either acquire an envelope and be secreted out of cells as an infectious virion or deliver the rcDNA into the nucleus to amplify the cccDNA pool, a process termed cccDNA intracellular amplification (15).
Although the reasons for the failure of current antiviral agents to cure chronic HBV infection after long-term therapy are not completely understood, clinical studies as well as studies in animal models suggest that the intrinsic stability of cccDNA is one of the key determining factors for viral persistence and outcomes of antiviral therapy (16–20). Therefore, development of antiviral agents to eliminate or functionally inactivate cccDNA should facilitate the cure of chronic hepatitis B (21). However, although recent studies indicated that several host cellular DNA repair proteins, such as tyrosyl-DNA phosphodiesterase 2 (TDP2) (22), DNA polymerase κ (Pol κ) (23), flap endonuclease 1 (FEN1) (24), and DNA ligases (25), are required for cccDNA synthesis in de novo infection and intracellular amplification, the molecular mechanism of cccDNA metabolism and structure organization in the nuclei of infected cells remains to be thoroughly investigated. These studies will identify and validate molecular targets for development of therapeutics that can eradicate or functionally inactivate cccDNA and, thus, cure chronic hepatitis B (26). To achieve this goal, we take a chemical genetics approach to identify host cellular metabolic enzymes required for HBV cccDNA biosynthesis with a newly developed cell-based assay that allows synchronized and rapid cccDNA synthesis (27). Our initial efforts revealed that the cellular DNA topoisomerase I (TOP1) and II (TOP2) are required for both de novo synthesis and intracellular amplification of cccDNA.
RESULTS
Characterization of cccDNA synthesis in HepAD38 cells.
HepAD38 is a HepG2-derived stable cell line in which HBV pgRNA transcription is controlled by a tetracycline (Tet)-inducible promoter and, thus, supports pgRNA transcription and DNA replication upon removal of Tet from the culture medium (28). In order to detect HBV cccDNA and all the possible biosynthesis intermediates, HepAD38 cells were cultured in the absence of Tet for 12 days to allow extensive HBV replication and cccDNA synthesis. Cellular DNA was extracted with the Hirt DNA extraction method, and HBV DNA species in the Hirt DNA preparations were detected by Southern blot hybridization with or without prior heating at 88°C to denature rcDNA species and/or digested with EcoRI to linearize cccDNA into a unit-length double-stranded linear DNA (dslDNA). As shown in Fig. 1A and B, the results indicated that the Hirt DNA preparation contains three major DNA species, supercoiled cccDNA, protein-free (PF) DNA, or deproteinized (DP) double-stranded linear DNA (DP-dslDNA) and relaxed-circular DNA (DP-rcDNA) (lanes 1). While the cccDNA is resistant to heat denaturalization, the heat denaturalization at 88°C converts DP-dslDNA and DP-rcDNA into single-stranded DNA (lanes 2). As expected, EcoRI digestion of heat-denatured Hirt DNA converts cccDNA to unit-length dslDNA (lanes 3). Moreover, to demonstrate the existence of the covalently closed negative-strand rcDNA species, or cc(−)rcDNA for short, a putative intermediate of cccDNA synthesis from DP-rcDNA (29), Hirt DNA preparations were digested with 3′ → 5′ exonucleases I and III (Exo I and Exo III, respectively) (lanes 4) to remove DNA species or strands with a free 3′ terminus, followed by EcoRI digestion (lanes 5) before electrophoresis. In agreement with a previous report (29), a DNA species that migrated faster than cccDNA with negative polarity and resistance to EcoRI digestion was revealed (lanes 4 and 5). The property of this DNA species is consistent with that of the covalently closed negative-strand DNA, or cc(−)DNA, derived from exonuclease digestion of gapped positive strand of cc(−)rcDNA. As expected, cccDNA was resistant to exonuclease digestion (lanes 4) and was converted into unit-length dslDNA by EcoRI digestion (lanes 5). Because EcoRI linearization of cccDNA after heat denaturalization of protein-free rcDNA and dslDNA increased Southern blot hybridization signal and resulted in more accurate quantification of cccDNA, we used this cccDNA validation method as our routine cccDNA assay in this study. Consistent with our previous reports (30, 31), upon removal of Tet from culture medium, HBV DNA replication intermediates gradually accumulated, and cccDNA became detectable at the sixth day after Tet removal and slowly increased thereafter (Fig. 1D).
FIG 1.

Characterization of protein-free HBV DNA species in HepAD38 cells. (A and B) HepAD38 cells cultured for 12 days in the absence of Tet. Hirt DNA was extracted and detected by Southern blot hybridization. The size markers of HBV DNA (in length) are indicated. Hirt DNA without prior treatment (lane 1), after heat denaturalization without (lane 2) or with subsequent EcoRI digestion (lane 3), or with Exo I and Exo III treatment without (lane 4) or with subsequent EcoRI digestion (lane 5) were resolved by agarose gel electrophoresis and transferred on nylon membranes. The membranes were hybridized with a full-length riboprobe specifically hybridizing to the negative strand (A) and positive strand (B) of HBV DNA. The red arrow indicates covalently closed circular negative-strand HBV DNA. (C) Schematic presentation of the experimental schedule. HepAD38 cells were maintained in culture in the presence of Tet and harvested at the indicated time postremoval of Tet. (D) HBV core DNA (upper panel) and Hirt DNA after heat denaturalization and EcoRI digestion (lower panel) were resolved by agarose gel electrophoresis and detected by Southern blot hybridization with a full-length riboprobe specifically hybridizing to negative-strand HBV DNA. Mitochondrial DNA (mtDNA) served as a loading control for Hirt DNA. Biological duplicate samples were obtained and analyzed for each of the indicated time points. rc, relaxed circular DNA; DP-rc, deproteinized rcDNA; *DP-rc, denatured deproteinized rcDNA; dsl, double-stranded linear DNA; ss, single-stranded DNA; ccc, covalently closed circular DNA; ccc*, EcoRI-linearized cccDNA.
Establishment of a synchronized and rapid cccDNA synthesis assay.
RNA interference and CRISPR/Cas9 gene editing technologies have been used to identify host cellular factors required for viral replication in general (32, 33) and HBV cccDNA biosynthesis in particular (23, 25). In order to reveal druggable molecular targets for pharmacological intervention of cccDNA metabolism and function, we intended to systematically test the effects of commercially available small molecular compounds that target cellular DNA metabolic enzymes on HBV cccDNA synthesis and maintenance. However, the slow kinetics of cccDNA synthesis in HepAD38 cells requires prolonged compound treatment that usually results in cytotoxicity. To overcome this problem, we took advantage of foscarnet, or phosphonoformate acid (PFA), a pyrophosphate analogue, to reversibly inhibit HBV DNA polymerase, thereby synchronizing HBV cccDNA synthesis (34). Although in duck hepatitis B virus (DHBV) replicating cells PFA treatment efficiently arrests DHBV DNA replication at a step immediately after the priming of minus-strand DNA synthesis (34, 35), PFA less effectively inhibits HBV DNA synthesis; in particular, PFA does not efficiently inhibit the synthesis of the minus strand of HBV DNA but predominantly arrests HBV replication at the single-strand DNA (27). In agreement with the prior observation, PFA treatment of HepAD38 cells arrested HBV DNA replication at the stage of full-length negative-strand DNA (Fig. 2B, core DNA panel, time 0). Upon addition of Tet to shut off pgRNA transcription from the HBV transgene within the host cellular chromosome and with removal of PFA to restore viral DNA synthesis, rcDNA was gradually accumulated, and cccDNA became detectable at 12 h and gradually increased in the following 12 h. As expected, synthesis of rcDNA and cccDNA can be completely inhibited by the HBV DNA polymerase inhibitor lamivudine (3TC). Therefore, the PFA arresting and releasing of HBV DNA replication in HepAD38 cells created a condition for synchronized and rapid cccDNA synthesis within a short period of time and, thus, allowed for screening of compounds that disrupt cccDNA synthesis or stability.
FIG 2.

A synchronized and rapid cccDNA synthesis assay in HepAD38 cells. (A) Schematic presentation of the experimental schedule. HepAD38 cells were cultured in the absence of Tet, and 2 mM PFA was added in the culture medium 2 days after Tet removal to arrest viral DNA synthesis. Four days later, while PFA was withdrawn, Tet was added back to culture medium to stop viral pgRNA transcription from the transgene. Cells were harvested at the indicated time points. (B) HBV core DNA and Hirt DNA after heat denaturalization at 88°C for 8 min and EcoRI digestion were resolved by agarose gel electrophoresis, and HBV DNA species, indicated on the right, were detected by Southern blot hybridization with a riboprobe specifically hybridizing to negative-strand DNA. Lane M, molecular size marker. (C) The amounts of HBV rcDNA and cccDNA were quantified by phosphorimager and plotted as fold change relative to the amount of the corresponding DNA species at time zero of PFA removal. Means and standard deviations from two biological duplicates are presented.
TOP inhibitors inhibit cccDNA amplification.
Using the above-described assay, we screened compounds that target DNA replication and repair enzymes as well as enzymes catalyzing posttranscriptional modifications of histones to identify cellular functions that are required for or regulate cccDNA synthesis (Fig. 3A). We consistently found that all of the human DNA TOP1 and TOP2 inhibitors tested, including topotecan (Fig. 3B and C), camptothecin (Fig. 3D and E), idarubicin (Fig. 3F and G), and doxorubicin (Fig. 3H and I), reduced the amounts of HBV cccDNA in a dose-dependent manner but did not affect the levels of core DNA and DP-rcDNA. As a positive control, 3TC treatment efficiently inhibited the production of DP-rcDNA and cccDNA under this experimental condition. The cytotoxicity of these compounds was tested under the same treatment condition with an extended incubation period of time, i.e., 48 h instead of 24 h, for the efficacy assay. As shown in Table 1, all TOP inhibitors significantly reduced HBV cccDNA at concentrations much lower than their maximal noncytotoxic concentrations. Moreover, as shown in Fig. 3, all of the TOP inhibitors tested did not apparently reduce HBV core DNA and cellular mitochondrial DNA (mtDNA). In addition, we further demonstrated that treatment of the cells with topotecan or doxorubicin started at 16 h after removal of PFA for only 8 h significantly inhibited the increase of cccDNA (Fig. 4). The results indicate that these compounds rapidly blocked cccDNA synthesis.
FIG 3.

TOP1 and TOP2 inhibitors reduce the level of cccDNA. (A) Schematic presentation of the experimental schedule. HepAD38 cells were cultured in the absence of Tet, and HBV DNA replication was arrested by PFA treatment between days 3 and 6 after Tet removal. The cells were immediately harvested (0 h) or cultured in the presence of Tet and absence of PFA and mock treated (untreated, UT) or treated with 10 μM 3TC or the indicated concentrations of DNA TOP poisons for 24 h. (B, D, F, and H) Intracellular HBV core DNA and Hirt DNA after heat denaturalization at 88°C for 8 min and EcoRI digestion were resolved by agarose gel electrophoresis, and HBV DNA species were detected by Southern blot hybridization with a riboprobe specifically hybridizing to negative-strand DNA. mtDNA served as a loading control of Hirt DNA analysis. (C, E, G, and I) Core DNA (including ssDNA and rcDNA), DP-rcDNA, and cccDNA were quantified by phosphorimager and normalized to the amount of mtDNA. The average level from two biological duplicates under compound treatment were plotted as the percentage of that in the mock-treated cells at 24 h post-PFA removal.
TABLE 1.
Effects of topoisomerase poisons on cccDNA synthesis
| Target and compound | Concn (μM)a | cccDNA (% of control) | Maximum nontoxic concn (μM)b |
|---|---|---|---|
| TOP2 | |||
| Doxorubicin | 1.00 | 22 | 2.0 |
| Idarubicin | 0.25 | 19 | 2.5 |
| TOP1 | |||
| Camptothecin | 1.00 | 37 | 2.0 |
| Topotecan | 0.25 | 30 | 2.5 |
Compound concentration used in screening test.
Cytotoxicity was determined by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay and visual inspection under microscopy after 48 h of compound treatment.
FIG 4.

TOP1 and TOP2 inhibitors block cccDNA synthesis. (A) Schematic presentation of the experimental schedule. HepAD38 cells were cultured in the absence of Tet, and 2 mM PFA was added to the culture medium 2 days after Tet removal to arrest viral DNA synthesis. Four days later, while PFA was withdrawn, Tet was added back to the culture medium to stop viral pgRNA transcription from the transgene. Cells were left untreated or treated with topotecan (TPT; 1 μM) or doxorubicin (Doxo; 1 μM) at 16 h after PFA removal and harvested at the indicated time points. (B) Hirt DNA after heat denaturalization at 88°C for 8 min and EcoRI digestion was resolved by agarose gel electrophoresis and HBV DNA species were detected by Southern blot hybridization with a riboprobe specifically hybridizing to negative-strand DNA. mtDNA served as a loading control. (C) The amounts of cccDNA were quantified by phosphorimager, normalized to the amount of mtDNA, and plotted as the percentage of that in the mock-treated (UT) cells harvested at 16 h post-PFA removal. Means and standard deviations (n = 4) are presented. *DP-rc, denatured deproteinized rc DNA; ccc*, EcoRI-linearized cccDNA.
Because all of the TOP inhibitors examined are mechanistically TOP poisons, they freeze TOP1 and TOP2, covalently attaching to the 3′ and 5′ ends of cleaved DNA and, thus, result in single-strand and double-strand DNA breaks, respectively (36, 37). It is, therefore, possible that the observed reduction of cccDNA amounts in the TOP poison-treated cells is not due to the inhibition of cccDNA synthesis but the result of the cleavage of already formed cccDNA. To distinguish between these two possibilities, we further examined the effects of these compounds on established cccDNA in HepAD38 cells. To this end, as illustrated in Fig. 5A and C, the cells were cultured for 48 h after removal of PFA to allow for establishment of a cccDNA pool and subsequently treated with various concentrations of topotecan or doxorubicin, as indicated in Fig. 5B or D, respectively, for an additional 24 h. The results demonstrated that treatment with both compounds did not apparently alter the levels of HBV core DNA and cccDNA. These results thus imply that both TOP1 and TOP2 poisons inhibited HBV cccDNA synthesis but did not induce the decay or cleavage of cccDNA.
FIG 5.

TOP1 and TOP2 inhibitors do not alter the level of preexisting cccDNA. (A and C) Schematic representation of the experimental schedules. (B and D) Intracellular HBV core DNA and Hirt DNA after heat denaturalization at 88°C for 8 min and EcoRI digestion were resolved by agarose gel electrophoresis, and HBV DNA species were detected by Southern blot hybridization with a riboprobe specifically hybridizing to negative-strand DNA. mtDNA served as a loading control of Hirt DNA analysis. For the data shown in panel D, treatment with 3TC alone was started at day 6, and the cells were harvested at day 9, whereas mock treatment or treatment with the indicated concentrations of doxorubicin was started at day 8, and the cells were harvested at day 9, as illustrated in panel C. The amounts of cccDNA were quantified by phosphorimager, normalized to the amount of the mtDNA. The average levels of cccDNA in compound-treated cells were denoted as the percentage of that in the mock-treated (UT) cells.
Moreover, it had been shown recently that treatment of cells with TOP1 or TOP2 poison can induce the cellular DNA damage response and subsequently activate an innate immune response to inhibit viral replication (38, 39). To rule out this possibility, we demonstrated that under our experimental conditions, treatment with both TOP1 and TOP2 poisons did not induce any detectable inflammatory cytokine response (Fig. 6). Taken together, the results presented above indicate that the TOP1 and TOP2 poisons most likely directly inhibit HBV cccDNA synthesis.
FIG 6.

TOP1 and TOP2 inhibitors did not induce prominent cytokines. HepAD38 cells were mock treated (UT) or treated with 250 nM camptothecin (CTP) or 250 nM doxorubicin (Doxo) for 6 h or 24 h. Intracellular IFN-β, interleukin-6 (IL-6), IL-29, IL-28A, IL-28B, and tumor necrosis factor alpha (TNF-α) transcripts were quantified by qRT-PCR assays and normalized to the level of β-actin mRNA. Means and standard deviations (n = 3) are presented.
TOP2 DNA binding and ATPase inhibitors also inhibit cccDNA amplification.
As illustrated in Fig. 7A, in addition to TOP2 poisons that inhibit the TOP2 catalytic cycle after DNA is cleaved but before DNA religation, resulting in double-stranded DNA breaks, TOP2 can be inhibited at other steps of its catalytic cycle with different biochemical and biological consequences (37, 40). For instance, inhibition of TOP2 binding to DNA by aclarubicin or competitive inhibition by merbarone of ATP binding to TOP2, which prevents strand passage, should not produce DNA damage (41). TOP2 can also be inhibited after strand passage is completed but before ATP hydrolysis and dissociation of amino terminal dimerization. ICRF-187 and ICRF-193 inhibit ATP hydrolysis and maintain the TOP2 structure as a closed clamp (42). To further investigate the role and molecular mechanism of TOPs in cccDNA synthesis, we tested the effects of additional TOP2 inhibitors and a TOP1 enzymatic inhibitor, β-lapachone (43), on HBV cccDNA synthesis. As shown in Fig. 7B to E, in addition to another TOP2 poison, mitoxantrone (MTX), both aclarubicin and merbarone reduced HBV cccDNA levels in a concentration-dependent manner at concentrations that did not affect the levels of mtDNA. The maximal noncytotoxic concentrations of aclarubicin and merbarone are 1,000 nM and 100 μM, respectively. However, treatment with the inhibitors of TOP2 release ICRF-193, ICRF-187, or etoposide as well as the TOP1 enzymatic inhibitor β-lapachone did not apparently inhibit HBV cccDNA synthesis (Fig. 7E). This result could be due to either the failure of these compounds to reach their effective concentrations in the cells under this assay condition or to the unique interaction between TOPs and HBV DNA to evade inhibition by these compounds. In summary, the results presented above suggest that inhibition of several distinct steps of the TOP2 catalytic cycle can efficiently reduce HBV cccDNA synthesis.
FIG 7.

Effects of distinct TOP1 and TOP2 inhibitors on HBV cccDNA synthesis. (A) Illustration of DNA TOP2 catalytic cycle. Briefly, TOP2 enzyme binds to the DNA molecule (step 1). In the presence of Mg++, two ATP molecules bind to the ATPase domain which results in its dimerization and cleavage of one double-stranded DNA (blue) (step 2). The second DNA molecule (orange) is transported through the break (step 3). Upon transport of the DNA segment through the break, one molecule of the ATP is hydrolyzed (step 4), followed by the religation of the cleaved DNA segment along with hydrolysis of another ATP molecule (step 5) and release of a DNA fragment (step 6). Compounds that inhibit each of these steps are indicated. (B to E) HepAD38 cells were cultured in the absence of Tet, and HBV DNA replication was arrested by PFA treatment between days 3 and 6 after Tet removal. The cells were immediately harvested (0 h) or cultured in the presence of Tet and absence of PFA and mock treated (UT) or treated with 10 μM 3TC or the indicated concentrations of aclarubicin and merbarone (B) or 500 nM ICRF-187, 500 nM etoposide (Etop), 500 nM mitoxantrone (MXT), 500 nM ICRF-193, 500 nM β-lapachone (β-Lap), and 50 μM merbarone (Merb) (E) for 24 h. Hirt DNA was resolved by agarose gel electrophoresis after heat denaturalization at 88°C for 8 min and EcoRI digestion. HBV DNA species were detected by Southern blot hybridization with a riboprobe specifically hybridizing to negative-strand DNA. mtDNA served as a loading control of Hirt DNA analysis. The amounts of cccDNA were quantified by phosphorimager and normalized to the amount of mtDNA. The levels of cccDNA in compound-treated cells were plotted (C and D) or represented as the percentage of that in the mock-treated cells (E).
TOP inhibitors disrupt de novo cccDNA synthesis in C3AhNTCP cells.
Having shown that TOP inhibitors inhibit cccDNA formation through the intracellular amplification pathway in HepAD38 cells, we next tested whether cccDNA synthesis from de novo HBV infection can also be inhibited. As shown in Fig. 8 and as anticipated, treatment of C3A cells expressing human NTCP (C3AhNTCP cells) with myrcludex B (MyrB), an acylated peptide derived from the HBV large envelope protein that blocks virus entry (44), completely inhibited HBV infection and consequently cccDNA synthesis. Interestingly, treatment of the cells starting at HBV infection for 36 h with the indicated TOP1 and/or TOP2 inhibitors significantly reduced the amounts of HBV cccDNA (Fig. 8B and C). These results thus suggest that both TOP1 and TOP2 are required for cccDNA synthesis in de novo HBV infection and the intracellular amplification pathway. Both pathways are essential for establishment and maintenance of a cccDNA pool in virally infected hepatocytes (45).
FIG 8.
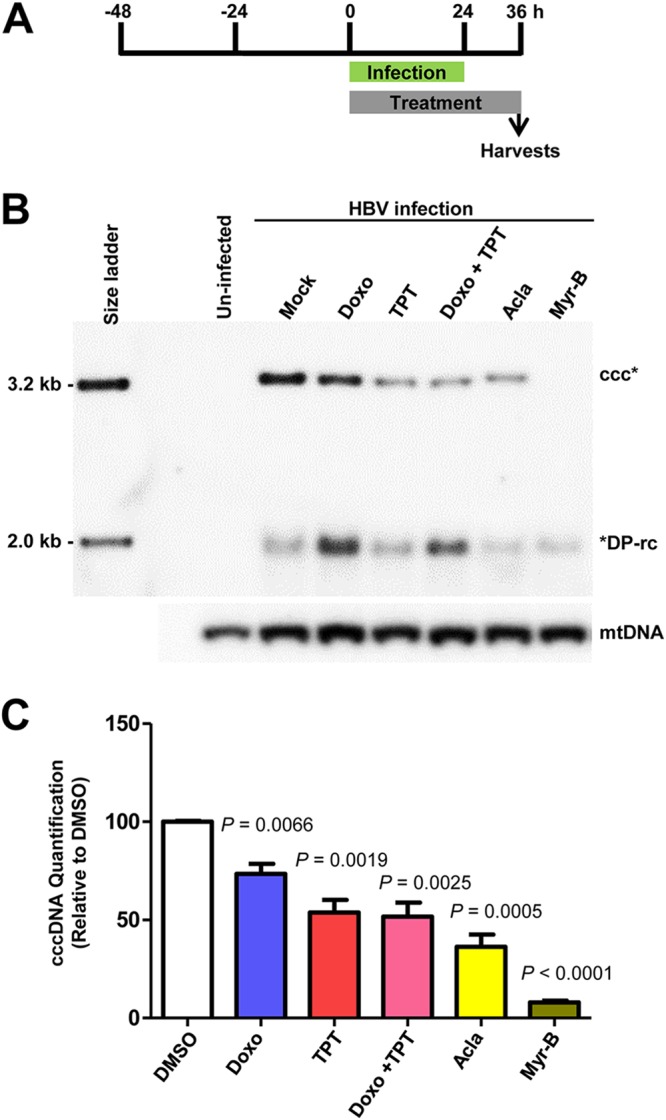
TOP1 and TOP2 inhibitors inhibited HBV cccDNA synthesis in de novo infection. (A) Schematic representation of the experimental schedule. C3AhNTCP cells were infected with HBV at an MOI of 250 genome equivalents for 24 h. The cells were mock treated or treated with 200 nM doxorubicin (Doxo), 200 nM topotecan (TPT), 200 nM doxorubicin and 200 nM topotecan (Doxo + TPT), 200 nM aclarubicin (Acla), or 1 μg/ml myrcludex B (Myr-B) starting from HBV infection for a total of 36 h. (B) Hirt DNA was resolved by agarose gel electrophoresis after heat denaturalization at 88°C for 8 min and EcoRI digestion. HBV DNA species were detected by Southern blot hybridization with a riboprobe specifically hybridizing to negative-strand DNA. mtDNA served as a loading control of Hirt DNA analysis. (C) cccDNAs were quantified by a phosphorimager. The data from three independent experiments are presented. P values calculated by Student's t test are presented.
TOP1 and TOP2 inhibitors block distinct steps of HBV cccDNA synthesis.
While it is very clear that the rcDNA in the infected virion particles or progeny mature nucleocapsids is the precursor of cccDNA synthesis from de novo infection or intracellular amplification, respectively, the DP-rcDNA had been postulated as a potential intermediate of cccDNA synthesis (31, 46, 47). Interestingly, during further characterization of PF rcDNA by exonuclease digestion, a covalently closed negative-strand rcDNA, or cc(−)rcDNA, was revealed (29) (Fig. 1A). Existence of this novel rcDNA species implies that the gap in negative-strand DNA in rcDNA was repaired before the gap in the positive strand (Fig. 9A). While the precursor-product relationship of DP-rcDNA, cc(−)rcDNA, and cccDNA has not been firmly established in the field, it will be interesting to dissect their relationship by inhibition of cccDNA synthesis with topoisomerase inhibitors. To this end, the effects of TOP1 and TOP2 inhibitors on this newly identified putative intermediate of cccDNA synthesis were determined. Due to the lower levels of cc(−)rcDNA under the synchronized and rapid cccDNA synthesis conditions, the numbers of cells for Hirt DNA extraction were doubled in this experiment compared with the experimental results presented in Fig. 1A. As shown in Fig. 9B to D, treatment of Hirt DNA preparations with Exo I and Exo III revealed a ladder of nine DNA species migrating between cccDNA and DP-rcDNA. All of these DNA species can be linearized into a single species of dslDNA by EcoRI digestion and thus represent the different topological isoforms of cccDNA (48, 49). Moreover, an additional DNA species migrating faster than the supercoiled cccDNA band was resistant to EcoRI digestion, and thus the cc(−)DNA was derived from exonuclease digestion of the gapped positive-strand of cc(−)rcDNA (Fig. 1A and 9). Interestingly, while TOP1 inhibitor treatment significantly reduced the amounts of cc(−)DNA and cccDNA (Fig. 9B and E), TOP2 inhibitor treatment decreased the amounts of cc(−)DNA to a lesser extent but significantly reduced the amounts of cccDNA (Fig. 9C to E). These results suggest that while TOP1 inhibitors disrupted cccDNA synthesis at a step in the repair of the negative-strand DNA gap, TOP2 inhibitors more efficiently inhibited the repair of the gap in positive-strand DNA and was less effective in suppressing repair of the gap in negative-strand DNA.
FIG 9.

TOP1 and TOP2 inhibitors disrupted distinct steps of HBV cccDNA synthesis. (A) Schematic illustration of cccDNA synthesis from DP-rcDNA via an intermediate, cc(−)rcDNA, and production of cc(−)DNA by exonuclease I and III digestion of cc(−)rcDNA. (B to E) HepAD38 cells were cultured in the absence of Tet, and HBV DNA replication was arrested by PFA treatment between days 3 and 6 after Tet removal. The cells were then cultured in the presence of Tet and absence of PFA and mock-treated (UT) or treated for 24 h with 500 nM camptothecin (CPT) (B), 500 nM doxorubicin (Doxo) (C), or 40 μM merbarone (Merb) (D). Hirt DNA without prior treatment, digested with Exo I and Exo III without or with subsequent EcoRI restriction, was resolved by agarose gel electrophoresis. HBV DNA species were detected by Southern blot hybridization with a riboprobe specifically hybridizing to negative-strand DNA. (E) The amounts of cc(−)DNA were quantified by a phosphorimager and normalized to the amount in mock (DMSO)-treated cells. Mean and standard derivations from six (camptothecin and doxorubicin) or three (merbarone) independent experiments are presented. P values calculated by Student's t test are presented.
Knockdown of TOP2 expression significantly reduces cccDNA synthesis.
While the pharmacological evidence obtained so far clearly indicated that TOP1 and TOP2 may have played distinct and nonredundant roles in HBV cccDNA synthesis, we desired to validate their roles with genetic approaches. However, due to their essential roles in cell proliferation and survival, we could only utilize small interfering (siRNA) knockdown technology to determine the role of TOP1, TOP2α, and TOP2β in cccDNA synthesis in HepAD38 cells. As shown in Fig. 10A, siRNA transfection of HepAD38 cells specifically reduced the levels of targeted TOP mRNA but not the mRNA of untargeted TOPs. The reduction in the respective TOP proteins in TOP siRNA transfected cells was confirmed by Western blot assays (Fig. 10B and D). Interestingly, while knocking down the expression of TOP2α and TOP2β, alone (Fig. 10C) or in combination (Fig. 10E), significantly reduced the level of cccDNA, transfection of TOP1 siRNA slightly, but statistically significantly, increased the level of cccDNA (Fig. 10C, E, and F). Whereas our results clearly demonstrated that both TOP2α and TOP2β play an essential role in HBV cccDNA synthesis, the role and mechanism of TOP1 in cccDNA synthesis remain to be further investigated.
FIG 10.

Knockdown of TOP2 mRNA reduced cccDNA formation. HepAD38 cells were mock transfected or transfected with the indicated siRNA and cultured in the absence of Tet and presence of PFA for 2 days. The cells were then cultured in the presence of Tet and absence of PFA for an additional 24 h. (A) Total cellular RNAs were extracted, and TOP mRNAs were quantified by qRT-PCR assay, normalized to the level of β-actin mRNA, and expressed as the ratio to the level of the respective TOP mRNA in cells transfected with a scrambled siRNA. (B and D) The levels of TOP proteins were determined by Western blot assays. TATA-binding protein (TBP) served as a loading control. (C and E) Hirt DNA was resolved by agarose gel electrophoresis after heat denaturalization at 88°C for 8 min and EcoRI digestion. HBV DNA species were detected by Southern blot hybridization with a riboprobe specifically hybridizing to negative-strand DNA. mtDNA served as a loading control of Hirt DNA analysis. (F) cccDNA was quantified by a phosphorimager. Means and standard deviations are presented (n = 8). P values calculated by Student's t test are presented.
DISCUSSION
Although a small fraction of HBV cccDNA can be synthesized from dslDNA via the nonhomologous end joining (NHEJ) DNA repair pathway (50), the vast majority of functional HBV cccDNA is synthesized from rcDNA (51, 52). Biochemically, conversion of rcDNA into cccDNA requires the involvement of at least four classes of cellular enzymes, including DNA repair nucleases to process the ends, DNA polymerases to fill in the gaps, DNA ligases to ligate the ends, and topoisomerases for the winding or unwinding of rcDNA and cccDNA. In order to assemble functional cccDNA minichromosomes, histones and their modification enzymes as well as chromatin structure remodelers are also required (15). It is thus conceivable that many cellular DNA metabolic proteins are involved in cccDNA biosynthesis, maintenance, and functioning. While recent studies identified a few cellular DNA repair enzymes, including TDP2, Pol κ, DNA ligases, and FEN1, that participate in cccDNA synthesis, the molecular pathways that repair rcDNA into cccDNA remain elusive (15, 21). Because many of these host cellular DNA metabolic enzymes are essential for cell proliferation and survival, genome-wide CRISPR/Cas9 gene knockout technology is not a suitable approach to identify host cellular proteins required for cccDNA synthesis and functioning. RNA interference (RNAi) screening of DNA repair genes for their roles in cccDNA synthesis has achieved only limited success (25). We report here the development of a rapid cccDNA synthesis assay that is suitable for targeted screening of small molecules, which disrupt the function of cellular DNA metabolic and epigenetic modification enzymes, to inhibit cccDNA synthesis (Fig. 2). Among approximately 200 compounds tested thus far, compounds that target several host cellular proteins have been discovered to modulate cccDNA synthesis, and their modes of action are under investigation. We report here that TOP1 and TOP2 poisons as well as TOP2 DNA binding and ATPase inhibitors significantly reduced the amounts of cccDNA (Fig. 3, 4, and 7). It was further demonstrated that all of these inhibitors also disrupted cccDNA synthesis during de novo HBV infection of C3AhNTCP cells (Fig. 8). Interestingly, we were able to show that TOP1 and TOP2 inhibitors rapidly inhibited cccDNA synthesis (Fig. 4), and while TOP1 inhibitors blocked the repair of the gap in negative-strand DNA, TOP2 inhibitors disrupted the repair of the gap in positive-strand DNA in the conversion of rcDNA to cccDNA (Fig. 9). Finally, using RNAi technology, we demonstrated that both TOP2α and TOP2β are required for cccDNA synthesis, but a contradictory result of TOP1 on cccDNA synthesis was revealed (Fig. 10).
DNA TOPs regulate the function of genomic DNA by changing its topology. Biochemically, DNA TOPs cleave the phosphodiester bond in a coordinated way and religate the ends of DNA to unwind or wind DNA to control the supercoiling (53) and resolve disordered DNA entanglements and knots (54, 55). Because the dynamics of topological conformation of genomic DNA is an integral part of its functions, such as replication, repair, transcription, chromatin assembly, remodeling, and segregation, DNA TOPs are thus essential for all living organisms (56, 57). Except for a few large DNA viruses that encode their own DNA TOPs (58–60), many DNA viruses, including herpesviruses (61–63), vaccinia virus (64), adenovirus (65), and polyomavirus (66, 67), recruit host cellular DNA TOPs for their replication and transcription. Moreover, it has been shown that both cellular TOP1 and TOP2 are associated with human immunodeficiency virus type 1 (HIV-1) or recruited to the viral DNA replication complex to promote reverse transcriptional viral DNA synthesis (68, 69). TOPs have also been demonstrated to play a role in the replication of other retroviruses (70). Thus far, even though the functional roles of TOPs in viral DNA replication and RNA transcription as well as the recruitment of TOPs to viral genome replication complex have been clearly demonstrated, the biochemical mechanism of TOPs to facilitate viral replication and transcription for most of these viruses remains to be illustrated.
Concerning the role of TOPs in HBV replication, an early study suggested that TOP1 can cleave DHBV rcDNA in vitro at specific sites of both negative and positive strands and linearize rcDNA (71). Based on this biochemical study, Pourquier and colleagues postulated that TOP1 may play a role in the circularization of negative-stranded DNA in cccDNA synthesis and viral DNA integration into the host cellular chromosome. Interestingly, our result that TOP1 poisons inhibited the production of covalently closed circular negative-strand DNA is consistent with this hypothesis (Fig. 9B). However, the slight increase in cccDNA levels in HepAD38 cells when TOP1 expression was reduced by siRNA knockdown is apparently contradictory to TOP1 having an essential role in cccDNA synthesis (Fig. 10). A possible explanation of these contradictory results is that while camptothecin treatment efficiently arrests TOP1-catalyzed negative-strand DNA religation and prevents its circularization, reduction of the amount of TOP1 protein by siRNA may result in the recruitment of alternative cellular enzymes or repair pathways that can more efficiently catalyze cccDNA synthesis. Nevertheless, despite the discrepancy between the results obtained from TOP1 inhibitor treatment and siRNA knockdown of TOP1 expression, the results indicate that TOP1 is involved in or regulates cccDNA synthesis. Further analyses of DNA repair intermediates under these specific experimental conditions should resolve the molecular basis of the discrepancy. In contrast to TOP1, both pharmacological and genetic evidence obtained in this study support that TOP2 plays an essential role in HBV cccDNA synthesis. Mechanistically, our results indicate that, unlike TOP1 that catalyzes negative-strand DNA circularization, TOP2 appears to be required for the circularization of both strands of rcDNA (Fig. 9B and E). In conclusion, our results provide evidence suggesting that TOP1 and TOP2 play nonredundant roles in HBV cccDNA synthesis. Further investigation to understand the biochemical mechanism of these cellular enzymes in cccDNA synthesis will advance our knowledge of HBV biology and establish a molecular basis for development of therapeutics to suppress cccDNA synthesis and transcriptional function and ultimately cure chronic hepatitis B.
MATERIALS AND METHODS
Cell culture.
HepAD38, a HepG2-derived cell line supporting HBV replication in a Tet-inducible manner, was obtained from Christoph Seeger at Fox Chase Cancer Center, Philadelphia, PA (28), and cultured in Dulbecco’s modified Eagle’s medium-F12 medium (DMEM/F12) medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 μg/ml tetracycline. The C3AhNTCP cell line, derived from C3A, a subclone of HepG2 (ATCC HB-8065), and stably expressing human NTCP (72), was maintained in DMEM/F12 medium supplemented with 10% FBS and 100 U/ml penicillin and 100 μg/ml streptomycin.
Chemical reagents.
Topoisomerase inhibitors doxorubicin (catalog no. S1208), camptothecin (S1288), etoposide (S1225), dexrazoxane (ICRF-187) HCl (S1222), and mitoxantrone 2HCl (S1889) were purchased from Selleckchem. Topotecan (HY-13768A) and idarubicin (HY-17381) were purchased from MedChemExpress (MCE). Aclarubicin (A-101-5) was obtained from GoldBio. Merbarone (M2070), ICRF-193 (I4659), and β-lapachone (L2037) were purchased from Sigma-Aldrich.
Analyses of HBV DNA.
HBV core DNA and Hirt DNA extraction from HepAD38 or C3AhNTCP cells as well as analyses by Southern blot hybridization was described previously (73–75). Human mitochondrial DNA, as a loading control for Hirt DNA, was also detected with Southern blot hybridization after stripping of HBV probes. Briefly, a 2,709-bp-long fragment of mtDNA from HepG2 cells was amplified with the primers HuMt NotI-F (ACACACGCGGCCGCCTGCTGGCATCACTATACTACTA) and HuMt NdeI-R (ACACACCATATGGATTGGTGGGTCATTATGTGTTG) by standard PCR and cloned into pGEM T Easy Vector (Promega). Recombinant plasmid was amplified in DH5α competent cells (Invitrogen), purified with Midiprep (Qiagen), and linearized with Sph1-HF (catalog no. R3182; New England Biolabs [NEB]). An [α-32P]UTP-labeled riboprobe was prepared by in vitro transcription of the linearized recombinant plasmid with RiboProbe System-SP6 (catalog no. P1420; Promega) and used for Southern blot hybridization of human mtDNA. An mtDNA fragment of 7,366 nucleotides (nt) in length can be detected in EcoRI-digested Hirt DNA preparations.
Restriction enzyme and exonuclease treatment of Hirt DNA.
For Southern blot detection of HBV cccDNA, Hirt DNA preparations were either left untreated or heated at 88°C for 8 min to denature DP-rcDNA and dslDNA into single-stranded DNA; cccDNA species were subsequently digested by EcoRI at 37°C for 1 h to convert them into unit-length double-stranded linear DNA (27). For detection of single-stranded closed circular rcDNA, Hirt DNA preparations were digested with exonuclease I (Exo I) (catalog no. M0293S; NEB) and exonuclease III (Exo III) (M0206S; NEB) for 2 h to remove DNA species with free 5′ and 3′ ends (29). After Exo I and Exo III digestion, the remaining DNA in reaction products was extracted by phenol and precipitated with isopropanol. If needed, the purified DNA samples could be digested by EcoRI to convert cccDNA into unit-length double-stranded linear DNA as described above.
siRNA knockdown in PFA-arrested HepAD38 cells.
HepAD38 cells were cultured in the absence of tetracycline (Tet) for 2 days; 2 mM PFA was added into culture medium, and cells were cultured for another 2 days. The cells were then reseeded into 12-well plates at a density of 4 × 105 cells per well and transfected with a 10 nM concentration of the indicated siRNA at 6 h postseeding with Lipofectamine RNAiMax (catalog no. 13778150; Invitrogen) (Fig. 10). The following human topoisomerase siRNAs were bought from OriGene: TOP1 human siRNA oligo duplex (locus 7150; catalog no. SR304897), TOP2A human siRNA oligo duplex (locus 7153; SR322074), and TOP2B Human siRNA oigo duplex (locus 7155; SR304899). At 48 h posttransfection, the cells were refreshed with DMEM/F12 medium supplemented with Tet to stop HBV pgRNA transcription from the transgene and without PFA to resume HBV DNA synthesis and cccDNA formation. The cells were harvested 24 h later for analyses of topoisomerase mRNA and HBV DNA.
Quantification of inflammatory cytokine topoisomerase mRNA by qRT-PCR assay.
Total cellular RNA was extracted by using TRIzol reagent (Invitrogen). cDNA was synthesized by using a SuperScript III Platinum one-step qRT-PCR kit (Invitrogen). Real-time PCR assays were performed using a LightCycler 480 II. Primer sequences for topoisomerase mRNA levels determined by quantitative reverse transcription-PCR (qRT-PCR) analyses are provided in Table 2. Primer sequences for analyses of cytokine mRNA were reported previously (72).
TABLE 2.
Sequence of the primers for quantitative PCR
| Species and target (NCBI GeneID) | Primer direction | Primer sequence |
|---|---|---|
| Human | ||
| TOP1 (7150) | Sense | GAACAAGCAGCCCGAGGATGAT |
| Antisense | TGCTGTAGCGTGATGGAGGCAT | |
| TOP2A (7153) | Sense | GTGGCAAGGATTCTGCTAGTCC |
| Antisense | ACCATTCAGGCTCAACACGCTG | |
| TOP2B (7155) | Sense | GGTCAGTTTGGAACTCGGCTTC |
| Antisense | AGGAGGTTGTCATCCACAGCAG | |
| TOP3A (7156) | Sense | GCATCGACTCTTTAACCACACGG |
| Antisense | CTCCACAGTGTCCAAGGCTTGA | |
| TOP3B (8940) | Sense | GATGCTGGAGAAGCAGACGAAC |
| Antisense | CTCTCCACCGTGACATAGTTGC | |
| TOP1MT (116447) | Sense | GACCTACAACGCCTCCATCACT |
| Antisense | TGCTCGCTGATGGTTGCAGAGA | |
| β-Actin | Sense | CACCATTGGCAATGAGCGGTTC |
| Antisense | AGGTCTTTGCGGATGTCCACGT | |
| HBV | ||
| Core DNA | Sense | GGCTTTCGGAAAATTCCTATG |
| Antisense | AGCCCTACGAACCACTGAAC | |
| cccDNA | Sense | GGGGCGCACCTCTCTTTA |
| Antisense | CCACCCAGGTAGCTAGAGTCATTAG |
Western blot assay.
Cells in a well of 12-well plate were lysed with 200 μl of NuPAGE SDS sample buffer (Thermo Fisher Scientific) supplemented with 2.5% 2-mercaptoethanol (Sigma). Cell lysate was subjected to denaturing gel electrophoresis with a NuPAGE 4% to 12% Bis-Tris gel and NuPAGE morpholinepropanesulfonic acid (MOPS)-SDS running buffer (Thermo Fischer Scientific). Proteins were transferred from the gel onto a polyvinylidene difluoride (PVDF) membrane using an iBlot 2 dry blotting system (Thermo Fischer Scientific). Membranes were blocked with Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBST) plus 5% nonfat milk for 1 h and incubated with the desired antibody overnight at 4°C. We used anti-TOP1 (catalog no. ab3825; Abcam), anti-TOP2α (sc-166934; Santa Cruz), and anti-TOP2β, (A300-949A; Bethyl Laboratories, Inc.) primary antibodies. After the membrane was washed with TBST, it was incubated with Li-Cor IRDye secondary antibodies. Membranes were again washed with TBST and imaged with a Li-Cor Odyssey system (Li-Cor Biotechnology).
HBV infection of C3AhNTCP cells.
For HBV infection, C3AhNTCP cells were seeded into collagen-coated 12-well plates at a density of 1.5 × 106 cells per well and cultured in complete DMEM medium containing 3% dimethyl sulfoxide (DMSO). One day later, the cells were infected with HBV prepared from HepAD38 cell culture medium at a multiplicity of infection (MOI) of 250 genome equivalents per cell in DMEM containing 4% polyethylene glycol (PEG) 8000. The inocula were removed at 24 h, and cells were washed three times with phosphate-buffered saline (PBS). The infected cultures were maintained in complete DMEM medium containing 3% DMSO until harvesting.
ACKNOWLEDGMENTS
We thank Eain A. Murphy for critical reading of and comments on the manuscript.
This work was supported by a grant from the National Institutes of Health, USA (AI113267), Arbutus Biopharma, Inc., and the Commonwealth of Pennsylvania through the Hepatitis B Foundation.
REFERENCES
- 1.Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 2.GBD 2013 Mortality and Causes of Death Collaborators. 2015. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385:117–171. doi: 10.1016/S0140-6736(14)61682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Block TM, Guo H, Guo JT. 2007. Molecular virology of hepatitis B virus for clinicians. Clin Liver Dis 11:685–706. vii. doi: 10.1016/j.cld.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dienstag JL. 2009. Benefits and risks of nucleoside analog therapy for hepatitis B. Hepatology 49:S112–S121. doi: 10.1002/hep.22920. [DOI] [PubMed] [Google Scholar]
- 5.Perrillo R. 2009. Benefits and risks of interferon therapy for hepatitis B. Hepatology 49:S103–S111. doi: 10.1002/hep.22956. [DOI] [PubMed] [Google Scholar]
- 6.Chang J, Guo F, Zhao X, Guo JT. 2014. Therapeutic strategies for a functional cure of chronic hepatitis B virus infection. Acta Pharm Sin B 4:248–257. doi: 10.1016/j.apsb.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang L, Zhao Q, Wu S, Cheng J, Chang J, Guo JT. 2017. The current status and future directions of hepatitis B antiviral drug discovery. Expert Opin Drug Discov 12:5–15. doi: 10.1080/17460441.2017.1255195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rehermann B, Ferrari C, Pasquinelli C, Chisari FV. 1996. The hepatitis B virus persists for decades after patients' recovery from acute viral hepatitis despite active maintenance of a cytotoxic T- lymphocyte response. Nat Med 2:1104–1108. doi: 10.1038/nm1096-1104. [DOI] [PubMed] [Google Scholar]
- 9.Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, Guo JT. 2013. Chronic hepatitis B: what should be the goal for new therapies? Antiviral Res 98:27–34. doi: 10.1016/j.antiviral.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. doi: 10.1016/0092-8674(82)90157-X. [DOI] [PubMed] [Google Scholar]
- 11.Wang GH, Seeger C. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J Virol 67:6507–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seeger C, Mason WS. 2015. Molecular biology of hepatitis B virus infection. Virology 479–480:672–686. doi: 10.1016/j.virol.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Falth M, Stindt J, Koniger C, Nassal M, Kubitz R, Sultmann H, Urban S. 2014. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146:1070–1083. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 15.Guo JT, Guo H. 2015. Metabolism and function of hepatitis B virus cccDNA: Implications for the development of cccDNA-targeting antiviral therapeutics. Antiviral Res 122:91–100. doi: 10.1016/j.antiviral.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sung JJ, Wong ML, Bowden S, Liew CT, Hui AY, Wong VW, Leung NW, Locarnini S, Chan HL. 2005. Intrahepatic hepatitis B virus covalently closed circular DNA can be a predictor of sustained response to therapy. Gastroenterology 128:1890–1897. doi: 10.1053/j.gastro.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 17.Laras A, Koskinas J, Dimou E, Kostamena A, Hadziyannis SJ. 2006. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology 44:694–702. doi: 10.1002/hep.21299. [DOI] [PubMed] [Google Scholar]
- 18.Wursthorn K, Lutgehetmann M, Dandri M, Volz T, Buggisch P, Zollner B, Longerich T, Schirmacher P, Metzler F, Zankel M, Fischer C, Currie G, Brosgart C, Petersen J. 2006. Peginterferon alpha-2b plus adefovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology 44:675–684. doi: 10.1002/hep.21282. [DOI] [PubMed] [Google Scholar]
- 19.Reaiche GY, Le Mire MF, Mason WS, Jilbert AR. 2010. The persistence in the liver of residual duck hepatitis B virus covalently closed circular DNA is not dependent upon new viral DNA synthesis. Virology 406:286–292. doi: 10.1016/j.virol.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 20.Allweiss L, Volz T, Giersch K, Kah J, Raffa G, Petersen J, Lohse AW, Beninati C, Pollicino T, Urban S, Lutgehetmann M, Dandri M. 2018. Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA in vivo. Gut 67:542–552. doi: 10.1136/gutjnl-2016-312162. [DOI] [PubMed] [Google Scholar]
- 21.Nassal M. 2015. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 22.Koniger C, Wingert I, Marsmann M, Rosler C, Beck J, Nassal M. 2014. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc Natl Acad Sci U S A 111:E4244–E4253. doi: 10.1073/pnas.1409986111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qi Y, Gao Z, Xu G, Peng B, Liu C, Yan H, Yao Q, Sun G, Liu Y, Tang D, Song Z, He W, Sun Y, Guo JT, Li W. 2016. DNA polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog 12:e1005893. doi: 10.1371/journal.ppat.1005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitamura K, Que L, Shimadu M, Koura M, Ishihara Y, Wakae K, Nakamura T, Watashi K, Wakita T, Muramatsu M. 2018. Flap endonuclease 1 is involved in cccDNA formation in the hepatitis B virus. PLoS Pathog 14:e1007124. doi: 10.1371/journal.ppat.1007124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Long Q, Yan R, Hu J, Cai D, Mitra B, Kim ES, Marchetti A, Zhang H, Wang S, Liu Y, Huang A, Guo H. 2017. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog 13:e1006784. doi: 10.1371/journal.ppat.1006784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu J, Cheng J, Tang L, Hu Z, Luo Y, Li Y, Zhou T, Chang J, Guo JT. 2018. Virological basis for the cure of chronic hepatitis B. ACS Infect Dis doi: 10.1021/acsinfecdis.8b00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo F, Zhao Q, Sheraz M, Cheng J, Qi Y, Su Q, Cuconati A, Wei L, Du Y, Li W, Chang J, Guo JT. 2017. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog 13:e1006658. doi: 10.1371/journal.ppat.1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. doi: 10.1128/AAC.41.8.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo J, Cui X, Gao L, Hu J. 2017. Identification of intermediate in hepatitis B virus CCC DNA formation and sensitive and selective CCC DNA detection. J Virol 91:e00539-17. doi: 10.1128/JVI.00539-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou T, Guo H, Guo JT, Cuconati A, Mehta A, Block TM. 2006. Hepatitis B virus e antigen production is dependent upon covalently closed circular (ccc) DNA in HepAD38 cell cultures and may serve as a cccDNA surrogate in antiviral screening assays. Antiviral Res 72:116–124. doi: 10.1016/j.antiviral.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puschnik AS, Majzoub K, Ooi YS, Carette JE. 2017. A CRISPR toolbox to study virus-host interactions. Nat Rev Microbiol 15:351–364. doi: 10.1038/nrmicro.2017.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe T, Kawaoka Y. 2015. Influenza virus-host interactomes as a basis for antiviral drug development. Curr Opin Virol 14:71–78. doi: 10.1016/j.coviro.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J Virol 77:1885–1893. doi: 10.1128/JVI.77.3.1885-1893.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670. doi: 10.1016/0092-8674(92)90599-8. [DOI] [PubMed] [Google Scholar]
- 36.Capranico G, Marinello J, Chillemi G. 2017. Type I DNA topoisomerases. J Med Chem 60:2169–2192. doi: 10.1021/acs.jmedchem.6b00966. [DOI] [PubMed] [Google Scholar]
- 37.Nitiss JL. 2009. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luthra P, Aguirre S, Yen BC, Pietzsch CA, Sanchez-Aparicio MT, Tigabu B, Morlock LK, Garcia-Sastre A, Leung DW, Williams NS, Fernandez-Sesma A, Bukreyev A, Basler CF. 2017. Topoisomerase II inhibitors induce DNA damage-dependent interferon responses circumventing Ebola virus immune evasion. mBio 8:e00368-17. doi: 10.1128/mBio.00368-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mlcochova P, Caswell SJ, Taylor IA, Towers GJ, Gupta RK. 2018. DNA damage induced by topoisomerase inhibitors activates SAMHD1 and blocks HIV-1 infection of macrophages. EMBO J 37:50–62. doi: 10.15252/embj.201796880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nitiss JL. 2009. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 9:327–337. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fortune JM, Osheroff N. 1998. Merbarone inhibits the catalytic activity of human topoisomerase IIalpha by blocking DNA cleavage. J Biol Chem 273:17643–17650. doi: 10.1074/jbc.273.28.17643. [DOI] [PubMed] [Google Scholar]
- 42.Andoh T, Ishida R. 1998. Catalytic inhibitors of DNA topoisomerase II. Biochim Biophys Acta 1400:155–171. doi: 10.1016/S0167-4781(98)00133-X. [DOI] [PubMed] [Google Scholar]
- 43.Li CJ, Averboukh L, Pardee AB. 1993. β-Lapachone, a novel DNA topoisomerase I inhibitor with a mode of action different from camptothecin. J Biol Chem 268:22463–22468. [PubMed] [Google Scholar]
- 44.Petersen J, Dandri M, Mier W, Lutgehetmann M, Volz T, von Weizsacker F, Haberkorn U, Fischer L, Pollok JM, Erbes B, Seitz S, Urban S. 2008. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat Biotechnol 26:335–341. doi: 10.1038/nbt1389. [DOI] [PubMed] [Google Scholar]
- 45.Ko C, Chakraborty A, Chou WM, Hasreiter J, Wettengel JM, Stadler D, Bester R, Asen T, Zhang K, Wisskirchen K, McKeating JA, Ryu WS, Protzer U. 2018. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J Hepatol 69:1231–1241. doi: 10.1016/j.jhep.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao W, Hu J. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81:6164–6174. doi: 10.1128/JVI.02721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo H, Mao R, Block TM, Guo JT. 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84:387–396. doi: 10.1128/JVI.01921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Newbold JE, Xin H, Tencza M, Sherman G, Dean J, Bowden S, Locarnini S. 1995. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol 69:3350–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu F, Campagna M, Qi Y, Zhao X, Guo F, Xu C, Li S, Li W, Block TM, Chang J, Guo JT. 2013. Alpha-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog 9:e1003613. doi: 10.1371/journal.ppat.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo H, Xu C, Zhou T, Block TM, Guo JT. 2012. Characterization of the host factors required for hepadnavirus covalently closed circular (ccc) DNA formation. PLoS One 7:e43270. doi: 10.1371/journal.pone.0043270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang W, Mason WS, Summers J. 1996. Covalently closed circular viral DNA formed from two types of linear DNA in woodchuck hepatitis virus-infected liver. J Virol 70:4567–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang W, Summers J. 1995. Illegitimate replication of linear hepadnavirus DNA through nonhomologous recombination. J Virol 69:4029–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gilbert N, Allan J. 2014. Supercoiling in DNA and chromatin. Curr Opin Genet Dev 25:15–21. doi: 10.1016/j.gde.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bocquet N, Bizard AH, Abdulrahman W, Larsen NB, Faty M, Cavadini S, Bunker RD, Kowalczykowski SC, Cejka P, Hickson ID, Thoma NH. 2014. Structural and mechanistic insight into Holliday-junction dissolution by topoisomerase IIIα and RMI1. Nat Struct Mol Biol 21:261–268. doi: 10.1038/nsmb.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kozuki T, Chikamori K, Surleac MD, Micluta MA, Petrescu AJ, Norris EJ, Elson P, Hoeltge GA, Grabowski DR, Porter ACG, Ganapathi RN, Ganapathi MK. 2017. Roles of the C-terminal domains of topoisomerase IIα and topoisomerase IIβ in regulation of the decatenation checkpoint. Nucleic Acids Res 45:5995–6010. doi: 10.1093/nar/gkx325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang JC. 2002. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 57.Hauk G, Berger JM. 2016. The role of ATP-dependent machines in regulating genome topology. Curr Opin Struct Biol 36:85–96. doi: 10.1016/j.sbi.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freitas FB, Frouco G, Martins C, Leitao A, Ferreira F. 2016. In vitro inhibition of African swine fever virus-topoisomerase II disrupts viral replication. Antiviral Res 134:34–41. doi: 10.1016/j.antiviral.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 59.Coelho J, Ferreira F, Martins C, Leitao A. 2016. Functional characterization and inhibition of the type II DNA topoisomerase coded by African swine fever virus. Virology 493:209–216. doi: 10.1016/j.virol.2016.03.023. [DOI] [PubMed] [Google Scholar]
- 60.Shuman S. 1998. Vaccinia virus DNA topoisomerase: a model eukaryotic type IB enzyme. Biochim Biophys Acta 1400:321–337. doi: 10.1016/S0167-4781(98)00144-4. [DOI] [PubMed] [Google Scholar]
- 61.Wu T, Wang Y, Yuan Y. 2014. Antiviral activity of topoisomerase II catalytic inhibitors against Epstein-Barr virus. Antiviral Res 107:95–101. doi: 10.1016/j.antiviral.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 62.Wang P, Rennekamp AJ, Yuan Y, Lieberman PM. 2009. Topoisomerase I and RecQL1 function in Epstein-Barr virus lytic reactivation. J Virol 83:8090–8098. doi: 10.1128/JVI.02379-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzalez-Molleda L, Wang Y, Yuan Y. 2012. Potent antiviral activity of topoisomerase I and II inhibitors against Kaposi's sarcoma-associated herpesvirus. Antimicrob Agents Chemother 56:893–902. doi: 10.1128/AAC.05274-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin YC, Li J, Irwin CR, Jenkins H, DeLange L, Evans DH. 2008. Vaccinia virus DNA ligase recruits cellular topoisomerase II to sites of viral replication and assembly. J Virol 82:5922–5932. doi: 10.1128/JVI.02723-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wong ML, Hsu MT. 1990. Involvement of topoisomerases in replication, transcription, and packaging of the linear adenovirus genome. J Virol 64:691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khopde S, Simmons DT. 2008. Simian virus 40 DNA replication is dependent on an interaction between topoisomerase I and the C-terminal end of T antigen. J Virol 82:1136–1145. doi: 10.1128/JVI.01314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khopde S, Roy R, Simmons DT. 2008. The binding of topoisomerase I to T antigen enhances the synthesis of RNA-DNA primers during simian virus 40 DNA replication. Biochemistry 47:9653–9660. doi: 10.1021/bi800825r. [DOI] [PubMed] [Google Scholar]
- 68.Kondapi AK, Satyanarayana N, Saikrishna AD. 2006. A study of the topoisomerase II activity in HIV-1 replication using the ferrocene derivatives as probes. Arch Biochem Biophys 450:123–132. doi: 10.1016/j.abb.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 69.Takahashi H, Matsuda M, Kojima A, Sata T, Andoh T, Kurata T, Nagashima K, Hall WW. 1995. Human immunodeficiency virus type 1 reverse transcriptase: enhancement of activity by interaction with cellular topoisomerase I. Proc Natl Acad Sci U S A 92:5694–5698. doi: 10.1073/pnas.92.12.5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Warren K, Warrilow D, Meredith L, Harrich D. 2009. Reverse transcriptase and cellular factors: regulators of HIV-1 reverse transcription. Viruses 1:873–894. doi: 10.3390/v1030873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pourquier P, Jensen AD, Gong SS, Pommier Y, Rogler CE. 1999. Human DNA topoisomerase I-mediated cleavage and recombination of duck hepatitis B virus DNA in vitro. Nucleic Acids Res 27:1919–1925. doi: 10.1093/nar/27.8.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo F, Tang L, Shu S, Sehgal M, Sheraz M, Liu B, Zhao Q, Cheng J, Zhao X, Zhou T, Chang J, Guo JT. 2017. Activation of stimulator of interferon genes in hepatocytes suppresses the replication of hepatitis B virus. Antimicrob Agents Chemother 61:e00771-17. doi: 10.1128/AAC.00771-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, Wei L, Chang J, Block TM, Guo JT. 2010. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol 84:9332–9340. doi: 10.1128/JVI.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo JT. 2013. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol 87:6931–6942. doi: 10.1128/JVI.00582-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cai D, Nie H, Yan R, Guo JT, Block TM, Guo H. 2013. A southern blot assay for detection of hepatitis B virus covalently closed circular DNA from cell cultures. Methods Mol Biol 1030:151–161. doi: 10.1007/978-1-62703-484-5_13. [DOI] [PMC free article] [PubMed] [Google Scholar]